

Abstract
Large molecular machines regulate daily cycles of transcriptional activity and help generate rhythmic behavior. In recent years, structural and biochemical analyses have elucidated a number of principles guiding the interactions of proteins that form the basis of circadian timing. In its simplest form, the circadian clock is composed of a transcription/translation feedback loop. However, this description elides a complicated process of activator recruitment, chromatin decompaction, recruitment of coactivators, expression of repressors, formation of a repressive complex, repression of the activators, and ultimately degradation of the repressors and reinitiation of the cycle. Understanding the core principles underlying the clock requires careful examination of molecular and even atomic level details of these processes. Here we review major structural and biochemical findings in circadian biology and make the argument that shared protein interfaces within the clockwork are critical for both the generation of rhythmicity and timing of the clock.
Keywords: rhythm, Cryptochrome, Period, transcription, Bmal1
Graphical Abstract
Structural and biochemical analyses have uncovered a number of key features of the proteins that form the basis of circadian timing in vertebrates. In particular, a repeating motif in protein-protein interactions within the clockwork is shared, competitive interfaces. Here we review major findings and make the case that competition at these sites between coactivators and repressors drives oscillatory gene expression and represents a key node for the regulation of periodicity within the clock.
A Transcription/Translation Feedback Loop
In biology, as in life, timing is everything. In particular, there are a preponderance of environmental challenges that occur with regularity and require innate timing systems to predict and respond in order to maintain a competitive advantage in the wild. An environmental timing challenge can be relatively simple, for instance needing to predict whether it will be light or dark at any given time while living near the equator, or it can be complex, as in the case of the short-lived marine midge, Clunio marinus, which must navigate both lunar and daily cycles to mate and oviposit during extreme low tides at various latitudes (Kaiser et al., 2016). The study of daily cycles, or circadian biology (circa meaning about and dian meaning day), has led to rapid advancement in our understanding of how these endogenous timing systems are generated and perpetuated even in the absence of environmental input.
Discovery of the activators
Although decades of behavioral research preceded, the story of the mammalian molecular clock begins with the discovery of a gene called Clock, which encodes a protein with a basic Helix-Loop-Helix (bHLH) domain and two tandem Per-Arnt-Sim (PAS) domains (King et al., 1997). Clock was discovered in a forward genetic mutagenesis screen searching for mice with aberrant period phenotypes (Vitaterna et al., 1994). The mutation identified in this screen, Clock-Δ19, is semi-dominant and maps to a 5’-splice donor site in intron 19 that causes skipping of exon 19 (King et al., 1997). Heterozygous carriers of the mutation have slightly elongated endogenous periods of 24.5 to 24.8 hours, while homozygous carriers range from roughly 27 to 30 hours (Table 1) (Vitaterna et al., 1994). Shortly after the discovery and cloning of Clock, a second bHLH-PAS gene, Bmal1, was cloned (Hogenesch et al., 1997) and shown to interact with CLOCK in a yeast two-hybrid screen (Gekakis et al., 1998). Furthermore, CLOCK and BMAL1 were shown to form a heterodimeric transcription factor, which activates transcription from E-box elements in the genome (Gekakis et al., 1998). This role as a transcriptional activator is dependent on an intact exon 19, as CLOCK-Δ19 was unable to induce target gene expression with BMAL1 (Gekakis et al., 1998). In concert, this work suggested that a key feature of the mammalian clock is transcriptional control of gene targets. This finding echoed previous work in fruit flies and the bread mold Neurospora crassa, which theorized that endogenous clocks in these organisms are composed of transcription/translation feedback loops in which positive regulators are repressed by products of their own gene targets (Aronson et al., 1994; Hardin et al., 1990).
Table 1.
Effects of deletion or mutagenesis of core clock components on period and amplitude.
| Clock Δ19/Δ19 | Activator | 1–4 h Long/ Arrhythmic |
Decreased | ENU-induced mutation in mice causing skipping of exon 19. Heterozygotes have slightly longer period length. Homozygotes have a substantially longer period. | (Katada and Sassone-Corsi, 2010; King et al., 1997)(Katada and Sassone-Corsi, 2010; King et al., 1997) |
| Clock −/− | Activator | 0.5 h Short | Decreased | Targeted deletion of exons 5–6 in mice. Null allele. | (Debruyne et al., 2006) |
| Bmal1 −/− | Activator | Arrhythmic | Targeted disruption of exons 4–5 containing bHLH in mice. Null allele. | (Bunger et al., 2000) | |
| Npas2 −/− | Activator | 0.2 h Short | No change | Targeted disruption of bHLH coding region in mice. | (Dudley et al., 2003) |
| Cry1 −/− | Repressor | 1 h Short | Decreased | Targeted disruption of Cry1 coding sequence in mice. | (van der Horst et al., 1999; Vitaterna et al., 1999) |
| Cry2 −/− | Repressor | 1 h Long | Decreased | Targeted disruption of Cry2 coding sequence in mice. | (van der Horst et al., 1999; Vitaterna et al., 1999) |
|
Cry1
−/−
/
Cry2 −/− |
Repressor | Arrhythmic | (van der Horst et al., 1999; Vitaterna et al., 1999) | ||
| Per1 ldc | Repressor | 0.5 h Short/ Arrhythmic |
Decreased | Targeted deletion of 3 kb of Per1 in mice. | (Bae et al., 2001) |
| Per2 ldc | Repressor | No change/ Arrhythmic |
Decreased | Targeted deletion of 1.4 kb of Per2 in mice. | (Bae et al., 2001) |
| Per3 −/− | Repressor | 0.5 h Short | No change | Targeted deletion of 1.6 kb of Per3 in mice. | (Shearman et al., 2000a) |
| Nr1d1 −/− | Repressor | 0.5 h Short | No change | Targeted deletion using Cre/loxp system. | (Cho et al., 2012) |
| Nr1d2 −/− | Repressor | No change | No change | Targeted deletion using Cre/loxp system. | (Cho et al., 2012) |
| Nr1d1 −/− /Nr1d2 −/− | Repressor | 2.5 h Short | Decreased | (Cho et al., 2012) | |
| Fbxl3 Afh/Afh | Turnover | 3 h Long | Decreased | C358S mutation in mice | (Godinho et al., 2007) |
| Fbxl3 Ovtm/ Ovtm | Turnover | 2.5 h Long | Decreased | I364T mutation in mice | (Siepka et al., 2007) |
|
Fbxl21
Psttm/
Psttm |
Turnover | 0.5 h Short | Increased | G149E mutation in mice | (Yoo et al., 2013) |
| Fbxl21 −/− | Turnover | No change | No change | Targeted deletion in mice. | (Hirano et al., 2013) |
| Csnk1δ T44A | Turnover | 0.5 h Short | Spontaneous mutation (T44A) in human family | (Xu et al., 2005) | |
| Csnk1ε Tau | Turnover | 4 h Short | Spontaneous mutation (R178C) in golden hamster | (Lowrey et al., 2000) |
A simple model takes shape
In the early 1970s, pioneering research in fruit flies by Konopka and Benzer identified several mutations that caused aberrant period phenotypes in eclosion and locomotor activity (Konopka and Benzer, 1971). All three mutations mapped to the same genetic locus and about a decade later the gene, period, was cloned by two labs (Bargiello et al., 1984; Bargiello and Young, 1984; Reddy et al., 1984; Zehring et al., 1984). Later work identified per as a negative regulator of its own expression (Hardin et al., 1990) along with a binding partner, TIMELESS (TIM) (Sehgal et al., 1995).
Concurrent with the discovery of the heterodimeric transcriptional activator CLOCK/BMAL1, a number of genes with homology to the Drosophila per gene were identified and cloned (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997; Sun et al., 1997; Tei et al., 1997; Zylka et al., 1998). Expression of this gene family (Period 1, 2, and 3) is oscillatory in an anatomical region of the brain called the suprachiasmatic nucleus (SCN) (Shearman et al., 1997; Sun et al., 1997; Tei et al., 1997; Zylka et al., 1998), which has been shown to function as a master pacemaker (Ralph et al., 1990). Moreover, all three PER proteins were shown to be capable of repressing the transcriptional activity of CLOCK and BMAL1 (Jin et al., 1999; Sangoram et al., 1998). The discovery that the positive arm of the Drosophila oscillator is composed of homologs of Clock and Bmal1, Clk and Cycle respectively, suggested that the mammalian circadian clock might be composed of the same components as the Drosophila clock (Allada et al., 1998; Rutila et al., 1998). However, unlike the molecular oscillator in Drosophila, PER proteins have a unique binding partner in mammalian clocks: CRYPTOCHROMEs (CRYs). Cry1 and Cry2 were originally cloned from human cDNAs before mouse homologs were identified (Hsu et al., 1996; Kobayashi et al., 1998). Originally thought to be a mammalian photoreceptor involved in light entrainment of the clock (Thresher et al., 1998), it quickly became apparent that CRYs are necessary components of a functioning clock with a direct, light-independent role in repression of CLOCK/BMAL1-mediated transcription (Griffin et al., 1999; Kume et al., 1999; van der Horst et al., 1999; Vitaterna et al., 1999). Thus, the core mechanism of the mammalian circadian clock is a transcription/translation feedback loop in which CLOCK and BMAL1 regulate the transcription of their repressors, PERs and CRYs (Figure 1).
Figure 1. Simple model of the mammalian transcription/translation feedback loop.

CLOCK and BMAL1 bind to E-boxes driving expression of their own repressors, CRYPTOCHROME and PERIOD. BMAL1 is also rhythmically expressed as a result of competitive binding at its promoter by the activator ROR and the repressor REV-ERB, which is under the control of CLOCK and BMAL1. Degradation of the repressors, CRY and PER, through interaction with FBXL3 and β–TrCP allows the cycle to begin again.
This simple model has been expanded to include an accessory loop in which Bmal1 expression is regulated by several members of the retinoic acid-related orphan receptor family: Rora, Rorb, Rorc, and Nr1d1 (Rev-erb α) and Nr1d2 (Rev-erb β). RORs function as positive regulators of Bmal1 expression while both REV-ERBs function as negative regulators, competing with the RORs for a binding site in the Bmal1 promoter (Guillaumond et al., 2005; Preitner et al., 2002; Sato et al., 2004; Ueda et al., 2002). Rev-erb α and β are in turn regulated by CLOCK and BMAL1 (Preitner et al., 2002). The REV-ERBs function redundantly in the core clock mechanism. Deletion of either Rev-erb α or β has a minimal effect on normal clock function, but deletion of both results in either very low amplitude rhythms with a period roughly 2.5 hours shorter than WT or outright locomotor arrhythmicity, reminiscent of Bmal1−/− mice (Table 1) (Cho et al., 2012; Liu et al., 2008; Preitner et al., 2002).
In the years since the outlines of the clock began to come together, the portrait has become more complex. A large number of additional clock components have been identified and characterized. Structures for several of the key proteins have been solved. Network dynamics of the SCN and their contribution to periodicity, rhythmicity, and robustness of the oscillator are better understood. Regardless, one of the most critical questions in trying to understand a timing system remains poorly understood: how is periodicity determined and what are the key nodes in the molecular network where the period can be tuned?
This review will largely ignore the contributions of the SCN network and post-transcriptional regulation to periodicity to focus on an oft-overlooked facet of the clock: structural dynamics and the formation and function of the repressive complex. In doing so, it will become clear that assembly of the repressive complex on the activators CLOCK and BMAL1 is not only integral to a functional oscillator, but central to its timing.
The activation complex
High-resolution structures of the bHLH-PAS domains of CLOCK and BMAL1 have provided insight into how CLOCK and BMAL1 heterodimerize and bind DNA (Huang et al., 2012; Wang et al., 2013). In addition to static structures, conformational dynamics of a protein interaction domain in the C-terminus of BMAL1 have been reported helping to elucidate a key structural mechanism in clock function (Gustafson et al., 2017; Xu et al., 2015). Beyond structural information, several labs have identified post-translational modifications on CLOCK and BMAL1 and additional components of the activator complex. Here we describe these findings and their implications for a mechanistic understanding of the clock.
The CLOCK/BMAL1 heterodimer
CLOCK is an 855 amino acid protein and its structure is defined principally by three major features: an N-terminal bHLH domain immediately succeeded by tandem PAS domains (PAS-A and PAS-B), and the exon 19 region in CLOCK’s disordered C-terminal domain (Figure 2A and B). Similarly, comprised of 626 amino acids, BMAL1 is also defined by its N-terminal bHLH domain and tandem PAS domains, though its disordered C-terminus contains a transactivation domain (TAD) (Figure 2A and C). The primary crystal structure of CLOCK and BMAL1 is composed of the bHLH and PAS domains of each protein (residues 26–384 of CLOCK and 62–447 of BMAL1) (Huang et al., 2012). Notably, all three of these structural features are heavily involved in the heterodimerization of CLOCK and BMAL1 with each domain interacting primarily with its associated partner such that both bHLH domains interact, both PAS-A domains interact, and both PAS-B domains interact. The interactions of the PAS domains are primarily driven by large patches of surface-exposed hydrophobic residues at each interface. The interface between the PAS-A domains is extensive, burying a surface area of nearly 2000 Å2 while the interface between the PAS-B domains is still substantial at roughly 700 Å2. As a result, the heterodimerization of CLOCK and BMAL1 is highly robust to mutations at these interfaces. Minimal effects on heterodimerization and transactivation were observed when single hydrophobic residues were substituted with charged residues at these interfaces. Critically, the two PAS interactions seen in this structure are highly divergent. The PAS-A domains adopt common PAS folds with several α helices surrounding the concave surface of a five-stranded antiparallel β sheet. An α helix from each PAS-A packs against the β sheet face of the opposing PAS-A (Figure 2D). In contrast to the symmetrical interaction between the PAS-A domains, the PAS-B domains interact at a single interface. The BMAL1 PAS-B domain sits atop CLOCK’s PAS-B domain forming an interaction between the concave β sheet surface of BMAL1’s PAS-B domain and an α helix from CLOCK’s PAS-B. The nature of this embrace leaves a substantial portion of CLOCK’s PAS-B domain exposed and available for an additional protein-protein interaction, which we shall return to shortly (Figure 2E).
Figure 2. Molecular architecture of the activators CLOCK and BMAL1.

(A) On the top is the crystal structure of the CLOCK/BMAL1 heterodimer, encompassing the bHLH and PAS domains of each protein (PDB: 4F3L). CLOCK is shown in peach and BMAL1 is shown in blue. Below is a cartoon rendering of the heterodimer based on the structure, but with additional disordered regions drawn in. (B) Using available structural data, the structure of CLOCK is rendered in graphical form with domains of interest labeled and numbered based on the amino acid sequence of CLOCK from Mus musculus. (C) The structure of BMAL1 is rendered in graphical form with domains of interest labeled and numbered based on the amino acid sequence of BMAL1 from Mus musculus. (D) The PAS-A domains of CLOCK and BMAL1 have a reciprocal interaction in which the first α-helix of each PAS-A domain binds the β-sheet interface of its partner. (E) The PAS-B domains of CLOCK and BMAL1 interact through the β-sheet interface of BMAL1 and an α-helix of CLOCK, leaving a significant portion of CLOCK’s PAS-B available for other protein-protein interactions. (F) Residues identified as important for interaction between the CLOCK PAS-B domain, primarily its HI loop, and CRY are highlighted on the PAS-B structure in blue.
Like other bHLH-PAS proteins, the bHLH domain is used primarily for mediating a direct interaction with target DNA (Wang et al., 2013). CLOCK and BMAL1 have been shown to interact with both canonical (CACGTG) and non-canonical (e.g. CCAATG, CATTGG, CATGTG, AACGTG) E-boxes (Hogenesch et al., 1998; Koike et al., 2012; Panda et al., 2002; Storch et al., 2002; Ueda et al., 2002; Yoo et al., 2005). Wang and colleagues solved a structure of the bHLH domains of CLOCK and BMAL1 bound to a canonical E-box DNA sequence (CACGTG) and found that basic helical regions insert into the major groove of DNA and residues from both CLOCK (R39, E43, R47) and BMAL1 (H77, E81, R85) have specific interactions with this motif (Wang et al., 2013). The authors go on to show that interaction with non-canonical E-boxes often requires an additional hydrophobic interaction between the BMAL1 residue I80 and a flanking thymine next to the E-box (e.g. ACACGTGT, E-box underlined). In addition to specifying DNA targets, the bHLH domains also stabilize the CLOCK/BMAL1 heterodimer through interactions between the two helices of each protein, which form a four-helical bundle (Huang et al., 2012; Wang et al., 2013). Although the bHLH domains can homodimerize, steric clashes resulting from CLOCK H84 or BMAL1 L125 render homodimers far less stable than the heterodimer, suggesting a mechanism for mutual recognition and preferential formation of the heterodimer (Wang et al., 2013).
CLOCK-specific features
The C-terminal region of CLOCK beyond the PAS-B domain is intrinsically disordered, but contains at least one region of critical importance to normal clock function. The exon 19 region of CLOCK (residues 514–564) falls within a glutamine-rich region of the C-terminus (King et al., 1997). CLOCK has one known paralog, NPAS2, which can serve as a secondary binding partner for BMAL1, rhythmically activating clock-controlled genes like Per1, Per2, and Cry1 (Reick et al., 2001). Interestingly, the exon 19 region of CLOCK represents the only region of sequence similarity between CLOCK and NPAS2 beyond the bHLH-PAS domains (King et al., 1997). Moreover, CLK proteins from Drosophila and the silk moth Antheraea pernyi (dCLK and apCLK respectively) both contain C-terminal sequences with homology to the exon 19 region of CLOCK (Chang et al., 2003; Lee et al., 2016). Deletion of these homologous sequences prevents repressive feedback from dPER and apPER. Lee and colleagues also showed that deletion of the exon 19 homology region in dCLK impairs interaction with dPER (Lee et al., 2016). This observation extended to the associated mouse proteins. In transiently transfected cells, CLOCK Δ19 showed weakened interaction with all three PER proteins compared with WT CLOCK.
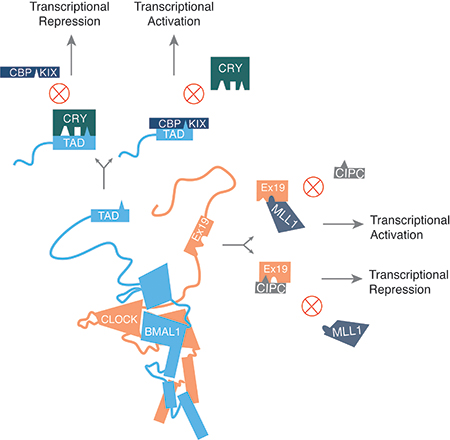
Substantial evidence exists to suggest that CLOCK and BMAL1 effect some of their role as transcription factors through epigenetic regulation, opening chromatin and recruiting several histone-modifying enzymes (DiTacchio et al., 2011; Katada and Sassone-Corsi, 2010; Menet et al., 2014; Nam et al., 2014). One such enzyme, Mixed Lineage Leukemia 1 (MLL1, encoded by Kmt2a), promotes transcriptional activity through its histone methyltransferase (HMT) activity (Katada and Sassone-Corsi, 2010). MLL1’s HMT activity is specialized for trimethylation of H3K4 (H3K4me3). Katada and Sassone-Corsi demonstrated that there is a circadian rhythm of H3K4me3 at the promoters of clock-controlled genes and MLL1 is essential for rhythmic expression of these genes. Moreover, though recruitment of MLL1 is circadian, its expression is not. Loss of MLL1 results in severely attenuated expression and blunted amplitude of Dbp and Per2 mRNA, two targets of CLOCK and BMAL1 (Table 2). The HMT activity of MLL1 is also regulated in a circadian manner by acetylation, controlled in part by the NAD+-dependent deacetylase SIRT1 (Sirt1) (Aguilar-Arnal et al., 2015). Notably, MLL1 physically interacts with WT CLOCK and BMAL1, but not with CLOCK Δ19 (Katada and Sassone-Corsi, 2010). In concert with the work characterizing the exon 19 region of CLOCK, these data suggest the intriguing possibility that the exon 19 region recruits MLL1 to the activator complex at the start of the active phase to orchestrate a cascade of epigenetic modifications opening chromatin and allowing transcription to begin. Since the exon 19 region has also been reported to interact with PER and an additional repressor, CLOCK-interacting protein, circadian (CIPC), it is likely that negative feedback at least partially entails sequestration of the exon 19 region by one or more repressors (Lee et al., 2016; Zhao et al., 2007). In fact, a competitive interaction interface shared by coactivators and repressors represents a recurring motif in the molecular architecture of the circadian clock and potentially represents a means of transmuting physical interactions into rhythms in gene expression.
Table 2.
Effects of depletion or deletion of ancillary clock components on period and amplitude.
| MLL1 | Activator | KO MEFs | Arrhythmic | (Katada and Sassone-Corsi, 2010) | |
| TRAP150 | Activator | siRNA in U2OS cells | 1 h Long | Decreased | (Lande-Diner et al., 2013) |
| JARID1a | Activator | KO MEFs | 1 h Short | Decreased | (DiTacchio et al., 2011) |
| LSD1 | Activator | S112A Knock-in mice | No change | Decreased | (Nam et al., 2014) |
| GAPVD1 | Repressor | siRNA in Bmal1-Luc immortalized (BLi) reporter cells | 1–2 h Long | No change | (Aryal et al., 2017) |
| DDB1 | Repressor | siRNA in BLi reporter cells | 1.5 h Short | No change | (Tamayo et al., 2015) |
| CUL4 | Repressor | siRNA in BLi reporter cells | 2 h Short | No change | (Tamayo et al., 2015) |
| MTA2 | Repressor | siRNA in U2OS cells | 2 h Short | Increased | (Kim et al., 2014) |
| CHD4 | Repressor | siRNA in U2OS cells | 1–1.5 h Long | Decreased | (Kim et al., 2014) |
| MBD2 | Repressor | siRNA in U2OS cells | 0.5 h Short | No change | (Kim et al., 2014) |
| SNF2H | Repressor | siRNA in U2OS cells | 0.5 h Long | Increased | (Kim et al., 2014) |
| BRG1 | Repressor | siRNA in U2OS cells | 1.5 h Long | Decreased | (Kim et al., 2014) |
| PSF | Repressor | shRNA in BLi reporter cells | 1 h Short | No change | (Duong et al., 2011) |
| SIN3A | Repressor | shRNA in BLi reporter cells | 1 h Short | No change | (Duong et al., 2011) |
| EZH2 | Repressor | shRNA in Per2-Luc or Bmal1-Luc reporter NIH3T3 cells | Arrhythmic | (Etchegaray et al., 2006) | |
| SUV39H | Repressor | siRNA in BLi reporter cells | 2 h Short | No change | (Duong and Weitz, 2014) |
| SETX | Repressor | siRNA in reporter fibroblasts | Arrhythmic | (Padmanabhan et al., 2012) | |
| RACK1 | Repressor | shRNA in BLi reporter cells | 0.5 h Short | No change | (Robles et al., 2010) |
| PKCalpha | Repressor | shRNA in BLi reporter cells | 0.5 h Short | No change | (Robles et al., 2010) |
| CIPC | Repressor | siRNA in NIH3T3 fibroblasts | 1 h Short | No change | (Zhao et al., 2007) |
| CHRONO | Repressor | shRNA in NIH3T3 fibroblasts and KO mice | 0.5–1.5 h Long | Decreased | (Anafi et al., 2014; Goriki et al., 2014) |
| NONO | Repressor | siRNA in NIH3T3 fibroblasts | 2 h Short | Decreased | (Brown et al., 2005) |
In addition to the exon 19 region, the disordered C-terminus of CLOCK also contains a glutamine-rich region with limited sequence similarity to ACTR, a member of the SRC family of histone acetyltransferases (HATs), and ESA1, a member of the MYST family of HATs (Figure 2B) (Doi et al., 2006). CLOCK was shown to have intrinsic HAT activity, which requires an intact Acetyl-coenzyme A (CoA) binding motif in this region (Doi et al., 2006). CLOCK’s HAT activity was shown to be essential to circadian regulation of Dbp and Per1, potentially through epigenetic regulation of H3K9 and H3K14. Beyond its role in histone acetylation, CLOCK also selectively acetylates its binding partner BMAL1 at residue K537 (Figure 2C) (Hirayama et al., 2007). Acetylation at this site is critical for recruitment of CRY1 to BMAL1 and for normal cycling behavior through a mechanism addressed in the text below.
While the PAS domains of CLOCK are important for mediating the heterodimerization of CLOCK and BMAL1, the PAS-B domain is also responsible for an interaction with CRY1. In a random mutagenesis screen of CLOCK, Sato and colleagues identified several residues in the PAS-B domain that were responsible for gating physical interaction with CRY1 (Sato et al., 2006). When mutated, residues G332, H360, W362, and E367 disrupted binding with CRY1 and rendered CLOCK resistant to CRY1-mediated repression, suggesting that a physical interaction at this interface is necessary for repressive feedback. Notably, the structure of the CLOCK and BMAL1 bHLH-PAS domains revealed that all of these residues are located on a part of the PAS-B domain that protrudes out from the rest of the structure, making them accessible for a protein-protein interaction (Figure 2F) (Huang et al., 2012).
BMAL1-specific features
The bHLH-PAS domains of BMAL1 are primarily used to mediate interactions with DNA and CLOCK, but outside of these domains, there is a single known structural feature of import in the disordered C-terminus of BMAL1: a transactivation domain with multiple binding partners. The transcriptional coactivators p300 (Ep 300) and CREB-binding protein (CBP, Crebbp) have been identified as members of the CLOCK/BMAL1 activator complex with potential roles in epigenetic regulation (through domain-specific HAT activity) and recruitment of transcription initiation complex machinery (Etchegaray et al., 2003; Takahata et al., 2000). In particular, p300/CBP enhances transcriptional activation when co-expressed with CLOCK/BMAL1, though this role is dependent on an interaction with the BMAL1 C-terminus (Etchegaray et al., 2003; Takahata et al., 2000). However, co-expression of CRY1 blocks this effect (Etchegaray et al., 2003). Consistent with these data, concurrent reports from two groups identified a region in the distal C-terminus that is critical for both BMAL1-mediated transactivation and CRY1-mediated feedback repression (Kiyohara et al., 2006; Sato et al., 2006). Mutation of BMAL1 A611 or G612 desensitizes BMAL1 to CRY1 repression and disrupts binding between the two proteins (Sato et al., 2006). Likewise, BMAL1 constructs truncated at residue 554, 608, and 619 all disrupt binding to CRY1 and result in weaker transactivation activity (Kiyohara et al., 2006). Taken together, these data suggest one or more protein interaction motifs in the final 25–50 residues of the BMAL1 C-terminus.
BMAL1 has a highly conserved paralog, BMAL2, which is nonetheless incapable of rescuing circadian rhythms in Bmal1−/- fibroblasts (Xu et al., 2015). Domain swapping rescue experiments demonstrated that a C-terminal region of BMAL1 with a binding motif (IxxLL) for the p300/CBP KIX domain is necessary for normal circadian transcriptional activity. Chemical-shift mapping of the BMAL1 TAD in the presence of CRY1’s coiled-coil domain and the CBP KIX domain showed that these two proteins share an overlapping interface on the BMAL1 TAD. Critically, substitutions in and around the IxxLL motif result in dramatic changes in periodicity (between 19 and 26 hours) in rescue assays. These changes in period are highly correlated with shifts in the affinity of CRY1 for the TAD with longer periods stemming from higher affinity interactions and shorter periods from lower affinity interactions (Xu et al., 2015), suggesting that the balance between activator and repressor at this interface is a major node of period regulation in the clock. It is worth noting that the BMAL1 TAD is an additional competitive interface within the core molecular clock machinery, comparable to CLOCK’s exon 19 region.
Further characterization of the C-terminal TAD revealed that there is a slow conformational switch between cis and trans isomers at a conserved Trp-Pro imide bond in the extreme C-terminus of BMAL1 (Gustafson et al., 2017). Substitution of an alanine at either position of the switch (W624 and P625) locks the TAD into the trans isomer. Trans-locked analogs used in cell-based rescue assays resulted in period shortening of up to 3 hours compared to rescues with WT BMAL1. However, there was no difference in affinity for either CRY1 or the CBP KIX domain with the cis or trans isomers, suggesting an alternate explanation underlying the shift in periodicity for trans-locked mutants. Isomerization of the switch in BMAL1’s TAD occurs over a slow timescale of minutes, but cyclophilins, a family of peptidyl prolyl isomerases, can catalyze the isomerization resulting in substantially faster interconversion. Antagonizing the activity of cyclophilins with specific inhibitors results in dose-responsive period lengthening in cycling cell assays, though this effect was reduced in the context of trans-locked TAD mutants. The mechanism underlying these effects on periodicity in the clock is not yet known, but clearly represents an important node for regulation of clock speed.
As stated previously, CLOCK can acetylate BMAL1 at K537 in BMAL1’s intrinsically disordered C-terminus (Hirayama et al., 2007). Characterization of the binding affinity between various CRY1/2 and BMAL1 TAD fragments revealed a potential role for this post-translational modification on BMAL1 (Czarna et al., 2011). The C-terminal region of CRY contains a structured α-helix (sometimes referred to as the coiled coil for historical reasons), which is highly conserved between CRY1 and CRY2, and an intrinsically disordered tail region, which is completely divergent between the two CRYs. Czarna and colleagues purified the C-terminal regions of CRY1 and CRY2, denominated CRY1CCT and CRY2CCT respectively, and measured the affinity of these fragments for a short (residues 577–625) and long (residues 490–625) fragment of the BMAL1 C-terminus. While both CCT constructs bind the short fragment of BMAL1 with ~10 μM affinity, there is a clear difference in affinity between CRY1CCT (~20–40 μM depending on method of analysis) and CRY2CCT (~10 μM) with the longer fragment of BMAL1, which contains the acetylated residue K537. Interestingly, substitution of a glutamine at K537 (K537Q), which mimics an acetylated lysine, results in a stronger affinity between CRY1CCT and the long fragment (~10 μM). Whether this observation is relevant in vivo is unknown, but it raises the question of how acetylation status at BMAL1 K537 might affect the circadian timing mechanism. The work of Xu and colleagues certainly suggests that the balance of affinity between CRY1 and the BMAL1 TAD is an important regulatory node for period length (Xu et al., 2015).
Components of the activator complex
In addition to the previously described components of the circadian activator complex MLL1 and p300/CBP, several other proteins have been identified with roles in the molecular clockwork. Thyroid hormone receptor-associated protein-150 (TRAP150, Thrap150) was identified in CLOCK/BMAL1 complexes (Lande-Diner et al., 2013). TRAP150 can function as a coactivator for certain nuclear receptors, but also has roles in RNA splicing and DNA repair. However, in the clock, its role appears to be confined to coactivation with CLOCK and BMAL1. Expression of TRAP150 is controlled in part by CLOCK and BMAL1 through an E-box in its promoter, with mRNA peaking at circadian time (CT) 4, early in the subjective day. TRAP150 physically associates with CLOCK and BMAL1 through an unknown interface and recruits the Mediator complex, a large protein complex that functions as a modulator of the RNA polymerase II preinitiation complex. Depletion of TRAP150 results in low amplitude, long period rhythms and a reduction in RNA polymerase II at E-box sites of clock-controlled genes (Table 2).
JumonjiC (JmjC) and ARID domain-containing histone lysine demethylase 1a (JARID1a, Kdm5a) has also been identified in activator complexes (DiTacchio et al., 2011). DiTacchio and colleagues identified circadian oscillations in histone modifications at histone 3 (H3) lysine 9 (H3K9Ac) and H3K4 (H3K4me3). As previously described, trimethylation at H3K4 is promoted by MLL1 (Katada and Sassone-Corsi, 2010), so the authors focused on the JmjC domain-containing H3K4me3 demethylase family as potential regulators of demethylation at this site. However, JARID1a was found to associate with CLOCK and BMAL1 during the positive phase of the circadian cycle and function as a coactivator for circadian target genes, enhancing the activity of CLOCK and BMAL1 (DiTacchio et al., 2011). This role is inconsistent with a function as a demethylase for H3K4me3 as this epigenetic marker is primarily associated with a poised chromatin state, available for active transcription (Wang et al., 2009). Subsequently, DiTacchio and colleagues demonstrated that JARID1a’s demethylase activity is not required in its role as a coactivator (DiTacchio et al., 2011). Rather it functions in the clock primarily as an antagonist of histone deacetylase 1 (HDAC1, Hdac1), repressing its activity and increasing acetylation of H3K9 at the Per2 E-box. Deletion of Jarid1a results in shorter periods and lower amplitude oscillations in clock-controlled gene expression (Table 2). Notably, related family members JARID1b (Kdm5b) and JARID1c (Kdm5c) do not serve as coactivators of CLOCK/BMAL1-mediated transcription, but function in a repressive capacity by reducing H3K4me3 modifications at the Per2 promoter.
Photic input to the clock through the activation complex appears to come in part from a single cascade in which Protein kinase C α (PKCα, Prkca) phosphorylates lysine-specific demethylase 1 (LSD1, KDM1A). Phosphorylated LSD1 subsequently helps to recruit CLOCK and BMAL1 to target E-boxes, functioning primarily as a coactivator (Nam et al., 2014). Though its mechanism of action is unknown, its demethylase activity is not involved, and phosphorylation by PKCα at S112 is necessary. Phosphorylation at this site also occurs rhythmically with a peak at CT8 in the liver, suggesting that it not only mediates photic input to the activation complex, but also functions in normal daily rhythms. However, knockin mice with S112A substitutions have relatively minor effects on overall clock function (Table 2). This LSD1 mutant shows attenuated binding with CLOCK and BMAL1 and less recruitment to target promoters, but at a behavioral level there is no change in period. The most significant behavioral effect is lower amplitude rhythms suggesting that LSD1 primarily reinforces normal clock function.
Finally, recent work characterizing the nuclear activator complex has demonstrated that CLOCK and BMAL1 are part of an ~750 kDa complex during the activation phase (Aryal et al., 2017). Aryal and colleagues immunoprecipitated this complex using an antibody specific to CLOCK and assessed its size and composition using a specialized blue native-agarose polyacrylamide gel. Surprisingly, most of the purported activator components identified previously (including MLL1, JARID1a, and TRAP150) did not appear as stoichiometric components of the stable ~750 kDa activator complex. The authors, however, report that these components are specifically co-immunoprecipitated with CLOCK and are detectable in one or more smaller, less frequently observed complexes, supporting a potential transient role in circadian regulation. The components of the stable activator complex were not reported. Thus, further characterization of the stable activator complex remains an area of active inquiry.
Stability of the activators
In contrast to the negative regulators, which will be discussed shortly, the mechanisms regulating the degradation of CLOCK and BMAL1 have been studied very little. Minimal information exists about the regulation of CLOCK’s stability, though there is some evidence to support a role for glycogen synthase kinase-3 β (GSK3β, GSK3B) in the cooperative phosphorylation of seven serine/threonine residues in the ten residue span from 427–437 (Spengler et al., 2009). Phosphorylation at these sites appears to destabilize CLOCK. Additionally, the exon 19 region of CLOCK may be necessary for phosphorylation at these sites, possibly through interaction with CIPC (Yoshitane et al., 2009). BMAL1 is the subject of regulation by a few factors. Phosphorylation of BMAL1 at different sites has been shown to have opposing effects on its stability. GSK3β has been shown to phosphorylate BMAL1 at S17 and T21, which leads to subsequent ubiquitylation and degradation (Sahar et al., 2010). Conversely, phosphorylation by Protein kinase C γ (PKCγ, Prkcg) at S269 stabilizes BMAL1 by promoting cleavage of ubiquitin and blocking polyubiquitylated chains from forming on BMAL1 (Zhang et al., 2012). Ubiquitylation of BMAL1 is mediated at least in part by ubiquitin protein ligase E3A (UBE3A, Ube3a) (Gossan et al., 2014; Shi et al., 2015). Decreased levels of UBE3A lengthen period and generally decrease rhythm amplitude in both in vitro and in vivo experiments, though the effect on periodicity is fairly modest. Perhaps the most interesting aspect of activator stability is the relationship between stability and transcriptional activity. Stratmann and colleagues observed that recruitment of BMAL1 to a Dbp promoter array is highly dynamic even in short time windows (100 min) at the peak of Dbp transcription (Stratmann et al., 2012). Critically, the authors found that treatment with a proteasome inhibitor stabilized BMAL1 on the Dbp promoter while also blocking transcriptional activation. This observation suggests that immediate degradation is imperative for BMAL1’s function as a transcriptional activator, consistent with the kamikaze model of transcriptional activation. Together, these data suggest several conclusions. First, more work is needed to identify regulatory components of activator stability and elucidate the mechanisms driving degradation. Second, it is surprising that these components have not been identified given the amount of effort that has been devoted to identifying molecular components of the circadian clock. Perhaps the lack of candidates reflects a comparatively minimal role for activator degradation in the generation of rhythmicity and the determination of periodicity.
The repressive complex
Our understanding of the molecular details of the circadian clock has accelerated in the last decade due in no small part to a wealth of data on the repressors CRY and PER. Structural details on CRYs and PERs have emerged through crystallographic and NMR studies. Mechanistic details of how the repressors are targeted and protected from degradation have also been revealed. Meanwhile, a number of different post-translational modifications have been identified and their roles elucidated to paint a picture of how the repressors are regulated. Finally, the size and scope of the circadian repressive complex has been characterized conveying a sense of both the mechanistic basis of repression and the contributions of different elements to timing.
Structural features of the Cry/Photolyase Family
CRYs belong to a family of proteins (the Cry/Photolyase Family (CPF)) with an ancestral role in DNA repair (Chaves et al., 2011). DNA Photolyases (PHLs) catalyze repair of UV-damaged DNA through a flavin adenine dinucleotide (FAD) cofactor (Kavakli et al., 2017). FAD is complexed in a large cavity in the globular domain that comprises most of the PHL (Park et al., 1995; Tamada et al., 1997). DNA lesions bind at this site and appose the FAD molecule, bringing the two components of the reaction into close proximity for a reduction of the cyclobutane pyrimidine dimer bond in the damaged DNA (Mees et al., 2004). This reaction is dependent on exposure to blue light and the reaction dynamics can be improved by a light-harvesting, variable secondary cofactor bound in a distal pocket on the other side of CPF proteins (Kavakli et al., 2017; Park et al., 1995; Tamada et al., 1997). Secondary cofactors, traditionally either methenyltetrahydrofolate (MTHF) or 8-hydroxy-5-deazaflavin (8-HDF), are complexed in different ways (Park et al., 1995; Tamada et al., 1997). While MTHF extends out of the secondary pocket, 8-HDF is fully enclosed in the cavity.
Though structurally related to PHLs, CRYs are functionally divergent (Park et al., 1995; Xing et al., 2013; Zoltowski et al., 2011). Although mammalian CRYs have been shown to bind FAD like PHLs (Xing et al., 2013), they possess no DNA repair activity (Ozgur and Sancar, 2003). Moreover, though CRYs exist broadly across the domains of life with a diversity of roles, scant evidence exists to suggest that any eukaryotic CRYs complex a secondary cofactor. For instance, despite several attempts to obtain a structure with a secondary cofactor, none of the existing CRY structures contain one, suggesting that CRYs have evolved to function without secondary cofactors (Brautigam et al., 2004; Czarna et al., 2013; Xing et al., 2013; Zoltowski et al., 2011).
On a functional level, animal CRYs can be grouped into two broad classes: type I and type II CRYs. Type I CRYs (also known as Drosophila-type Cry, insect-like Cry, or Cry-d (Rubin et al., 2006)) function as circadian photoreceptors with an ancillary role in the molecular clockwork of Drosophila and a number of other insects (Emery et al., 1998; Stanewsky et al., 1998; Yuan et al., 2007). Although type I CRYs have been shown to function as direct repressors of CLK and CYC in some peripheral tissues (Krishnan et al., 2001), their primary role is as a photic input to the clock (Emery et al., 1998; Stanewsky et al., 1998). Indeed, type I CRYs bind the Drosophila repressor TIM in a light-dependent interaction and mediate its degradation (Koh et al., 2006). Type II CRYs (also known as mammalian-type Cry, vertebrate-like Cry, and Cry-m (Rubin et al., 2006)) function primarily as direct repressors of CLOCK and BMAL1 in vertebrates (Shearman et al., 2000b) and CLK and CYC in a subset of insects (Chang et al., 2003; Rubin et al., 2006; Yuan et al., 2007). Notably, insect clock architectures can be grouped into three cohorts: (1) a Drosophila-like clock in which CLK and CYC are repressed by PER and TIM with photic input from a type I CRY, (2) a vertebrate-like clock in which CLK and CYC are repressed by PER and a type II CRY, and (3) an integrated clock in which both a type I and type II CRY are present and functional, providing both a photic input to the system and direct repressive input to CLK and CYC. Surprisingly, of the organisms that have been characterized so far, the Drosophila-like architecture is least characteristic of insect clocks, as most adopt either a vertebrate-like (bees, ants, red flour beetles) or integrated architecture (monarch butterflies, silk moths, mosquitos) (Chang et al., 2003; Ingram et al., 2012; Rubin et al., 2006; Yuan et al., 2007). It is not yet known how an integrated architecture works at a structural level, but it is worth noting that at least a subset of insects with vertebrate-like or integrated architectures express a version of CYC that contains a C-terminal TAD with significant homology to mammalian BMAL1, unlike the truncated version of CYC found in Drosophila (Chang et al., 2003; Rubin et al., 2006; Zhang et al., 2017). These findings suggest that type II CRYs function as direct repressors of CLOCK and BMAL1/CYC in part due to their ability to sequester the BMAL1/CYC TAD as in mice (Xu et al., 2015). Adding further confusion, most vertebrates have at least two type II CRYs, traditionally called CRY1 and CRY2. While the two are structurally quite similar (Czarna et al., 2013; Kobayashi et al., 1998; Xing et al., 2013), there are several key functional differences that will be discussed shortly.
On a structural level, CRYs have a stereotyped architecture consisting primarily of two major domains: a globular photolyase homology region (PHR) and a highly variable intrinsically disordered C-terminal tail (Figure 3A) (Czarna et al., 2013; Xing et al., 2013; Zoltowski et al., 2011). The PHR contains several structural features of note: two cavities on opposite sides of the protein where PHLs ancestrally bound FAD (FAD-binding pocket) and the secondary cofactor (secondary pocket) and a C-terminal α-helix, which is often referred to as the coiled coil (CC) helix due to structural characteristics common to coiled coils (Figure 3A and B) (Chaves et al., 2006). Furthermore, the PHR can be divided into an N-terminal α/β domain connected to a C-terminal α-helical domain by a flexible interdomain linker (Figure 3B) (Czarna et al., 2013; Xing et al., 2013). The surface area of the secondary pocket is made up of residues from both the α/β domain and the α-helical domain, while the FAD-binding pocket is entirely associated with the α-helical domain (Figure 3B). The CC helix is notable for its role as a high traffic interface for protein-protein interactions in type II CRYs (Czarna et al., 2011; Nangle et al., 2014; Schmalen et al., 2014; Xing et al., 2013; Xu et al., 2015) and for its potential role in nuclear localization of type II CRYs (Chaves et al., 2006). Additionally, several flexible loops are associated with protein-protein interactions including the serine and interface loops, associated with binding to PER2 and FBXL3 (F-box/LRR-repeat protein 3)/PER2 respectively (Figure 3B) (Nangle et al., 2014; Schmalen et al., 2014; Xing et al., 2013). Finally, the intrinsically disordered tails of CRYs are highly divergent and represent the clearest region of departure between CRY1 and CRY2 (Figure 3B and C).
Figure 3. CRYPTOCHROME domain architecture.

(A) On the left, the CRY1 structure (PDB: 5T5X) is colored to show the α/β domain (spearmint) and the α-helical domain (blue). On the right, two graphical renderings of the CRY structure based on the actual crystal structures of CRY1 at left. Important features of the protein are labeled, including two cavities with roles in protein-protein interactions and a superficial structural feature (CC helix) that functions as a shared interface for competitive protein-protein interactions. (B) The structure of CRY1/2 is rendered in graphical form with features of interest labeled and numbered based on the amino acid sequences of CRY1 and CRY2 from Mus musculus. The first set of numbers refer to CRY1’s sequence and the second set in any pair refers to CRY2. CRY1 and CRY2 share the same basic structure outside of the variable C-terminal tail. Outside of the cavities, which are covered in the main text, CRY is notable for several flexible loops (the serine loop, the phosphate loop, and the interface loop), which function to some extent in physical interactions with other proteins. The serine loop and interface loop are both involved in binding to PER2. Additionally, the serine loop, along with α4, α15, and α16 contribute residues to the surface of the secondary pocket that play a role in binding to CLOCK’s HI loop. (C) An alignment of the CC helix and tail of murine CRYs. The alignment was made using Clustal Omega and visualized using ESPript 3 (Robert and Gouet, 2014). Although the CC helix is nearly identical, the tails of each CRY are highly divergent.
Regulation of CRY stability
Circadian periodicity is highly subject to the dynamics of CRY degradation. Degradation of CRY is primarily driven by interactions with two different Skp1-Cul1-F-box (SCF) E3 ubiquitin ligase complexes: SCFFBXL3 and SCFFBXL21 (Busino et al., 2007; Godinho et al., 2007; Hirano et al., 2013; Siepka et al., 2007; Yoo et al., 2013). Mutations in F-box/LRR-repeat protein 3 (FBXL3, Fbxl3) that disrupt binding to CRYs result in significant period lengthening in vivo (26–27 h) due to the stabilization of CRY (Table 1) (Busino et al., 2007; Godinho et al., 2007; Siepka et al., 2007). Surprisingly, the opposite phenotype is present in mice with a mutation or deletion of F-box/LRR-repeat protein 21 (FBXL21, Fbxl21) (Table 1) (Hirano et al., 2013; Yoo et al., 2013). Although both complexes possess E3 ligase activity, SCFFBXL21 ubiquitylate CRY less efficiently than SCFFBXL3 (Hirano et al., 2013; Yoo et al., 2013). However, FBXL21 acts as an antagonist of FBXL3 due to its stronger physical interaction with CRY, effectively stabilizing CRY in the presence of FBXL3. Interestingly, while FBXL21 interacts with CRY in both the cytoplasm and nucleus of cells, FBXL3’s interaction is entirely nuclear. Thus, FBXL21 functions as the primary E3 ligase for CRY in the cytoplasm whereas in the nucleus it plays a protective role against the primary nuclear E3 ligase FBXL3. Finally, FBXL3 was shown to ubiquitylate eleven lysine residues on CRY1, while FBXL21 targets a single site, K11, whose side chain forms the back wall of CRY’s secondary pocket cavity (Yoo et al., 2013).
CRY stability is also potentially regulated in vivo by several kinases. Adenosine monophosphate-activated protein kinase (AMPK) phosphorylates CRY1 at S71 and S280 (Lamia et al., 2009). Substitution of an alanine at either site to block phosphorylation stabilized CRY1 and substitution of an aspartate to mimic phosphorylation destabilized CRY1. However, a recent dataset containing effects of substitutions in a large cohort of CRY1 serines and threonines presented a contradictory assessment of the role of these AMPK targets (Ode et al., 2017). In contrast to the previous report, Ode and colleagues found that substitution of an aspartate at S71 significantly increased the half-life of CRY1, but made it a significantly less potent repressor of CLOCK and BMAL1. Perhaps these divergent observations reflect the effects of unknown factors in the cellular environment. CRY1’s tail is also the target of post-translational regulation at S588 (Gao et al., 2013; Papp et al., 2015). S588 is phosphorylated both rhythmically and in response to DNA damage by an unknown kinase. Phosphorylation at this site stabilizes CRY1 by antagonizing its interaction with FBXL3 and promoting an interaction with the deubiquitinase Herpes virus associated ubiquitin-specific protease (HAUSP, Usp7).
Nuclear localization of CRY
In their work on CRY1 and CRY2, Li and colleagues suggest that the balance between nuclear and cytoplasmic CRY might be determinative for periodicity (Li et al., 2016). Nuclear import mechanisms for CRYs are still poorly understood, but work from Hirayama et al. suggests that CRY1 and CRY2 have a conserved nuclear localization sequence (NLS) spanning residues 265–282 and 283–300 respectively (Hirayama et al., 2003). CRY1 and CRY2 also appear to contain a less conserved bipartite NLS in their C-terminal tails that requires an intact CC helix (Chaves et al., 2006). The CC helix, the N-terminal NLS, and the C-terminal NLS are each sufficient to direct CRY to the nucleus and at least one is necessary (Chaves et al., 2006). Members of the Importin α/β family (in particular KPNB1) have been implicated in nuclear localization of CRY2, but CRY1 nuclear entry appears to be primarily mediated through an alternative mechanism (Lee et al., 2015; Sakakida et al., 2005). Interestingly, while CRYs are efficiently translocated to the nucleus on their own, the rate of PER nuclear accumulation is significantly increased in the presence of CRY (Lee et al., 2001; Ollinger et al., 2014; Sakakida et al., 2005; Yagita et al., 2002). Modulating the rate of nuclear import of PER and CRY has clear effects on period, suggesting that it could be an important regulator of the overall timing mechanism, though a more thorough understanding of the mechanisms driving nuclear import of the repressors is warranted.
Protein-protein interactions of CRY
Of all of the core clock proteins, the greatest wealth of structural information belongs to CRY. To date, three structures of the CRY1 PHR and five structures of the CRY2 PHR have been solved with various cofactors and binding partners (Czarna et al., 2013; Michael et al., 2017; Nangle et al., 2013; Nangle et al., 2014; Schmalen et al., 2014; Xing et al., 2013). Additionally, interactions between CRY and the BMAL1 TAD have been characterized in depth by a series of biophysical experiments (Czarna et al., 2011; Gustafson et al., 2017; Xu et al., 2015). Finally, CRYs have been subjected to substantial mutagenic analysis through which residues involved in periodicity, repression, and protein-protein interactions have been identified (Froy et al., 2002; Gao et al., 2013; Khan et al., 2012; Lamia et al., 2009; Li et al., 2016; McCarthy et al., 2009; Michael et al., 2017; Nangle et al., 2014; Ode et al., 2017; Ozber et al., 2010; Rosensweig et al., 2018; Sanada et al., 2004; Schmalen et al., 2014; Xing et al., 2013; Yoo et al., 2013). From this bounty, a few major observations have been gleaned.
First, CRY’s CC helix is a widely shared interface for protein-protein interactions (Figure 6A). Comparison of a structure of the CRY2/FBXL3 complex to a structure of the CRY2/PER2 CRY-binding domain (CBD) complex illuminates an oft-described observation that PER2 stabilizes CRY (Nangle et al., 2014; Xing et al., 2013). Based on these structures, PER2 and FBXL3 have overlapping binding interfaces at the CC helix, suggesting that interaction with CRY is mutually exclusive. PER2 adopts a sinuous, elongated interface with CRY1 and CRY2, wrapping from just above the secondary pocket of CRY to the CC helix before swooping below the helix and coming back up the other side next to the FAD-binding pocket (Nangle et al., 2014; Schmalen et al., 2014). In a curious twist, CRY and PER2 chelate a zinc ion in an intermolecular zinc finger when bound and disruption of this interface destabilizes their interaction, suggesting the potential for redox sensitivity in the core clock mechanism. FBXL3 embraces the CC helix primarily through a curved β-sheet domain, though it also penetrates deep into the FAD-binding pocket with its C-terminal tail (Xing et al., 2013). In fact, its final residue is a tryptophan and the side-chain mimics the aromatic rings of the flavin moiety usually found in this pocket. FBXL3 activity can be antagonized by a small molecule (KL001) that stabilizes CRY and lengthens the period in cycling cells (Hirota et al., 2012). Comparison of the FBXL3 complex to structures of CRY2 with either FAD or KL001 bound suggest that both molecules stabilize CRY by binding in the FAD-binding pocket and blocking FBXL3’s tail from entering the cavity (Nangle et al., 2013; Xing et al., 2013). In addition to FBXL3 and PER2, the CC helix also participates in an interaction with the BMAL1 TAD in a way that is likely to be competitive with both FBXL3 and PER2 (Czarna et al., 2011; Xu et al., 2015). How these interactions are integrated in a dynamic time-keeping mechanism is not fully understood, but likely to be highly informative in understanding the driving molecular features of periodicity.
Figure 6. Competitive protein-protein interactions drive clock function.

(A) CRY’s CC helix is involved in three mutually exclusive protein-protein interactions with FBXL3, PER, and BMAL1. The competitive interaction is depicted here along with the likely outcome of any given interaction. (B) Competitive interactions at two particular interfaces are potential drivers of oscillatory gene expression. The transcriptional repressor CRY competes with the transcriptional coactivator CBP to bind BMAL1’s TAD. As CRY levels build up throughout the activation phase of the clock, CRY supplants CBP at this interface causing a switch to a more repressive phase of the cycle. Similarly, MLL1 and CIPC both bind to CLOCK’s exon 19 region, likely in a mutually exclusive way. Interaction with MLL1 is critical for transcriptional activation at clock-regulated genes due to its role in chromatin decompaction. CIPC (and possibly PER proteins) functions as a repressor in part due to its ability to bind and sequester the exon 19 region of CLOCK. When the interaction partner at BMAL1’s TAD or CLOCK’s exon 19 switches, it allows for a transition in the phase of gene expression within the clock cycle.
Emerging evidence also points toward a second major interface at CRY’s secondary pocket. A screen for mutations that would weaken CRY’s repressive capacity identified three residues along a helix forming one boundary of the secondary pocket (McCarthy et al., 2009). Characterization of these mutants (CRY1 E103K, G106R, and R109Q) revealed that they not only weaken CRY1’s repressive capacity, but also block CRY1’s ability to drive rhythms in a rescue assay and disrupt binding between CRY and the CLOCK/BMAL1 complex (Nangle et al., 2014; Rosensweig et al., 2018). Further support for the secondary pocket as a critical protein-protein interface comes from computational docking combined with biochemical characterization of purified CRY1 and CLOCK PAS-B proteins (Michael et al., 2017). Finally, our lab demonstrated that disruption of the secondary pocket interface can have profound effects on periodicity, producing changes in periodicity in cycling cell assays of up to five hours, largely due to abrogated binding between CRY1 and the CLOCK/BMAL1 heterodimer (Rosensweig et al., 2018). This finding is in strong agreement with the results of Xu and colleagues in their study of the interaction between CRY1 and the BMAL1 TAD (Xu et al., 2015). Together, these data strongly support a role for the secondary pocket as a binding site for the CLOCK PAS-B domain and suggest that modulation of affinity between the repressors and activators can play a substantial role in determining periodicity.
However, the requirements for formation of a repressive complex are still somewhat contentious in the field. Chen and colleagues found that constitutive expression of Cry1 did not disrupt rhythms in cycling fibroblasts whereas constitutive expression of either Per1 or Per2 did (Chen et al., 2009). They demonstrated that co-IP of CRY1 with CLOCK and BMAL1 was barely above baseline levels, but the addition of PER2 made this interaction significantly more robust. PER2 also co-immunoprecipitated with CLOCK and BMAL1 without CRY1, suggesting that CRY1 requires PER2 to form a stable complex with CLOCK and BMAL1, but not the reverse. In contrast, work from Ye and colleagues suggests that CRY1 has a direct interaction with CLOCK and BMAL1 even in the absence of PER proteins (Ye et al., 2011). ChIP analysis of the Per1 and Per2 promoters in various KO cell lines showed that CRY1 is bound at target promoters even in the absence of endogenous PER. Moreover, CRY1 is competent to repress CLOCK/BMAL1-mediated transcriptional activation without PER. Further work suggested the additional conclusion that CRY and PER function in completely different modes of repression (Ye et al., 2014). CRY binds CLOCK and BMAL1 and directly represses their activity as a “blocking-type” repressor while PER physically removes the complex from DNA as a “displacement-type” repressor. Additionally, work from our lab supports the notion that CRY1 is capable of forming a repressive complex with CLOCK and BMAL1 in the absence of PER or when point mutations have been introduced to disrupt binding to PER2 (Nangle et al., 2014; Rosensweig et al., 2018). However, we observed that co-expression of PER2 stabilizes this interaction and is even able to drive formation of a stable complex in certain cases in which point mutations have weakened the interaction between CRY1 and CLOCK and BMAL1 (Rosensweig et al., 2018). Ultimately, the preponderance of evidence suggests that CRY is directly binding CLOCK and BMAL1 without the express need for PER, but the nature of PER’s role in this interaction is still up for debate.
Taking all of these protein-protein interactions into account, it is easy to view CRY as a nexus bridging multiple components of the circadian complex. Whether the implicit allostery involved in such an intricate web of interactions is a key principle of rhythm generation or merely coincidental is an area for future research. However, what is clear from data collected on the BMAL1 TAD’s interaction with CRY1 is that modulation of these competitive interfaces is likely to be determinative in matters of periodicity (Xu et al., 2015).
CRY1 and CRY2
Broadly speaking, CRY1 and CRY2 play the same role in the mammalian clock, functioning as indispensable and direct repressive components (Kume et al., 1999; Shearman et al., 2000b; van der Horst et al., 1999; Vitaterna et al., 1999). However, the details of their respective roles are far murkier. Genetic knock-out (KO) models demonstrate that deletion of both Cry1 and Cry2 results in arrhythmic locomotor behavioral rhythms (Table 1) (van der Horst et al., 1999; Vitaterna et al., 1999). Cry1-/- and Cry2-/- mice maintain rhythmic locomotor activity, but the former cohort have short endogenous free-running rhythms (~22.5 h) while the latter have long periods (~24.5 h) compared to WT mice (Table 1) (van der Horst et al., 1999; Vitaterna et al., 1999). There is evidence that CRY1 plays a more dominant role in the clock than CRY2. SCN explants from Cry1-/- mice maintain oscillatory expression of a PER2::LUC (Luciferase) reporter, while explants from peripheral tissues and dispersed fibroblast cultures did not (Liu et al., 2007). Under the same conditions Cry2-/- tissues and fibroblasts remained rhythmic suggesting that intercellular coupling within the SCN manifests a protective role against weak cell autonomous rhythms. Moreover, it supports the conclusion that Cry1 is more indispensable than Cry2 in the clock. In further support of this conclusion, work from Khan and colleagues found that CRY2 was a weaker repressor of CLOCK/BMAL1-mediated transcription (Khan et al., 2012).
The stark divergence in functional character between CRY1 and 2 is surprising given the level of conservation between the two at a structural level. CRY1 and 2 are 66.4% identical and 76.7% similar across an entire alignment, but excluding their completely divergent tails, the PHR domains are 77.4% identical and 88% similar. Of the residues that diverge in the PHR, the largest cluster is a group of superficial residues in the α/β domain, though there is no substantial dataset to date that implicates this particular region of CRY as an area of importance in normal CRY function (Figure 4A). Alignment of apo structures of CRY1 and CRY2 also suggests that they are highly similar with a root mean square deviation (RMSD) of 0.493 Å and an all-atom RMSD of 2.162 Å (Czarna et al., 2013; Xing et al., 2013).
Figure 4. Divergence between CRY1 and CRY2.

(A) Two views of the CRY2 PHR structure (PDB: 4I6E) with all of the residues diverging between CRY1 and CRY2 labeled in blue. The vast majority of divergence is in one particular region of the α/β domain shown on the right.
(B) The seven divergent residues at the secondary pocket are shown in blue on a surface representation of CRY2 (PDB: 4I6E).
Due to the high degree of structural conservation, several theories have arisen to explain the divergent periodicity characteristics observed. One possibility is that there are intrinsic differences in stability between CRY1 and CRY2 that drive distinctly periodic output. In fact, a recent report suggests that there is in fact an inherent difference in stability between the two proteins (Li et al., 2016). Based on the in vivo phenotypes of Fbxl3 mutants, one would expect that stabilization of CRY would lead to longer periods (Godinho et al., 2007; Siepka et al., 2007). However, Li and colleagues found that CRY2 is actually more stable than CRY1, which is inconsistent with the notion that their intrinsic periodicity characteristics stem from stability differences (Li et al., 2016). Further complicating this hypothesis is data from Ode and colleagues examining a large group of serine and threonine residues in CRY1 and their role in both stability and periodicity (Ode et al., 2017). A wide range of periods (ranging from 20–34 h) were observed in cell-based rescue assays with various mutants, but there was little correlation between period in the rescue assay and half-life of the protein. In fact, this data reflects an emergent view in circadian biology that the quality of a protein is just as important as the quantity in determining period (Larrondo et al., 2015).
Another possible explanation is that the phase of expression of Cry1 and Cry2 plays a role in their unique periodicities. Cry2 expression is regulated primarily by CLOCK and BMAL1 through E-box elements, but Cry1 expression relies on both E-box elements in its promoter and a Rev-Erb/ROR-binding element (RRE) in one of its introns (Ueda et al., 2002; Ueda et al., 2005; Ukai-Tadenuma et al., 2011). As a result, peak Cry1 expression is delayed compared to Cry2, Per1, and Per2, lengthening the period of a circadian luciferase reporter in cell-based rescue assays (Ukai-Tadenuma et al., 2011). Lending credence to this theory, circadian chromatin immunoprecipitation sequencing analysis of core clock proteins in the mouse liver identified concurrent peak DNA occupancy for CRY2, PER1, and PER2 in the early evening, but CRY1’s peak occupancy occurred in the late night/early morning forming a late repressive complex on its own with CLOCK and BMAL1 (Koike et al., 2012). In contrast, it was recently demonstrated that PER2::LUC rhythms could be rescued in Cry1-/-/Cry2-/- SCN explants following viral transduction of a plasmid expressing either Cry1 or Cry2 under the control of Cry1’s promoter, but not the intronic RRE (Edwards et al., 2016). Despite the fact that Cry1 and Cry2 were expressed under the control of the same promoter element, the rescues displayed period phenotypes characteristic of the previously described single KO mice (i.e. Cry2 rescues had short periods and Cry1 rescues had long periods). Rescues in which Cry expression was driven by the Bmal1 promoter (i.e. anti-phase to the normal phase of expression) resulted in low amplitude, erratic rhythms. Ultimately these results suggest that while phasing plays an important role in a robust oscillator, the distinct periodicity differences observed in CRY1- or CRY2-driven rhythms appear to be intrinsic to the proteins themselves.
Recently several labs have begun to unravel this mystery. The identification of the secondary pocket as a potential binding site for CLOCK PAS-B allowed for reassessment of data from Khan and colleagues on the domains that differentiate CRY1 and CRY2 as repressors (Khan et al., 2012; Michael et al., 2017; Rosensweig et al., 2018). An α-helix at one edge of the secondary pocket has subtly diverged between CRY1 and CRY2, maintaining amino acid characteristics like charge and size without preserving the exact side chains (Figure 4B). Individually these changes have a minimal effect on core circadian function (Rosensweig et al., 2018). However, in combination, they can have a dramatic effect on periodicity, which is due primarily to changes in affinity between CRY and the CLOCK/BMAL1 heterodimer. Ultimately, divergence at this site explains much of the periodicity differences observed in Cry1-/- and Cry2-/- knockouts. Chimeric analysis in rescue assays also supports a role for the divergent C-terminal tails of CRY1 and CRY2 in specifying periodicity and together with the secondary pocket architecture provides a structural basis for determining periodicity. It remains to be seen how the C-terminal tail functions in determining periodicity. Additionally, more work is needed to determine how phase of expression and protein stability are integrated with intrinsic protein characteristics to set periodicity.
Structural features of PER proteins
In sharp contrast with CLOCK, BMAL1, and CRY, structural biology is markedly more challenging in the case of PER proteins. This is due in large part to the fact that PER proteins are large (~1100–1300 residues), intrinsically disordered proteins with just a few structured domains (Albrecht et al., 1997; Sun et al., 1997; Tei et al., 1997; Zylka et al., 1998). Like CLOCK and BMAL1, PER1/2/3 have a set of N-terminal tandem PAS domains (PAS-A and PAS-B), which are primarily used to mediate protein-protein dimerization interactions (Figure 5A and B). Just beyond the PAS domains is a region (from residue 450–763 in PER2) that interacts with the F-box protein β-transducing repeat-containing protein (β-TrCP) and casein kinase 1δ/ε (CKIδ/ε), known as the casein kinase-binding domain (CKBD) (Figure 5B) (Eide et al., 2005). C-terminal to this interaction domain is a disordered proline-rich region and at the extreme C-terminus a roughly 100 amino acid binding interface for CRY1/2 known as the Cry-binding domain (CBD) (Figure 5B) (Nangle et al., 2014; Schmalen et al., 2014). PER3’s C-terminus is highly divergent compared to PER1 and PER2 and lacks a functional CBD (Zylka et al., 1998). Scattered throughout PER2 are multiple, functional nuclear export sequences (NES) as well as a single, bipartite nuclear localization sequence (NLS) roughly in the middle of the protein (Figure 5B) (Yagita et al., 2002).
Figure 5. PERIOD domain architecture.

(A) The PAS domain homodimer structures of PER1, PER2, and PER3 respectively. Overall structures are very similar. Each protein homodimerizes in an orthogonal orientation. The PAS-A and PAS-B domains of one PER1 subunit are circled in the figure on the left. N and C termini are labeled for each monomer of each PER complex. (B) Using available structural data, the structure of PER2 is rendered in graphical form with domains of interest labeled and numbered based on the amino acid sequence of PER2 from Mus musculus. The region between the PAS-B domain and the CRY-Binding Domain (CBD) is shown as a flexible region, which reflects the fact that little structural information is available within this region. Within this region is the loosely defined Casein Kinase-Binding Domain (CKBD) and the even more loosely defined Proline-Rich Domain (PRD). No major roles have been ascribed to the PRD, but the CKBD contains several serines that are phosphorylated by casein kinase 1δ/ε. These serines (labeled here) are distal sites for regulation of PER stability. S478 is recognized by β-TrCP to target PER for degradation. S659, S662, S665, S668, and S671 are serially phosphorylated by CK1δ/ε and stabilize PER. Substitution of a glycine (S659G) at a homologous site in human PER2 is a known cause of Familial Advanced Sleep Phase Syndrome (FASPS) (Toh et al., 2001). CRY binds to PER at the distal C-terminal end of the protein beyond the PRD. PER1 and PER2 share a similar domain architecture, but PER3 diverges beyond the PAS domains and lacks a CBD altogether. Finally, three nuclear export sequences (NES) and a bipartite nuclear localization sequence (NLS) have been identified and validated in PER2 (Yagita et al., 2002).
At present, structures of the PAS domains of all three PERs have been solved as well as structures of the PER2 CBD in complex with CRY1 and CRY2 (Hennig et al., 2009; Kucera et al., 2012; Nangle et al., 2014; Schmalen et al., 2014). The PAS domains of PER1, 2, and 3 all participate in homodimer interactions, primarily mediated by an antiparallel β-sheet interface between PAS-B domains and a conserved tryptophan moiety on a PAS-B loop (W448PER1, W419PER2, W359PER3) that is partially buried in the homodimer (Figure 5A) (Hennig et al., 2009; Kucera et al., 2012). The role of homodimer formation is not yet clear, though disruption of the homodimer interface hastens the mobility of PER2 (but not PER1) in cells (Kucera et al., 2012). PERs have been shown to form large protein complexes in both the cytoplasm and nucleus often containing multiple PER proteins (Aryal et al., 2017). One possibility is that homo- and heterodimerization interactions through the PAS domains are mediating complex formation. PER2’s elongated interface with CRY buries 2800 Å2 of solvent accessible surface area, which informs previously observed high affinity interactions between the two (Nangle et al., 2014). However, understanding the contribution of the interaction between the two proteins to the overall mechanism of the clock is complicated by the fact that the interface on CRY overlaps with FBXL3 and BMAL1.
Functional and structural divergence in the mammalian PER family is less obvious than CRY1 and CRY2. PER1 and PER2 are 43% identical and 54% similar; PER1 and PER3 are 33% identical and 45% similar; and PER2 and PER3 are 33% identical and 46% similar. Overall the level of divergence is consistent with a protein family defined primarily by a few structural motifs connected by long intrinsically disordered regions under weak selective pressure. However, despite fairly substantial structural divergence, PER1 and PER2 appear to play redundant roles in the clock. Per1-/- and Per2-/- mice display reduced circadian amplitude and very minor period differences in light/dark cycles and drift into arrhythmicity after several weeks in constant darkness (Table 1) (Bae et al., 2001). Per1-/-/Per2-/- double KO mice on the other hand are arrhythmic immediately after transition to constant darkness (Table 1). PER3 is functionally dissimilar from PER1 and 2. Per3-/- mice maintain circadian amplitude, but have faster endogenous clocks (~0.5 h short) (Table 1) (Shearman et al., 2000a). Moreover, compound mutants Per1-/-/Per3-/- and Per2-/-/Per3-/- displayed behavioral phenotypes consistent with single KOs (Per1-/- or Per2-/-) (Bae et al., 2001). Thus, PER3 appears to be a superfluous clock component.
Regulation of PER stability
PER protein stability has proven to be every bit as potent a regulator of periodicity as CRY stability. The stability of this repressor first came to the forefront of circadian biology through a spontaneous mutation with a major effect on periodicity (Ralph and Menaker, 1988). A single male golden hamster from the breeding supplier Charles River was identified with an abnormal free-running period of 22 h (Ralph and Menaker, 1988). After this hamster was bred into a colony, it was discovered that a single mutation (tau) causes the behavioral phenotype and functions semi-dominantly. Homozygous carriers have dramatically shortened endogenous rhythms of 20 h (Table 1). After an extensive and laborious process, Lowrey and colleagues identified the mutation as an allele of casein kinase I epsilon (CKIε, Csnk1e) with a substitution at a highly conserved residue (R178C) (Lowrey et al., 2000). Although CKIε tau binds PER1 and PER2 comparably to WT, it phosphorylates PER less efficiently. Further characterization in null and knock-in mice revealed that CKIε tau functions essentially as a gain-of-function mutation, accelerating PER protein turnover (Meng et al., 2008). Null mutants are behaviorally inert, likely due to redundant activity in the form of closely related family member CKIδ (Csnk1d).
Extensive characterization of CKIδ/ε’s interaction with PER has identified a region of PER that gates interaction with CKIδ/ε (Akashi et al., 2002; Eide et al., 2005; Lee et al., 2004). In PER2, the region from residue 450–763 is broadly where CKIδ/ε binds, but two short segments within this region (582–606 and 731–756) appear to be especially critical for mediating this interaction (Eide et al., 2005). Furthermore, the CKIδ/ε binding domain (CKBD) of PER3 has diverged from that of PER1 and PER2, weakening its interaction with CKIδ/ε (Lee et al., 2004).
Interaction with and phosphorylation by CKIδ/ε is regulated in part by phosphorylation at a priming site (S662hPER2/S659mPER2) associated with familial advanced sleep phase syndrome (FASPS) (Toh et al., 2001). Recent work suggests that this priming site is in fact also phosphorylated by CKIδ/ε, with a preference for CKIε and a particular splice variant of CKIδ (CKIδ2), which has an extreme C-terminus that is more like CKIε (Fustin et al., 2018; Narasimamurthy et al., 2018). Nearby serines are subsequently phosphorylated following priming, stabilizing PER2. In contrast, phosphorylation by CKIδ/ε at S478mPER2 recruits the E3 ligase complex SCFβ-TRCP, which ubiquitylates PERs and directs them to the proteasome for degradation (Figure 5B) (Shirogane et al., 2005; Zhou et al., 2015). Interestingly, the interplay between these two phosphorylation sites is regulated by temperature and a unique multi-stage decay process (Zhou et al., 2015). Ambient temperature can bias one pathway over the other and result in acceleration or deceleration of PER2 turnover, potentially suggesting a mechanism for temperature compensation in the mammalian clock. Further support for the idea that temperature compensation is mediated at this node comes from a biophysical and biochemical study of CKIδ, which demonstrated a role for temperature sensitivity in substrate and product binding (Shinohara et al., 2017). Temperature plays a substantial role in regulating the processivity of CKIδ, which may have an effect especially on multi-site phosphorylation dynamics. However, additional work is needed to understand how this might connect to the interplay between the two temperature-dependent PER phosphorylation/degradation pathways identified by Zhou and colleagues.
In addition to being rhythmically phosphorylated (Lee et al., 2001), PER is also rhythmically acetylated and deacetylated in vivo in part by the NAD+-dependent deacetylase, SIRT1 (Asher et al., 2008). SIRT1 rhythmically associates with CLOCK, BMAL1, and PER, deacetylating PER in the process and promoting its degradation. Due to the fact that this process is NAD+-dependent, it represents a functional input from metabolism to the clock. Furthermore, it builds the case along with the temperature-sensitive regulation of PER described above that various external conditions converge on PER, regulating its stability through post-translational modifications and thereby affecting the timing of the clock.
Clearly the combined regulation of repressor stability is a significant factor in the timing of the mammalian clock, but the overall concentration of these proteins is secondary to their function. Understanding function at a mechanistic level will provide key insight into why concentration matters for timing.
The role of PER in the mammalian clock
Despite relatively detailed accounting of regulatory sites, protein interaction domains, and cellular mobility, one of the outstanding questions in circadian biology remains what does PER do? Though reports on the requirement of PER for assembly of a stable repressive complex differ (Chen et al., 2009; Ye et al., 2014; Ye et al., 2011), one detail is clear: CRY is a strong repressor of CLOCK/BMAL1 with or without PER and the reverse is not true (Koike et al., 2012; Shearman et al., 2000b; Ye et al., 2011). These reports implicitly suggest that CRY must be able to bind CLOCK and BMAL1 and mediate its repressive effects without PER. Furthermore, characterization of nuclear localization of PER and CRY suggests that CRY nuclear import is insensitive to PER, but nuclear import of PER is highly regulated by CRY and CKIδ/ε (Lee et al., 2015; Ollinger et al., 2014; Sakakida et al., 2005; Yagita et al., 2002). Again, these data support a critical, PER-insensitive role for CRYs in the clock, but do little to explain PER’s function.
One possible and surprising role for PER may be as a circadian derepressor (Akashi et al., 2014). Coexpression of small amounts of PER1 and PER2 attenuate transcriptional repression by CRY in a dose-dependent manner. This response requires both the presence of CRY and an intact PER CBD, which precludes PER3 from playing a role in derepression. And perhaps most importantly, full-length PER2 and a small fragment containing the CBD are both able to block CRY1 from binding to CLOCK and BMAL1. This finding is perhaps not entirely shocking in the context of known interaction domains on CRY, but it strongly suggests that the PER2 CBD and BMAL1 TAD are in competition. Based on the conflicting data in the literature about PER’s role and the assembly of the repressive complex on the activators, a clear study of the kinetics of various domain associations would be valuable in understanding what interactions are important and when.
A final possible role for PER is that of a large scaffolding protein necessary for the assembly of a sizeable repressive complex. Work from a number of groups has identified over 20 different protein components of the repressive complex (detailed in the next section) with various roles and effects on the function of the clock. Some estimates put the size of this complex at roughly 2 MDa (Aryal et al., 2017; Kim et al., 2015). Notably, the heterogeneity of this complex is an open question, but at least one consistent, stable complex of this size can be purified (Aryal et al., 2017). Moreover, PER proteins have an architecture consistent with other scaffolding proteins (i.e. intrinsically disordered with several discrete structured elements throughout), which suggests a major potential role in the assembly of this repressive complex(Gustafson and Partch, 2015). In support of this notion, Aryal and colleagues reported several different repressive complexes assembling in different subcellular locations: a larger (1.1 MDa) and smaller (0.9 MDa) cytoplasmic complex and a single 2 MDa nuclear complex (Aryal et al., 2017). The smaller cytoplasmic complex contains CRY1, CRY2, PER1, PER2, and CK1δ, while the larger cytoplasmic complex adds PER3 and a guanine nucleotide exchange factor for Rab GTPases involved in cytoplasmic trafficking called GAPVD1 (GTPase-activating protein and VPS9 domain-containing protein 1, Gapvd1). The nuclear complex contains a number of proteins described in the following section, but also contains all of the proteins from the larger cytoplasmic complex aside from GAPVD1. These data suggest an ordinal sequence of events in which the core repressors CRYs and PERs form the basis of a major repressive complex in the cytoplasm, then enter the nucleus and begin assembling other components until the complex doubles in size and becomes highly stable.
Buried within this discussion of PER’s role is an implicit suggestion gaining traction within the field. While PER is the enigmatic alpha protein of the molecular clock, CRY appears to be the journeyman doing the primary work of repression in vertebrate systems.
Components of the repressive complex and other repressors
The identification of repressive complex components has been an ongoing process for over a decade and, based on the findings, supports the idea that these components can be coarsely sorted into two basic groups: (1) epigenetic regulators and (2) repressors without a clear mechanism of action.
Epigenetic regulators have primarily been identified through immunoprecipitation of PER complexes followed by mass spectrometry (Brown et al., 2005; Duong et al., 2011; Duong and Weitz, 2014; Kim et al., 2014; Padmanabhan et al., 2012; Tamayo et al., 2015). Studies of complex members indicate a progression of epigenetic regulators throughout the active and repressive phases of the clock ultimately leading to cycles of decompaction and compaction of chromatin (Aguilar-Arnal et al., 2013). CLOCK and BMAL1 actually participate in their own repression by associating with the adaptor protein WD repeat-containing protein 76 (WDR76, Wdr76), which recruits DNA damage binding protein 1 (DDB1, Ddb1) and Cullin-4 (CUL4, Cul4) (Tamayo et al., 2015). DDB1-CUL4 then monoubiquitinates histone 2B (H2B) at the promoter of CLOCK/BMAL1 targets, which serves to recruit PER complexes during the repressive phase. siRNA-mediated knock down of DDB1 or CUL4 shortens the period in cycling fibroblasts (Table 2). Early in the repressive phase, members of the Mi-2-nucleosome remodeling and deacetylase (NuRD) transcriptional co-repressor complex are split between CLOCK/BMAL1 and CRY/PER (Kim et al., 2014). Chromodomain-helicase-DNA-binding protein 4 (CHD4, Chd4), metastasis-associated 1 family member 2 (MTA2, Mta2), and sucrose nonfermenting protein 2 homolog (SNF2H, Smarca5) and BRG1 (Smarca4) (two members of the SWI/SNF family) are bound to the activators and depletion of CHD4, SNF2H, or BRG1 results in modest period lengthening (Table 2). The repressive complex on the other hand is bound to Methyl-CpG-binding domain protein 2 (MBD2, Mbd2), RbAp48 (Rbbp4), GATA zinc finger domain containing 2A (GATAD2a, Gatad2a), and HDAC1/2 and depletion of MBD2 shortens the period in cycling cells (Table 2). Only through interaction of the activator and repressor complexes are all of the NuRD components reconstituted to function as a repressor of CLOCK and BMAL1. The repressive complex also includes polypyrimidine tract-binding protein-associated-splicing factor (PSF, Sfpq), which recruits the SIN3A (Sin3a)-HDAC1 complex during the repressive phase and modulates the acetylation state at H3K9 and H4K5 (Duong et al., 2011). Depletion of either PSF or SIN3A results in period shortening (Table 2). Rhythms in H3K27 di- and trimethylation are regulated in part by the polycomb group protein EZH2 (Ezh2), which associates with the repressive complex and functions in concert with CRY as a co-repressor (Etchegaray et al., 2006). shRNAs targeted to EZH2 result in arrhythmicity of Per2-Luc and Bmal1-Luc rhythms (Table 2). Several other histone methyltransferases have also been identified in the repressive complex including WD repeat-containing protein 5 (WDR5, Wdr5) and HP1γ (Heterochromatin protein 1 homolog gamma, Cbx3)-SUV39H (Suppression of variation 3–9 homolog 1, Suv39h1) (Brown et al., 2005; Duong and Weitz, 2014). The details of WDR5’s role in the clock are scant, but depletion resulted in the loss of an antiphase rhythm in H3K4 and H3K9 methylation (Table 2) (Brown et al., 2005). The HP1γ-SUV39H complex promotes di- and trimethylation of H3K9 in later phases of repression (Duong and Weitz, 2014). Interestingly, HDAC1 is recruited to deacetylate H3K9 early in the repressive phase and followed later by HP1γ-SUV39H. Moreover, the authors report that these repressive complex components appear to be part of distinct complexes separated in a spatiotemporal manner, suggesting either that PER complexes are fluid, with components joining and leaving, or that unique complexes form throughout the repressive phase and interact in a phase-specific manner with CLOCK and BMAL1. However, a more recent report from the same group suggests immunoprecipitated nuclear PER complexes are highly stable and uniform in their composition (Aryal et al., 2017). One possible explanation is that transient complexes are too labile to be captured and purified like the stable 2 MDa complex.
In addition to histone modifiers, the repressive complex also contains a number of helicases involved in transcriptional termination (Ddx5, Dhx9, and Setx) (Padmanabhan et al., 2012). During the repressive phase, SETX (Senataxin) in particular is recruited to termination sites on clock-controlled genes where it blocks transcriptional termination and causes a buildup of RNAP II, thus providing an additional layer of transcriptional regulation. Depletion of SETX causes arrhythmicity in cycling fibroblasts (Table 2).
Beyond epigenetic regulators, several other co-repressors have been identified. Receptor for activated C kinase-1 (RACK1, Rack1) recruits PKCα to CLOCK and BMAL1 where PKCα phosphorylates BMAL1, repressing its activity (Robles et al., 2010). As discussed previously, CLOCK and BMAL1 are also repressed by CIPC (Zhao et al., 2007). CIPC forms a coiled-coil complex with the exon 19 region of CLOCK through a 62 amino acid region of its C-terminus with a surprising 1:2 stoichiometry (Hou et al., 2017). Mutation of the exon 19 interface has the effect of diminishing CLOCK’s potency as a transcriptional activator while also disrupting CIPC’s ability to repress the residual activity (Hou et al., 2017). Together, these data suggest that CIPC shares a competitive interface with a transcriptional coactivator, but may function primarily in the regulation of CLOCK at tandem E-boxes or E-boxes in distal regions of the genome brought together through inter- and intra-chromosomal interactions (Figure 6B). Depletion of CIPC shortens the period (Table 2) (Zhao et al., 2007). In addition to CRY, BMAL1 has an additional negative regulator interacting with its TAD: CHRONO (Computationally highlighted repressor of the network oscillator, Ciart, also known as Gm129) (Anafi et al., 2014; Annayev et al., 2014; Goriki et al., 2014). Concurrent reports from three different labs suggest that CHRONO binds to BMAL1 and represses the transcriptional activity of CLOCK and BMAL1. Though the mechanism is not entirely clear, it appears to disrupt BMAL1’s interaction with CBP and antagonize circadian acetylation of H3K9 at CLOCK/BMAL1 targets, potentially through an interaction with HDAC1. Chrono-/- mice had modestly lengthened locomotor behavioral rhythms in constant darkness (Table 2) (Goriki et al., 2014). Not to be confused with CHRONO, a protein called NONO (Non-POU domain-containing octamer-binding protein, Nono) is also found in PER complexes and may in fact function as a weak antagonist of the repressors of CLOCK and BMAL1 (Brown et al., 2005). siRNA’s targeted to Nono result in period shortening (Table 2).
Concluding thoughts
The wealth of structural analysis that has been performed on mammalian clock proteins has opened windows into the function of the clock. Ultimately, building a model for how a molecular machine works requires an understanding of how the components fit together, which interactions are dynamic, and what the kinetics of those dynamic states are. In the case of the clock, regulation is occurring at many levels, but focusing on the key interactions might tell us about the most important aspect of a timing mechanism: measuring time.
To that end, there are a few key components that appear to drive the clock. (1) CLOCK and BMAL1 must form a stable complex that binds to DNA targets. (2) CLOCK and BMAL1 must be able to recruit the necessary transcriptional machinery and cofactors for chromatin decompaction. (3) CRY and PER must bind and sequester the relevant domains of CLOCK and BMAL1 mediating recruitment of cofactors. (4) There must be a mechanism in place to remove the repressors from the system and allow the cycle to begin anew.
In accounting for the various components of the activator and repressor complexes, it becomes clear that many of them have relatively minor effects on periodicity or overall clock function based on knockdown and knockout studies (Table 2). In most cases these components appear to serve in making the clock more robust. In contrast, the most dramatic effects on periodicity and clock function come from stabilizing or destabilizing the repressors (Busino et al., 2007; Godinho et al., 2007; Hirano et al., 2013; Meng et al., 2008; Ralph and Menaker, 1988; Siepka et al., 2007; Yoo et al., 2013) or changing the nature of the interaction at a competitive interface between the repressors and activators (Katada and Sassone-Corsi, 2010; King et al., 1997; Xu et al., 2015). In many ways, these two features of periodicity are two sides of the same coin.
Clearly the intrinsic stability of PER and CRY proteins in a cellular milieu plays a significant role in the timing of the clock. One approach to this idea is to think of how changes in PER and CRY stability affect dynamics at competitive structural interfaces (Figure 6B). For instance, if the dissociation constant measured for interaction between CRY1 and the BMAL1 TAD is ~10 μM (Czarna et al., 2011) and the dissociation constant for the CBP KIX domain is ~2 μM (Xu et al., 2015), stabilizing CRY1 is likely to raise the concentration of CRY1 above the threshold necessary for a competitive interaction with the BMAL1 TAD for a longer interval, thus extending the length of the repressive phase of the clock. Likewise, if PER proteins are competing with MLL1 to interact with the CLOCK exon 19 region, then changing the rate of nuclear import is a means of raising the effective concentration of PER and changing the dynamic of the competition at this interface.
Indeed, experiments in which CRY is exogenously expressed in a cell-based rescue assay may help to demonstrate some of these principles. We observed that increasing the amount of Cry rescue vector used in our transfection has a substantial effect on the period of Per2-Luc rhythms (Rosensweig et al., 2018). Smaller amounts of plasmid resulted in longer periodicity, while larger amounts resulted in shorter periodicity. However, it should be noted that Cry1 and Cry2 each generated periodicities within a range consistent with their intrinsic properties. One way of interpreting these data is to assume that CRY1 and CRY2 have innate characteristics that generate specific periods within a circadian system. By transfecting more or less plasmid, we are altering the accumulation dynamics within the system. Within a certain range of CLOCK/BMAL1 concentrations, increasing the number of copies of Cry should increase the rate at which CRY accumulates, artificially pushing CRY levels above a critical threshold necessary for feedback repression more quickly. In principle, the different ranges of periodicity generated by Cry1 and Cry2 reflect an intrinsic difference in the nature of the competitive interactions of both CRYs. Although the two CRYs are likely to accumulate at roughly the same rate (especially at higher concentrations of plasmid), differences in on and off rates could easily generate the intrinsic periodicities observed in these assays.
Perhaps the most important finding for circadian biology in the last decade from a structural biology perspective is how densely populated the molecular clock is by competitive interactions (Figure 6A and B). A carefully calibrated balance in each of these interactions drives both timing and function. Of course, it follows that comprehensive models of timing within the clock will need to integrate these competitive protein interaction dynamics with measurements of protein stability and mobility to gain insight into how the molecules of the clock achieve precision in timing.
Acknowledgments
The writing of this review was supported by NIH grants F31 NS089241 and T32 HL007909 (C.R.) and R01 GM112991, R01 GM111387 and R01 AG045795 (C.B.G.)
Abbreviations
- 8-HDF
8-hydroxy-5-deazaflavin
- AMPK
Adenosine monophosphate-activated protein kinase
- bHLH
basic Helix-Loop-Helix
- β-TrCP
β-transducing repeat-containing protein
- BMAL1
Brain and muscle Arnt-like 1
- CBD
CRY-binding domain
- CBP
CREB-binding protein
- CC Helix
Coiled coil helix
- CHD4
Chromodomain-helicase-DNA-binding protein 4
- CHRONO
Computationally highlighted repressor of the network oscillator
- CIPC
CLOCK-interacting protein, circadian
- CKBD
Casein kinase-binding domain
- CKIδ/ε
Casein kinase 1δ/ε
- CLK
CLOCK
- CPF
Cry/Photolyase Family
- CRY
Cryptochrome
- CUL4
Cullin-4
- CYC
Cycle
- DDB1
DNA damage binding protein 1
- FAD
Flavin adenine dinucleotide
- FBXL3
F-box/LRR-repeat protein 3
- FBXL21
F-box/LRR-repeat protein 21
- GAPVD1
GTPase-activating protein and VPS9 domain-containing protein 1
- GATAD2a
GATA zinc finger domain containing 2A
- GSK3β
Glycogen synthase kinase-3 β
- HAT
Histone acetyltransferase
- HAUSP
Herpes virus associated ubiquitin-specific protease
- HDAC1
Histone deacetylase 1
- HMT
Histone methyltransferase
- HP1γ
Heterochromatin protein 1 homolog gamma
- JARID1a
JumonjiC (JmjC) and ARID domain-containing histone lysine demethylase 1a
- KD
Knockdown
- KI
Knock-in
- KO
Knockout
- LSD1
Lysine-specific demethylase 1
- LUC
Luciferase
- MBD2
Methyl-CpG-binding domain protein 2
- MLL1
Mixed lineage leukemia 1
- MTA2
Metastasis-associated 1 family member 2
- MTHF
Methenyltetrahydrofolate
- NAD
Nicotinamide adenine dinucleotide
- NES
Nuclear export sequence
- NLS
Nuclear localization sequence
- NONO
Non-POU domain-containing octamer-binding protein
- NPAS2
Neuronal PAS domain protein 2
- NuRD
Mi-2-nucleosome remodeling and deacetylase complex
- PAS
PER-ARNT-SIM
- PER
Period
- PHL
Photolyase
- PHR
Photolyase homology region
- PKCα
Protein kinase C α
- PKCγ
Protein kinase C γ
- PSF
Polypyrimidine tract-binding protein-associated-splicing factor
- RACK1
Receptor for activated C kinase-1
- RMSD
Root mean square deviation
- ROR
Retinoic acid-related orphan receptor
- RRE
Rev-Erb/ROR-binding element
- SCF
Skp1-Cul1-F-box
- SCN
Suprachiasmatic nucleus
- SETX
Senataxin
- SNF2H
Sucrose nonfermenting protein 2 homolog
- SUV39H
Suppression of variation 3–9 homolog 1
- TAD
Transactivation domain
- TIM
Timeless
- TRAP150
Thyroid hormone receptor-associated protein-150
- UBE3A
Ubiquitin protein ligase E3A
- WDR5
WD repeat-containing protein 5
- WDR76
WD repeat-containing protein 76
Footnotes
Statement of Competing Financial Interests
All authors declare no competing financial or other interests.
Data Accessibility
N/A
Citations
- Aguilar-Arnal L, Hakim O, Patel VR, Baldi P, Hager GL, and Sassone-Corsi P (2013). Cycles in spatial and temporal chromosomal organization driven by the circadian clock. Nature structural & molecular biology 20, 1206–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Arnal L, Katada S, Orozco-Solis R, and Sassone-Corsi P (2015). NAD(+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nature structural & molecular biology 22, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi M, Okamoto A, Tsuchiya Y, Todo T, Nishida E, and Node K (2014). A positive role for PERIOD in mammalian circadian gene expression. Cell reports 7, 1056–1064. [DOI] [PubMed] [Google Scholar]
- Akashi M, Tsuchiya Y, Yoshino T, and Nishida E (2002). Control of intracellular dynamics of mammalian period proteins by casein kinase I epsilon (CKIepsilon) and CKIdelta in cultured cells. Molecular and cellular biology 22, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht U, Sun ZS, Eichele G, and Lee CC (1997). A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell 91, 1055–1064. [DOI] [PubMed] [Google Scholar]
- Allada R, White NE, So WV, Hall JC, and Rosbash M (1998). A mutant Drosophila homolog of mammalian Clock disrupts circadian rhythms and transcription of period and timeless. Cell 93, 791–804. [DOI] [PubMed] [Google Scholar]
- Anafi RC, Lee Y, Sato TK, Venkataraman A, Ramanathan C, Kavakli IH, Hughes ME, Baggs JE, Growe J, Liu AC, et al. (2014). Machine learning helps identify CHRONO as a circadian clock component. PLoS biology 12, e1001840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annayev Y, Adar S, Chiou YY, Lieb JD, Sancar A, and Ye R (2014). Gene model 129 (Gm129) encodes a novel transcriptional repressor that modulates circadian gene expression. The Journal of biological chemistry 289, 5013–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson BD, Johnson KA, Loros JJ, and Dunlap JC (1994). Negative feedback defining a circadian clock: autoregulation of the clock gene frequency. Science 263, 1578–1584. [DOI] [PubMed] [Google Scholar]
- Aryal RP, Kwak PB, Tamayo AG, Gebert M, Chiu PL, Walz T, and Weitz CJ (2017). Macromolecular Assemblies of the Mammalian Circadian Clock. Molecular cell 67, 770–782 e776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, and Schibler U (2008). SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328. [DOI] [PubMed] [Google Scholar]
- Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, and Weaver DR (2001). Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron 30, 525–536. [DOI] [PubMed] [Google Scholar]
- Bargiello TA, Jackson FR, and Young MW (1984). Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature 312, 752–754. [DOI] [PubMed] [Google Scholar]
- Bargiello TA, and Young MW (1984). Molecular genetics of a biological clock in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 81, 2142–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA, Smith BS, Ma Z, Palnitkar M, Tomchick DR, Machius M, and Deisenhofer J (2004). Structure of the photolyase-like domain of cryptochrome 1 from Arabidopsis thaliana. Proceedings of the National Academy of Sciences of the United States of America 101, 12142–12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Ripperger J, Kadener S, Fleury-Olela F, Vilbois F, Rosbash M, and Schibler U (2005). PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 308, 693–696. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, and Bradfield CA (2000). Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, and Pagano M (2007). SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science 316, 900–904. [DOI] [PubMed] [Google Scholar]
- Chang DC, McWatters HG, Williams JA, Gotter AL, Levine JD, and Reppert SM (2003). Constructing a feedback loop with circadian clock molecules from the silkmoth, Antheraea pernyi. The Journal of biological chemistry 278, 38149–38158. [DOI] [PubMed] [Google Scholar]
- Chaves I, Pokorny R, Byrdin M, Hoang N, Ritz T, Brettel K, Essen LO, van der Horst GT, Batschauer A, and Ahmad M (2011). The cryptochromes: blue light photoreceptors in plants and animals. Annual review of plant biology 62, 335–364. [DOI] [PubMed] [Google Scholar]
- Chaves I, Yagita K, Barnhoorn S, Okamura H, van der Horst GT, and Tamanini F (2006). Functional evolution of the photolyase/cryptochrome protein family: importance of the C terminus of mammalian CRY1 for circadian core oscillator performance. Molecular and cellular biology 26, 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Schirmer A, Lee Y, Lee H, Kumar V, Yoo SH, Takahashi JS, and Lee C (2009). Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Molecular cell 36, 417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, et al. (2012). Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature 485, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarna A, Berndt A, Singh HR, Grudziecki A, Ladurner AG, Timinszky G, Kramer A, and Wolf E (2013). Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell 153, 1394–1405. [DOI] [PubMed] [Google Scholar]
- Czarna A, Breitkreuz H, Mahrenholz CC, Arens J, Strauss HM, and Wolf E (2011). Quantitative analyses of cryptochrome-mBMAL1 interactions: mechanistic insights into the transcriptional regulation of the mammalian circadian clock. The Journal of biological chemistry 286, 22414–22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne JP, Noton E, Lambert CM, Maywood ES, Weaver DR, and Reppert SM (2006). A clock shock: mouse CLOCK is not required for circadian oscillator function. Neuron 50, 465–477. [DOI] [PubMed] [Google Scholar]
- DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, and Panda S (2011). Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science 333, 1881–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi M, Hirayama J, and Sassone-Corsi P (2006). Circadian regulator CLOCK is a histone acetyltransferase. Cell 125, 497–508. [DOI] [PubMed] [Google Scholar]
- Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, and McKnight SL (2003). Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science 301, 379–383. [DOI] [PubMed] [Google Scholar]
- Duong HA, Robles MS, Knutti D, and Weitz CJ (2011). A molecular mechanism for circadian clock negative feedback. Science 332, 1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HA, and Weitz CJ (2014). Temporal orchestration of repressive chromatin modifiers by circadian clock Period complexes. Nature structural & molecular biology 21, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MD, Brancaccio M, Chesham JE, Maywood ES, and Hastings MH (2016). Rhythmic expression of cryptochrome induces the circadian clock of arrhythmic suprachiasmatic nuclei through arginine vasopressin signaling. Proceedings of the National Academy of Sciences of the United States of America 113, 2732–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, and Virshup DM (2005). Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Molecular and cellular biology 25, 2795–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery P, So WV, Kaneko M, Hall JC, and Rosbash M (1998). CRY, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity. Cell 95, 669–679. [DOI] [PubMed] [Google Scholar]
- Etchegaray JP, Lee C, Wade PA, and Reppert SM (2003). Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421, 177–182. [DOI] [PubMed] [Google Scholar]
- Etchegaray JP, Yang X, DeBruyne JP, Peters AH, Weaver DR, Jenuwein T, and Reppert SM (2006). The polycomb group protein EZH2 is required for mammalian circadian clock function. The Journal of biological chemistry 281, 21209–21215. [DOI] [PubMed] [Google Scholar]
- Froy O, Chang DC, and Reppert SM (2002). Redox potential: differential roles in dCRY and mCRY1 functions. Current biology : CB 12, 147–152. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Kojima R, Itoh K, Chang HY, Ye S, Zhuang B, Oji A, Gibo S, Narasimamurthy R, Virshup D, et al. (2018). Two Ck1delta transcripts regulated by m6A methylation code for two antagonistic kinases in the control of the circadian clock. Proceedings of the National Academy of Sciences of the United States of America 115, 5980–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Yoo SH, Lee KJ, Rosensweig C, Takahashi JS, Chen BP, and Green CB (2013). Phosphorylation of the cryptochrome 1 C-terminal tail regulates circadian period length. The Journal of biological chemistry 288, 35277–35286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, and Weitz CJ (1998). Role of the CLOCK protein in the mammalian circadian mechanism. Science 280, 1564–1569. [DOI] [PubMed] [Google Scholar]
- Godinho SI, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, et al. (2007). The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science 316, 897–900. [DOI] [PubMed] [Google Scholar]
- Goriki A, Hatanaka F, Myung J, Kim JK, Yoritaka T, Tanoue S, Abe T, Kiyonari H, Fujimoto K, Kato Y, et al. (2014). A novel protein, CHRONO, functions as a core component of the mammalian circadian clock. PLoS biology 12, e1001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, Glossop N, Zhang YQ, Fukada Y, and Meng QJ (2014). The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic acids research 42, 5765–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EA Jr., Staknis D, and Weitz CJ (1999). Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science 286, 768–771. [DOI] [PubMed] [Google Scholar]
- Guillaumond F, Dardente H, Giguere V, and Cermakian N (2005). Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. Journal of biological rhythms 20, 391–403. [DOI] [PubMed] [Google Scholar]
- Gustafson CL, Parsley NC, Asimgil H, Lee HW, Ahlbach C, Michael AK, Xu H, Williams OL, Davis TL, Liu AC, et al. (2017). A Slow Conformational Switch in the BMAL1 Transactivation Domain Modulates Circadian Rhythms. Molecular cell 66, 447–457 e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson CL, and Partch CL (2015). Emerging models for the molecular basis of mammalian circadian timing. Biochemistry 54, 134–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin PE, Hall JC, and Rosbash M (1990). Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 343, 536–540. [DOI] [PubMed] [Google Scholar]
- Hennig S, Strauss HM, Vanselow K, Yildiz O, Schulze S, Arens J, Kramer A, and Wolf E (2009). Structural and functional analyses of PAS domain interactions of the clock proteins Drosophila PERIOD and mouse PERIOD2. PLoS biology 7, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, and Fukada Y (2013). FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 152, 1106–1118. [DOI] [PubMed] [Google Scholar]
- Hirayama J, Nakamura H, Ishikawa T, Kobayashi Y, and Todo T (2003). Functional and structural analyses of cryptochrome. Vertebrate CRY regions responsible for interaction with the CLOCK:BMAL1 heterodimer and its nuclear localization. The Journal of biological chemistry 278, 35620–35628. [DOI] [PubMed] [Google Scholar]
- Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, and Sassone-Corsi P (2007). CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 450, 1086–1090. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh DK, Brenner DA, et al. (2012). Identification of small molecule activators of cryptochrome. Science 337, 1094–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu YZ, Pray-Grant M, Perdew GH, and Bradfield CA (1997). Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. The Journal of biological chemistry 272, 8581–8593. [DOI] [PubMed] [Google Scholar]
- Hogenesch JB, Gu YZ, Jain S, and Bradfield CA (1998). The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proceedings of the National Academy of Sciences of the United States of America 95, 5474–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Su L, Pei J, Grishin NV, and Zhang H (2017). Crystal Structure of the CLOCK Transactivation Domain Exon19 in Complex with a Repressor. Structure 25, 1187–1194 e1183. [DOI] [PubMed] [Google Scholar]
- Hsu DS, Zhao X, Zhao S, Kazantsev A, Wang RP, Todo T, Wei YF, and Sancar A (1996). Putative human blue-light photoreceptors hCRY1 and hCRY2 are flavoproteins. Biochemistry 35, 13871–13877. [DOI] [PubMed] [Google Scholar]
- Huang N, Chelliah Y, Shan Y, Taylor CA, Yoo SH, Partch C, Green CB, Zhang H, and Takahashi JS (2012). Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 337, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram KK, Kutowoi A, Wurm Y, Shoemaker D, Meier R, and Bloch G (2012). The molecular clockwork of the fire ant Solenopsis invicta. PloS one 7, e45715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, and Reppert SM (1999). A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell 96, 57–68. [DOI] [PubMed] [Google Scholar]
- Kaiser TS, Poehn B, Szkiba D, Preussner M, Sedlazeck FJ, Zrim A, Neumann T, Nguyen LT, Betancourt AJ, Hummel T, et al. (2016). The genomic basis of circadian and circalunar timing adaptations in a midge. Nature 540, 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada S, and Sassone-Corsi P (2010). The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nature structural & molecular biology 17, 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavakli IH, Baris I, Tardu M, Gul S, Oner H, Cal S, Bulut S, Yarparvar D, Berkel C, Ustaoglu P, et al. (2017). The Photolyase/Cryptochrome Family of Proteins as DNA Repair Enzymes and Transcriptional Repressors. Photochemistry and photobiology 93, 93–103. [DOI] [PubMed] [Google Scholar]
- Khan SK, Xu H, Ukai-Tadenuma M, Burton B, Wang Y, Ueda HR, and Liu AC (2012). Identification of a novel cryptochrome differentiating domain required for feedback repression in circadian clock function. The Journal of biological chemistry 287, 25917–25926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kwak PB, Gebert M, Duong HA, and Weitz CJ (2015). Purification and analysis of PERIOD protein complexes of the mammalian circadian clock. Methods in enzymology 551, 197–210. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kwak PB, and Weitz CJ (2014). Specificity in circadian clock feedback from targeted reconstitution of the NuRD corepressor. Molecular cell 56, 738–748. [DOI] [PubMed] [Google Scholar]
- King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, et al. (1997). Positional cloning of the mouse circadian clock gene. Cell 89, 641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyohara YB, Tagao S, Tamanini F, Morita A, Sugisawa Y, Yasuda M, Yamanaka I, Ueda HR, van der Horst GT, Kondo T, et al. (2006). The BMAL1 C terminus regulates the circadian transcription feedback loop. Proceedings of the National Academy of Sciences of the United States of America 103, 10074–10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Kanno S, Smit B, van der Horst GT, Takao M, and Yasui A (1998). Characterization of photolyase/blue-light receptor homologs in mouse and human cells. Nucleic acids research 26, 5086–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Zheng X, and Sehgal A (2006). JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science 312, 1809–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka RJ, and Benzer S (1971). Clock mutants of Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America 68, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan B, Levine JD, Lynch MK, Dowse HB, Funes P, Hall JC, Hardin PE, and Dryer SE (2001). A new role for cryptochrome in a Drosophila circadian oscillator. Nature 411, 313–317. [DOI] [PubMed] [Google Scholar]
- Kucera N, Schmalen I, Hennig S, Ollinger R, Strauss HM, Grudziecki A, Wieczorek C, Kramer A, and Wolf E (2012). Unwinding the differences of the mammalian PERIOD clock proteins from crystal structure to cellular function. Proceedings of the National Academy of Sciences of the United States of America 109, 3311–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, and Reppert SM (1999). mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205. [DOI] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, et al. (2009). AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 326, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande-Diner L, Boyault C, Kim JY, and Weitz CJ (2013). A positive feedback loop links circadian clock factor CLOCK-BMAL1 to the basic transcriptional machinery. Proceedings of the National Academy of Sciences of the United States of America 110, 16021–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrondo LF, Olivares-Yanez C, Baker CL, Loros JJ, and Dunlap JC (2015). Circadian rhythms. Decoupling circadian clock protein turnover from circadian period determination. Science 347, 1257277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, and Reppert SM (2001). Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107, 855–867. [DOI] [PubMed] [Google Scholar]
- Lee C, Weaver DR, and Reppert SM (2004). Direct association between mouse PERIOD and CKIepsilon is critical for a functioning circadian clock. Molecular and cellular biology 24, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Cho E, Kang DH, Jeong EH, Chen Z, Yoo SH, and Kim EY (2016). Pacemaker-neuron-dependent disturbance of the molecular clockwork by a Drosophila CLOCK mutant homologous to the mouse Clock mutation. Proceedings of the National Academy of Sciences of the United States of America 113, E4904–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Jang AR, Francey LJ, Sehgal A, and Hogenesch JB (2015). KPNB1 mediates PER/CRY nuclear translocation and circadian clock function. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xiong W, and Zhang EE (2016). The ratio of intracellular CRY proteins determines the clock period length. Biochemical and biophysical research communications 472, 531–538. [DOI] [PubMed] [Google Scholar]
- Liu AC, Tran HG, Zhang EE, Priest AA, Welsh DK, and Kay SA (2008). Redundant function of REV-ERBalpha and beta and non-essential role for Bmal1 cycling in transcriptional regulation of intracellular circadian rhythms. PLoS genetics 4, e1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, Buhr ED, Singer O, Meeker K, Verma IM, et al. (2007). Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell 129, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, and Takahashi JS (2000). Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EV, Baggs JE, Geskes JM, Hogenesch JB, and Green CB (2009). Generation of a novel allelic series of cryptochrome mutants via mutagenesis reveals residues involved in protein-protein interaction and CRY2-specific repression. Molecular and cellular biology 29, 5465–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mees A, Klar T, Gnau P, Hennecke U, Eker AP, Carell T, and Essen LO (2004). Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 306, 1789–1793. [DOI] [PubMed] [Google Scholar]
- Menet JS, Pescatore S, and Rosbash M (2014). CLOCK:BMAL1 is a pioneer-like transcription factor. Genes & development 28, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng QJ, Logunova L, Maywood ES, Gallego M, Lebiecki J, Brown TM, Sladek M, Semikhodskii AS, Glossop NR, Piggins HD, et al. (2008). Setting clock speed in mammals: the CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron 58, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael AK, Fribourgh JL, Chelliah Y, Sandate CR, Hura GL, Schneidman-Duhovny D, Tripathi SM, Takahashi JS, and Partch CL (2017). Formation of a repressive complex in the mammalian circadian clock is mediated by the secondary pocket of CRY1. Proceedings of the National Academy of Sciences of the United States of America 114, 1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, Boo K, Kim D, Han DH, Choe HK, Kim CR, Sun W, Kim H, Kim K, Lee H, et al. (2014). Phosphorylation of LSD1 by PKCalpha is crucial for circadian rhythmicity and phase resetting. Molecular cell 53, 791–805. [DOI] [PubMed] [Google Scholar]
- Nangle S, Xing W, and Zheng N (2013). Crystal structure of mammalian cryptochrome in complex with a small molecule competitor of its ubiquitin ligase. Cell research 23, 1417–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangle SN, Rosensweig C, Koike N, Tei H, Takahashi JS, Green CB, and Zheng N (2014). Molecular assembly of the period-cryptochrome circadian transcriptional repressor complex. eLife 3, e03674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimamurthy R, Hunt SR, Lu Y, Fustin JM, Okamura H, Partch CL, Forger DB, Kim JK, and Virshup DM (2018). CK1delta/epsilon protein kinase primes the PER2 circadian phosphoswitch. Proceedings of the National Academy of Sciences of the United States of America 115, 5986–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ode KL, Ukai H, Susaki EA, Narumi R, Matsumoto K, Hara J, Koide N, Abe T, Kanemaki MT, Kiyonari H, et al. (2017). Knockout-Rescue Embryonic Stem Cell-Derived Mouse Reveals Circadian-Period Control by Quality and Quantity of CRY1. Molecular cell 65, 176–190. [DOI] [PubMed] [Google Scholar]
- Ollinger R, Korge S, Korte T, Koller B, Herrmann A, and Kramer A (2014). Dynamics of the circadian clock protein PERIOD2 in living cells. Journal of cell science 127, 4322–4328. [DOI] [PubMed] [Google Scholar]
- Ozber N, Baris I, Tatlici G, Gur I, Kilinc S, Unal EB, and Kavakli IH (2010). Identification of two amino acids in the C-terminal domain of mouse CRY2 essential for PER2 interaction. BMC molecular biology 11, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, and Sancar A (2003). Purification and properties of human blue-light photoreceptor cryptochrome 2. Biochemistry 42, 2926–2932. [DOI] [PubMed] [Google Scholar]
- Padmanabhan K, Robles MS, Westerling T, and Weitz CJ (2012). Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science 337, 599–602. [DOI] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, and Hogenesch JB (2002). Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109, 307–320. [DOI] [PubMed] [Google Scholar]
- Papp SJ, Huber AL, Jordan SD, Kriebs A, Nguyen M, Moresco JJ, Yates JR, and Lamia KA (2015). DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Kim ST, Sancar A, and Deisenhofer J (1995). Crystal structure of DNA photolyase from Escherichia coli. Science 268, 1866–1872. [DOI] [PubMed] [Google Scholar]
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, and Schibler U (2002). The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260. [DOI] [PubMed] [Google Scholar]
- Ralph MR, Foster RG, Davis FC, and Menaker M (1990). Transplanted suprachiasmatic nucleus determines circadian period. Science 247, 975–978. [DOI] [PubMed] [Google Scholar]
- Ralph MR, and Menaker M (1988). A mutation of the circadian system in golden hamsters. Science 241, 1225–1227. [DOI] [PubMed] [Google Scholar]
- Reddy P, Zehring WA, Wheeler DA, Pirrotta V, Hadfield C, Hall JC, and Rosbash M (1984). Molecular analysis of the period locus in Drosophila melanogaster and identification of a transcript involved in biological rhythms. Cell 38, 701–710. [DOI] [PubMed] [Google Scholar]
- Reick M, Garcia JA, Dudley C, and McKnight SL (2001). NPAS2: an analog of clock operative in the mammalian forebrain. Science 293, 506–509. [DOI] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic acids research 42, W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles MS, Boyault C, Knutti D, Padmanabhan K, and Weitz CJ (2010). Identification of RACK1 and protein kinase Calpha as integral components of the mammalian circadian clock. Science 327, 463–466. [DOI] [PubMed] [Google Scholar]
- Rosensweig C, Reynolds KA, Gao P, Laothamatas I, Shan Y, Ranganathan R, Takahashi JS, and Green CB (2018). An evolutionary hotspot defines functional differences between CRYPTOCHROMES. Nature communications 9, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin EB, Shemesh Y, Cohen M, Elgavish S, Robertson HM, and Bloch G (2006). Molecular and phylogenetic analyses reveal mammalian-like clockwork in the honey bee (Apis mellifera) and shed new light on the molecular evolution of the circadian clock. Genome research 16, 1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutila JE, Suri V, Le M, So WV, Rosbash M, and Hall JC (1998). CYCLE is a second bHLH-PAS clock protein essential for circadian rhythmicity and transcription of Drosophila period and timeless. Cell 93, 805–814. [DOI] [PubMed] [Google Scholar]
- Sahar S, Zocchi L, Kinoshita C, Borrelli E, and Sassone-Corsi P (2010). Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PloS one 5, e8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakida Y, Miyamoto Y, Nagoshi E, Akashi M, Nakamura TJ, Mamine T, Kasahara M, Minami Y, Yoneda Y, and Takumi T (2005). Importin alpha/beta mediates nuclear transport of a mammalian circadian clock component, mCRY2, together with mPER2, through a bipartite nuclear localization signal. The Journal of biological chemistry 280, 13272–13278. [DOI] [PubMed] [Google Scholar]
- Sanada K, Harada Y, Sakai M, Todo T, and Fukada Y (2004). Serine phosphorylation of mCRY1 and mCRY2 by mitogen-activated protein kinase. Genes to cells : devoted to molecular & cellular mechanisms 9, 697–708. [DOI] [PubMed] [Google Scholar]
- Sangoram AM, Saez L, Antoch MP, Gekakis N, Staknis D, Whiteley A, Fruechte EM, Vitaterna MH, Shimomura K, King DP, et al. (1998). Mammalian circadian autoregulatory loop: a timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron 21, 1101–1113. [DOI] [PubMed] [Google Scholar]
- Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, Naik KA, FitzGerald GA, Kay SA, and Hogenesch JB (2004). A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 43, 527–537. [DOI] [PubMed] [Google Scholar]
- Sato TK, Yamada RG, Ukai H, Baggs JE, Miraglia LJ, Kobayashi TJ, Welsh DK, Kay SA, Ueda HR, and Hogenesch JB (2006). Feedback repression is required for mammalian circadian clock function. Nature genetics 38, 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalen I, Reischl S, Wallach T, Klemz R, Grudziecki A, Prabu JR, Benda C, Kramer A, and Wolf E (2014). Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell 157, 1203–1215. [DOI] [PubMed] [Google Scholar]
- Sehgal A, Rothenfluh-Hilfiker A, Hunter-Ensor M, Chen Y, Myers MP, and Young MW (1995). Rhythmic expression of timeless: a basis for promoting circadian cycles in period gene autoregulation. Science 270, 808–810. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Jin X, Lee C, Reppert SM, and Weaver DR (2000a). Targeted disruption of the mPer3 gene: subtle effects on circadian clock function. Molecular and cellular biology 20, 6269–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, et al. (2000b). Interacting molecular loops in the mammalian circadian clock. Science 288, 1013–1019. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF Jr., and Reppert SM (1997). Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron 19, 1261–1269. [DOI] [PubMed] [Google Scholar]
- Shi SQ, Bichell TJ, Ihrie RA, and Johnson CH (2015). Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Current biology : CB 25, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, et al. (1997). Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell 91, 1043–1053. [DOI] [PubMed] [Google Scholar]
- Shinohara Y, Koyama YM, Ukai-Tadenuma M, Hirokawa T, Kikuchi M, Yamada RG, Ukai H, Fujishima H, Umehara T, Tainaka K, et al. (2017). Temperature-Sensitive Substrate and Product Binding Underlie Temperature-Compensated Phosphorylation in the Clock. Molecular cell 67, 783–798 e720. [DOI] [PubMed] [Google Scholar]
- Shirogane T, Jin J, Ang XL, and Harper JW (2005). SCFβ-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. The Journal of biological chemistry 280, 26863–26872. [DOI] [PubMed] [Google Scholar]
- Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, and Takahashi JS (2007). Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell 129, 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler ML, Kuropatwinski KK, Schumer M, and Antoch MP (2009). A serine cluster mediates BMAL1-dependent CLOCK phosphorylation and degradation. Cell cycle 8, 4138–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanewsky R, Kaneko M, Emery P, Beretta B, Wager-Smith K, Kay SA, Rosbash M, and Hall JC (1998). The cryb mutation identifies cryptochrome as a circadian photoreceptor in Drosophila. Cell 95, 681–692. [DOI] [PubMed] [Google Scholar]
- Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, and Weitz CJ (2002). Extensive and divergent circadian gene expression in liver and heart. Nature 417, 78–83. [DOI] [PubMed] [Google Scholar]
- Stratmann M, Suter DM, Molina N, Naef F, and Schibler U (2012). Circadian Dbp transcription relies on highly dynamic BMAL1-CLOCK interaction with E boxes and requires the proteasome. Molecular cell 48, 277–287. [DOI] [PubMed] [Google Scholar]
- Sun ZS, Albrecht U, Zhuchenko O, Bailey J, Eichele G, and Lee CC (1997). RIGUI, a putative mammalian ortholog of the Drosophila period gene. Cell 90, 1003–1011. [DOI] [PubMed] [Google Scholar]
- Takahata S, Ozaki T, Mimura J, Kikuchi Y, Sogawa K, and Fujii-Kuriyama Y (2000). Transactivation mechanisms of mouse clock transcription factors, mClock and mArnt3. Genes to cells : devoted to molecular & cellular mechanisms 5, 739–747. [DOI] [PubMed] [Google Scholar]
- Tamada T, Kitadokoro K, Higuchi Y, Inaka K, Yasui A, de Ruiter PE, Eker AP, and Miki K (1997). Crystal structure of DNA photolyase from Anacystis nidulans. Nature structural biology 4, 887–891. [DOI] [PubMed] [Google Scholar]
- Tamayo AG, Duong HA, Robles MS, Mann M, and Weitz CJ (2015). Histone monoubiquitination by Clock-Bmal1 complex marks Per1 and Per2 genes for circadian feedback. Nature structural & molecular biology 22, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tei H, Okamura H, Shigeyoshi Y, Fukuhara C, Ozawa R, Hirose M, and Sakaki Y (1997). Circadian oscillation of a mammalian homologue of the Drosophila period gene. Nature 389, 512–516. [DOI] [PubMed] [Google Scholar]
- Thresher RJ, Vitaterna MH, Miyamoto Y, Kazantsev A, Hsu DS, Petit C, Selby CP, Dawut L, Smithies O, Takahashi JS, et al. (1998). Role of mouse cryptochrome blue-light photoreceptor in circadian photoresponses. Science 282, 1490–1494. [DOI] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, and Fu YH (2001). An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 291, 1040–1043. [DOI] [PubMed] [Google Scholar]
- Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, et al. (2002). A transcription factor response element for gene expression during circadian night. Nature 418, 534–539. [DOI] [PubMed] [Google Scholar]
- Ueda HR, Hayashi S, Chen W, Sano M, Machida M, Shigeyoshi Y, Iino M, and Hashimoto S (2005). System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nature genetics 37, 187–192. [DOI] [PubMed] [Google Scholar]
- Ukai-Tadenuma M, Yamada RG, Xu H, Ripperger JA, Liu AC, and Ueda HR (2011). Delay in feedback repression by cryptochrome 1 is required for circadian clock function. Cell 144, 268–281. [DOI] [PubMed] [Google Scholar]
- van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, et al. (1999). Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 398, 627–630. [DOI] [PubMed] [Google Scholar]
- Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, and Takahashi JS (1994). Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 264, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, Hitomi K, Thresher RJ, Ishikawa T, Miyazaki J, et al. (1999). Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proceedings of the National Academy of Sciences of the United States of America 96, 12114–12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wu Y, Li L, and Su XD (2013). Intermolecular recognition revealed by the complex structure of human CLOCK-BMAL1 basic helix-loop-helix domains with E-box DNA. Cell research 23, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, and Zhao K (2009). Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W, Busino L, Hinds TR, Marionni ST, Saifee NH, Bush MF, Pagano M, and Zheng N (2013). SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature 496, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Gustafson CL, Sammons PJ, Khan SK, Parsley NC, Ramanathan C, Lee HW, Liu AC, and Partch CL (2015). Cryptochrome 1 regulates the circadian clock through dynamic interactions with the BMAL1 C terminus. Nature structural & molecular biology 22, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, and Fu YH (2005). Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644. [DOI] [PubMed] [Google Scholar]
- Yagita K, Tamanini F, Yasuda M, Hoeijmakers JH, van der Horst GT, and Okamura H (2002). Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. The EMBO journal 21, 1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Selby CP, Chiou YY, Ozkan-Dagliyan I, Gaddameedhi S, and Sancar A (2014). Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes & development 28, 1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Selby CP, Ozturk N, Annayev Y, and Sancar A (2011). Biochemical analysis of the canonical model for the mammalian circadian clock. The Journal of biological chemistry 286, 25891–25902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Ko CH, Lowrey PL, Buhr ED, Song EJ, Chang S, Yoo OJ, Yamazaki S, Lee C, and Takahashi JS (2005). A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proceedings of the National Academy of Sciences of the United States of America 102, 2608–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, Kornblum I, Kumar V, Koike N, Xu M, et al. (2013). Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 152, 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitane H, Takao T, Satomi Y, Du NH, Okano T, and Fukada Y (2009). Roles of CLOCK phosphorylation in suppression of E-box-dependent transcription. Molecular and cellular biology 29, 3675–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Metterville D, Briscoe AD, and Reppert SM (2007). Insect cryptochromes: gene duplication and loss define diverse ways to construct insect circadian clocks. Molecular biology and evolution 24, 948–955. [DOI] [PubMed] [Google Scholar]
- Zehring WA, Wheeler DA, Reddy P, Konopka RJ, Kyriacou CP, Rosbash M, and Hall JC (1984). P-element transformation with period locus DNA restores rhythmicity to mutant, arrhythmic Drosophila melanogaster. Cell 39, 369–376. [DOI] [PubMed] [Google Scholar]
- Zhang L, Abraham D, Lin ST, Oster H, Eichele G, Fu YH, and Ptacek LJ (2012). PKCgamma participates in food entrainment by regulating BMAL1. Proceedings of the National Academy of Sciences of the United States of America 109, 20679–20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Markert MJ, Groves SC, Hardin PE, and Merlin C (2017). Vertebrate-like CRYPTOCHROME 2 from monarch regulates circadian transcription via independent repression of CLOCK and BMAL1 activity. Proceedings of the National Academy of Sciences of the United States of America 114, E7516–E7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WN, Malinin N, Yang FC, Staknis D, Gekakis N, Maier B, Reischl S, Kramer A, and Weitz CJ (2007). CIPC is a mammalian circadian clock protein without invertebrate homologues. Nature cell biology 9, 268–275. [DOI] [PubMed] [Google Scholar]
- Zhou M, Kim JK, Eng GW, Forger DB, and Virshup DM (2015). A Period2 Phosphoswitch Regulates and Temperature Compensates Circadian Period. Molecular cell 60, 77–88. [DOI] [PubMed] [Google Scholar]
- Zoltowski BD, Vaidya AT, Top D, Widom J, Young MW, and Crane BR (2011). Structure of full-length Drosophila cryptochrome. Nature 480, 396–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Shearman LP, Weaver DR, and Reppert SM (1998). Three period homologs in mammals: differential light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of brain. Neuron 20, 1103–1110. [DOI] [PubMed] [Google Scholar]