

Abstract
Production of 1-butene, a major monomer in polymer industry, is dominated by homogeneous protocols via ethylene dimerization. Homogeneous catalysts can achieve high selectivity but require large amounts of activators and solvents, and exhibit poor recyclability; in turn, heterogeneous systems are robust but lack selectivity. Here we show how the precise engineering of metal–organic frameworks (MOFs) holds promise for a sustainable process. The key to the (Ru)HKUST-1 MOF activity is the intrapore reactant condensation that enhances ethylene dimerization with high selectivity (> 99% 1-butene) and high stability (> 120 h) in the absence of activators and solvents. According to spectroscopy, kinetics, and modeling, the engineering of defective nodes via controlled thermal approaches rules the activity, while intrapore ethylene condensation accounts for selectivity and stability. The combination of well-defined actives sites with the concentration effect arising from condensation regimes paves the way toward the development of robust MOF catalysts for diverse gas-phase reactions.
Subject terms: Catalytic mechanisms, Heterogeneous catalysis, Metal-organic frameworks
The search for robust heterogeneous catalysts for the production of linear α-olefins is still a challenge as they do not show well-defined sites and lack the high selectivity reported for homogeneous counterparts. Here the authors show how the precise engineering of metal–organic frameworks (MOFs) holds promise for a sustainable process.
Introduction
Linear α-olefins, derived from light alkene oligomerization reactions, are broadly used as lubricants and co-monomers in polymer synthesis, and represent one of few examples for which homogeneous catalysts1 are still employed in bulk chemical production. Transition-metal compounds selectively convert light alkenes into industrially-relevant products2, but their limited recyclability and excessive use of solvent and activators present environmental and economic limitations. The search for robust heterogeneous catalysts is still a challenge as they do not show well-defined sites and lack the high selectivity reported for homogeneous counterparts.
Metal–organic frameworks (MOFs)3 hold promise to get the best ingredients from both worlds, i.e., selective and well-defined catalytic sites distributed on porous solids4–6. While typical pristine MOFs are hardly reactive during ethylene dimerization, recent advances in defect-engineering synthetic methods enable tailoring a broad range of lattice defects that enhance catalytic activity7–11. However, there are several challenges to render these materials into practice in large-scale processes. For instance, remarkable ethylene dimerization turnovers were obtained using Ni-containing MOFs12–20, but they still require large amounts of activators (up to 500 equivalents per active metal) to generate the active species. Moreover, the Ni-MOF catalysts undergo fast deactivation12. Given the narrow channels in MOFs, we propose the use of intrapore reactant condensation21 to provide catalyst stability during gas-phase reactions at mild temperatures.
Here, we present heterogeneous catalysis and defect synthesis methods for (Ru)HKUST-1 that allow controlled defect engineering and present unique activity, selectivity, and stability in ethylene dimerization. The thermal engineering of defects yields catalytically active species with remarkable activity in the absence of activators, high selectivity to 1-butene (>99% selectivity), and long-time stability (>120 h) under intrapore ethylene condensation regime. We prove that active Ru–H sites created through economic and versatile thermal approaches behave the same as those prepared via conventional ligand-engineered methods. Kinetic, spectroscopic, and theoretical methods provide new insights on the nature and concentration of catalytic sites and demonstrate the kinetic consequences of intrapore reactant condensation on the stability of MOF catalysts. The exquisite control over well-defined active sites combined with intrapore liquid regime opens new research opportunities to develop heterogeneous porous systems that work at wider temperature ranges than those under liquid phase.
Results
Ligand-engineered defective MOF catalysts
In order to tailor an active and stable MOF catalyst for ethylene dimerization, we chose the (Ru)-HKUST-1 material, which is amenable to include lattice defective sites without compromising the framework structure (Fig. 1)7,22. Previously, defective (Ru)HKUST-1, prepared via controlled in-synthesis incorporation of coordinatively deficient pyridine-3,5-dicarboxylate ligands, led to the formation of robust metal–hydride species upon H2 exposure at 150 °C23,24. Inspired by these results, we propose a new experimental set up where such defective MOFs can efficiently catalyze ethylene dimerization in the absence of activators.
Fig. 1.
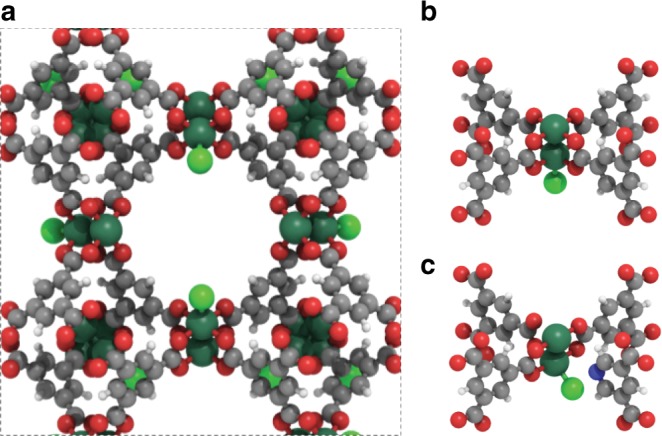
Structure of (Ru)HKUST-1 metal–organic framework. a Periodic structure of pristine (Ru)HKUST-1 with a pore size of 1.5 nm. b Paddle-wheel node of pristine (Ru)HKUST-1. c Paddle-wheel node of defective (Ru)HKUST-1. (Ru = dark green, Cl = green, O = red, N = blue, C = grey, H = white)
Here we use direct catalytic evidences to learn on ethylene dimerization site requirements in (Ru)HKUST-1 MOFs containing 25% of pyridine-3,5-dicarboxylate linkers, namely MOFL25. Figure 2a shows ethylene dimerization turnover frequencies (TOFs) vs. time on stream at 50 °C using different pre-treated MOFL25. As discussed later, we consider one active Ru atom per defective node to calculate TOF values. The reaction pressure (4.2 MPa) and temperature (50 °C) conditions in Fig. 2a lead to the remarkable stabilization of active species, facilitated by the intrapore condensation of ethylene reactants within MOFL25, as discussed in next sections. After N2 treatment at 200 °C to remove labile adsorbates (purple line), MOFL25 exhibits dimerization activity after an induction period with 99% selectivity to 1-butene (Fig. 2a). Slow induction periods reflect the limited capacity of ethylene to form active Ru–H species at dimerization conditions (50 °C). Interestingly, further exposure to H2 at 150 °C (blue line) produces a catalyst with initial rates one order of magnitude higher than those found previously24, while maintaining high selectivity (Supplementary Fig. 7). The catalytic differences in Fig. 2a are attributed to the facile formation of Ru–H species in ligand-engineered open metal sites under H2 atmosphere at 150 °C (vide infra). The generation of Ru–H species is optimized at 150 °C in H2 without involving the degradation of the porous framework (see N2 physisorption in Supplementary Fig. 2). Higher pre-treatment temperatures (200 °C in H2) lead to complete MOF degradation into metallic Ru particles (Supplementary Fig. 4). The catalytic role of Ru–H species is supported by quenching under oxidizing environments (O2 at 150 °C, orange line in Fig. 2a), leading to inactive Ru species and induction periods in contact with ethylene. The results in Fig. 2a show compelling evidences on the role of Ru–H species in ligand-engineered defective MOFs to achieve stable alkene dimerization turnovers in the absence of co-catalysts and solvents.
Fig. 2.

Catalytic activity and formation of the Ru–H active site in MOFL25. a Ethylene dimerization TOFs at 50 °C and 4.2 MPa ethylene after activation of MOFL25 in N2 at 200 °C, in N2 at 200 °C + H2 at 150 °C, and in N2 at 200 °C + H2 at 150 °C + O2 at 150 °C. Active Ru corresponds to one metal atom per defective node. b IR spectra after subsequent treatments for MOFL25: N2 at 200 °C, H2 at 150 °C, and CO at 30 °C. All spectra are shown after N2 flushing
To evaluate the (Ru)HKUST-1 pre-treatment effects in ethylene dimerization (Fig. 2a), we turn to IR spectroscopy. Figure 2b shows in-situ IR spectroscopic data of MOFL25 after successive gas pre-treatments. The two bands at 2059 and 1978 cm−1 after N2 and H2 (purple and blue lines) are characteristic of Ru–H species, as reported in literature23. Periodic density functional theory (DFT) calculations evidence the facile formation of Ru–H species via a heterolytic H–H bond breaking process under H2 atmosphere (Supplementary Fig. 27)25–27. After CO exposure (green line), MOFL25 exhibits two additional intense stretching vibrations at 2082 and 2015 cm−1, which correspond to single carbonyl species bonded to Ru2+ and Ru+δ. The latter were previously identified as partially reduced Ru (0 < δ < 2)23,28. Moreover, a C = O stretching band appears at 1656 cm−1, which is assigned to Ru(II)–formyl species29 coming from CO reduction via Ru–H. Even after N2 purging at 30 °C, these CO-adsorbed species are stable, suggesting the remarkable binding strength of CO to Ru species. At 30 °C, IR spectra rule out the presence of either sym-/asymmetric Ru(CO)2 gem-dicarbonyl or Ru2(CO) bridged-carbonyl species, as compared to −80 °C23,30. DFT-computed C = O frequencies of 1950–2050 cm−1 for CO and 1600–1700 cm−1 for formyl are in line with experimental data (Supplementary Fig. 28). MOFL25 treated at 200 °C in N2 also shows the bands that correspond to Ru–H species, and their presence is also confirmed by the initial dimerization activity and the induction period in contact with ethylene at 50 °C (Fig. 2a). These evidences suggest that active Ru–H species within the defects of (Ru)HKUST-1 can also be formed via thermal approaches in contact with N2 as discussed in the next section.
Thermal-engineered defective MOF catalysts
Based on the above-mentioned catalytic and spectroscopic evidences for MOFL25 (Fig. 2), together with a potential partial decarboxylation of linkers at high temperature31, we next examine the generation of defects and Ru–H sites via a controlled thermal approach. We start from pristine (Ru)HKUST-1 without any pyridine-3,5-dicarboxylate linkers, namely MOFL0, and evaluate thermal pre-activation effects on ethylene dimerization activity (Fig. 3a). MOFL0 treated at 150 °C in N2 shows an induction period in contact with ethylene at 50 °C and 4.2 MPa. After thermal pre-activation at 200 °C in N2, MOFL0 exhibits greater initial rates than when treated at 150 °C, while maintaining selectivity. Temperature effects on ethylene dimerization activity are optimized for MOFL0 pre-treated at 300 °C, as the initial rates abruptly decrease when MOFL0 is pre-treated at 400 °C in N2. Such catalytic differences inspired us to perform analogous pre-treatment thermal experiments on MOFL0 via in-situ TGA-MS, XRD, and IR. TGA analyses (Fig. 3b) quantify the number of generated defects (blue line), XRD determines the MOF structural stability (Fig. 3c), and IR experiments (Fig. 3d) evaluate the nature of the resulting defective site as a function of temperature. The proposed mechanism for the thermal generation of defects is supported by MS (Fig. 3b), which identifies the composition of released species.
Fig. 3.

Thermal treatment effects on activity, surface species, and structure of MOFL0. a Ethylene dimerization rates at 50 °C and 4.2 MPa ethylene after MOFL0 thermal pre-treatment. b Thermogravimetric analysis (TGA) coupled to mass-spectrometry (MS) for MOFL0. c XRD data for MOFL0 in N2. d DRIFT spectra of MOFL0 in N2
N2-treatments up to 200 °C incorporate only ca. 5% of defects in MOFL0 (purple area, Fig. 3b) and induce carboxylate protonation, as indicated by the significant appearance of a new band in IR at 1698 cm−1 (purple line, Fig. 3d), assigned to the stretching of free carboxylic acids. Heating from 200 to 300 °C increases the defect concentration up to 38% on MOFL0 (orange area, Fig. 3b). In this range, a progressive attenuation of IR signals occurs for both symmetric carboxylates [νsym(COO)] and free carboxylic acids due to further release of CO2, while signals attributed to phenyl rings [ν(C = C)] remain constant (orange line, Fig. 3d). Moreover, intense IR signals appear between 2100–1900 cm−1, which are assigned to Ru–CO and Ru–H stretching bands. Similar bands were observed in MOFL25 at 200 °C (Fig. 2b), which contains a combination of ligand-engineered (25.0%) and thermal-engineered (8.2%) defects (Supplementary Fig. 13). The presence of Ru–CO and Ru–H species at defective Ru sites is supported by the simultaneous release of CO2 and CO at 300 °C (Fig. 3b), which can originate from the catalytic rupture of formic acid32. These evidences suggest a consequent reduction of Ru atoms with temperature to compensate the charge over the MOF node, which is consistent with the release of HCl (Fig. 3b) and elemental analysis (Supplementary Table 2). A similar metal reduction via dehalogenation was reported for Fe-containing MOFs33. XPS data (Supplementary Fig. 20) are also consistent with the reduction of Ru atoms and the dehalogenation process when MOFL0 is treated above 200 °C in N2. Nevertheless, in-situ XRD data indicate the remarkable structural stability of the thermal-engineered MOFL0 structure and the absence of Ru nanoparticles up to 300 °C (orange line, Fig. 3c).
N2-treatments at 400 °C (green area, Fig. 3b) lead to nearly complete MOF decarboxylation (up to 95%) and the consequent reduction of Ru–CO and Ru–H stretching bands (green line, Fig. 3d). These effects are consistent with the sintering of metallic Ru nanoclusters into larger crystalline domains at 400 °C, as supported by the appearance of a broad signal centered at 42 theta degree in XRD (green line, Fig. 3c). XPS data also support the advanced degree of Ru reduction (Supplementary Table 3) at 400 °C, which leads to the fusion of phenyl rings into a carbonaceous matrix31.
The differences between the number of engineered defects and active Ru–H species explain the activity trends observed in Fig. 3a as a function of thermal pre-treatment in N2. Compared to the more limited in-synthesis ligand engineering procedures, the thermal approach reveals a general and straightforward route to create active sites from as-synthesized MOFL0 without H2 activation. The active sites are the same in both cases but the thermal treatment, when properly controlled, is a much more appealing technique to design defect sites. Next, we will compare this versatile procedure to conventional ligand engineering approaches as a function of defect content.
Role of defects in selective ethylene dimerization
In order to evaluate the influence of the type and number of defects on catalytic activity, we prepared and tested a series of ligand- and thermal-engineered MOFs. For ligand-engineered samples (% of defects): MOFL10 (20.4%), MOFL25 (33.3%), and MOFL50 (51.0%). For thermal-engineered samples (% of defects): MOFL0−200 (5.0%), MOFL0−250 (13.4%), MOFL0−300 (38%), MOFL0−350 (71.4%), and MOFL0−400 (95.0%).
Figure 4a, b plot ethylene dimerization TOFs per total Ru in the MOF and per active Ru sites, respectively, as a function of measured defect contents for ligand- (blue) and thermal-engineered (green) MOFs. Figure 4a shows a volcano shape with a maximum TOF regardless of the method employed to create defects. The increase in activity parallels the higher concentration of active sites (the active Ru/total Ru fraction increases), but there is a maximum to the incorporation of active sites without compromising the mechanical stability. When defects are above 40%, partial amorphization for ligand-engineered samples (SBET decreased from 974 m2/g for MOFL25 to 668 m2/g for MOFL50, Supplementary Fig. 1) causes smaller dimerization TOFs. Such decrease in TOFs for thermal-engineered catalysts is also attributed to the aggregation of Ru into larger inactive particles (Fig. 3c). This latter hypothesis is confirmed by testing the catalytic activity of Ru nanoparticles embedded on a carbonaceous matrix prepared at 500 °C under N2 (Supplementary Fig. 24), which show no ethylene dimerization products. Figure 4b displays a plateau at low defect content, suggesting that the nature of active sites in both ligand- and thermal-engineered MOFs is the same and that they behave as single ensemble catalysts. In-situ FTIR data indicate the presence of bound ethyl intermediates when MOF catalysts are exposed to ethylene (Supplementary Fig. 10). The optimized MOFL25 catalyst presents a TOF of ca. 200 h−1, which is comparable to related gas-phase systems under flow conditions: 1614, 25213, and 157012 h−1 (Supplementary Table 4).
Fig. 4.

Effects of ligand- and thermal-engineered defects on catalytic activity. a Turnover frequencies per total Ru as a function of measured defects via ligand (blue) and thermal (green) approaches. b Turnover frequencies per active Ru as a function of measured defects via ligand (blue) and thermal (green) approaches. c Computed reaction mechanism and energy profile for ethylene dimerization via ligand- (L, blue) and thermal-engineered (T, green) defective MOFs
To get insights on the reaction mechanism and selectivity to dimers, we turn to computational modeling at periodic DFT level (see Computational Details). We use a unit cell of ca. 26 × 26 × 26 Å3, which prevents a spurious concentration of defect sites when computing periodic images. Benchmark calculations indicate that PBE-D2 is suitable to describe the electronic structure of (Ru)HKUST-1 (Supplementary Table 5). We computed the ethylene dimerization via a Cossee–Arlman mechanism15,34,35 using a ligand-engineered defective node. Figure 4c shows the model L-1 used in the simulations (Supplementary Fig. 26) and the reaction profile in blue. From the active Ru–H species 3, ethylene binds to Ru forming 5 with an adsorption energy of 0.89 eV. Species 5 undergoes insertion via TS5 with a relative energy barrier of 0.26 eV. The resulting ethyl species binds a second ethylene as in 7, which inserts into the Ru–C bond via TS7 with an energy barrier of 0.91 eV. Such C–C bond formation appears to be the rate-determining step, where ethyl intermediates are potential resting states of the catalytic cycle in line with in-situ FTIR (Supplementary Fig. 10). The butyl intermediate (8) quickly undergoes β-H elimination through a barrierless process via TS8 to form 1-butene (9). Direct desorption of 1-butene via 10 takes only 1.09 eV; alternatively, at high ethylene pressures, ethylene can directly substitute 1-butene from 9 to 1136. To evaluate entropic contributions, we estimate the relative Gibbs energies for the C–C bond formation step, 7 to TS7, and the alkene desorption process, 9–10. The resulting Gibbs (electronic) energy barriers are 0.91 (0.91) and 0.68 (1.09) eV, respectively, which further supports the C–C bond formation as the rate-determining step of the catalytic cycle. We also studied the reaction mechanism using a thermal-engineered defective node. Figure 4c shows the model used in the calculations T-1 (Supplementary Fig. 26) and the reaction profile in green. Similar energies were observed, with a barrier of 0.93 eV (cf. 0.91 eV) for the C–C bond-forming step and 1.14 eV (cf. 1.09 eV) for alkene desorption. The kinetic resemblance between ligand- and thermal-engineered active sites toward dimerization is in line with previous experimental results (Fig. 4a, b).
The dimerization mechanism in Fig. 4c takes place on one metal atom, which allows defining TOFs per Ru atom per defective node (Figs. 2a and 4b). To further explore the role of the second metal, we prepared a periodic structure with a node containing only one Ru atom (Supplementary Fig. 30) and computed the desorption of 1-butene. Interestingly, we found a significantly higher value of 1.74 eV, compared to 1.09 eV for the bimetallic node. It seems that the second metal plays a key electronic role favoring the alkene desorption. Bader analyses located positive charges of +0.95 |e−| on Ru in the bimetallic model and +1.22 |e−| on Ru in the monometallic model. The higher charge of the latter is in line with the more energy-demanding desorption step. To further highlight the unique nature of the bimetallic active site, we attached mononuclear Ru and Ni species to bipyridine linkers in (Zr)UiO-67-(Bpy)37,38. However, under our activator-free conditions, no dimerization products were experimentally observed. These results explain how the active site controls the dimerization selectivity via: (i) the slower rate of ethylene insertion into Ru–alkyl groups (ca. 0.9 eV via TS7) compared to chain termination (barrierless step via TS8)15 and (ii) facile alkene desorption (ca. 1.1 eV from 9 to 10). The first one is due to the engineered defect (via ligand or thermal approaches), and the second one arises from the bimetallic nature of the MOF node. Such facile desorption is further enhanced in the presence of a solvating environment, as shown below.
Stability of MOF catalysts under intrapore condensation
After characterization and reactivity studies, we next evaluate the stability of these catalysts during ethylene dimerization with respect to reaction conditions. Figure 5a shows ethylene dimerization TOFs for H2-activated MOFL25 as a function of systematic changes in temperature (at constant 4.2 MPa) and ethylene pressure (at constant 50 °C). Stable TOFs are achieved at temperatures below 60 °C (at 4.2 MPa) or pressures above 3.3 MPa (at 50 °C). However, high temperatures (>60 °C, 4.2 MPa) or low pressures (<3.6 MPa, 50 °C) lead to fast deactivation rates. Such deactivation occurs via the irreversible binding of oligomer products in secondary reactions of primary dimer products21, as evidenced by the curved shape of ethylene dimerization rate data at high ethylene conversion (100 °C in Fig. 5a).
Fig. 5.

Stability of MOF catalysts. a Turnover frequencies per active Ru as a function of temperature at 4.2 MPa ethylene and pressure at 50 °C. b First-order deactivation constants, derived from ethylene pressure changes in (a), as a function of ethylene relative saturation pressure (P/Psat) within MOFL25. c Long-term stability during ethylene dimerization at 4.2 MPa ethylene and 50 °C for MOFL25, MOFL10, and MOFL0
Figure 5b shows first-order deactivation constants (kd) as a function of ethylene relative saturation pressures (P/Psat). Deactivation is accurately measured by kd within small changes in conversion for MOFL25. Figure 5b shows a continuous increase in kd values below P/Psat of 0.4, whereas deactivation is undetectable at P/Psat above 0.4. Such reaction conditions and the narrow micropores of MOFL25 (1.5 nm diameter) suggest that active site stabilization is related to the intrapore condensation of ethylene molecules. We use N2 uptakes as surrogate to estimate ethylene intrapore condensation at reaction conditions (Supplementary Fig. 3)21. These analyses indicate that kinetically-relevant pores are liquid-filled at P/Psat above 0.4. Condensation within MOF pores also leads to very high selectivity for C4 among products (>99%) and 1-butene (primary C4 product, Supplementary Fig. 6), as liquid ethylene promotes the desorption of bound alkenes by substitution and prevents the formation of larger oligomers. To support such claim, we estimated the Gibbs energy difference associated with the ligand exchange from 9 to 11 (Fig. 4c) using reported entropies in gas and liquid phases39. The electronic energy difference to exchange 1-butene by ethylene is endothermic by 0.20 eV. The resulting Gibbs energy differences in low-pressure (ethylene in gas phase) and high-pressure (ethylene in liquid phase to model intrapore condensation) are +0.01 and −0.33 eV, respectively. The exoergicity of the process only under intrapore condensation conditions further proves the benefits of our experimental protocol.
Although MOFL25 starts to deactivate at P/Psat below 0.4, it can be regenerated in H2 at 150 °C (Supplementary Fig. 7). Such mild conditions are more feasible than high temperatures and oxidizing environments used in typical catalyst regeneration procedures. This suggests that the regeneration mechanism proceeds through Ru-catalyzed hydrogenation of bound alkenes rather than oligomer-desorption pathways.
The long-term stable rates, enabled by controlled intrapore ethylene condensation, indicate that the crystalline and porous structure of MOFs is maintained during catalytic conditions or regeneration steps. Such enhanced stability is independent of the synthetic protocol to create catalytic sites. Figure 5c shows turnover numbers (TONs) under intrapore liquid ethylene regime for ligand- (MOFL25 and MOFL10 H2-activated at 150 °C) and thermal-engineered (MOFL0 pre-treated at 300 °C in N2) catalysts, with values of 13,000. Our TON results outstand over reported Ru-based MOF catalysts24 and are comparable to the 16,000 reported for state-of-the-art Ni-MOF catalysts12. Both ligand and thermal approaches lead to materials with remarkable stability, showing a mean life of one order of magnitude larger than Ni-MOF systems requiring co-catalysts or organic solvents12.
In summary, we have developed a method for tailoring MOF defects via thermal-based protocols, resulting in defective nodes that behave as well-defined active sites for ethylene dimerization with remarkable activity (TOF of ca. 200 h−1) and high selectivity (99% to 1-butene). Moreover, we demonstrate that working under reactant intrapore condensation regime within thermal-engineered defective MOFs provides outstanding stability (>120 h) to these catalytic systems (TON of 13,000). Such enhanced stability represents a significant advancement in the field of synthesis and performance of porous catalysts for ethylene dimerization reactions. Due to the absence of co-catalysts and solvents, such intrapore condensation effects in narrow-pore MOFs open exciting opportunities for the design of competitive procedures in terms of process intensification, safety, and production costs in other reactions involving gaseous reactants and structured catalysts.
Methods
MOF synthesis
(Ru)HKUST-1 containing 0% (MOFL0), 10% (MOFL10), 25% (MOFL25), and 50% (MOFL50) of ligand-engineered defects were prepared by combining the appropriate mixture of organic ligands 1,3,5-benzenetricarboxylic acid and pyridine-3,5-dicarboxylic acid with Ru precursor [Ru2(CH3COO)4Cl],under solvothermal conditions at 160 °C according to literature23. In a typical synthesis, 1 g of Ru precursor, 600 mg of the selected mixture of organic ligand, 5 mL of acetic acid and 25 mL of H2O were loaded into a 100-mL autoclave and heated at 160 °C for 24 h. The resulting powder was thoroughly washed with H2O in a filtration funnel and purified in a Soxhlet apparatus with MeOH. All the samples were evacuated at 150 °C under vacuum.
Ru precursor [Ru2(CH3COO)4Cl] was prepared by mixing 10 g of RuCl3·xH2O (40 wt.%Ru) and 12 g of LiCl(anhydrous), previously evacuated at 80 °C overnight in a vacuum oven, with 70 mL of acetic anhydride and 350 mL of glacial acetic acid. The mixture was stirred and refluxed for 2–4 days until the solution turned into reddish color. After cooling down, the Ru precursor was collected by filtration and was thoroughly washed with acetone.
Ethylene dimerization reactivity tests
Ethylene dimerization rates to butene products were measured in a high-pressure tubular reactor integrated in a fully-automated lab-scale reaction unit. As-prepared MOF samples (50 mg) were diluted with inert SiO2 (1:20 mass, Davisil-62, Sigma-Aldrich). Activation tests on ligand-engineered MOF samples were carried out in a flow of inert (N2, 2.0 cm3 g−1 s−1, 99.999%, Air Liquide) and oxidizing (O2, 2.0 cm3 g−1 s−1) or reducing environment (H2, 99.9%, 2.0 cm3 g−1 s−1) depending on the defect formation temperature and conditions. Kinetic isotope effects were evaluated using D2 (99%, Air Liquide) as activator instead of H2. Thermal-engineered MOF samples were treated in N2 for 1 h at each reaction temperature. Prior to each catalytic test, the reactor was flushed with N2 before cooling to reaction temperature, initially pressurized in N2 until desired setpoint and ethylene (99.9%, Air Liquide) was introduced at 10–20 mol g−1 h−1. Temperatures were measured with a K-type thermocouple (K-type, Omega) and controlled electronically. Pressure was electronically controlled with a servometer. The concentrations of ethylene and products were measured by flame ionization detection after chromatography separation (Agilent 6890; HP-5 methyl silicone). First-order deactivation constants (kd) were measured from rate values (r) in a specific time interval (t):
| Eq. 1 |
N2 sorption isotherms
MOF samples were analyzed in a Micromeritics ASAP (Accelerated Surface Area and Porosimetry) 2020 System. Samples were weighted into tubes with seal frits and degassed under vacuum (<500 µm Hg) with heating. Samples were initially heated at 150 °C and held for 4 h, and finally cooled to room temperature and backfilled with N2. The samples were re-weighted before analysis. The analysis adsorptive was N2 at 77 K. A multi-point BET surface area was determined from 6 measurements at relative pressures (P / Psat) ranging from 0.05 to 0.30. Single point adsorption total pore volume was measured near saturation pressure (Psat ≈ 770 mmHg). Adsorption average pore width was also calculated. Pore size distribution plot was determined by Horvath-Kawazoe method using the Cylinder Pore Geometry (Saito-Foley) with Cheng-Yang Correction.
X-ray diffraction
XRD was used to study the crystalline structure of the MOF catalyst. XRD patterns were recorded using a Panalytical Empyrean X-ray diffractometer with Cu Kα radiation (λ = 1.54778 Å). The samples were prepared by filling the holder with the dry powder. Crystalline phase stability was investigated using an XRK900 high temperature oven chamber. Sample was first heated in the chamber from 25 to 800 °C with a heating rate of 3 °C/min. Diffraction patterns were measured throughout the whole heat treatment using Cu Kα X-ray radiation with a wavelength of 1.5418 Å and a 2θ range of 4.5°–60°. Each pattern was measured for 4 min using a step size and count time of 2θ = 0.0263° and 147 s/step, respectively.
Fourier Transform Infrared Spectroscopy
In-situ FTIR analyses were carried out in a diffuse reflectance cell (Harrick) using a Bruker Vertex V70 module equipped with a liquid-nitrogen cooled MCT detector. Samples were sieved into fine powders (<30 μm particle size) and treated using same MOF activation procedures as described in main text in N2 or H2. Temperature was controlled with a Harrick module. CO adsorption experiments were carried out using 1% CO in He between −80 and 30 °C, but given the remarkable binding strength of CO onto Ru–H species, the spectra after CO adsorption are shown at 30 °C. Ethylene was also introduced at 50 °C at same reaction conditions but at atmospheric pressure. All IR spectra are measured in N2 to avoid signal-disturbance for gas-phase H2 or CO.
H2-temperature programmed reduction
H2-TPR measurements of MOF catalysts were performed using an AutoChem II 2920 reactor (Micromeritics) equipped with a built-in TCD detector, and with the reactor downstream connected to a benchtop quadrupole mass spectrometer (TA). To mimic the MOF activation procedures as described in manuscript in N2 or H2, 100 mg sample was first degassed at 200 °C for 1 h in He (50 mL/min), and then treated at 150 °C for 1 h with H2 (50 mL/min). The TCD calibration was performed with H2 concentrations varied from 0 to 100% in Ar and a background test with an empty reactor at the same reaction condition was performed to create a baseline.
Thermogravimetric analyses (TGA) and mass spectrometry (MS)
TGA-MS analyses of the MOF catalysts were performed using a TA Q500 unit coupled to a benchtop quadrupole mass spectrometer (TA).
XPS measurements
X-ray Photoelectron Spectroscopy (XPS) was performed using a monochromatized Al Kα source (hν = 1486.6 eV), operated at 225 W, on a Kratos Axis Ultra DLD with a pass energy for narrow scan spectra of 20 eV, corresponding to an instrument resolution of ~600 meV. Survey spectra were collected with a pass energy of 80 eV. Spectral fitting was performed using Casa XPS analysis software. Spectral positions were corrected by shifting the primary C 1 s core level position to 285.0 eV, and curves were fitted with quasi-Voigt lines following Shirley background subtraction.
Computational details
All calculations reported in the manuscript were performed at periodic DFT level using the Vienna Ab-initio Simulation Package (VASP)40,41. The PBE functional42 was used together with Grimme’s D2 dispersion scheme43 and modified parameters for transition metals44. Core electrons were described by projector augmented wave (PAW)45 and valence electrons in plane waves with a kinetic energy cutoff of 450 eV. The Brillouin zone was sampled at the Γ-point through the Monkhorst–Pack method46. Transition states were located with the climbing image nudged elastic band method47. The assessment of the minima and transition states was performed by diagonalizing the numerical Hessian matrix obtained by ±0.015 Å displacements. Selected cluster calculations were computed at the DFT level using Gaussian09 for benchmark purposes (Supplementary Table 5). All inputs and final structures can be found in the ioChem-BD repository48,49.
Supplementary information
Acknowledgements
This work was financially supported by the research fund from Basque Government (IT993–16) and RTI international funds. M.A.O. and N.L. acknowledge BSC-RES for generous computer resources (RES-QCM-2018–2–0020). M.A.O. acknowledges the “Juan de la Cierva–Incorporación” programme from MINECO (IJCI-2016–29762) and the “Beatriu de Pinós” programme from AGAUR (2017-BP-00039).
Author contributions
I.A.T. contributed to dimerization reactions; M.O.B. and I.G. contributed to IR measurements; I.L., M.S., and M. L. contributed to MOF synthesis and characterization (XRD, TGA-MS, and XPS); M.A.O. contributed to DFT computation and N. L. contributed to the manuscript writing. All authors contributed to the design of experiments and modeling.
Data availability
The data that support this article and other findings are available from the corresponding authors upon request. The computational data can be freely accessed through the ioChem-BD repository (10.19061/iochem-bd-1-83).
Competing interests
The authors declare no competing interests.
Footnotes
Journal peer review information: Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors equal contribution: Iker Agirrezabal-Telleria, Ignacio Luz, Manuel A. Ortuño.
Contributor Information
Iker Agirrezabal-Telleria, Email: iker.aguirrezabal@ehu.eus.
Manuel A. Ortuño, Email: mortuno@iciq.es
Mustapha Soukri, Email: msoukri@rti.org.
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-019-10013-6.
References
- 1.Gollwitzer A, Dietel T, Kretschmer WP, Kempe R. A broadly tunable synthesis of linear α-olefins. Nat. Comm. 2017;8:1226. doi: 10.1038/s41467-017-01507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen C. Designing catalysts for olefin polymerization and copolymerization: beyond electronic and steric tuning. Nat. Rev. Chem. 2018;2:6–14. doi: 10.1038/s41570-018-0003-0. [DOI] [Google Scholar]
- 3.Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM. The chemistry and applications of metal-organic frameworks. Science. 2013;341:1230444. doi: 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- 4.Rogge SMJ, et al. Metal–organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 2017;46:3134–3184. doi: 10.1039/C7CS00033B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui X, Li W, Ryabchuk P, Junge K, Beller M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 2018;1:385–397. doi: 10.1038/s41929-018-0090-9. [DOI] [Google Scholar]
- 6.Luz I, Llabres i Xamena FX, Corma A. Bridging homogeneous and heterogeneous catalysis with MOFs: “Click” reactions with Cu-MOF catalysts. J. Catal. 2010;276:134–140. doi: 10.1016/j.jcat.2010.09.010. [DOI] [Google Scholar]
- 7.Fang Z, Bueken B, De Vos DE, Fischer RA. Defect-engineered metal–organic frameworks. Angew. Chem. Int. Ed. 2015;54:7234–7254. doi: 10.1002/anie.201411540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dissegna E, Epp K, Heinz WR, Kieslich G, Fischer RA. Defective metal–organic frameworks. Adv. Mater. 2018;30:1704501. doi: 10.1002/adma.201704501. [DOI] [PubMed] [Google Scholar]
- 9.Canivet J, Vandichel M, Farrusseng D. Origin of highly active metal–organic framework catalysts: defects? Defects!. Dalton Trans. 2016;45:4090–4099. doi: 10.1039/C5DT03522H. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, et al. Impact of synthesis parameters on the formation of defects in HKUST‐1. Eur. J. Inorg. Chem. 2017;5:925–931. doi: 10.1002/ejic.201601239. [DOI] [Google Scholar]
- 11.Sholl DS, Lively RP. Defects in metal–organic frameworks: challenge or opportunity? J. Phys. Chem. Lett. 2015;6:3437–3444. doi: 10.1021/acs.jpclett.5b01135. [DOI] [PubMed] [Google Scholar]
- 12.Madrahimov ST, et al. Gas-phase dimerization of ethylene under mild conditions catalyzed by MOF materials containing (bpy)NiII complexes. ACS Catal. 2015;5:6713–6718. doi: 10.1021/acscatal.5b01604. [DOI] [Google Scholar]
- 13.Li Z. Schweitzer, et al., Sintering-resistant single-site nickel catalyst supported by metal–organic framework. J. Am. Chem. Soc. 2016;138:1977–1982. doi: 10.1021/jacs.5b12515. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, et al. Beyond the active site: Tuning the activity and selectivity of a metal−organic framework-supported Ni catalyst for ethylene dimerization. J. Am. Chem. Soc. 2018;140:11174–11178. doi: 10.1021/jacs.8b06006. [DOI] [PubMed] [Google Scholar]
- 15.Metzger ED, Brozek CK, Comito RJ, Dincă M. Selective dimerization of ethylene to 1-butene with a porous catalyst. ACS Cent. Sci. 2016;2:148–153. doi: 10.1021/acscentsci.6b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji P, et al. Transformation of metal–organic framework secondary building units into hexanuclear Zr-alkyl catalysts for ethylene polymerization. J. Am. Chem. Soc. 2017;139:11325–11328. doi: 10.1021/jacs.7b05761. [DOI] [PubMed] [Google Scholar]
- 17.Metzger ED, Comito RJ, Hendon CH, Dincă M. Mechanism of single-site molecule-like catalytic ethylene dimerization in Ni-MFU-4l. J. Am. Chem. Soc. 2017;139:757–762. doi: 10.1021/jacs.6b10300. [DOI] [PubMed] [Google Scholar]
- 18.Canivet J, Aguado S, Schuurman Y, Farrusseng D. MOF-supported selective ethylene dimerization single-site catalysts through one-pot postsynthetic modification. J. Am. Chem. Soc. 2013;135:4195–4198. doi: 10.1021/ja312120x. [DOI] [PubMed] [Google Scholar]
- 19.Mlinar AN, et al. Selective propene oligomerization with nickel(II)-based metal−organic frameworks. ACS Catal. 2014;4:717–721. doi: 10.1021/cs401189a. [DOI] [Google Scholar]
- 20.Metzger ED, et al. Highly selective heterogeneous ethylene dimerization with a scalable and chemically robust MOF catalyst. ACS Sustain. Chem. Eng. 2019;7:6654–6661. doi: 10.1021/acssuschemeng.8b05703. [DOI] [Google Scholar]
- 21.Agirrezabal-Telleria I, Iglesia E. Stabilization of active, selective, and regenerable Ni-based dimerization catalysts by condensation of ethene within ordered mesopores. J. Catal. 2017;352:505–514. doi: 10.1016/j.jcat.2017.06.025. [DOI] [Google Scholar]
- 22.Marx S, Kleist W, Baiker A. Synthesis, structural properties, and catalytic behavior of Cu-BTC and mixed-linker Cu-BTC-PyDC in the oxidation of benzene derivatives. J. Catal. 2016;281:76–87. doi: 10.1016/j.jcat.2011.04.004. [DOI] [Google Scholar]
- 23.Kozachuk O, et al. Multifunctional, defect-engineered metal–organic frameworks with ruthenium centers: sorption and catalytic properties. Angew. Chem. Int. Ed. 2014;53:7058–7062. doi: 10.1002/anie.201311128. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, et al. Ruthenium metal–organic frameworks with different defect types: Influence on porosity, sorption, and catalytic properties. Chem. Eur. J. 2016;22:14297–14307. doi: 10.1002/chem.201602641. [DOI] [PubMed] [Google Scholar]
- 25.Bullock RM, Helm ML. Molecular electrocatalysts for oxidation of hydrogen using earth-abundant metals: Shoving protons around with proton relays. Acc. Chem. Res. 2015;48:2017–2026. doi: 10.1021/acs.accounts.5b00069. [DOI] [PubMed] [Google Scholar]
- 26.Fiorio JL, López N, Rossi LM. Gold–ligand-catalyzed selective hydrogenation of alkynes into cis-alkenes via H2 heterolytic activation by frustrated Lewis Pairs. ACS Catal. 2017;7:2973–2980. doi: 10.1021/acscatal.6b03441. [DOI] [Google Scholar]
- 27.Fiorio JL, et al. Accessing frustrated Lewis pair chemistry through robust Gold@ N-doped carbon for selective hydrogenation of alkynes. ACS Catal. 2018;8:3516–3524. doi: 10.1021/acscatal.8b00806. [DOI] [Google Scholar]
- 28.Noei H, et al. CO adsorption on a mixed-valence ruthenium metal–organic framework studied by UHV-FTIR spectroscopy and DFT calculations. J. Phys. Chem. C. 2013;117:5658–5666. doi: 10.1021/jp3056366. [DOI] [Google Scholar]
- 29.Smith, G., Cole-Hamilton, D. J., Thornton-Pett, M. & Hursthouse, M. B. The preparation and characterisation of cationic formyl complexes of ruthenium(II), crystal and molecular structure of the complex trans-bis[1,2-bis(diphenylphosphino)ethane-PP’]carbonyl (deuterioformyl)ruthenium(II) hexafluoroantimonate-dichloromethane(1/1). J. Chem. Soc., Dalton Trans. 0, 2501–2507 (1983).
- 30.Matsubu JC, Yang VN, Christopher P. Isolated metal active site concentration and stability control catalytic CO2reduction selectivity. J. Am. Chem. Soc. 2015;137:3076–3084. doi: 10.1021/ja5128133. [DOI] [PubMed] [Google Scholar]
- 31.Lee KJ, Lee JH, Jeoung S, Moon HR. C Transformation of metal–organic frameworks/coordination polymers into functional nanostructured materials: experimental approaches based on mechanistic insights. Acc. Chem. Res. 2017;50:2684–2692. doi: 10.1021/acs.accounts.7b00259. [DOI] [PubMed] [Google Scholar]
- 32.Winter K, Barton D. The thermal decomposition of benzoic acid. Can. J. Chem. 1970;48:3797–3801. doi: 10.1139/v70-641. [DOI] [Google Scholar]
- 33.Yoon JW, et al. Controlled reducibility of a metal–organic framework with coordinatively unsaturated sites for preferential gas sorption. Angew. Chem. Int. Ed. 2010;49:5949–5952. doi: 10.1002/anie.201001230. [DOI] [PubMed] [Google Scholar]
- 34.Bernales V, et al. Computationally guided discovery of a catalytic cobalt-decorated metal–organic framework for ethylene dimerization. J. Phys. Chem. 2016;C120:23576–23583. [Google Scholar]
- 35.Pellizzeri S, et al. Catalytic descriptors and electronic properties of single-site catalysts for ethene dimerization to 1-butene,Catal. Today. 2018;312:149–157. doi: 10.1016/j.cattod.2018.02.024. [DOI] [Google Scholar]
- 36.Ye J, Gagliardi L, Truhlar DG, Cramer CJ. Single Ni atoms and Ni4 clusters have similar catalytic activity for ethylene dimerization. J. Catal. 2017;354:278–286. doi: 10.1016/j.jcat.2017.08.011. [DOI] [Google Scholar]
- 37.Øien S, et al. Probing reactive platinum sites in UiO-67 zirconium metal–organic frameworks. Chem. Mater. 2015;27:1042–1056. doi: 10.1021/cm504362j. [DOI] [Google Scholar]
- 38.Gonzalez MI, Oktawiec J, Long JR. Ethylene oligomerization in metal–organic frameworks bearing nickel(ii) 2,2′-bipyridine complexes. Faraday Discuss. 2017;201:351–367. doi: 10.1039/C7FD00061H. [DOI] [PubMed] [Google Scholar]
- 39.NIST. NIST Chemistry WebBook, SRD 69, https://webbook.nist.gov/chemistry/.
- 40.Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54:11169–11186. doi: 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- 41.Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996;6:15–50. doi: 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- 42.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 43.Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 44.Almora-Barrios N, Carchini G, Błoński P, López N. Costless derivation of dispersion coefficients for metal surfaces. J. Chem. Theory Comput. 2014;10:5002–5009. doi: 10.1021/ct5006467. [DOI] [PubMed] [Google Scholar]
- 45.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59:1758–1775. doi: 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- 46.Monkhorst Hendrik J., Pack James D. Special points for Brillouin-zone integrations. Physical Review B. 1976;13(12):5188–5192. doi: 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- 47.Henkelman G, Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000;113:9978–9985. doi: 10.1063/1.1323224. [DOI] [Google Scholar]
- 48.Alvarez-Moreno M, et al. Managing the computational chemistry big data problem: the ioChem-BD platform. J. Chem. Inf. Model. 2015;55:95–103. doi: 10.1021/ci500593j. [DOI] [PubMed] [Google Scholar]
- 49.Ortuño, M. A. Database ioChem-BD, 10.19061/iochem-bd-1-83.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support this article and other findings are available from the corresponding authors upon request. The computational data can be freely accessed through the ioChem-BD repository (10.19061/iochem-bd-1-83).