

Abstract
Background
Studying patients with rare Mendelian diabetes has uncovered molecular mechanisms regulating β‐cell pathophysiology. Previous studies have shown that Class IIa histone deacetylases (HDAC4, 5, 7, and 9) modulate mammalian pancreatic endocrine cell function and glucose homeostasis.
Methods
We performed exome sequencing in one adolescent nonautoimmune diabetic patient and detected one de novo predicted disease‐causing HDAC4 variant (p.His227Arg). We screened our pediatric diabetes cohort with unknown etiology using Sanger sequencing. In mouse pancreatic β‐cell lines (Min6 and SJ cells), we performed insulin secretion assay and quantitative RT‐PCR to measure the β‐cell function transfected with the detected HDAC4 variants and wild type. We carried out immunostaining and Western blot to investigate if the detected HDAC4 variants affect the cellular translocation and acetylation status of Forkhead box protein O1 (FoxO1) in the pancreatic β‐cells.
Results
We discovered three HDAC4 mutations (p.His227Arg, p.Asp234Asn, and p.Glu374Lys) in unrelated individuals who had nonautoimmune diabetes with various degrees of β‐cell loss. In mouse pancreatic β‐cell lines, we found that these three HDAC4 mutations decrease insulin secretion, down‐regulate β‐cell‐specific transcriptional factors, and cause nuclear exclusion of acetylated FoxO1.
Conclusion
Mutations in HDAC4 disrupt the deacetylation of FoxO1, subsequently decrease the β‐cell function including insulin secretion, resulting in diabetes.
Keywords: diabetes, FoxO1, HDAC4 mutations, pancreatic β‐cells
1. INTRODUCTION
About 3% of diabetic patients involve single‐gene mutations (Mendelian) that may also cause type 2 diabetes (Yang & Chan, 2016). More than twenty genes highly expressed in pancreatic β‐cells have been identified within these monogenic subtypes (Alkorta‐Aranburu et al., 2014). Recently, two national surveys revealed that most patients with monogenic diabetes are likely to be unrecognized and misdiagnosed as type 1 or type 2 diabetes (Delvecchio et al., 2017; Johansson et al., 2017). Genetic diagnosis leads to improved treatment, better prediction of disease prognosis and progression, genetic counseling, and possibly prevention. The benefit of a molecular genetic diagnosis has been shown in the patients with monogenic diabetes caused by a mutation in the ABCC8, KCNJ11, HNF1A, or HNF4A; all these patients were sensitive to sulfonylureas, which greatly improved glycemic control and quality of life (Hattersley & Patel, 2017). However, patients with a mutation in GCK typically do not require pharmacological intervention (Ajjan & Owen, 2014). Therefore, identification of novel disease‐related loci may provide further opportunities to derive new drug targets.
Histone deacetylases (HDACs) have a broad impact on the development of human disease by regulating histone modification and gene transcription (Mathias, Guise, & Cristea, 2015; Mielcarek, Zielonka, Carnemolla, Marcinkowski, & Guidez, 2015). Class II HDAC4, 5, 7, and 9 modulate endocrine cell function and glucose homeostasis in skeletal muscles, adipose tissue, and liver (Daneshpajooh et al., 2017; Lenoir et al., 2011; Mathias et al., 2015; Mihaylova et al., 2011). In addition, HDAC4 was also found to be a key regulator controlling the pancreatic β/δ lineage during embryogenesis (Lenoir et al., 2011). Makinistoglu et al. found that in osteoblast‐specific Hdac4 gene‐deleted (/) mice, HDAC4 regulates not only the spatial learning and memory, male fertility, and appetite, but also glucose metabolism through its expression in osteoblasts. They observed that the insulin content and β‐cell area in the pancreas, as well as insulin levels and insulin secretion in the circulation blood, were significantly decreased in Hdac4osb / compared with the control littermates (Makinistoglu & Karsenty, 2015), indicating that HDAC4 regulates insulin content, secretion, and sensitivity through direct or indirect pathways. In humans, HDAC4 was found to be expressed in the pancreas as well, and associated with type 2 diabetes (Rani et al., 2017).
We report three pediatric hyperglycemic patients harboring individual HDAC4 (OMIM: 605314) mutations. These HDAC4 variants decreased the insulin secretion and down‐regulated the key β‐cells transcriptional factors of pancreatic β‐cells. Furthermore, deacetylation of FoxO1 was inhibited by these HDAC4 mutations resulting in FoxO1 acetylation and nuclear exporting, therefore decreasing β‐cell functions.
2. PATIENTS AND METHODS
2.1. Ethical compliance
The Charité committee on human subjects’ research approved the study (EA‐No EA2/054/11) and written informed consent was obtained.
2.2. Patients
All individuals with hyperglycemia tested negative for β‐cell autoantibodies and developed diabetes before the age of 18 years. They were recruited from Charité and international diabetes centers. We excluded mutations in known genes causing monogenic diabetes (GCK, HNF4A, HNF1A, HNF1B, ABCC8, KCNJ11, and INS) by Sanger sequencing.
2.3. Variants discovery with exome sequencing and Sanger sequencing
We conducted exome sequencing in one patient and his healthy parents and analyzed the sequencing results with our reported pipeline (Kuhnen et al., 2014) for rare de novo variants (Table S1) against the available databases using the Agilent SureSelect Human All Exon Kit (Agilent SureSelect v4, 50 Mb) and next‐generation sequencing (Hiseq, Illumina, USA). We confirmed the HDAC4 (NG_009235.1) mutation (p.H227R) by Sanger sequencing only in this patient, but not in his parents and other 200 healthy controls (Applied Biosystems, USA). In addition, through screening our diabetic cohort of 94 patients with the unknown origin for HDAC4 mutations (Table S2), we detected two other individual heterozygous HDAC4 mutations in two unrelated families (p.D234N & p.E374K).
2.4. Mutagenesis and transfection
The human Flag‐tagged HDAC4 cDNA (pcDNA3 N‐Flag) was kindly provided by Prof. Jens Fielitz of Charite. We sequenced the cDNA clones before the experiments, confirming the HDAC4 cDNA and the tagged Flag sequences. We performed mutagenesis with the primers (Table S2) designed for all the three mutations (p.H227R, p.D234N, p.E374K) according to the manufacturer's instruction (QuikChange II Site‐Directed Mutagenesis Kit, Agilent Technologies, USA). We extracted plasmid DNA using Qiagen kits: QIAprep® Spin Miniprep Kit (minipreps) and QIAfilterTM Plasmid Maxi Kit (Maxipreps) according to the manufacturer's instruction (QIAGEN, Germany). We transfected the HDAC4 constructs and the empty vector into the SJ pancreatic β‐cells (Jia et al., 2015) using AmaxaTM Cell Line NucleofectorTM Kit V and AmaxaTM NucleofectorTM 2b device with transfection efficiency of 80%–90% (Lonza, Switzerland, Figure S1a), and transfected the HDAC4 constructs into MIN6 cells with lipofectamine 2000 reagents with transfection efficiency of 70% or more (#11668027, Thermofisher, USA, Figure S1b). All the constructs were screened with Sanger sequencing to ensure of the only pointed mutations before further investigation.
2.5. Insulin secretion assay
The glucose‐sensitive mouse β‐cell line SJ β‐cells were cultured and insulin‐secretion assay was performed as reported earlier (Jia et al., 2015). In brief, insulin secreted into medium and from whole cell lysates was quantified with Mouse High Range Insulin ELISA kit, according to the manufacturer's protocol (Alpco, USA). Cells were incubated for 30 min with low (3.3 mmol/L) or high (16.7 mmol/L) glucose concentration. We normalized secreted insulin by the total cellular protein. All the values were expressed in relation to cells transfected with wild‐type human HDAC4.
2.6. Quantitative RT‐PCR
We checked the mRNA expression levels of key markers for pancreatic β‐cells using SYBR® Green chemistry (Applied Biosystems, USA) with the according primers (Table S3). Key factors for β‐cells function and maintenance include Pdx1, Neurod1, Foxa2, Hnf1α, Hnf1β, Hnf4α, Nkx6.1, MafA, FoxO1, Scl2a2, Abcc8, Kcnj11, Ins1, Ins2, and Gck.
2.7. Immunoblotting (IB), immunofluorescence (IF), and immunoprecipitation (IP)
Immunoblotting and immunofluorescence staining were carried out as previously described (Jing et al., 2011). Immunoprecipitation was performed using Dynabeads Protein A Immunoprecipitation Kit according to the manufacturer's protocol (#10006D, Thermofisher, USA). Antibodies used in this study: HDAC4 (#7628, Cell signaling, Germany), FoxO1 (#2880, Cell signaling, Germany), Pdx1 (sc‐14664, Santa Cruz, USA), acetylated lysine (#9441, Cell signaling Germany), α‐Tubulin (#T6199, Sigma, USA), and total histone H3 (#4499, Cell signaling, Germany) (Table S4).
We used GraphPad Prism 5.0 software (GraphPad Software). Differences between three or more groups were assessed by 1‐way ANOVA with Dunnett's correction. p < 0.05 were considered significant.
3. RESULTS
3.1. Patients with defective insulin secretion harbor HDAC4 mutations
Our first patient developed a large pubic abscess at the age of 15 years. Hyperglycemia and elevated HbA1c were noticed (Figure 1a, left and Table 1). He received multiple daily insulin injections but discontinued insulin after an intended weight loss (Figure 1a, left). Insulin was then reinstituted after an increase in HbA1C. At the age of 19 years, he skipped insulin injections and developed ketoacidosis (Figure 1a, left and Table 1).
Figure 1.
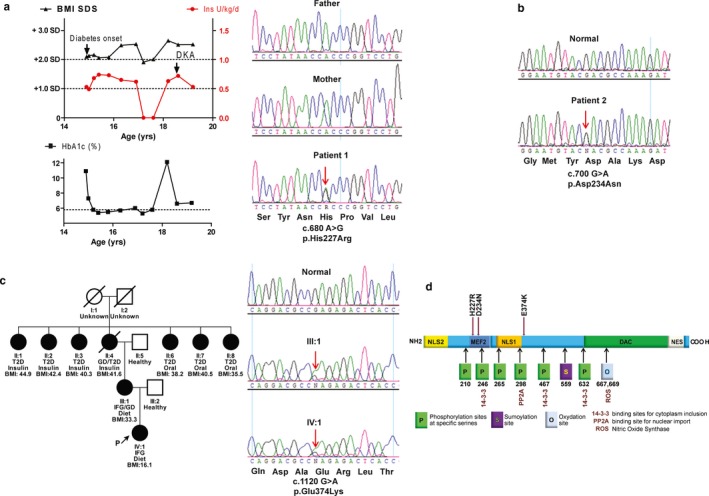
Mutations in HDAC4 are present in pediatric hyperglycemic patients. a. The disease progress of the first patient and the de novo HDAC4 mutation (p.H227R) found in the affected patient. DKA: diabetic ketoacidosis. b. Sporadic patient 2 carried one heterozygous mutation (p.D234N) in HDAC4. c. The studied Turkish family presented as early onset of diabetes (age 30–40 years) and obesity carried one heterozygous mutation (p.E374K) in HDAC4. Solid symbols illustrate affected individuals, open symbols illustrate unaffected individuals. Squares illustrate male subjects and circles illustrate female subjects; a diagonal line through a symbol represents a deceased individual; arrow indicates the index patient. Under the symbols: diagnosis, treatment, and body mass index (BMI) of the affected individuals were shown vertically. T2D: type 2 diabetes, GD: gestational diabetes, Oral: oral drug administration, IFG: impaired fasting glucose d. Schematic representation of the HDAC4 protein domains and its established binding sites (MEF2s), nuclear localization signal (NLS1, NLS2) in the N‐terminal part; a highly conserved deacetylase domain, a nuclear export signal (NES) at the C‐terminal end, three 14‐3‐3 binding sites, and other regulatory sites (phosphorylation, Sumoylation or oxidation sites, binding sites for nuclear import, as well as nitric oxide synthase). The locations and character of the HDAC4 mutations, modified from Mielcarek et al (Mielcarek et al., 2015)
Table 1.
Clinical characteristics of patients with heterozygous HDAC4 mutations
| Patient ID | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Gender | Male | Male | Female |
| Ethnicity | German | German | Turkish |
| Family history | Parents healthy | Unknown | Mother diabetes (diet), grandmother mother side diabetes (insulin) |
| Diabetes onset (years) | 14.9 | 8.6 | 6.3 (IFG) |
| Initial glucose (mg/dl) | 293 (random) | >400 (random) | 110 (fasting) |
| Initial HbA1c (%) | 10.4 | 11.0 | 5.9 |
| HOMA‐IR | n.a. | n.a. | 2.6 (elevated for prepubertal) |
| BMI (kg/m2) | 30.1 | 18.2 | 16.1 |
| BMI‐SDS | +2.3 | +0.6 | 0.48 |
| Antibodiesa | Negative | Negative | Negative |
| Diabetes treatment | Initial MDI insulin, off insulin age 17.2–18.4, continued NPH insulin alone | Insulin (pump), no remission | Diet |
| g.Pos (Hg19) | 2:240078401 T>C | 2:240078381 C>T | 2:240056115 C>T |
| c.posb | c.680A>G | c. 700 G>A | c. 1120 G>A |
| Zygosity | Heterozygous | Heterozygous | Heterozygous |
| Ref.SNP (MAF) | Novel | rs549858743 (0.0002)c | rs370963321 (3.13e−5) |
| Function change | p.H227R | p.D234N | p.E374K |
| Predictiond | Disease causing | Disease causing | Disease causing |
AA, amino acid: MAF, minor allele frequency from ExAc database; NA, not available; IFG, impaired fasting glucose; g.Pos, genomic position; c.Pos, coding position.
Antibodies tested: IA‐2, GAD65, IAA.
It is novel in the European population.
Classified as pathogenic in Polyphen, SIFT, Mutation Taster, GERP++, and LRT.
We performed exome sequencing in the patient and his healthy parents. From the 34 de novo heterozygous variants, eight variants were functionally predicted to be disease causing by multiple in‐silico predictive algorithms (Table S1). We focused on HDAC4 due to its specific expression pattern and roles in pancreatic islet development (Lenoir et al., 2011), gluconeogenesis, and insulin resistance (Mathias et al., 2015; Mihaylova et al., 2011). We confirmed the novel heterozygous HDAC4 mutation solely in the patient (Hg19, chr2: 240,078,401, T > C, H227R > p.H227R, novel in the gnomAD database, Figure 1a, right, and Table 1), but not in his parents and in additional 200 healthy controls. The mutation was not observed in 138,496 alleles in the gnomAD database and is predicted to probably damage the protein structure, function, protein–protein interaction, or to interfere with the conservation with multiple lines of computational algorithms (Table 1). Therefore, this variant is likely pathogenic to the glucose metabolism according to the American College of Medical Genetics and Genomics (ACMG) guideline of the sequence variants (Richards et al., 2015). After screening our cohort of 94 patients for HDAC4 mutations (Table S2), we identified two more HDAC4 mutations in two unrelated hyperglycemic individuals (Figure 1b,c), these two variants were not detected in the 200 healthy individuals either.
The second patient had hyperglycemia (> 400 mg/dl) and a highly elevated HbA1c level (Table 1) at the age of 8.6 years. Multiple daily insulin injections were begun. He carried an HDAC4 mutation (Hg19: chr2: 240078381, G>A. p.H234N, rs549858743, MAF in the European population: 0/126708 in the gnomAD database, Figure 1b), which was not found in any other patients of our cohort. Unfortunately, his parents were not available for genetic testing.
The third patient came from a large Türkish kindred and was investigated because of high diabetes prevalence in this family and her persistent fasting hyperglycemia. She exhibited slightly increased fasting glucose (110–120 mg/dl) and HbA1c levels when she was 6 years old (Table 1). Her now 33‐year‐old mother was obese (BMI: 33.3 kg/m2), had increased fasting glucose (110–120 mg/dl) since puberty, gestational diabetes during pregnancy, but was managed since then with diet alone (Figure 1c). The maternal grandmother had gestational diabetes that progressed to type 2 diabetes at the age of 32 years. She had required insulin and died at the age of 49 years from myocardial infarction. Overall, six sisters of the maternal grandmother were diagnosed with type 2 diabetes at ages between 30 and 40 years, of whom three had been treated with insulin (for 3–10 years) and three were treated with oral antihyperglycemic drugs. All were obese (BMI: 35–45 kg/m2). We found one heterozygous HDAC4 mutation in this family (Figure 1c and Table 1), which is relatively rare in the European population (Hg 19: chr2: 240056115 G>A, E374K>p.E374K, rs370963321, MAF in the European population: 1/27,424 in the gnomAD database).
All three detected variants localize at the N‐terminal region of the HDAC4 protein, which harbors cleavage and phosphorylation sites (Mielcarek et al., 2015), suggesting this region as an important regulatory HDAC4 domain (Figure 1d).
3.2. HDAC4 mutations disrupt insulin secretion and down‐regulate transcription factors
We transfected wild‐type and the three mutated HDAC4 constructs into glucose‐sensitive mouse SJ β‐cells (Jia et al., 2015). Insulin secretion was significantly decreased by the three HDAC4 mutations compared to wild‐type HDAC4 in both low (3.3 mmol/L) and high glucose (16.7 mmol/L) levels (Figure 2a). To examine the effects of each of the HDAC4 mutations on the expression of key factors in β‐cells (Table S3), we performed quantitative RT‐PCR and found that expression of FoxO1, Pdx1, Neurod1, and Gck was significantly down‐regulated by each of the three HDAC4 mutations, compared to wild‐type HDAC4 (Figure 2b). For Hnf4a and Foxa2, down‐regulation could only be achieved by the two HDAC4 p.H227R and p.D234N mutations, but not by the p.E374K mutation, corresponding to the milder phenotypes in the third patient’s family. The regulation of the transcription factors of FoxO1, Pdx1, Neurod1, and glucokinase, Gck, by the HDAC4 mutations were repeatedly detected irrespective of transfection times (48 and 72 hr) or cell batches.
Figure 2.
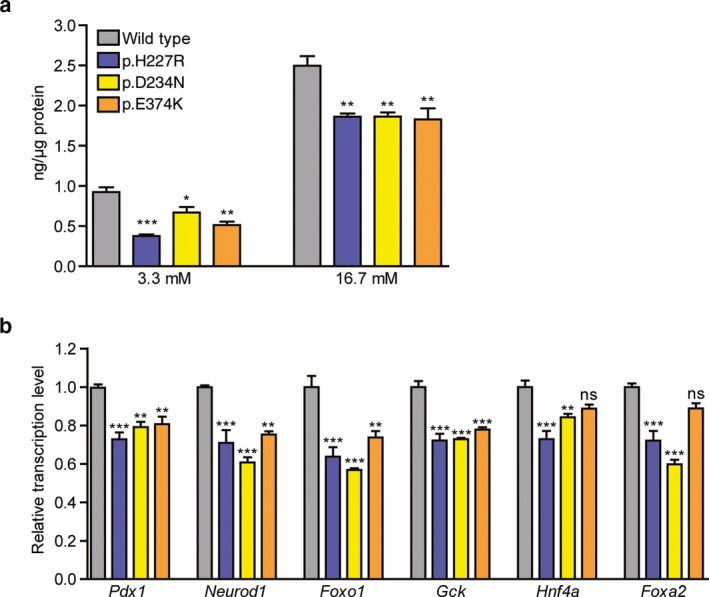
HDAC4 mutations impair β‐cell function. a. Insulin secretion in mouse SJ β‐cells was suppressed by transfection with three mutated HDAC4 at both low (3.3 mmol/L) and high glucose levels (16.7 mmol/L) compared to that in the cells transfected with wild‐type HDAC4. Technical replications =4, biological replications =4. b. Pancreatic β‐cell‐specific transcriptional and regulatory factors were down‐regulated by the mutated HDAC4. Statistical analyses were performed using GraphPad Prism 5.0 software. *: p < 0.05, **:p < 0.01, ***: p < 0.001, ns: not significant
3.3. HDAC4 mutations inhibit nuclear FoxO1 translocation
FoxO1 is translocated into the nuclear when deacetylated by SIRT1, induces the NeuroD1 and MafA expression and thus protects against pancreatic β failure (Kitamura et al., 2005). To investigate whether or not cellular translocation of FoxO1 is critically influenced by our diabetes‐associated HDAC4 mutations, we transfected wild‐type and three mutated HDAC4s (p.H227R, p.D234N, p.E374K) into mouse MIN6 β‐cells and examined FoxO1 with immunostaining. We found that the three HDAC4 mutations promoted both HDAC4 and FoxO1 nuclear exclusion, compared to wild‐type HDAC4 (Figure 3a). To further validate the FoxO1 cellular translocation, we separated cytosol proteins from the nuclear in MIN6 cells. Subsequently, immunoblotting specifically demonstrated that all three HDAC4 mutations prevented translocation of FoxO1 from a cytoplasmic into a nuclear fraction in the mouse β‐cells (Figure 3b). The experiments were replicated with different transfection time as well as with different batches of MIN6 and SJ β‐cell lines (Figure S2a,b), respectively. However, the PDX1 cellular location was not affected by the HDAC4 mutations in pancreatic β‐cells (Figure S2c).
Figure 3.
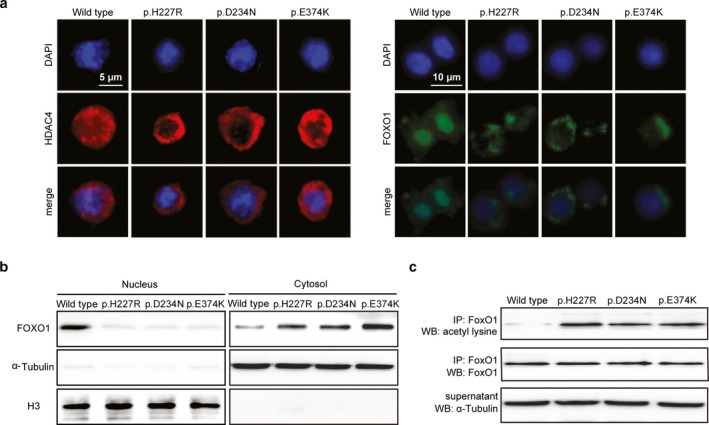
HDAC4 mutations export FoxO1 from nuclear to cytosol through increasing levels of acetylation in Min6 cells. a. Immunostaining of DAPI (blue), HDAC4(red), and FoxO1 (green) in MIN6 cells that were transfected with wild‐type (WT) or three mutated HDAC4 (p.H227R, p.D234N, and p.E374K). Both HDAC4 (left) and FoxO1 (right) were translocated into the cell cytosol in the mutated HDAC4 transfected cells compared to the wild type of HDAC4 transfected cells. b. Immunoblotting with antibodies against FoxO1, α‐Tubulin, and Histone 3 (H3) of lysates separately extracted from nuclear (left) and cytosol (right) of MIN6 cells that were transfected with wild‐type (WT) or three mutated HDAC4 (p.H227R, p.D234N and p.E374K). c. Immunoblotting with antibodies against acetylated‐lysine FoxO1, and α‐Tubulin using anti‐FoxO1 immunoprecipitation lysates of MIN6 cells that are transfected with wild‐type (WT) or three mutated HDAC4 (p.H227R, p.D234N, and p.E374K)
3.4. HDAC4 mutations specifically fail to deacetylate FoxO1 in pancreatic β‐cells
Acetylated FoxO1 is primarily cytoplasmic, when fully deacetylated, it lingers in the nuclear and protects β‐cell function (Kim‐Muller et al., 2016). To investigate whether or not the HDAC4 mutations fail to deacetylate FoxO1, we performed immunoprecipitation with specific antibodies against FoxO1 and measured acetylated‐lysine level by western blot analysis. Interestingly, we observed that acetylated FoxO1 dramatically increased if cells were exposed to diabetes‐associated HDAC4 mutations, compared to wild‐type HDAC4, while there was no much change in total FoxO1 protein level (Figure 3c). These results indicate that FoxO1 is a deacetylation target of HDAC4 and diabetes‐associated HDAC4 mutations show a selective impaired ability to deacetylate FoxO1 in pancreatic β‐cells.
4. DISCUSSION
We detected a de novo HDAC4 p.H227R variant by exome sequencing in a diabetic child and afterward detected two young hyperglycemic individuals harboring independent HDAC4 variants (p.D234N, p.E374K). Though the latter two variants have been shown in the population of Asian in both gnomAD and ExAC databases (p.D234N: 13/18870 in East Asian, 17/277220 in all populations; p.E374K: 139/30660 in South Asian; 149/242078 in all populations), the frequencies of which are quite rare in the European population (In non‐Finnish European D234N: 0/126708; p.E374K: 4/109696), which are very likely due to population heterogeneity or the possibility of incomplete penetrance. The incomplete penetrance for severe Mendelian childhood disorders is likely more common than previously believed, even with homozygous variants in the candidate genes with strong confidence for the relevant disorders (Chen et al., 2016). This state‐of‐affair could be due to the combination of factors, such as expression patterns of normal alleles, epigenetic modification, genetic drift, and specific population genetic variant modifiers, or environmental modifiers (Cooper, Krawczak, Polychronakos, Tyler‐Smith, & Kehrer‐Sawatzki, 2013).
The utility of publicly available databases such as gnomAD or ExAC as controls has limitations since both these databases contain many type 2 diabetes cohorts (http://gnomad.broadinstitute.org/about). Most cases of monogenic diabetes were often misdiagnosed as having type 2 or type 1 diabetes (Delvecchio et al., 2017; Johansson et al., 2017). Furthermore, Mendelian diabetes mutations were previously thought to be highly penetrant, but were found to have reduced or incomplete penetrance, and therefore can be identified more frequently in age‐matched normoglycemic individuals compared with other highly penetrant mutations (Patel et al., 2017). The reduced penetrance of the mutation is important to genetic counseling of at‐risk relatives undergoing predictive tests because the chance of presenting with diabetes is no longer based simply on the odds of inheriting the mutation. Not all heterozygous persons will develop diabetes, and those that do are likely to have a later onset. Therefore, individuals harboring the specific mutation could be encouraged to perform preventive strategies such as healthier lifestyle. We have a similar scenario of our third hyperglycemia family. The index child showed slight hyperglycemia; her mother and mother‐side grandmothers all showed either gestational diabetes and/or early‐onset type 2 diabetes. Even though the HDAC4 p.E374K variant in this family has a relatively high allele frequency of the South Asian, the allele frequency in European is quite rare, indicating both population heterogeneity and the incomplete penetrance of this variant to the hyperglycemia.
Our functional analysis revealed that the three detected HDAC4 variants could disrupt the mature β‐cells to secrete insulin in both statuses of basic and high glucose concentration. The decreased β‐cell function was due to the down‐regulation of the vital transcriptional or glucose transport factors by the HDAC4 variants. So far, there are only a few studies focusing on the association for HDAC4 and the function of the pancreas and/or islets (Lenoir et al., 2011; Makinistoglu & Karsenty, 2015). During embryogenesis, overexpression of HDAC4 and HDAC5 showed a decreased pool of insulin‐producing β‐cells and somatostatin‐producing δ‐cells (Lenoir et al., 2011). In contrast, the osteoblast‐specific Hdac4 knock‐out mice showed decreased β‐cell area as well as insulin contents in the pancreas, and decreased insulin secretion in the blood (Makinistoglu & Karsenty, 2015). Recently, HDAC4 was found to be one of the protective genes for type 2 diabetes in human (Rani et al., 2017).
How HDAC4 operates to influence insulin secretion in the pancreas is a question that requires further detail investigation. From this report, we showed that the post‐transcriptional regulation (deacetylation) of FoxO1 was affected by the variants of HDAC4 in patients, the endogenous HDAC4 expression and its enzyme activity remained unchanged (data not shown). The HDAC4 variants, therefore, very likely cause the FoxO1 deacetylation failure, results in FoxO1 acetylation and nuclear exclusion, therefore, disrupts β‐cell function and maintenance (Figure 3). The diverse severity of clinical features; from severe hyperglycemia (p.H227R & p.D234N) to the modest IFG (p.E374K) matches their responding functional abnormality from different variants (Figure 2ab). All the above‐mentioned evidence supports that these pathogenic variants cause hyperglycemia according to the guideline of The American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015).
HDAC4 haploinsufficiency was reported to result in a severe brachydactyly mental retardation syndrome. The syndrome includes a variety of clinical features, such as brachydactyly E, developmental delays, behavioral problems, and obesity in 80% of the patients. However, the glucose levels were not reported (Williams et al., 2010). Mihaylova et al. found that HDAC4/5 recruit HDAC3 resulting in promoter induction of gluconeogenesis enzymes such as G6Pase, controls the FOXO acetylation in hepatocytes and liver; contribute to the hyperglycemic phenotype of type 2 diabetic rodent models in their insulin‐resistant states (Mihaylova et al., 2011). Similarly, in skeletal muscle cells, HDAC4 down‐regulates genes involved in energy expenditure, results in insulin resistance and type 2 diabetes (Fang et al., 2016). Moreover, Hdac4 suppressed transcription of Glut4, a key protein for glucose uptake in adipocytes (Henriksson et al., 2015), and disruption of Hdac4 in macrophages is sufficient to promote insulin resistance and obesity (Luan et al., 2014). From this report, obesity is present in two of the three hyperglycemic families, and the detected HDAC4 variants are missense variants instead of HDAC4 haploinsufficiency. Our first patient was severely obese and his glucose levels were improved with the loss of weight. For the third family, the mother and seven mother‐side grandmothers of the index patient are present as severe obesity and type 2 diabetes, even though the index patient is currently slim very likely due to the young age, she showed slightly insulin resistance (Table 1). The pathogenic roles of HDAC4 in hyperglycemia, and/or, obesity should be thus systematically investigated in the near future.
Except for its critical regulatory role of insulin signaling in liver (Langlet et al., 2017), FoxO1 maintains β‐cell mass (Kitamura et al., 2002; Okamoto et al., 2006), β‐cell identity, and proper β‐cell function (Kawamori et al., 2006; Talchai, Xuan, Kitamura, DePinho, & Accili, 2012). Impaired FoxO activity causes MODY‐like diabetes through the reduced metabolic flexibility of β‐cells (Kim‐Muller et al., 2014). FoxO1 transcriptional activity is regulated by a complex array of post‐translational modifications, one of the key events in regulating FoxO1 activity in β‐cells is the acetylation of specific lysine residues. When fully acetylated, FoxO1 exports from nuclear to the cytoplasm, whereas fully deacetylated, it imports into the nuclear and increases its transcriptional activity. In addition, the acetylation of FoxO1 increases the levels of its phosphorylation, resulting in its cytoplasm translocation and down‐regulation of the expression of the target genes (Matsuzaki et al., 2005).
Deacetylated FoxO1 protects β‐cell function by limiting mitochondrial lipid utilization through decreasing fatty acid oxidation and enhancing insulin secretion in the context of diabetes (Kim‐Muller et al., 2016); which is in consist in the findings that the levels of acetylated FoxO1 are markedly increased in pancreatic tissues of diabetic, compared to healthy rats (Ding et al., 2014). We found elevated levels of acetylated FoxO1 in the pancreatic β‐cells exposed to the HDAC4 mutations compared to the wild type, indicating the failure of FoxO1 deacetylation in the diabetic patients. (Zhang et al., 2016) revealed that FoxO1 plays an important role in regulating β‐cell compensation for insulin resistance to maintain euglycemia in obesity; FoxO1 acts in parallel with Pdx1 in nuclear of β‐cells and colocalize in human fetal islets, in contrast to the assertion that FoxO1 counteracts Pdx1 by promoting Pdx1 nuclear exclusion in β‐cells (Kawamori et al., 2006). In this report, we found dramatically increasing levels of acetylation FoxO1 and obvious nuclear export of both HDAC4 and FoxO1, but the location of Pdx1 remains unchanging, indicating the HDAC4 mutations disrupt the deacetylation of FoxO1, but not the pdx1. We therefore hypothesize FoxO1 is deacetylated not only by Sirt1, Sirt2, and Sirt6, but also by HDAC4 in the pancreas (Daitoku et al., 2004; Kim‐Muller et al., 2016; Kitamura et al., 2005; Song, Wang, Ka, Bae, & Park, 2016). The regulation of FoxO1 to β‐cell compensation to overnutrition and obesity (Zhang et al., 2016) was thereafter disrupted by the FoxO1 deacetylation failure in the diabetic patients harboring HDAC4 mutations. How these mutations cause deacetylation failure of the FoxO1 and further affects the β‐cell function changing should be investigated in the subsequent studies.
In summary, we detected three HDAC4 variants (de novo, and with incomplete penetrance) in three separate hyperglycemic families with/without obesity. In pancreatic β‐cells, these variants were found to decrease the insulin secretion and down‐regulate the pancreatic β‐cells key factors. In the mechanism, the β‐cells dysfunction might be caused by the FoxO1 deacetylation failure due to the HDAC4 mutations. The functional evidence in combination with what was reported in previous animal model studies: the FoxO1 deacetylation affects the function of β‐cells, clearly shows that the HDAC4 mutations are likely pathogenic of childhood hyperglycemia, and the HDAC4 variants cause childhood hyperglycemia with at least limited or even moderate evidence (Strande et al., 2017). Our study also suggests that HDAC4‐mediated FoxO1 deacetylation could represent a novel target for pharmacotherapy, not only of decreasing glucose‐sensitive insulin secretion but also for insulin resistance. However, the complex interplay of histone modifications and hyperglycemia is required for further comprehensive investigation.
CONFLICT OF INTEREST
The authors have no interest conflicts.
Supporting information
ACKNOWLEDGMENTS
We thank Friedrich C. Luft for his assistance. We appreciate all the studied individuals. We thank Nadine Wittstruck, Jeannette Mothes, and Mathias Gerhard for their technical support. The ECRC and the Berlin Institute of Health (BIH) supported Klemens Raile. This work was supported by European Society for Paediatric Endocrinology (ESPE) to Klemens Raile and Khalid Hussain (ESPE research unit grant 2012‐2014); and the Foundation “Das zuckerkranke Kind” to Maolian Gong and Klemens Raile.
Gong M, Yu Y, Liang L, et al. HDAC4 mutations cause diabetes and induce β‐cell FoxO1 nuclear exclusion. Mol Genet Genomic Med. 2019;7:e602 10.1002/mgg3.602
REFERENCES
- Ajjan, R. A. , & Owen, K. R. (2014). Glucokinase MODY and implications for treatment goals of common forms of diabetes. Current Diabetes Reports, 14(12), 559 10.1007/s11892-014-0559-0 [DOI] [PubMed] [Google Scholar]
- Alkorta‐Aranburu, G. , Carmody, D. , Cheng, Y. W. , Nelakuditi, V. , Ma, L. , Dickens, J. T. , … Del Gaudio, D. (2014). Phenotypic heterogeneity in monogenic diabetes: The clinical and diagnostic utility of a gene panel‐based next‐generation sequencing approach. Molecular Genetics and Metabolism, 113(4), 315–320. 10.1016/j.ymgme.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R. , Shi, L. , Hakenberg, J. , Naughton, B. , Sklar, P. , Zhang, J. , … Friend, S. H. (2016). Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nature Biotechnology, 34(5), 531–538. 10.1038/nbt.3514 [DOI] [PubMed] [Google Scholar]
- Cooper, D. N. , Krawczak, M. , Polychronakos, C. , Tyler‐Smith, C. , & Kehrer‐Sawatzki, H. (2013). Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Human Genetics, 132(10), 1077–1130. 10.1007/s00439-013-1331-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daitoku, H. , Hatta, M. , Matsuzaki, H. , Aratani, S. , Ohshima, T. , Miyagishi, M. , … Fukamizu, A. (2004). Silent information regulator 2 potentiates Foxo1‐mediated transcription through its deacetylase activity. Proceedings of the National Academy of Sciences, 101(27), 10042–10047. 10.1073/pnas.0400593101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshpajooh, M. , Bacos, K. , Bysani, M. , Bagge, A. , Ottosson Laakso, E. , Vikman, P. , … Ling, C. (2017). HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia, 60(1), 116–125. 10.1007/s00125-016-4113-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvecchio, M. , Mozzillo, E. , Salzano, G. , Iafusco, D. , Frontino, G. , Patera, P. I. , … Barbetti, F. (2017). Monogenic Diabetes Accounts for 6.3% of Cases Referred to 15 Italian Pediatric Diabetes Centers During 2007 to 2012. The Journal of Clinical Endocrinology & Metabolism, 102(6), 1826–1834. 10.1210/jc.2016-2490 [DOI] [PubMed] [Google Scholar]
- Ding, H. , Zhu, T. , Yin, X. , Liu, J. , Zhang, L. , Bernier, M. , & Zhao, R. (2014). Pyrrolidine dithiocarbamate protects pancreatic beta‐cells from oxidative damage through regulation of FoxO1 activity in type 2 diabetes rats. Acta Biochimica Et Biophysica Sinica (Shanghai), 46(7), 582–589. 10.1093/abbs/gmu034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, M. , Fan, Z. , Tian, W. , Zhao, Y. , Li, P. , Xu, H. , … Xu, Y. (2016). HDAC4 mediates IFN‐gamma induced disruption of energy expenditure‐related gene expression by repressing SIRT1 transcription in skeletal muscle cells. Biochimica Et Biophysica Acta, 1859(2), 294–305. 10.1016/j.bbagrm.2015.11.010 [DOI] [PubMed] [Google Scholar]
- Hattersley, A. T. , & Patel, K. A. (2017). Precision diabetes: Learning from monogenic diabetes. Diabetologia, 60(5), 769–777. 10.1007/s00125-017-4226-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson, E. , Sall, J. , Gormand, A. , Wasserstrom, S. , Morrice, N. A. , Fritzen, A. M. , … Goransson, O. (2015). SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. Journal of Cell Science, 128(3), 472–486. 10.1242/jcs.153932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, S. , Ivanov, A. , Blasevic, D. , Muller, T. , Purfurst, B. , Sun, W. , … Birchmeier, C. (2015). Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic beta‐cell function. EMBO Journal, 34(10), 1417–1433. 10.15252/embj.201490819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing, H. , Kase, J. , Dorr, J. R. , Milanovic, M. , Lenze, D. , Grau, M. , … Lee, S. (2011). Opposing roles of NF‐kappaB in anti‐cancer treatment outcome unveiled by cross‐species investigations. Genes & Development, 25(20), 2137–2146. 10.1101/gad.17620611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, B. B. , Irgens, H. U. , Molnes, J. , Sztromwasser, P. , Aukrust, I. , Juliusson, P. B. , … Njolstad, P. R. (2017). Targeted next‐generation sequencing reveals MODY in up to 6.5% of antibody‐negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia, 60(4), 625–635. 10.1007/s00125-016-4167-1 [DOI] [PubMed] [Google Scholar]
- Kawamori, D. , Kaneto, H. , Nakatani, Y. , Matsuoka, T. A. , Matsuhisa, M. , Hori, M. , & Yamasaki, Y. (2006). The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX‐1 through its intracellular translocation. Journal of Biological Chemistry, 281(2), 1091–1098. 10.1074/jbc.M508510200 [DOI] [PubMed] [Google Scholar]
- Kim‐Muller, J. Y. , Kim, Y. J. , Fan, J. , Zhao, S. , Banks, A. S. , Prentki, M. , & Accili, D. (2016). FoxO1 deacetylation decreases fatty acid oxidation in beta‐cells and sustains insulin secretion in diabetes. Journal of Biological Chemistry, 291(19), 10162–10172. 10.1074/jbc.M115.705608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim‐Muller, J. Y. , Zhao, S. , Srivastava, S. , Mugabo, Y. , Noh, H. L. , Kim, Y. R. , … Accili, D. (2014). Metabolic inflexibility impairs insulin secretion and results in MODY‐like diabetes in triple FoxO‐deficient mice. Cell Metabolism, 20(4), 593–602. 10.1016/j.cmet.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura, T. , Nakae, J. , Kitamura, Y. , Kido, Y. , Biggs, W. H. III , Wright, C. V. , … Accili, D. (2002). The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. Journal of Clinical Investigation, 110(12), 1839–1847. 10.1172/JCI16857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura, Y. I. , Kitamura, T. , Kruse, J. P. , Raum, J. C. , Stein, R. , Gu, W. , & Accili, D. (2005). FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metabolism, 2(3), 153–163. 10.1016/j.cmet.2005.08.004 [DOI] [PubMed] [Google Scholar]
- Kuhnen, P. , Turan, S. , Frohler, S. , Guran, T. , Abali, S. , Biebermann, H. , … Krude, H. (2014). Identification of PENDRIN (SLC26A4) mutations in patients with congenital hypothyroidism and "apparent" thyroid dysgenesis. Journal of Clinical Endocrinology and Metabolism, 99(1), E169–E176. 10.1210/jc.2013-2619 [DOI] [PubMed] [Google Scholar]
- Langlet, F. , Haeusler, R. A. , Linden, D. , Ericson, E. , Norris, T. , Johansson, A. , … Accili, D. (2017). Selective inhibition of FOXO1 activator/repressor balance modulates hepatic glucose handling. Cell, 171(4), 824.e818–835.e818. 10.1016/j.cell.2017.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir, O. , Flosseau, K. , Ma, F. X. , Blondeau, B. , Mai, A. , Bassel‐Duby, R. , … Scharfmann, R. (2011). Specific control of pancreatic endocrine beta‐ and delta‐cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes, 60(11), 2861–2871. 10.2337/db11-0440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan, B. , Goodarzi, M. O. , Phillips, N. G. , Guo, X. , Chen, Y. D. , Yao, J. , … Montminy, M. (2014). Leptin‐mediated increases in catecholamine signaling reduce adipose tissue inflammation via activation of macrophage HDAC4. Cell Metabolism, 19(6), 1058–1065. 10.1016/j.cmet.2014.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinistoglu, M. P. , & Karsenty, G. (2015). The class II histone deacetylase HDAC4 regulates cognitive, metabolic and endocrine functions through its expression in osteoblasts. Molecular Metabolism, 4(1), 64–69. 10.1016/j.molmet.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias, R. A. , Guise, A. J. , & Cristea, I. M. (2015). Post‐translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Molecular & Cellular Proteomics, 14(3), 456–470. 10.1074/mcp.O114.046565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki, H. , Daitoku, H. , Hatta, M. , Aoyama, H. , Yoshimochi, K. , & Fukamizu, A. (2005). Acetylation of Foxo1 alters its DNA‐binding ability and sensitivity to phosphorylation. Proceedings of the National Academy of Sciences, 102(32), 11278–11283. 10.1073/pnas.0502738102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek, M. , Zielonka, D. , Carnemolla, A. , Marcinkowski, J. T. , & Guidez, F. (2015). HDAC4 as a potential therapeutic target in neurodegenerative diseases: A summary of recent achievements. Frontiers in Cellular Neuroscience, 9, 42 10.3389/fncel.2015.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova, M. M. , Vasquez, D. S. , Ravnskjaer, K. , Denechaud, P. D. , Yu, R. T. , Alvarez, J. G. , … Shaw, R. J. (2011). Class IIa histone deacetylases are hormone‐activated regulators of FOXO and mammalian glucose homeostasis. Cell, 145(4), 607–621. 10.1016/j.cell.2011.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, H. , Hribal, M. L. , Lin, H. V. , Bennett, W. R. , Ward, A. , & Accili, D. (2006). Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. J Clin Invest, 116(3), 775–782. 10.1172/JCI24967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, K. A. , Kettunen, J. , Laakso, M. , Stancakova, A. , Laver, T. W. , Colclough, K. , … Weedon, M. N. (2017). Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nature Communications, 8(1), 888 10.1038/s41467-017-00895-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani, J. , Mittal, I. , Pramanik, A. , Singh, N. , Dube, N. , Sharma, S. , … Ramachandran, S. (2017). T2DiACoD: a gene atlas of type 2 diabetes mellitus associated complex disorders. Scientific Reports, 7(1), 6892 10.1038/s41598-017-07238-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. ; Committee, Acmg Laboratory Quality Assurance . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, M. Y. , Wang, J. , Ka, S. O. , Bae, E. J. , & Park, B. H. (2016). Insulin secretion impairment in Sirt6 knockout pancreatic beta cells is mediated by suppression of the FoxO1‐Pdx1‐Glut2 pathway. Scientific Reports, 6, 30321 10.1038/srep30321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strande, N. T. , Riggs, E. R. , Buchanan, A. H. , Ceyhan‐Birsoy, O. , DiStefano, M. , Dwight, S. S. , … Berg, J. S. (2017). Evaluating the clinical validity of gene‐disease associations: an evidence‐based framework developed by the clinical genome resource. American Journal of Human Genetics, 100(6), 895–906. 10.1016/j.ajhg.2017.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai, C. , Xuan, S. , Kitamura, T. , DePinho, R. A. , & Accili, D. (2012). Generation of functional insulin‐producing cells in the gut by Foxo1 ablation. Nature Genetics, 44(4), 406–412, S401. 10.1038/ng.2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, S. R. , Aldred, M. A. , Der Kaloustian, V. M. , Halal, F. , Gowans, G. , McLeod, D. R. , … Elsea, S. H. (2010). Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. American Journal of Human Genetics, 87(2), 219–228. 10.1016/j.ajhg.2010.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , & Chan, L. (2016). Monogenic diabetes: what it teaches us on the common forms of type 1 and type 2 diabetes. Endocrine Reviews, 37(3), 190–222. 10.1210/er.2015-1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Kim, D. H. , Xiao, X. , Lee, S. , Gong, Z. , Muzumdar, R. , … Dong, H. H. (2016). FoxO1 plays an important role in regulating beta‐cell compensation for insulin resistance in male mice. Endocrinology, 157(3), 1055–1070. 10.1210/en.2015-1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials