

Abstract
Background
Nonsyndromic cleft lip and/or palate is one of the most common human birth defects worldwide that affects the lip and/or palate. The incidence of clefts varies among populations through ethnic, race, or geographical differences. The focus on Malay nonsyndromic cleft lip and/or palate (NSCL/P) is because of a scarce report on genetic study in relation to this deformity in Malaysia. We are interested to discuss about the genes that are susceptible to cause orofacial cleft formation in the family.
Methods
Genome‐wide linkage analysis was carried out on eight large extended families of NSCL/P with the total of 91 individuals among Malay population using microarray platform. Based on linkage analyses findings, copy number variation (CNV) of LPHN2, SATB2, PVRL3, COL21A1, and TOX3 were identified in four large extended families that showed linkage evidence using quantitative polymerase chain reaction (qPCR) as for a validation purpose. Copy number calculated (CNC) for each genes were determined with Applied Biosystems CopyCallerTM Software v2.0. Normal CNC of the target sequence expected was set at two.
Results
Genome‐wide linkage analysis had discovered several genes including TOX3 and COL21A1 in four different loci 4p15.2‐p16.1, 6p11.2‐p12.3, 14q13‐q21, and 16q12.1. There was significant decreased, p < 0.05 of SATB2, COL21A1, and TOX3 copy number in extended families compared to the normal controls.
Conclusion
Novel linkage evidence and significant low copy number of COL21A1 and TOX3 in NSCLP family was confirmed. These genes increased the risks toward NSCLP formation in that family traits.
Keywords: cleft lip and/or palate, copy number, genetic, linkage, microarray
1. INTRODUCTION
Nonsyndromic cleft lip with or without palate (NSCLP) is a malformation without any other signs or symptoms of atypical condition such as abnormal physical appearance or psychological disease (Wallace, Arellano, & Gruner, 2011). Cleft lip and/or palate are one of the most common human embryonic disorder reported in Western countries and the second most common birth deformities among the babies (Muhamad & Azzaldeen, 2012; Shaw, Croen, & Curry, 1991). It arises in about one per 500 to 1,000 live births through ethnic and geographic differences (Muhamad & Azzaldeen, 2012; Murray, 2002). Birth prevalence and incidence of orofacial clefts vary among populations based on ethnic background (Cooper, Ratay, & Marazita, 2006).
Linkage analysis of multiplex families and association studies using either case–control or family‐based designs have become the primary methods for identifying potential genes for CL/P (Zeiger et al., 2003). Linkage studies have successfully revealed several different candidate loci studied in different populations. For example, a consanguineous family with 17 members was reported to have a homozygous 237‐kb deletion at locus 1p31 among the family members that cause cleft lip (Yıldırım, Kerem, Köroğlu, & Tolun, 2014). There are a lot of studies that revealed the causative genes to nonsyndromic cleft lip palate (NSCLP) formation such as TGF family, FGF family, cleft lip, and palate have been associated transmembrane protein 1 (CLPTM1), special AT‐rich sequence‐binding protein 2 (SATB2), and small ubiquitin‐like modifier 1 (SUMO1) and IRF6 (Carter et al., 2010; Nie, Luukko, & Kettunen, 2006; Pauws & Stanier, 2007; Scapoli et al., 2005).
Furthermore, multiple genome‐wide association study (GWAS) and relative extension studies had identified 22 susceptible loci in NSCLP including IRF6 at 1q32 locus (Beaty et al., 2013; Leslie et al., 2016; Ludwig et al., 2016; Yu et al., 2017). Recent genomic study carried out among the cleft palate (CPO) of African population found two novel loci on chromosome 2 near CTNNA2 and on chromosome 19 in SULT2A1 (Butali et al., 2018). In addition, the emergence of next generation sequencing accelerates the finding of new loci associated with orofacial cleft (Cai et al., 2017). Copy number changes (CNC) analysis in exome sequencing has identified two CNC in ADH7 and AHR in two multiplex families from Honduran population, whereas these two genes play role in craniofacial development (Cai et al., 2017).
To date, there is scarce report on genetic study of NSCLP in Malaysia, particularly on the Malay race as a majority of Malaysian population. Previously, some common genes such as MSX1, IRF6, and TGF‐β have been reported to be associated with NSCLP formation but many more uncommon genes could be elucidated in this study. In addition, identification of specific mutation and causal genes could be a major contribution in understanding the pathogenesis of orofacial clefts and might aid in developing preventive strategies. Plus, these identifications would be helpful in screening high risk individuals for genetic counseling purpose.
2. MATERIALS AND METHODS
2.1. Subjects
Eight large extended Malay families with three–four generations consists of 91 individuals were included. Healthy individuals with no cleft lip and/or palate phenotype, no family history of clefts or syndromic diseases were included and acts as a normal control. All the blood withdrawal was undertaken during the home visit or appoinment at clinic. They have been referred to the craniofacial specialist before the blood was taken. This study was approved by the Research Ethics Committee (Human) of the Universiti Sains Malaysia, Health Campus, Malaysia; Reference No.: USMKK/PPP/JEPeM [258.3.(3)] and written informed consent was obtained from the participants or their parent/guardian.
2.2. Microarray analysis
DNA was extracted from 200 μl of heparinized peripheral blood for microarray analysis using Illumina Infinium HumanLinkage‐24 Beadchip. All the DNA samples were arrayed at St George's Hospital, University of London, using the Illumina technology.
2.3. Validation with Copy Number Variation (CNV) assay using quantitative PCR (QPCR)
2.3.1. Gene selection
Four large extended families that showed suggestive or significant linkage evidence were selected for the validation step. Commercialized human DNA control was used as a calibrator, healthy individuals as normal controls (C1‐C6), and targeted samples including both the affected and unaffected family members.
2.4. Taqman® copy number assay selection
The SNPs that showed suggestive or significant linkage were identified using National Center for Biotechnology Information (NCBI) database. Linkage interval for chromosome position or target information for the selected SNPs were entered into Assay Search Tool – Single Tube TaqMan® Assays from Life Technologies website (https://www.lifetechnologies.com/) to obtain specific primer‐probe pair. Five selected sequences were Homo sapiens SATB2 NCBI location: Chr.2:200134223–200335989; cytoband: 2q33.1d [Hs07541174_cn], TOX3 NCBI location: Chr.16:52503999; cytoband: 16q12.1 [Hs03924205_cn], COL21A1 NCBI location: Chr.6:56247500; cytoband: 6p12.1 [Hs00783345_cn], LPHN2 NCBI location: Chr.1:81800443; cytoband: 1p31.1 [Hs00381445_cn], and PVRL3 NCBI location: Chr.3:104871745; cytoband: 3q13.11 [Hs03228815_cn].
2.4.1. Quantitative PCR (qPCR)
qPCR was performed using TaqMan® Genotyping Master Mix for absolute quantitation of copy number for Latrophilin 2 (LPHN2; OMIM: 607018), SATB2 (OMIM: 608148), Poliovirus Receptor‐Related 3 (PVRL3; OMIM: 607147), alpha chain of type XX1 collagen (COL21A1; OMIM: 610002), and TOX High Mobility Group Box Family Member 3 (TOX3; OMIM: 611416). RNase P as an endogenous control and negative control (NTC). NTC is a reaction mixture without DNA template used as a negative control. qPCR was run using ABI 7,500 system.
2.4.2. CNV analysis
Copy number calculated (CNC) for the genes were determined with Applied Biosystems CopyCaller™ Software v2.0. Number of copies of the target sequence expected in the majority of samples was set at two. Samples were reviewed to achieve optimal experimental conditions where samples are of high quality, copy number, and reference assays have amplified and sample replicates have similar CT and ΔCT values. Accepted copy number calls should have confidence value > 95% and Z‐score < 1.75.
2.5. Bioinformatics analysis
2.5.1. Linkage analysis
Linkage analyses were carried out using Easy Linkage Plus v5.08 software (Hoffmann & Lindner, 2005). It allows multipoint simulation studies, detection of Mendelian/non‐Mendelian genotyping errors, and Hardy–Weinberg equilibrium (HWE). Due to limited pedigree capabilities of linkage programs supported by Easy Linkage software and limited pedigree plots are available from Gene Hunter software, Easy Linkage Plus extends the Gene Hunter plots by showing marker names and their genetic position (Hoffmann & Lindner, 2005). Gene Hunter‐Multipoint Linkage Analysis v2.1r5 was used to do multipoint parametric and nonparametric analysis under both dominant and recessive mode of inheritance as previously described by Shah, Salahshourifar, Sulong, Wan Sulaiman, and Halim (2016).
2.6. Statistical analysis
Statistical analysis of data was performed using Mann–Whitney test with SPSS Statistics (SPSS 22, SPSS, Inc., USA). Statistical significance was determined for p value <0.05 and all data were expressed as mean ± standard deviation (SD).
3. RESULTS
3.1. Linkage analysis results
Two human genetic maps markers were used; Illumina 6K Linkage 24 deCODE and AFFY 100k Marshfield Human Sex‐Averaged for comparison purposes. All markers were in Hardy–Weinberg Equlibrium (HWE) with p value <0.05 and the overall call rate was >98% for all samples. Both nonparametric and parametric analyses were carried out and came out with NPL and LOD score respectively. From eight extended families tested, there were four families showed positive linkage either suggestive or significant linkage at different loci. Significant linkage was only detected in Family 100 with NPL score ≥ 3.6 meanwhile other families had suggestive linkage with NPL score between 2.2 and 3.5. However, LOD score was only detected in two families; Family 100 and Family 99 with suggestive linkage (Table 1).
Table 1.
Protein coding genes identified in four families that showed linkage of evidence after microarray analysis. Genes highlighted in bold were selected for a validation
| Family | Linkage interval | SNP | NPL | LOD | Genes of interest in linkage interval |
|---|---|---|---|---|---|
| 50 | 1p31.1–p34.2 | rs697590 | 2.40 | – | PTPRF, AKR1AI, SSBP3, NFIA, LPHN2 |
| 2q34–q36.3 | rs1851328 | 2.49 | – | ERBB4, ABCA12, STK36, IHH, PAX3, SATB2 | |
| 58 | 6p11.2–p12.3 | rs1925154 | 3.17 | – | REL, COL21A1, DST |
| 16q12.1 | rs1420533 | 2.56 | – | TOX3, RBL2, RPGRIP1L, FTO | |
| 100 | 1p31.1–p31.3 | rs400382 | *3.64 | 2.16 | LPHN2, GFPT1 |
| 2q31.1–q35 | rs1002207 | *3.69 | 2.18 | ZNF533, DNAJC10, FSIP2, SATB2, SUMO1, SP3, HOXD4, HOXD3, HOXD2, MSTN, NRP2, ABCA12, ERBB4 | |
| 99 | 3q13.3–q13.33 | rs1398748 | – | 2.03 | COX17, GSK3B, FSTL1, HEG1, ZNF, CD200, BOC, CBLB, BBX, IFT57, PVRL3 |
*Significant NPL score detected in Family 100 at two loci. *Sequences: SATB2 NCBI location: Chr.2:200134223–200335989; cytoband: 2q33.1d; TOX3 NCBI location: Chr.16:52503999; cytoband: 16q12.1; COL21A1 NCBI location: Chr.6:56247500; cytoband: 6p12.1; LPHN2 NCBI location: Chr.1:81800443; cytoband: 1p31.1 and PVRL3 NCBI location: Chr.3:104871745; cytoband: 3q13.11.
All the SNPs that lay within the suggestive and significant linkage intervals were employed to identify the genes of interest using GeneBank and GeneDistiller 2014 database. Table 1 showed several protein coding genes identified from the single nucleotide polymorphisms (SNPs) of nonparametric linkage (NPL) and logarithm of odds (LOD) score. Five selected candidate genes were chosen for validation; LPHN2, SATB2, PVRL3, COL21A1, and TOX3 based on literature review that is related to any craniofacial deformities or embryonic development.
3.2. Validation of LPHN2 and SATB2 copy number
Validation of LPHN2 by copy number variation (CNV) assay was done as a continuous finding of suggestive linkage found at 1p31 region in Family 100. From the findings, copy number calculated (CNC) for each individual in the Family 100 including affected and unaffected members showed CNC similar to control group, CNC > 2.50 or 2.49 ≥ CNC ≥1.50. Scatter plot data showed resemble distribution of CNC value between Family 100 and normal controls, that both having more than two copy numbers, depicted by a reference line at copy number of 2 (Figure 1). Statistical analysis had found no significant difference of copy number of LPHN2 (p > 0.05) in Family 100 (2.49 ± 0.45) compared to the control group (2.46 ± 0.61), means that both groups had normal copy number for LPHN2. Therefore, LPHN2 is regulated normally and not susceptible to NSCLP malformation.
Figure 1.
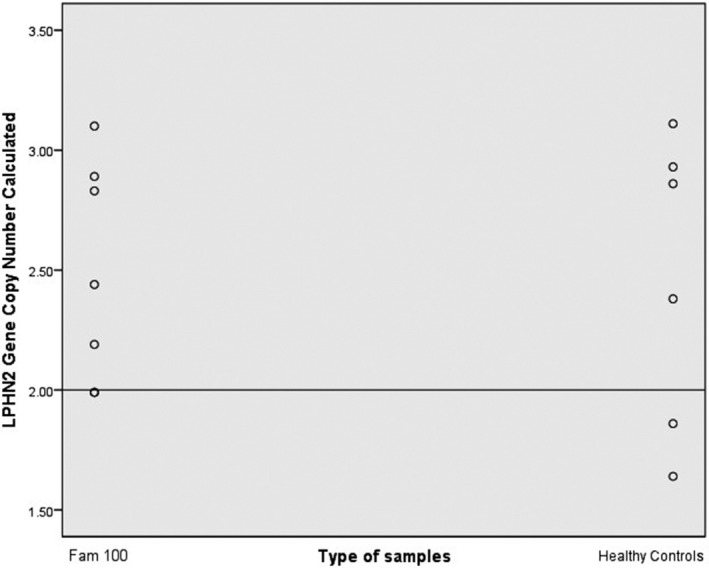
Scatter plot indicated LPHN2 copy number value in Family 100 in comparison to the normal controls. The straight line depicted the calibrator set value at 2
Copy number of SATB2 was determined for two families; Family 100 and Family 58. Scatter plot of the CNC data has shown distinct distribution of copy number between the affected NSCLP families from the normal control groups. Data plots were scattered below copy number of 2 in the affected families meanwhile normal group had higher than 2 copy numbers (Figure 2). There was a significant decreased in SATB2 copy number in both Family 50 (1.90 ± 0.12) and Family 100 (1.87 ± 0.24), p < 0.05 compared to the normal controls. Therefore, this indicates SATB2 copy loss was confirmed among the families with strong family history of NSCLP cases.
Figure 2.

SATB2 copy number in Family 50 and 100 in comparison to the healthy controls. The straight line depicted the calibrator set value at 2
3.3. Validation of COL21A1 copy number
Copy number for COL21A1 was determined for Family 58 as it showed suggestive linkage in this family. Commercial human control that acts as calibrator had CNC < 1.50 (CNC = 1.28). Meanwhile, all the six healthy normal controls (C1‐C6) tested had copy number of 2.49 ≥ CNC ≥1.50. In total, all family members including two affected members (58‐F and 58–2‐3) had gene copy number within 2.49 ≥ CNC ≥1.50 (Figure 3). These findings indicated the COL21A1 copy number was similar in both normal controls and NSCLP family.
Figure 3.

(a) Bars indicated copy number calculated (CNC) of COL21A1 in Family 58 (purple bars), normal control (blue bars), and calibrator (the first blue bar from right). (b) Scatter plot indicated copy number value of COL21A1 in Family 58 in comparison to the healthy controls. The straight line depicted the calibrator set value at 2
However, statistical analysis using scatter plot data had shown a distinct distribution of COL21A1 copy number between normal controls and Family 58. As depicted by the line at two copy numbers, gene copy number among the family members were lower than two compared to the normal controls that had copy number more than two (Figure 3). A significant decreased, p < 0.05 (p = 0.03) of COL21A1 copy number in Family 58 (1.86 ± 0.13) compared to the normal controls (2.16 ± 0.24) was confirmed.
3.4. Validation of TOX3 copy number
TOX3 copy number was detected from Family 58 which had shown suggestive linkage on 16q12.1 region through genome‐wide linkage analysis. Commercial human control (calibrator) had CNC < 1.50 (CNC = 1.36) for TOX3. However, most of normal controls had CNC > 2.50. In Family 58, all family members including two affected members (58‐F and 58–2‐3) had 2.49 ≥ CNC ≥1.50 (Figure 4). None of them has reached the CNC > 2.50.
Figure 4.

(a) Bars indicated copy number calculated (CNC) of TOX3 in Family 58 (blue bars), normal control (green bars), and calibrator (the first green bar from right). (b) Scatter plot indicated copy number value of TOX3 in Family 58 in comparison to the normal controls. The straight line depicted the calibrator set value at 2
Scatter plot data showed a distinct distribution of TOX3 copy number between normal controls and Family 58, whereas low TOX3 copy number was found in affected Family 58 (Figure 4). There was a significant decreased, p < 0.05 (p = 0.006) of TOX3 copy number in Family 58 (1.91 ± 0.26) compared to the normal controls (2.60 ± 0.34).
3.5. Validation of PVRL3 copy number
Copy number of PVRL3 in Family 99 was predicted which had shown suggestive linkage on 3q13.3 region through genome‐wide linkage analysis. All the normal controls (C1–C6) and Family 99 expressed 2.49 ≥ CNC ≥1.50. Statistical analysis through scatter plot had found similar PVRL3 copy number distribution between normal controls and Family 99 (Figure 5). Statistical significance had shown there was no significant difference, p > 0.05 (p = 0.55) of PVRL3 copy number in Family 99 (1.96 ± 0.13) compared to the normal controls (2.01 ± 0.17). This indicated that PVRL3 was not as a risk factor to the NSCLP formation in a family linkage analysis.
Figure 5.

Scatter plot indicated copy number value of PVRL3 in Family 99 in comparison to the normal controls. The straight line depicted the calibrator set value at 2
4. DISCUSSION
4.1. Nonsignificant copy number gain or loss in LPHN2 and PVRL3
Despite linkage evidence has been attained through linkage analysis in Family 100, and detect LPHN2 as a target gene, copy number analysis did not show any positive finding on LPHN2 associated with clefting. The nonfunctional role of LPHN2 in NSCLP formation was confirmed since nonsignificant copy number gain or loss in the family has been detected in comparison to the normal control. The detection of linkage at 1p31 region for LPHN2 at the first level of microarray linkage analysis might reflect to the one individual in the family who had a high arch palate phenotype, but not the other NSCLP affected members. This is supported by the previous finding that has detected a deletion at 1p31 region that was associated with craniofacial abnormalities, particularly high‐arched palate (Beleza‐Meireles, Clayton‐Smith, & Tassabehji, 2014).
Linkage evidence at 3q13.3 region of NSCLP family identified candidate gene PVRL3. Early assumption that the selected NSCLP family had a lower copy number of PVRL3 compared to the normal individuals was not significantly proven. A normal detection of PVRL3 copy number in the NSCLP family reflected the finding by Sözen, Hecht, and Spritz (2008) that found no evidence of sequence variations in exons, intron, and noncoding sequences of PVRL3 among North American Caucasian population (Sözen et al., 2008). On contrary, other nectin‐family paralogues, PVR, PVRL1, and PVRL2, have been identified as candidate genes and play a role in etiology of NSCLP via genome‐wide linkage and association studies (Sözen et al., 2008; Warrington et al., 2006).
4.2. Copy number loss of known SATB2
As anticipated, absolute copy number loss of SATB2 in Family 50 and Family 100 was significantly attained in the family with affected members of nonsyndromic clefting compared to the normal individuals with no history of orofacial cleft. This finding plausibly explained the importance role of SATB2 in normal craniofacial development, which any loss of it may cause cleft defects. Similar to our finding, it has been found that SATB2 lay within the deleted region, with low copy number of SATB2 was detected in a patient with cleft compared to the both parents and control (Urquhart, Black, & Clayton‐Smith, 2009). Since very low significant copy number loss in NSCLP family compared to the normal individuals were detected, we expect that a microdeletion on 2q32‐q33 region might occur. However, microdeletion could not be identified with absolute qPCR due to the limitation in the power to determine DNA copy number (Sebat et al., 2004).
SATB2 is known to play a role in orofacial cleft particularly the cleft palate, involving the mutation at the chromosomal 2q32‐q33 region (FitzPatrick et al., 2003). Previous reports had identified the 2q32‐q33 region was critical for normal palatogenesis, whereas haploinsufficiency caused significant cleft palate defect (Brewer, Holloway, Zawalnyski, Schinzel, & FitzPatrick, 1998; 1999). SATB2 loss was reported to be associated with increased cell death at the developing jaw primordial including palate, therefore hindered regional development and finally caused craniofacial defects (Britanova et al., 2006). The loss of SATB2 gene copy number in the family of affected cleft indicated the risk and susceptibility of SATB2 to cause orofacial cleft in the family trait.
4.3. Novel finding on COL21A1 copy number
The novel low copy number of 6p12.2 region in the affected family members has confirmed COL21A1 as one of the contributing genes to the NSCLP formation. This copy number finding has strengthened the linkage evidence detected previously via microarray. COL21A1 functions in maintaining the integrity of ECM. It has been reported that COL21A1 is a part and the smallest of FACIT family of collagen that is expressed in tissues expressing muscle phenotype such as skeletal muscle. It contains abundant ECM and enriched collagen I (Fitzgerald & Bateman, 2001). This co‐expression of collagen XXI and collagen I in tissues and muscles have important role in the organization of interstitial collagen fibrils by connecting it to other matrix components or cells (Chou, & Li, 2002; Fitzgerald, & Bateman, 2001). Therefore, detection of COL21A1 low copy number in NSCLP family possibly due to low concentration COL21A1 in DNA samples.
This condition might affect normal collagen activity in maintaining the tissue integrity of the lip and/or palate muscle and cause cleft lip deformity. As this gene has never been discussed or claimed to have a role in clefting, our finding is the first report of COL21A1 at 6p12.2 region that might be associated with nonsyndromic orofacial cleft. Similarly, one study on six Amish patients with multiplex human syndrome with cleft lip and palate anomaly has attained the candidate locus at a similar chromosome 6p12.2‐p12 but they harbored ICK missense mutation (Lahiry et al., 2009).
Since the evidence for linkage to this region was only detected in one of eight NSCLP families, this data could still provide an important significant output in a gene discovery to the occurrence of NSCLP, as it is known to be heterogeneous. Previously, two different studies has proposed that major clefting locus might be located at 6p and mutation in several genes such as COL2A1, COL11A1, and COL11A2 caused syndrome that was associated with cleft palate, Robin sequence, and micrognathia (Melkoniemi et al., 2003; Murray, 1995). On the other hand, it has also been found that the COL2A1, COL11A1, and COL11A2 were associated and influence the risk of nonsyndromic forms of cleft palate (Jugessur et al., 2009; Nikopensius et al., 2010). No one has reported the role of COL21A1 in association with nonsyndromic orofacial cleft so far but the mutation at the 6p region has been attained before.
Different gene copy number among individuals and populations reflects to the gene expression variation (Stranger et al., 2007). Many studies have reported that the change in gene copy number has led the cells modifying transcription process and change the expression level (Prestel, Feller, & Becker, 2010; Stranger et al., 2007). Hence, evidence for linkage between COL21A1 and potential cleft phenotype has been confirmed in this family. Loss of one copy of COL21A1 was clearly observed on the family members compared to the normal controls, so then we concluded that this COL21A1 might give a minor contribution to the NSCLP formation and could trigger the cleft occurrence in the family tree. This novel finding would help to shed light on the regulation of COL21A1 on the orofacial cleft formation.
4.4. Novel finding of low TOX3 copy number
Lower TOX3 copy number at 16q12.1 has been achieved through CNV analysis in one affected family, Family 58. Currently, it has never been reported that the TOX3 was susceptible to nonsyndromic orofacial cleft occurrence. Therefore, this would be the first in which a finding revealed dysregulation of TOX3 in a family member of affected NSCLP, supporting the data of our microarray linkage analysis. Previous finding has found a deletion on similar locus of 16q12.1‐q13 on a patient of Pierre Robin sequence with median cleft palate (Schuffenhauer et al., 1992). Second, two different studies have also been reported to have mutation on 16q12.1‐q12.2 in syndromic Japanese patients with different phenotypes, and another one Germany patient with cleft of the soft palate anomaly, similarly TOX3 was one of the protein‐coding genes detected (Morisada et al., 2014; Shoukier et al., 2012).
Deletion on 16q12 region has been extensively studied in decades in relation to syndromic cases with various phenotypes and one of the craniofacial anomalies identified was high‐arched palate or cleft of the soft palate (Callen et al., 1993; Chang et al., 2010; Elder, Ferguson, & Lockhart, 1984; Shoukier et al., 2012). Therefore, there is high possibility that this mutation also occurred in nonsyndromic cleft cases but the specific mechanism of action of TOX3 confers craniofacial deformity risk was unclear. Through literatures, it has been discussed that the mutation of TOX3 was associated with breast cancer susceptibility (Jones et al., 2013; Udler et al., 2010). In addition, studies have been done in investigating the metastasis of breast cancer cells to bone.
The mechanism of TOX3 in triggering CL/P formation was unknown. But, it was likely that mutation of TOX3 interact with impaired FGFs in developing this deformity, which meant both of them deficit during the bone fusion in pre‐developmental stage. This was supported by our study on cellular level, whereas significant FGF down‐regulation was proven interfered normal embryonic craniofacial developmental process via FGF signaling (unpublished data). We hypothesized that reduced induction of TOX3 triggering FGF dysregulation in progression of lip and palate fusion and finally drive to the abnormal growth.
Validation through copy number analysis has strengthened the previous linkage evidence outcomes. Having significantly low copy number of these genes; COL21A1 and TOX3 in each selected NSCLP families compared to normal groups has increased the risks toward NSCLP formation in their family traits.
CONFLICT OF INTEREST
The authors declare that there are no competing interests.
ACKNOWLEDGMENTS
We thank the voluntary patients and families for their full co‐operation given. This work was supported by Research University (RU) Grant: 1001/PPSP/812083 and student was funded by a scholarship SLAB/SLAI KPM/USM.
Mohamad Shah NS, Sulong S, Wan Sulaiman WA, Halim AS. Two novel genes TOX3 and COL21A1 in large extended Malay families with nonsyndromic cleft lip and/or palate. Mol Genet Genomic Med. 2019;7:e635 10.1002/mgg3.635
REFERENCES
- Beaty, T. H. , Taub, M. A. , Scott, A. F. , Murray, J. C. , Marazita, M. l. , Schwender, H. , … Ruczinski, I. (2013). Confirming genes influencing risk to cleft lip with/without cleft palate in a case–parent trio study. Human Genetics, 132(7), 771–781. 10.1007/s00439-013-1283-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beleza‐Meireles, A. , Clayton‐Smith, J. , & Tassabehji, M. (2014). Oculo‐Auriculo‐vertebral spectrum: A review of the literature. Journal of Genetic Syndromes and Genetic Therapy, 5(211), 2. [DOI] [PubMed] [Google Scholar]
- Brewer, C. , Holloway, S. , Zawalnyski, P. , Schinzel, A. , & FitzPatrick, D. (1998). A chromosomal deletion map of human malformations. The American Journal of Human Genetics, 63(4), 1153–1159. 10.1086/302041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, C. M. , Leek, J. P. , Green, A. J. , Holloway, S. , Bonthron, D. T. , Markham, A. F. , & FitzPatrick, D. R. (1999). A locus for isolated cleft palate, located on human chromosome 2q32. The American Journal of Human Genetics, 65(2), 387–396. 10.1086/302498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova, O. , Depew, M. J. , Schwark, M. , Thomas, B. L. , Miletich, I. , Sharpe, P. , & Tarabykin, V. (2006). Satb2 haploinsufficiency phenocopies 2q32‐q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. The American Journal of Human Genetics, 79(4), 668–678. 10.1086/508214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butali, A. , Mossey, P. A. , Adeyemo, W. L. , Eshete, M. A. , Gowans, L. J. , Busch, T. D. , ... Laurie, C. C. (2018). Genomic analyses in african populations identify novel risk loci for cleft palate. Human Molecular Genetics. 10.1093/hmg/ddy402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. I. , Patterson, K. E. , Reinier, F. , Keesecker, S. E. , Blue, E. , Bamshad, M. , & Haddad, J. (2017). Copy number changes identified using whole exome sequencing in nonsyndromic cleft lip and palate in a Honduran population. Birth Defects Research, 109(16), 1257–1267. 10.1002/bdr2.1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen, D. F. , Eyre, H. , Lane, S. , Shen, Y. , Hansmann, I. , Spinner, N. , … Wauters, J. (1993). High resolution mapping of interstitial long arm deletions of chromosome 16: Relationship to phenotype. Journal of Medical Genetics, 30(10), 828–832. 10.1136/jmg.30.10.828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, T. C. , Molloy, A. M. , Pangilinan, F. , Troendle, J. F. , Kirke, P. N. , Conley, M. R. , ... Doyle, A. (2010). Testing reported associations of genetic risk factors for oral clefts in a large Irish study population. Birth Defects Research Part A, 88(2), 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. F. , Li, L. H. , Wang, C. H. , Tsai, F. J. , Chen, T. C. , Wu, J. Y. , ... Tsai, A. C. H. (2010). Identification of a submicroscopic 3.2 Mb chromosomal 16q12. 2–13 deletion in a child with short stature, mild developmental delay, and craniofacial anomalies, by high‐density oligonucleotide array‐a recognizable syndrome. American Journal of Medical Genetics Part A, 152(9), 2365–2371. [DOI] [PubMed] [Google Scholar]
- Chou, M.‐Y. , & Li, H.‐C. (2002). Genomic organization and characterization of the human type XXI collagen (COL21A1) gene. Genomics, 79(3), 395–401. 10.1006/geno.2002.6712 [DOI] [PubMed] [Google Scholar]
- Cooper, M. E. , Ratay, J. S. , & Marazita, M. L. (2006). Asian oral‐facial cleft birth prevalence. The Cleft Palate‐Craniofacial Journal, 43(5), 580–589. 10.1597/05-167 [DOI] [PubMed] [Google Scholar]
- Elder, F. , Ferguson, J. , & Lockhart, L. (1984). Identical twins with deletion 16q syndrome: Evidence that 16q12. 2–q13 is the critical band region. Human Genetics, 67(2), 233–236. 10.1007/BF00273010 [DOI] [PubMed] [Google Scholar]
- Fitzgerald, J. , & Bateman, J. F. (2001). A new FACIT of the collagen family: COL21A11. FEBS Letters, 505(2), 275–280. 10.1016/S0014-5793(01)02754-5 [DOI] [PubMed] [Google Scholar]
- FitzPatrick, D. R. , Carr, I. M. , McLaren, L. , Leek, J. P. , Wightman, P. , Williamson, K. , ... Markham, A. F. (2003). Identification of SATB2 as the cleft palate gene on 2q32–q33. Human Molecular Genetics, 12(19), 2491–2501. 10.1093/hmg/ddg248 [DOI] [PubMed] [Google Scholar]
- Hoffmann, K. , & Lindner, T. H. (2005). easyLINKAGE‐Plus – Automated linkage analyses using large‐scale SNP data. Bioinformatics, 21(17), 3565–3567. 10.1093/bioinformatics/bti571 [DOI] [PubMed] [Google Scholar]
- Jones, J. O. , Chin, S.‐F. , Wong‐Taylor, L.‐A. , Leaford, D. , Ponder, B. A. , Caldas, C. , & Maia, A. T. (2013). TOX3 mutations in breast cancer. PLoS ONE, 8(9), e74102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur, A. , Shi, M. , Gjessing, H. K. , Lie, R. T. , Wilcox, A. J. , Weinberg, C. R. , … Bille, C. (2009). Genetic determinants of facial clefting: Analysis of 357 candidate genes using two national cleft studies from Scandinavia. PLoS ONE, 4(4), e5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry, P. , Wang, J. , Robinson, J. F. , Turowec, J. P. , Litchfield, D. W. , Lanktree, M. B. , … Hegele, R. A. (2009). A multiplex human syndrome implicates a key role for intestinal cell kinase in development of central nervous, skeletal, and endocrine systems. The American Journal of Human Genetics, 84(2), 134–147. 10.1016/j.ajhg.2008.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie, E. J. , Carlson, J. C. , Shaffer, J. R. , Feingold, E. , Wehby, G. , Laurie, C. A. , … Resick, J. (2016). A multi‐ethnic genome‐wide association study identifies novel loci for non‐syndromic cleft lip with or without cleft palate on 2p24. 2, 17q23 and 19q13. Human Molecular Genetics, 25(13), 2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, K. U. , Ahmed, S. T. , Böhmer, A. C. , Sangani, N. B. , Varghese, S. , Klamt, J. , … Aldhorae, K. A. (2016). Meta‐analysis reveals genome‐wide significance at 15q13 for nonsyndromic clefting of both the lip and the palate, and functional analyses implicate GREM1 as a plausible causative gene. PLoS Genetics, 12(3), e1005914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkoniemi, M. , Koillinen, H. , Männikkö, M. , Warman, M. L. , Pihlajamaa, T. , Kääriäinen, H. , … Ala‐Kokko, L. (2003). Collagen XI sequence variations in nonsyndromic cleft palate, Robin sequence and micrognathia. European Journal of Human Genetics, 11(3), 265 10.1038/sj.ejhg.5200950 [DOI] [PubMed] [Google Scholar]
- Morisada, N. , Sekine, T. , Ishimori, S. , Tsuda, M. , Adachi, M. , Nozu, K. , … Iijima, K. (2014). 16q12 microdeletion syndrome in two Japanese boys. Pediatrics International, 56(5), e75‐e78. [DOI] [PubMed] [Google Scholar]
- Muhamad, A. H. & Azzaldeen, A. (2012). Genetic of non‐syndromic cleft lip and palate. Nature Genetics, 1, 510 10.4172/scientificreports.510 [DOI] [Google Scholar]
- Murray, J. C. (1995). Face facts: Genes, environment, and clefts. American Journal of Human Genetics, 57(2), 227‐232. [PMC free article] [PubMed] [Google Scholar]
- Murray, J. (2002). Gene/environment causes of cleft lip and/or palate. Clinical Genetics, 61(4), 248–256. 10.1034/j.1399-0004.2002.610402.x [DOI] [PubMed] [Google Scholar]
- Nie, X. , Luukko, K. , & Kettunen, P. (2006). FGF signalling in craniofacial development and developmental disorders. Oral Diseases, 12(2), 102–111. 10.1111/j.1601-0825.2005.01176.x [DOI] [PubMed] [Google Scholar]
- Nikopensius, T. , Jagomägi, T. , Krjutškov, K. , Tammekivi, V. , Saag, M. , Prane, I. , … Metspalu, A. (2010). Genetic variants in COL2A1, COL11A2, and IRF6 contribute risk to nonsyndromic cleft palate. Birth Defects Research Part A: Clinical and Molecular Teratology, 88(9), 748–756. 10.1002/bdra.20700 [DOI] [PubMed] [Google Scholar]
- Pauws, E. , & Stanier, P. (2007). FGF signalling and SUMO modification: New players in the aetiology of cleft lip and/or palate. TRENDS in Genetics, 23(12), 631–640. 10.1016/j.tig.2007.09.002 [DOI] [PubMed] [Google Scholar]
- Prestel, M. , Feller, C. , & Becker, P. B. (2010). Dosage compensation and the global re‐balancing of aneuploid genomes. Genome Biology, 11(8), 216 10.1186/gb-2010-11-8-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapoli, L. , Palmieri, A. , Martinelli, M. , Pezzetti, F. , Carinci, P. , Tognon, M. , & Carinci, F. (2005). Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. The American Journal of Human Genetics, 76(1), 180–183. 10.1086/427344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuffenhauer, S. , Callen, D. F. , Seidel, H. , Shen, Y. , Lederer, G. , & Murken, J. (1992). De novo interstitial deletion 16 (q12. 1q13) of paternal origin in a 10‐year‐old boy. Clinical Genetics, 42(5), 246–250. 10.1111/j.1399-0004.1992.tb03249.x [DOI] [PubMed] [Google Scholar]
- Sebat, J. , Lakshmi, B. , Troge, J. , Alexander, J. , Young, J. , Lundin, P. , … Navin, N. (2004). Large‐scale copy number polymorphism in the human genome. Science, 305(5683), 525–528. 10.1126/science.1098918 [DOI] [PubMed] [Google Scholar]
- Shah, N. S. M. , Salahshourifar, I. , Sulong, S. , Wan Sulaiman, W. A. , & Halim, A. S. (2016). Discovery of candidate genes for nonsyndromic cleft lip palate through genome‐wide linkage analysis of large extended families in the Malay population. BMC Genetics, 17(1), 39 10.1186/s12863-016-0345-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, G. M. , Croen, L. A. , & Curry, C. J. (1991). Isolated oral cleft malformations: Associations with maternal and infant characteristics in a California population. Teratology, 43(3), 225–228. 10.1002/tera.1420430306 [DOI] [PubMed] [Google Scholar]
- Shoukier, M. , Wickert, J. , Schröder, J. , Bartels, I. , Auber, B. , Zoll, B. , … Burfeind, P. (2012). A 16q12 microdeletion in a boy with severe psychomotor delay, craniofacial dysmorphism, brain and limb malformations, and a heart defect. American Journal of Medical Genetics Part A, 158(1), 229–235. 10.1002/ajmg.a.34387 [DOI] [PubMed] [Google Scholar]
- Sözen, M. A. , Hecht, J. T. , & Spritz, R. A. (2008). Lack of mutations in the PVRL3 gene in North American caucasians with non‐syndromic cleft lip/palate. Genetics and Molecular Biology, 31(3), 649–650. 10.1590/S1415-47572008000400008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger, B. E. , Forrest, M. S. , Dunning, M. , Ingle, C. E. , Beazley, C. , Thorne, N. , … Dermitzakis, E. T. (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science, 315(5813), 848–853. 10.1126/science.1136678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udler, M. S. , Ahmed, S. , Healey, C. S. , Meyer, K. , Struewing, J. , Maranian, M. , … Dunning, A. M. (2010). Fine scale mapping of the breast cancer 16q12 locus. Human Molecular Genetics, 19(12), 2507–2515. 10.1093/hmg/ddq122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquhart, J. , Black, G. C. , & Clayton‐Smith, J. (2009). 4.5 Mb microdeletion in chromosome band 2q33. 1 associated with learning disability and cleft palate. European Journal of Medical Genetics, 52(6), 454–457. [DOI] [PubMed] [Google Scholar]
- Wallace, G. H. , Arellano, J. M. , & Gruner, T. M. (2011). Non‐syndromic cleft lip and palate: Could stress be a causal factor? Women and Birth, 24(1), 40–46. [DOI] [PubMed] [Google Scholar]
- Warrington, A. , Vieira, A. , Christensen, K. , Orioli, I. M. , Castilla, E. E. , Romitti, P. A. , & Murray, J. C. (2006). Genetic evidence for the role of loci at 19q13 in cleft lip and palate. Journal of Medical Genetics, 43(6), e26–e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yıldırım, Y. , Kerem, M. , Köroğlu, Ç. , & Tolun, A. (2014). A homozygous 237‐kb deletion at 1p31 identified as the locus for midline cleft of the upper and lower lip in a consanguineous family. European Journal of Human Genetics, 22(3), 333–337. 10.1038/ejhg.2013.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Zuo, X. , He, M. , Gao, J. , Fu, Y. , Qin, C. , … Bian, Z. (2017). Genome‐wide analyses of non‐syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. NatureCommunications, 8, 14364 10.1038/ncomms14364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiger, J. S. , Hetmanski, J. B. , Beaty, T. H. , VanderKolk, C. A. , Wyszynski, D. F. , Bailey‐Wilson, J. E. , … McIntosh, I. (2003). Evidence for linkage of nonsyndromic cleft lip with or without cleft palate to a region on chromosome 2. European Journal of Human Genetics, 11(11), 835 10.1038/sj.ejhg.5201052 [DOI] [PubMed] [Google Scholar]