

Abstract
Background
Inherited epimutations of Mismatch Repair (MMR) genes are responsible for Lynch Syndrome (LS) in a small, but well defined, subset of patients. Methylation of the MSH2 promoter consequent to the deletion of the upstream EPCAM gene is found in about 1%–3% of the LS patients and represents a classical secondary, constitutional and tissue‐specific epimutation. Several different EPCAM deletions have been reported worldwide, for the most part representing private variants caused by an Alu‐mediated recombination.
Methods
712 patients with suspected LS were tested for MMR mutation in our Institute. EPCAM deletions were detected by multiplex ligation‐dependent probe amplification (MLPA) and then defined by Long‐Range polymerase chain reaction (PCR)/Sanger sequencing. A comprehensive molecular characterization of colorectal cancer (CRC) tissues was carried out by immunohistochemistry of MMR proteins, Microsatellite Instability (MSI) assay, methylation specific MLPA and transcript analyses. In addition, somatic deletions and/or variants were investigated by MLPA and next generation sequencing (NGS).
Results
An EPCAM deletion was found in five unrelated probands in Italy: variants c.556‐490_*8438del and c.858+1193_*5826del are novel; c.859‐1430_*2033del and c.859‐670_*530del were previously reported. All probands were affected by CRC at young age; tumors showed MSI and abnormal MSH2/MSH6 proteins expression. MSH2 promoter methylation, as well as aberrant in‐frame or out‐of‐frame EPCAM/MSH2 fusion transcripts, were detected in CRCs and normal mucosae.
Conclusion
An EPCAM deletion was the causative variant in about 2% of our institutional series of 224 LS patients, consistent with previously estimated frequencies. Early age and multiple CRCs was the main clinical feature of this subset of patients.
Keywords: deletion, EPCAM, epigenetics, Lynch Syndrome, methylation, MSH2
1. INTRODUCTION
Pathogenic variants in MLH1, MSH2, MSH6 and PMS2 Mismatch Repair (MMR) genes (OMIM: 120436, 609309, 600678 and 600259) are causative of Lynch Syndrome (LS), an autosomal dominant condition that confers elevated risk of developing colorectal cancer (CRC), endometrial cancer (EC) and several other types of cancer. Typical phenotypic features of LS tumors are Microsatellite Instability (MSI) and loss of MMR protein expression (Lynch et al., 2009).
Biallelic inactivation of one of MMR genes, as a result of combined germline and second hit somatic mutations, facilitates carcinogenesis. Nonsense, missense, frameshift and splicing variants, as well as deletions of one or more exons, are responsible for loss of function of the inherited, predisposing MMR allele. In addition, some so‐called epimutations are reported. Epimutation is referred as a heritable change that does not alter the DNA sequence but affects gene expression via DNA methylation or histone modification. In LS, constitutional epimutations are mainly reported for the MLH1 gene, either as primary event arisen de novo in gametogenesis or as secondary event, caused by an in cis heritable genetic variant (Cini et al., 2015; Hitchins, 2013).
As about the MSH2 gene, a constitutional tissue‐specific promoter methylation can be inherited as secondary epimutation, due to a deletion involving the last exons of the EPCAM gene (OMIM: 185535) that maps upstream MSH2. As a consequence, EPCAM is brought close to MSH2 and, in EpCAM expressing tissues, a read‐through EPCAM/MSH2 fusion transcript is generated while the native MSH2 promoter is hypermethylated and silenced (Kovacs, Papp, Szentirmay, Otto, & Olah, 2009; Ligtenberg et al., 2009). This complex event represents the first hit in a well‐defined subset of LS patients (1%–3% of the total) (Kuiper et al., 2011) that are typified by the expression of aberrant EPCAM/MSH2 fusion transcripts in normal and tumor colon tissues (Kovacs et al., 2009; Ligtenberg et al., 2009).
Several different EPCAM deletions have been described worldwide, in some cases as recurrent/founder mutations (Dymerska et al., 2017; Eguchi et al., 2016; Mur et al., 2014; Spaepen et al., 2013). It has been established that the mechanism underlying these rearrangements is an Alu‐mediated deletion, involving different highly homologous Alu sequences that are interspersed in intronic and intergenic regions within the EPCAM/MSH2 locus (Tutlewska, Lubinski, & Kurzawski, 2013).
Here we report on the identification and characterization of four different EPCAM deletions in five unrelated Italian LS families, and their epigenetic effect on the MSH2 locus.
2. MATERIALS AND METHODS
From 1995 to 2017, DNA from a total of 712 unrelated cancer patients was collected in a diagnostic setting by the Functional Oncogenetics and Oncogenomics Laboratory at the CRO National Cancer Institute (Aviano, Italy). Patients were enrolled for genetic testing because of personal and family history consistent with LS and/or evidence of high MSI (MSI‐H) and/or negative for MMR protein according to immunohistochemistry (IHC). Diagnosis of LS was confirmed by MMR and EPCAM genetic testing in 224/712 patients.
The genetic testing protocol and use of DNA samples for research purposes was approved by the Local Independent Ethics Committee (CRO‐15‐1997). Written informed consent was obtained from all participants of the present study.
Mismatch Repair protein IHC was performed on Formalin‐Fixed and Paraffin‐Embedded (FFPE) sections, stained on the Ventana BenchMark Ultra platform (Ventana Medical Systems, Inc., Oro Valley AZ). MLH‐1 (M1), MSH2 (G219‐1129), MSH6 (44) and PMS2 (EPR3947) (Roche Diagnostics, Indianapolis, IN) monoclonal antibodies were used to qualitatively identify human MMR proteins. Lesions were considered negative for protein expression when a complete absence of nuclear staining was evident in tumor cells with concomitant nuclear staining of adjacent normal epithelial and stromal cells.
Genetic screening for MMR genes was carried out by Sanger sequencing of coding sequences (MSH2: NM_000251.2, MLH1: NM_000249.3, MSH6: NM_000179.2) and with SALSA® multiplex ligation‐dependent probe amplification (MLPA)® probemixes P003 (MLH1/MSH2), P072 (MSH6) and P248 (MLH1/MSH2 confirmation), followed by copy number variation analysis with Coffalyser.net (MRC‐Holland, Amsterdam, Holland).
Long‐Range polymerase chain reactions (PCRs) for breakpoints detection were performed with Expand™ Long Template PCR System (Merck KGaA, Darmstadt, Germany). Shorter PCRs including breakpoints were obtained with GoTaq (Promega, Madison, WI) (primers and conditions available upon request). Polymerase chain reaction products were purified from agarose gel with the Wizard® SV Gel and PCR Clean‐Up System (Promega), then sequenced using a modified BigDye® cycle sequencing protocol (30x 94°C 30″, 55°C 30″, 60°C 2′), to prevent secondary structures due to Alu‐mediated hybridization.
Tumor and normal mucosa methylation statuses were assessed using Methylation Specific‐MLPA (MS‐MLPA) with SALSA® MLPA® ME011 MMR genes probemix (MRC‐Holland). Blood DNA was used as a control.
Mutation analysis of tumor DNA was performed on the two available frozen CRC samples with a TruSeq Custom Amplicon (Illumina, San Diego, CA) next generation sequencing (NGS) panel, including coding sequences of MSH2, MSH6 and MLH1 genes. Libraries were run on an Illumina MiSeq platform, with a mean coverage >4,000 reads. MLPA analysis for loss of heterozygosity (LOH) detection was carried out on frozen and FFPE tumor samples with the SALSA® MLPA® probemix P072‐C1 (MSH6).
Aberrant transcripts were investigated in RNAs extracted from frozen or FFPE samples. In brief, RNA from tumor and/or normal mucosa of carriers was purified with the TRIzol™ Reagent (ThermoFisher Scientific, Waltham, MA) or the RNeasy FFPE kit (Qiagen, Hilden, Germany) according to manufacturer's protocols, and cDNA was retrotranscribed using the SuperScript™ III Reverse Transcriptase (ThermoFisher Scientific) and random primers. The presence of fusion transcripts was evaluated by amplification and direct sequencing of PCRs covering different regions between exon 4 or 7 of the EPCAM gene and exon 1 or 2 or 3 of MSH2 (primers and details upon request). The reference sequences NM_002354.2 and NG_012352.2 (EPCAM) and NM_000251.2 and NG_007110.2 (MSH2) were used for reporting aberrant transcripts and genomic deletions.
3. RESULTS
Of the 224 patients tested positive for LS in our laboratory from 1995 to 2017, five unrelated patients, all Italians, carried a partial deletion of the EPCAM gene, involving exons 6‐9 (families AV114 and UD18) or exons 8‐9 (families PD31, AV182 and PD78), as assessed by MLPA analyses. No other pathogenic variants were found in MSH2 and MSH6 genes of these patients.
All probands were diagnosed with MSI‐H CRC at young age (34–50 years), and had a family history of CRC. Immunohistochemistry, performed on a representative tumor of the proband, revealed abnormal MSH2 and MSH6 expression (Table 1). Notably, besides tumors with clear‐cut loss of nuclear MSH2/MSH6 staining, one tumor retained focal MSH2 expression and some showed heterogeneous MSH2 and/or MSH6 cytoplasmic staining (Figure 1a).
Table 1.
EPCAM deletion carriers
| Family ID | Patient ID | Sex | Clinical phenotype | IHC MSH2/MSH6 | MSI | MSH2 methylation | Somatic LOH |
|---|---|---|---|---|---|---|---|
| PD31 | CFS394 | F | CRC 45 yb, 55 y | H | |||
| CFS395a | M | CRC 43, 68 yb, duodenum 62 y, larynxc 67 y | −/Cytoplasmic | H | 33%(N) 23%(T) | No | |
| CFS890 | M | Healthy 37 y | |||||
| CFS892 | F | Healthy 39 y | |||||
| CFS918 | F | CRC 32 y, breastc 47 y | |||||
| CFS919 | F | Healthy 44 y | |||||
| CFS920 | M | Healthy 57 y | |||||
| AV114 | CFS396a | F | CRC 36, 54 yb, 54 y | Cytoplasmic/cytoplasmic | H | 29%(N) 50%(T) | EPCAM‐MSH2‐MSH6 loss |
| CFS487 | M | CRC 42 yb | Cytoplasmic/cytoplasmic | H | Not evaluable | EPCAM‐MSH2‐MSH6 loss | |
| CFS488 | F | Healthy 35 y | |||||
| CFS516 | M | CRC 25 yb | H | ||||
| UD18 | CFS825a | M | CRC 39, 53 yb | −/− | H | 23%(T) | No |
| CFS913 | M | Healthy 19 y | |||||
| CFS914 | M | Healthy 21 y | |||||
| AV182 | CFS1043a | M | CRC 50, 50, 63 yb | −/− | H | 23%(T) | EPCAM loss |
| PD78 | CFS1475a | M | CRC 34 yb | Focal/+ | H | 23%(T) | No |
| CFS1541 | M | Healthy 39 y |
CRC, colorectal cancer; H, MSI‐H; IHC, immunoistochemistry; LOH, loss of heterozygosity; LS, Lynch Syndrome; MSI, microsatellite instability; N, normal mucosa; T, tumor; y, years (age of tumor onset, if affected, and age at registration, if healthy).
Probands.
Tumors tested.
Tumors not included in LS spectrum.
Figure 1.
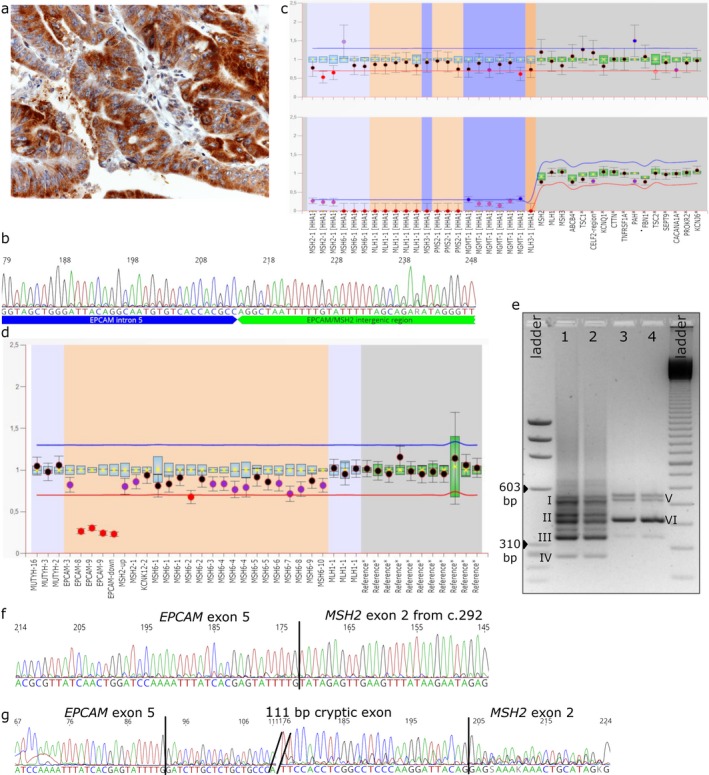
Representative molecular characterization of patient CFS396 carrying the EPCAM Del_16.5. (a) Immunohistochemistry showing that MSH2 protein is exclusively expressed in the cytoplasm of the tumor cells (H&E counterstain, O.M. 400x). (b) Sanger sequence of breakpoint. (c) Coffalyser analysis of methylation specific‐MLPA in tumor DNA, displaying MSH2 promoter methylation. (d) Coffalyser analysis of MLPA in tumor DNA, in which partial loss of heterozygosity of EPCAM is evidenced, due to a large somatic deletion involving also MSH2 and MSH6 genes. (e) Agarose‐gel electrophoresis showing aberrant EPCAM/MSH2 fusion transcripts amplified from tumor (lane 1–2) and normal mucosa (lanes 3–4) cDNAs; DNA ladders: ΦX174 DNA‐Hae III Digest (left) and 100 bp ladder (right). Roman numerals indicate the main PCR products that were sequenced. (f) Sanger sequence of an in‐frame fusion transcript (corresponding to amplicon III). (g) Sanger sequence of an out‐of‐frame fusion transcript (amplicon I and V)
Twenty relatives from four families were tested for EPCAM deletion: eight healthy subjects (age 19–57 years) and four patients affected by early‐onset CRC (age 25–45 years) tested positive at MLPA for the deletion detected in the proband. Clinical and molecular data of all carriers are listed in Table 1.
Long range PCRs from carriers produced smaller amplicons compared with wild‐type controls. Characterization of the breakpoints (Figure S1) revealed four different deletions, Del_4.9, Del_16.5 (Figure 1b), Del_2.6, Del_11.5 (Table 2), all residing in repetitive sequences and involving two Alu regions with very high homology (79%–88%); a microhomology sequence (from 9 to 36 bp) was identified for all the described deletions. Families AV114 and UD18 carried the same deletion.
Table 2.
Summary of EPCAM deletions
| Family ID | Deletion ID | Deleted EPCAM exons | Annotationa | Distance from MSH2 (kb) | Size (bp) | Alu family | Microhomology region (bp) | Max sequence homology (%) |
|---|---|---|---|---|---|---|---|---|
| PD31 | Del_4.9 | 8‐9 | c.859–1430_*2033del | 13 | 4,909 | Sx/Sq2 | 14 | 84% for 211/250 |
|
AV114 UD18 |
Del_16.5 | 6‐9 | c.556–490_*8438del | 7 | 16,588 | Sp/Sx | 9 | 82% for 196/239 |
| AV182 | Del_2.6 | 8‐9 | c.859–670_*530del | 15 | 2,648 | Sx1/Sx4 | 19 | 79% for 246/312 |
| PD78 | Del_11.5 | 8‐9 | c.858 + 1193_*5826del | 10 | 11,492 | Sp/Sp | 36 | 88% for 260/296 |
EPCAM reference sequence NM_002354.2.
Mismatch Repair gene methylation status was ascertained in five tumors (CFS395T, CFS396T, CFS825T, CFS1043T, CFS1475T) and two normal mucosae (CFS395N and CFS396N). MSH2 promoter was methylated, at different percentages (23%–50%), in both normal and tumor tissues (Table 1 and Figure 1c).
No pathogenic variant acting as a second hit was found by NGS screening in either MSH2 or other MMR genes in the DNA from CFS395T and CFS396T tumors. MLPA performed on available tumor samples highlighted a variegate pattern ranging from retention of the second EPCAM allele to LOH of various extent and amplitude (up to MSH2 and MSH6 loci) (Table 1 and Figure 1d).
Several EPCAM‐MSH2 fusion transcripts were detected by PCR in normal mucosa and in CRC samples CFS395T, CFS396T and CFS1475T (Table S1 and Figure 1e), but not in wild‐type controls (not shown).
Sequencing of PCR products evidenced at least five different aberrant transcripts originated from Del_16.5: three in‐frame and two creating a frameshift with a premature stop codon in exon 2 (Figure 1f‐g shows a representative case). Also Del_4.9 and Del_11.5 yielded different in‐frame and out‐of‐frame EPCAM/MSH2 fusion transcripts (Table 1).
4. DISCUSSION
An increasing body of evidence supports the relevance of epigenetic modifications in the pathogenesis of hereditary syndromes. The elucidation of the primary event causing the epigenetic change is crucial to ascertain the actual secondary nature of the epimutation and to optimize cascade testing in family members. In the context of LS testing, the importance of epimutations analysis is universally recognized and EPCAM deletion is a well‐known primary event. In the last 10 years many different variants of this gene have been described; however, construction of a complete and curated public database for EPCAM deletions is still in progress (https://databases.lovd.nl/shared/genes/EPCAM), and the actual frequency of EPCAM mutations in LS is poorly defined. In particular, no precise estimate of EPCAM variant incidence in the Italian LS population is available so far. Our institutional database includes 224 molecularly confirmed LS unrelated families, five of which with EPCAM deletion (>2%), a frequency consistent with literature data (Tutlewska et al., 2013).
All EPCAM deletions reported worldwide are bona fide Alu recombination‐mediated variants, as they are the four deletions presented here. With the exception of few founder mutations identified in the Netherlands and Poland (Dymerska et al., 2017; Niessen et al., 2009), in general deletions in the EPCAM gene are rare, sometimes private, variants with no recombination hotspot sites. Two of the four deletions (Del_16.5 and Del_11.5) that we describe here are novel, not recorded in any databases or literature. The Del_2.6 variant was identical to that reported in the Netherlands (Kuiper et al., 2011). The Del_4.9 variant was first described as founder mutation in Dutch population and in the large American Family R (Kuiper et al., 2011; Ligtenberg et al., 2009; Lynch et al., 2011; Niessen et al., 2009). Interestingly, the Del_4.9 variant described here and that previously reported (Ligtenberg et al., 2009) (GenBank: FJ347525.1) had different breakpoints, although involving the same Alu sequences, and our family had no known Dutch ancestry.
For three out of four deletions, we had evidence in both normal and tumor colon tissues, of EPCAM/MSH2 fusion transcripts. These were concomitant to MSH2 promoter methylation, which acted as a first hit for gene inactivation.
In all EPCAM deleted tumors analyzed, MSH2/MSH6 IHC nuclear expression was always abnormal. In three cases the loss of nuclear MSH2 staining was compatible with a large somatic deletion of the locus. A peculiarity was the MSH2 cytoplasmic staining detected in two cases. This aberrant localization is similar to that described in a previous report of a larger deletion involving both EPCAM and the first two exons of MSH2 (Sekine et al., 2017), and is consistent with the production of the alternative in‐frame fusion transcripts, predicted to be translated into chimeric proteins. It is tempting to speculate that the fusion with EPCAM alters the intrinsic capability of MSH2 to form a functional complex with MSH6, which is responsible for MSH2/MSH6 nuclear shuttling (Gassman et al., 2011).
Somatic EPCAM/MMR second hits could be identified only in three out of six EPCAM deletion‐associated tumors. The sub‐optimal quality of FFPE DNA from samples CFS825T and CFS1475T prevented NGS testing, so somatic MSH2 point‐mutations cannot be excluded. Instead, MSH2 deficiency remains unexplained in sample CFS395T, in which both MLPA and NGS analyses were performed, but neither LOH nor DNA sequence variants were detected.
EPCAM deletion carriers typically show high penetrance of early onset CRC and a CRC cumulative risk up to 75%; compared to MSH2 mutation carriers rarely develop multiple tumors of different histotypes. Moreover, the cumulative risk of EC in EPCAM deletion carriers is lower (12% vs. 51%), although this risk is slightly increased for deletions more proximal to the MSH2 locus (Kempers et al., 2011). According to previous genotype‐phenotype correlations, even in our small dataset EC is absent and the most frequent tumor is CRC, with 16 CRCs in eight affected carriers. Notably, one duodenal tumor was also recorded, confirming a previous suggestion of increased risk (Kempers et al., 2011), but the data are insufficient for correlating this clinical phenotype to particular EPCAM genotypes. Given the presence of multiple metachronous CRCs, the gastro‐intestinal prevention protocols for EPCAM deletion carriers should be focused on colon‐rectum, but surveillance of small intestine is also warranted. It is worth noting that EC occurrence, even if rare, cannot be excluded, and the gynecologic surveillance is still advised for all women with an EPCAM deletion.
This report underscores the importance of EPCAM genetic screening in LS patients, especially in the presence of an atypical pattern of MSH2 protein expression in tumors. In perspective, a thorough molecular characterization of the breakpoints might help in genetic counseling by laying down the basis for improved tumor risk estimates.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
Study supported by Associazione Federica per la Vita and the Italian Ministry of Health, Ricerca Corrente, Line 1, Project “Genetics of hereditary/familial tumors”. The authors thank Mauro Cassaro (Pathology, AULSS6 Euganea Regione Veneto) for his contribution on IHC.
Cini G, Quaia M, Canzonieri V, et al. Toward a better definition of EPCAM deletions in Lynch Syndrome: Report of new variants in Italy and the associated molecular phenotype. Mol Genet Genomic Med. 2019;7:e587 10.1002/mgg3.587
REFERENCES
- Cini, G. , Carnevali, I. , Quaia, M. , Chiaravalli, A. M. , Sala, P. , Giacomini, E. , … Viel, A. (2015). Concomitant mutation and epimutation of the MLH1 gene in a Lynch syndrome family. Carcinogenesis, 36(4), 452–458. 10.1093/carcin/bgv015 [DOI] [PubMed] [Google Scholar]
- Dymerska, D. , Gołębiewska, K. , Kuświk, M. , Rudnicka, H. , Scott, R. J. , & Billings, R. , … Kurzawski, G. (2017). New EPCAM founder deletion in Polish population. Clinical Genetics, 92(6), 649–653. 10.1111/cge.13026 [DOI] [PubMed] [Google Scholar]
- Eguchi, H. , Kumamoto, K. , Suzuki, O. , Kohda, M. , Tada, Y. , Okazaki, Y. , & Ishida, H. (2016). Identification of a Japanese Lynch syndrome patient with large deletion in the 3’ region of the EPCAM gene. Japanese Journal of Clinical Oncology, 46(2), 178–184. 10.1093/jjco/hyv172 [DOI] [PubMed] [Google Scholar]
- Gassman, N. R. , Clodfelter, J. E. , McCauley, A. K. , Bonin, K. , Salsbury, F. R. , & Scarpinato, K. D. (2011). Cooperative nuclear localization sequences lend a novel role to the N‐terminal region of MSH6. PLoS ONE, 6(3), e17907 10.1371/journal.pone.0017907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchins, M. P. (2013). The role of epigenetics in Lynch syndrome. Familial Cancer, 12(2), 189–205. 10.1007/s10689-013-9613-3 [DOI] [PubMed] [Google Scholar]
- Kempers, M. J. E. , Kuiper, R. P. , Ockeloen, C. W. , Chappuis, P. O. , Hutter, P. , Rahner, N. , … Ligtenberg, M. J. L. (2011). Risk of colorectal and endometrial cancers in EPCAM deletion‐positive Lynch syndrome: A cohort study. The Lancet Oncology, 12(1), 49–55. 10.1016/S1470-2045(10)70265-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, M. E. , Papp, J. , Szentirmay, Z. , Otto, S. , & Olah, E. (2009). Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Human Mutation, 30(2), 197–203. 10.1002/humu.20942 [DOI] [PubMed] [Google Scholar]
- Kuiper, R. P. , Vissers, L. E. L. M. , Venkatachalam, R. , Bodmer, D. , Hoenselaar, E. , Goossens, M. , … Ligtenberg, M. J. L. (2011). Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Human Mutation, 32(4), 407–414. 10.1002/humu.21446 [DOI] [PubMed] [Google Scholar]
- Ligtenberg, M. J. L. , Kuiper, R. P. , Chan, T. L. , Goossens, M. , Hebeda, K. M. , Voorendt, M. , … Hoogerbrugge, N. (2009). Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nature Genetics, 41(1), 112–117. 10.1038/ng.283 [DOI] [PubMed] [Google Scholar]
- Lynch, H. T. , Lynch, P. M. , Lanspa, S. J. , Snyder, C. L. , Lynch, J. F. , & Boland, C. R. (2009). Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clinical Genetics, 76(1), 1–18. 10.1111/j.1399-0004.2009.01230.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, H. T. , Riegert‐Johnson, D. L. , Snyder, C. , Lynch, J. F. , Hagenkord, J. , Boland, C. R. , … Ligtenberg, M. J. L. (2011). Lynch syndrome‐associated extracolonic tumors are rare in two extended families with the same EPCAM deletion. The American Journal of Gastroenterology, 106(10), 1829–1836. 10.1038/ajg.2011.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mur, P. , Pineda, M. , Romero, A. , del Valle, J. , Borràs, E. , Canal, A. , … Capellá, G. (2014). Identification of a founder EPCAM deletion in Spanish Lynch syndrome families. Clinical Genetics, 85(3), 260–266. 10.1111/cge.12152 [DOI] [PubMed] [Google Scholar]
- Niessen, R. C. , Hofstra, R. M. W. , Westers, H. , Ligtenberg, M. J. L. , Kooi, K. , Jager, P. O. J. , … Sijmons, R. H. (2009). Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes, Chromosomes & Cancer, 48(8), 737–744. 10.1002/gcc.20678 [DOI] [PubMed] [Google Scholar]
- Sekine, S. , Ogawa, R. , Saito, S. , Ushiama, M. , Shida, D. , Nakajima, T. , … Sugano, K. (2017). Cytoplasmic MSH2 immunoreactivity in a patient with Lynch syndrome with an EPCAM–MSH2 fusion. Histopathology, 70(4), 664–669. 10.1111/his.13104 [DOI] [PubMed] [Google Scholar]
- Spaepen, M. , Neven, E. , Sagaert, X. , De Hertogh, G. , Beert, E. , Wimmer, K. , … Brems, H. (2013). EPCAM germline and somatic rearrangements in lynch syndrome: Identification of a novel 3′EPCAM deletion. Genes, Chromosomes and Cancer, 52(9), 845–854. 10.1002/gcc.22080 [DOI] [PubMed] [Google Scholar]
- Tutlewska, K. , Lubinski, J. , & Kurzawski, G. (2013). Germline deletions in the EPCAM gene as a cause of Lynch syndrome – literature review. Hereditary Cancer in Clinical Practice, 11(1), 9 10.1186/1897-4287-11-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials