

Abstract
Background
Autosomal recessive congenital ichthyoses (ARCI) have been associated with different phenotypes including: harlequin ichthyosis (HI), congenital ichthyosiform erythroderma (CIE), and lamellar ichthyosis (LI). While pathogenic variants in all ARCI genes are associated with LI and CIE phenotypes, the unique gene associated with HI is ABCA12. In HI, the most severe ARCI form, pathogenic variants in both ABCA12 gene alleles usually have a severe impact on protein function. The presence of at least one non‐truncating variant frequently causes a less severe congenital ichthyosis phenotype (LI and CIE).
Methods
We report the case of a 4‐year‐old Ecuadorian boy with a severe skin disease. Genetic diagnosis was performed by NGS. In silico predictions were performed using Alamut software v2.11. A review of the literature was carried out to identify all patients carrying ABCA12 splice‐site and missense variants, and to explore their genotype‐phenotype correlations.
Results
Genetic testing revealed a nonsense substitution, p.(Arg2204*), and a new missense variant, p.(Val1927Leu), in the ABCA12 gene. After performing in silico analysis and a comprehensive review of the literature, we conclude that p.(Val1927Leu) affects a well conserved residue which could either disturb the protein function or alter the splicing process, both alternatives could explain the severe phenotype of our patient.
Conclusion
This case expands the spectrum of ABCA12 reported disease‐causing variants which is important to unravel genotype‐phenotype correlations and highlights the importance of missense variants in the development of HI.
Keywords: ABCA12 gene, Autosomal recessive congenital ichthyoses (ARCI), congenital ichthyosiform erythroderma (CIE), harlequin ichthyosis (HI), lamellar ichthyosis (LI), splice‐site pathogenic variant
1. INTRODUCTION
Autosomal recessive congenital ichthyoses (ARCIs) are a heterogeneous disease that can present with a wide range of phenotypes including harlequin ichthyosis, (HI), congenital ichthyosiform erythroderma (CIE), and lamellar ichthyosis (LI). HI is the most severe form of congenital ichthyoses (Fischer, 2009; Oji et al., 2010). Neonates are born encased in a thick skin that not only restricts their movements, but also distorts their facial features, averting their lips and eyelids. Although newborns frequently die within the first few days of life, some of them survive, and their skin eventually resembles severe CIE or LI. ARCI is a genetically heterogeneous condition that can be caused by pathogenic variants in at least 12 genes including TGM1 (OMIM #190195), ABCA12 (OMIM #607800), NIPAL4 (OMIM #609383), CYP4F22 (OMIM #611495), ALOX12B (OMIM #603741), ALOXE3 (OMIM #607206), LIPN (OMIM #613924), PNPLA1 (OMIM #612121), CERS3 (OMIM #615276), SDR9C7 (OMIM #609769), SULT2B1 (OMIM #604125), and CASP14 (OMIM #605848) (Fischer, 2009; Grall et al., 2012; Heinz et al., 2017; Kirchmeier, Zimmer, Bouadjar, Rösler, & Fischer, 2017; Lefèvre et al., 2003, 2006; Radner et al., 2013; Shigehara et al., 2016).
ABCA12 encodes a keratinocyte‐associated lipid transporter. Pathogenic variants in ABCA12 are known to cause the three major phenotypes of ARCI: HI, LI, and CIE. Genotype‐phenotype correlations have been established in ABCA12 associated disorders: homozygotes or compound heterozygotes with truncating ABCA12 variants generally lead to an HI phenotype while homozygous missense variants usually cause a milder phenotype (Akiyama, 2010).
Here we report a boy suffering from HI with compound heterozygous disease‐causing variants in ABCA12, one truncating mutation: nonsense variant c.6610C>T, p.(Arg2204*), and a novel missense variant, not previously reported: c.5779G>T, p.(Val1927Leu). The location of the new disease‐causing variant (first nucleotide of exon 39) suggests it can potentially alter the splicing process. In order to understand the effect of ABCA12 splice‐site and missense pathogenic variants, a literature search was performed.
2. CASE REPORT
The patient is a 4‐year‐old boy who was the third child of apparently non consanguineous parents from Manta, Manabí, Ecuador. There was no family history of congenital ichthyosis. Gestational age was approximately 7 months. After delivery the baby was placed in an incubator, where he spent one month. His mother mentioned that at birth he had several characteristics related to a harlequin fetus: thick large fissures over the whole body, flattened nose and ears, respiratory distress and feeding difficulties that required supplemental tube feeding; although he suffered from these complications he was able to breastfeed when he left the hospital. He also had toe blisters soon after birth that converted in toes synechia, affecting his gait. During the neonatal period the patient only received topical treatments.
Physical examination revealed: ectropion, eclabium, nasal hypoplasia, rudimentary external ears, dental hypoplasia, erythema, inflammation of the gums, and almost complete alopecia (Figure 1a). He presented generalized scales on an erythrodermal background with abundant fissures (Figure 1c). Upper‐extremities showed a high degree of retraction at finger joints, giving a claw hand aspect (Figure 1d). There were nail deformities, abundant fissures in bending sites and palmoplantar hyperkeratosis (Figure 1b). During the clinical examination the patient showed sensitivity and irritability, due to the pain caused by the fissures, when he moved. After obtaining informed consent, blood extraction was performed in the affected child, his parents, and his healthy sisters. Genomic DNA was isolated from peripheral blood cells using standard procedures in the Biomolecular Laboratory located in Cuenca, Ecuador and sent to the Fundación Pública Galega de Medicina Xenómica in Spain, where genetic diagnosis was carried out. Ethical approval was obtained and all research was performed in accordance with the principles of the Declaration of Helsinki. Three micrograms of patient's genomic DNA were enriched using SureSelect (Agilent Technologies) following the manufacturer's protocol. The target resequencing library was then sequenced on a SOLiD 5500xl (Life Technologies). Color space reads were mapped to the GRCh37/hg19 reference genome using LifeScope software version 2.5.1 (Life Technologies). Finally, variants were identified using GATK version 2.1 (Genome Analysis Toolkit, Broad Institute) and LifeScope version 2.5.1 and annotated with ANNOVAR version 2012Mar08. In silico prediction of potential variant effects on splicing were computed by using MaxEnt, NNSPLICE, and Splice Site Finder. Missense prediction analyses were performed by using Align GV‐GD, SIFT, and Mutation Taster. All these algorithms are integrated in the Alamut® Visual 2.11 software (Interactive Biosoftware, Rouen, France). The review of the existing literature on splice‐site and missense ABCA12 mutations was carried out by taking into consideration each of all carrier patients reported to date.
Figure 1.
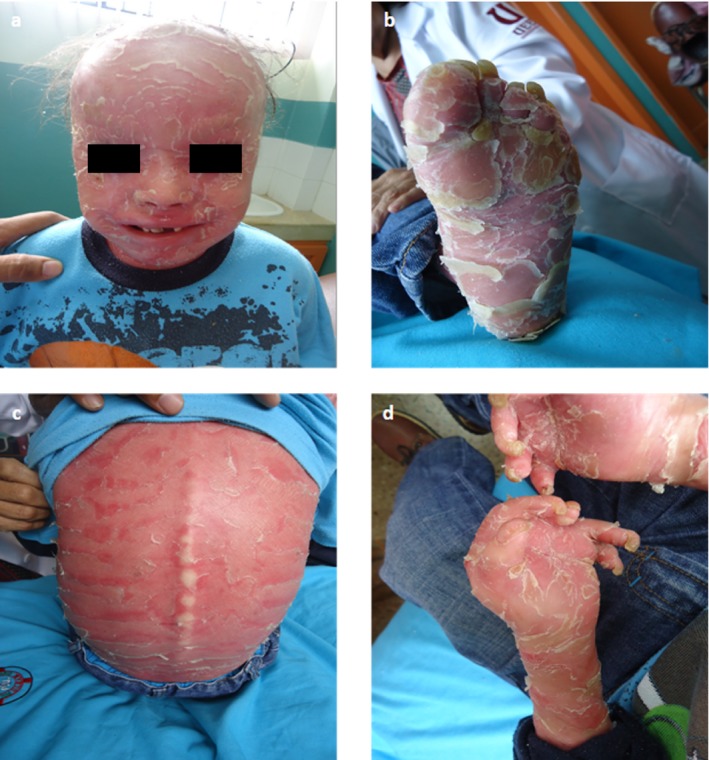
Clinical features of the patient: (a) Severe ectropion and almost complete alopecia, (b) Nail deformities and palmoplantar hyperkeratosis of the feet, (c) Patient's back showing large scales on an erythrodermic background, (d) Upper extremities severely affected. Retraction at finger joints
3. RESULTS
A total of 18 variants were identified in the patient's ABCA12 gene (NM 173076.2, NP_775099). Sixteen were filtered out while two putative ABCA12 variants in heterozygous state were prioritized by its location in the gene, the type of change they originated, and the frequency in 1000G project (http://www.1000genomes.org/): (a) a transition from C to T in exon 44: c.6610C>T; p.(Arg2204*) that leads to a nonsense substitution; it is located in the second transmembrane domain of the ABCA12 protein (Figure 2b,c). It has been previously identified in homozygous state in an African American patient that was born at 36 weeks of gestation, and died at 6 months of age from septicemia (Kelsell et al., 2005), (b) a transversion from G to T in exon 39: c.5779G>T; p.(Val1927Leu) that leads to a new missense substitution in a highly conserved amino acid Val1927 (Figure 2b,c). This novel variant, previously reported neither in HGMD nor Clinvar nor GnomAD, is located one nucleotide upstream of the canonical splicing acceptor site. The variant was predicted to have a deleterious effect (Align GV‐GD: Class C25, SIFT: Deleterious, Mutation‐Taster: Disease causing) and to also affect the splicing process (a total decrease in the score of the natural acceptor site of 59.0%, MaxEnt: −41.6%, NNSPLICE: −76.4%, and Splice Site Finder:‐8.5%). Taking all the evidence together, we classified ABCA12: c.5779G>T; p. (Val19227Leu) as likely pathogenic according to ACMG guidelines (Richards et al., 2015).
Figure 2.
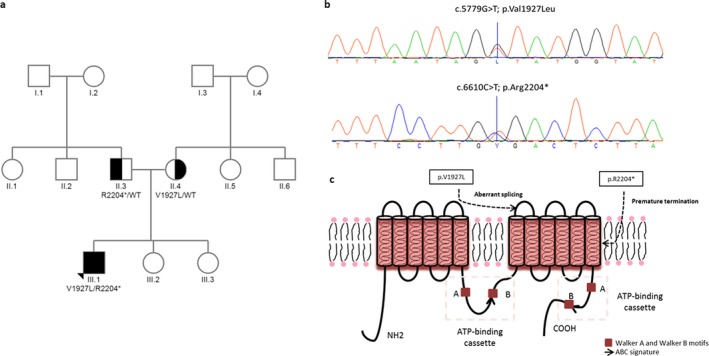
Pedigree of patient's family, electropherograms of both mutations and their location in the ABCA12 protein. (a) The patient (III:1) was a compound heterozygote for two ABCA12 mutations, a novel splice site mutation p.(Val1927Leu) and the nonsense mutation p.(Arg2204*). His parents were heterozygous carriers, (b) Electropherograms of both heterozygous mutations identified in the proband, (c) Representation of the ABCA12 protein structure and the location of the two identified mutations
Segregation analysis of the variants in the family shows that the father of the patient is carrier of the ABCA12 c.6610C>T mutation, and the mother of the c.5779G>T mutation. None of the sisters are the carriers of any of these variants (Figure 2a).
4. DISCUSSION
Pathogenic variants in ABCA12 have been described in ARCI including HI, CIE, and LI. HI shows the most severe phenotype, associated exclusively with ABCA12 mutations. Homozygous or compound heterozygous missense ABCA12 variants are frequently linked to LI and to a lesser extent CIE, whereas the majority of pathogenic variants associated with HI are homozygous or compound heterozygous nonsense and frameshift substitutions. Missense variants in combination with truncating mutations, including splice‐site variants, can be found in both CIE and HI (Akiyama, 2010). In this report we describe an Ecuadorian HI patient who harbors two different types of mutations in ABCA12. One is a nonsense variant which creates a premature codon. The second variant leads to a missense substitution in a conserved residue of the protein that is predicted to alter the splicing process by different algorithms. As both missense and splice‐site variants could lead to HI, any of these mechanisms could be affecting the pathogenicity of the variant. To better understand the implication of these type of variants in the development of the different ARCI subtypes, we performed a literature review of all ABCA12 missense and splice‐site mutation carrier patients and their associated phenotypes. Thirty patients carrying ABCA12 splice‐site variants were found (Table 1). Seven are homozygous carriers, and from these, those with pathogenic variants affecting the consensus splice‐sites and its surroundings, are classified as HI (patients 7, 11, 12, 18, 25, and 27) (Akiyama et al., 2005; Goldsmith et al., 2013; Hellström‐Pigg et al., 2016; Kelsell et al., 2005; Sheth, Bhavsar, Patel, Joshi, & Sheth, 2018; Thomas et al., 2008). Only two HI patients were compound heterozygous carriers of two different splice‐site variants (patients 24 and 28) (Esperón‐Moldes et al., 2018; Washio et al., 2017). Interestingly, the homozygous carrier of the synonymous variant c.3456G>A, p.(Ser1152=) (patient 10), located in the middle of the exon 24, shows a CIE phenotype. This milder phenotype could be explained by the fact that this mutation does not alter a consensus site but deregulates the expression of common transcripts; in this case a decrease in the expression of the wild type transcript and an increase in one minor transcript is observed (Goldsmith et al., 2013). Ten out of the 30 patients were compound heterozygous carriers of one ABCA12 splice‐site variant affecting the consensus splice‐site and a second truncating variant including eight nonsense (patients 1, 4, 8, 9, 13, 17, 22, and 30) and two frameshift (patients 5 and 23); (Akiyama et al., 2005, 2007; Diociaiuti et al., 2016; Hellström‐Pigg et al., 2016; Kelsell et al., 2005; Loo, Batilando, Tan, & Koh, 2018; Scott et al., 2013; Takeichi, Sugiura, Matsuda, Kono, & Akiyama, 2013; Thomas et al., 2006; Tourette et al., 2012; Umemoto et al., 2011) all these patients were diagnosed with HI at birth (with exception of patient 13 of whom there was not available phenotypic information). However, there are still few data of patients carrying a combination of splice‐site and missense variants; from the eight patients reported to date, four showed CIE (patients 3, 15, 19, 26) (Bochner et al., 2017; Esperón‐Moldes et al., 2018; Fukuda et al., 2012), and one presented HI (patient‐16) (Hellström‐Pigg et al., 2016).
Table 1.
ARCI splice‐site variant carrier patients described to date and bioinformatic prediction of variant outcomes
| Patient | Splice‐site mutation | Location/predicted splicing defect | Splicing prediction scoresa | Status | Second mutation | Phenotype | Ethnicity | Sex | Observations | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MaxEnt | NNSPLICE | SSF | ||||||||||
| 1 | c.1062–3_1074del; p.(Leu355Lysfs*12) | Acceptor splice site of exon 10 (skip exon 10:−100%) | −100.0% | −100.0% | −100.0% | het | c.5005C>T; p.(Gln1669*) | HI | Japanese | Female | Systemic retinoids from postnatal day 6. Skin dramatically improved during infancy | Takeichi et al. (2013) |
| 2 | c.1287 + 2 T>G | Donor splice site of intron 11(skip of exon 11:−100%) | −100.0% | −100.0% | −100.0% | het | c.4139A>G; p.(Asn1380Ser) | Not specified | Spanish | Not reported | – | Esperón‐Moldes et al. (2018) |
| 3 | c.1287 + 2 T>G | Donor splice site of intron 11(skip of exon 11:−100%) | −100.0% | −100.0% | −100.0% | het | c.4139A>G; p.(Asn1380Ser) | CIE | Spanish | Male | Patient presented with small and whitish scales, erythroderma, and palmoplantar keratoderma | Esperón‐Moldes et al. (2018) |
| 4 | c.1782G>A; p.(Glu594=) | Exonic substitution exon 14 (change at donor site: −95.2%) | −100.0% | −90.3% | 15.7% | het | c.596G>A, p.(Trp199*) | HI‐like | Scandinavian | No reported | Patient presented with collodion membrane at birth, ectropion, anhidrosis, and palmoplantar keratoderma | Hellström‐Pigg et al. (2016) |
| 5 | c.2332 + 2 T>G | Donor splice site of intron 17 (skip exon 17:−100%) | −100.0% | −100.0% | −100.0% | het | Exon 8 deletion | HI | British | Female | Neonatal mild hypothermia. Treated with systemic retinoids. Alive at 4 years of age | Kelsell et al. (2005); Scott et al. (2013); Thomas et al. (2006) |
| 6 | c.3295–1G>A | Acceptor splice site of intron 23 (skip exon 24:−100%) | −100.0% | −100.0% | −100.0% | het | unknown | HI | Malaysian | No reported | – | Numata et al. (2015) |
| 7 | c.3295–2A>G | Acceptor splice site intron 23(skip exon 24:−100%) | −100.0% | −100.0% | −100.0% | hom | – | HI | Japanese | Male | Survived infancy. Alive at publication (expresses some mutated ABCA12 protein) | Akiyama et al. (2005) |
| 8 | c.3295–2A>G | Acceptor splice site intron 23 (skip exon 24:−100%) | −100.0% | −100.0% | −100.0% | het | c.5848C>T, p.(Arg1950*) | HI | Japanese | Male | Died 3 days after birth | Akiyama et al. (2005) |
| 9 | c.3295–2A>G | Acceptor splice site intron 23(skip exon 24:−100%) | −100.0% | −100.0% | −100.0% | het | c.4543C>T; p.(Arg1515*) | HI | Japanese | Female | Systemic retinoids. Improved clinical symptoms at the age of 1 year and 7 months | Umemoto et al. (2011) |
| 10 | c.3456G>A; p.(Ser1152=) | Exonic substitution exon 24 (creates a novel acceptor splice site with similar scores as native site) | – | – | – | hom | – | CIE | Arab muslims | Female | Closely related parents. Several additional members of the family with similar condition | Goldsmith et al. (2013) |
| 11 | c.3829 + 1G>A | Donor splice site intron 26 (skip of exon 26:−100%) | −100.0% | −100.0% | −100.0% | hom | – | HI | unknown ethnicity | Not reported | – | Thomas et al. (2008) |
| 12 | c.3829 + 1G>A | Donor splice site intron 26 (skip of exon 26:−100%) | −100.0% | −100.0% | −100.0% | hom | – | HI | Scandinavian | Not reported | Patient presented with collodion membrane at birth, ectropion, anhidrosis, and palmoplantar keratoderma | Hellström‐Pigg et al. (2016) |
| 13 | c.4579 + 5G>A | Substitution in intron 30 (Change at donor site: −58.5%) | −67.2% | −49.9% | −16.0% | het | c.459 T>G, p.(Tyr153*) | ARCI | Italian | Female | Alive (6 years‐old) at examination | Diociaiuti et al. (2016) |
| 14 | c.5125_5128del; p.(Asp1709Thrfs*4) | Exonic deletion exon 33 (skip of exon 33:−100%) | −100.0% | −100.0% | −100.0% | het | unknown | HI | Syrian | Not reported | – | Thomas et al. (2008) |
| 15 | c.5128 + 3A>G | Substitution in intron 33 (change at donor site:–80.5%) | −74.3% | −86.7% | −5.8% | het | c.4139A>G, p.(Asn1380 Ser) | CIE | Japanese | Male | Alive (4 months) at publication | Fukuda et al. (2012) |
| 16 | c.5128 + 3A>G | Substitution in intron 33 (change at donor site:–80.5%) | −74.3% | −86.7% | −13.4% | het | c.3265G>T, p.(Val1089Phe) | HI | Scandinavian | Not reported | Patient presented with collodion membrane at birth, ectropion, anhidrosis, and palmoplantar keratoderma | Hellström‐Pigg et al. (2016) |
| 17 | c.5129–1G>T | Acceptor splice site of intron 33 (skip of exon 34:−100%) | −100.0% | −100.0% | −100.0% | het | c.7444C>T, p.(Arg2482*) | Harlequin fetus | French | Female | The fetus died at 31 weeks and 5 days gestation | Tourette et al. (2012) |
| 18 | c.5381 + 3_5381+4del | Deletion close to the donor splice site of exon 34 (skip of exon 34:−100.0%) | −100.0% | −100.0% | −36.6% | hom | – | HI |
Irish/Polish mother Italian/German father |
Not reported | – | Thomas et al. (2008) |
| 19 | c.5381 + 5G>A | Substitution in intron 34 near donor consensus (change at donor site: −95.5%) | −100.0% | −91.0% | −16.5% | het | c.4139A>G; p.(Asn1380Ser) | CIE | Spanish | Male | 8 months old baby with small and whitish scales on an erythrodermic background | Esperón‐Moldes et al. (2018) |
| 20 | c.5690G>C; p.(Arg1897Thr) | Exonic substitution exon 37 (change at donor site:−85.3%) | −100.0% | −70.6% | −16.2% | het | unknown | HI | Eritrean/Jamaican | No reported | – | Thomas et al. (2008) |
| 21b | c.5778 + 2 T>C | Donor splice site of intron 38 (skip of exon 38:−100%) | −100.0% | −100.0% | −0.5% | het | c.2956C>T, p.(Arg986Trp) | Not specified | Palestinian Armenian and Palestinian Catholic | Male | The child presented congenital exfoliative erythroderma, hypotrichosis, severe nail dystrophy, and failure to thrive | Bochner et al. (2017) |
| 22 | c.5779G>T; p.(Val1927Leu) | Exonic substitution exon 39 (change at acceptor site: −59.0%) | −41.6% | −76.4% | −8.5% | het | c. 6610C>T, p.(Arg2204*) | HI | Ecuadorian | Male | Alive (4 years old) at publication | This report |
| 23 | c.5884G>A; p.(Gly1962Ser) | Exonic substitution exon 39(change at donor site: −99%) | −100.0% | −97.9% | −16.4% | het | c.6858del; p.(Phe2286Leufs*6) | HI | Chinese | Female | Alive (5 months old) at publication | Loo et al. (2018) |
| 24 | c.5884 + 4_5884+5 del | Deletion close to the donor splice site of exon 39 (skip of exon 39: −69.2%) | −55.5% | −83.0% | −7.2% | het | c.7239G>A; p.(Leu2413=) | HI | Japanese | Male | Alive (2 years old) at publication | Washio et al. (2017) |
| 25 | c.5939 + 4A>G | Substitution in intron 40, near donor consensus (change at donor site: −49.9% | −41.5% | −58.3% | −12.5% | hom | – | HI | Gujarati, Indian | Female | Alive newborn at examination. She succumbed to septicemia 4 days after birth | Sheth et al., (2018) |
| 26 | c.5940–1G>C | Acceptor splice site of intron 40 (skip of exon 41: −100.0%) | −100.0% | −100.0% | −100.0% | het | c.2956C>T, p.(Arg986Trp) | CIE | Japanese | Female | Alive (9 years old) at publication. Younger sister suffered from severe skin symptoms, complications, and died | Fukuda et al. (2012) |
| 27 | c.6233 + 1G>T | Donor splice site intron 42 (skip of exon 42: −100.0%) | −100.0% | −100.0% | −100.0% | hom | – | HI | Iranian | Not reported | Septicemia. Died at age 4 months | Kelsell et al. (2005) |
| 28 | c.6394–2A>G | Acceptor splice site of intron 43 (skip of exon 44:−100%) | −100.0% | −100.0% | −100.0% | het | c.7436G>A; p.(Arg2479Lys) | HI | Spanish | Female | Alive (9 years old) at examination. The patient shows a CIE phenotype. | Esperón‐Moldes et al. (2018) |
| 29 | c.7105–22_7105–4 del | Deletion in intron 47 close to the acceptor splice site of exon 48 (skip of exon 48:−100%). | −100.0% | −99.9% | −100.0% | het | c.6941 T>C; p.(Ile2314Thr) | Not specified | Spanish | Not reported | – | Esperón‐Moldes et al. (2018) |
| 24 | c.7239G>A; p.(Leu2413=) | Exonic substitution exon 48 (change at donor site: −51.2%) | −44.9% | −57.4% | −14.1% | het | c.5884 + 4_5884+5del | HI | Japanese | Male | Alive (2 years old) at publication | Washio et al. (2017) |
| 30 | c.7436G>A; p.(Arg2479Lys) | Exonic substitution exon 50 (change at donor site: −99.5%) | −100.0% | −99.1% | −15.5% | het | c.3746C>A, p.(Ser1249*) | HI | French | Male | Died soon after birth | Akiyama et al. (2007) |
| 28 | c.7436G>A; p.(Arg2479Lys) | Exonic substitution exon 50 (change at donor site: −99.5%) | −100.0% | −99.1% | −15.5% | het | c.6394–2A>G | HI | Spanish | Female | Alive (9 years old) at examination. The patient shows a CIE phenotype. | Esperón‐Moldes et al. (2018) |
GenBank reference sequence (NM 173076.2, NP_775099)
ARCI: autosomal recessive congenital ichthyosis; CIE: congenital ichthyosiform erythroderma; het: heterozygous; HI: harlequin ichthyosis; HI‐like: CIE patients with ultrastructural findings resembling those detected in previous HI case; hom: homozygous; SSF: Splice Site Finder.
Percentages of variation predicted by Alamut at consensus splice‐sites.
Note that this patient shows an atypical ARCI phenotype (including severe hair and nail manifestations) and he also carries two additional heterozygous mutations in the CAPN12 gene [c.1511C>A; p.(P504Q), c.1090_1129del; p.(Val364Lysfs*11)].
We also identified a total of sixty‐three ABCA12 missense carrier patients. As shown in Table 2, most of HI patients bear at least one truncating variant in one of the two alleles (Patients 32, 34, 35, 41, 42, 50, 67, 79, 80, 81, 83, 86, 89, 91–93) (Akiyama et al., 2006, 2007; Esperón‐Moldes et al., 2018; Hellström‐Pigg et al., 2016; Kelsell et al., 2005; Loo et al., 2018; Numata et al., 2015; Peterson, Lofgren, Bremmer, & Krol, 2013; Scott et al., 2013; Tanahashi, Sugiura, Sato, & Akiyama, 2016; Xie et al., 2016). Two HI patients were described as carriers of missense variants in both alleles (Patients 31 and 47), however, the variants identified in patient 31; ABCA12: c.130C>G; p.(Arg44Gly) and c.2033A>G p.(Asn678Ser) (Scott et al., 2013) could be not the causative variants assuming that almost all algorithms predict a non‐deleterious effect and considering that a heterozygous known TGM1 mutation: c.401A>G; p.(Tyr134Cys) was also detected in this same patient; in the case of patient 47, described as carrier of a pathogenic variant c.3535G>A; p.(Gly1179Arg) in homozygous state, the zigosity needs to be confirmed. Interestingly, we did not find any difference between the type of mutations in patients with moderate and severe HI phenotypes. As previously reported, CIE patients carry at least one missense variant in combination with other missense, nonsense, splice‐site and frameshift mutations, while almost all LI patients are carriers of missense mutations in both alleles. Exceptions are two LI cases (patients 57 and 64) who harbor nonsense and frameshift variants. Interestingly these two patients did not show a more severe phenotype compared to other LI patients who carried missense mutations in both alleles (Akiyama et al., 2008; Bučková et al., 2016; Chao, Aleshin, Goldstein, Worswick, & Hogeling, 2018; Esperón‐Moldes et al., 2018; Fukuda et al., 2012; Hellström‐Pigg et al., 2016; Israeli et al., 2013; Lefèvre et al., 2003; Loo et al., 2018; Murase et al., 2018; Natsuga et al., 2007; Nawaz et al., 2012; Numata et al., 2015; Sakai et al., 2009; Scott et al., 2013; Shimizu et al., 2013; Sitek et al., 2018; Thomas et al., 2008; Wada et al., 2017; Wakil et al., 2016). The majority of the genotype‐phenotype associations found in these patients are in accordance with the correlations previously established by Akiyama, with some few exceptions as previously stated (Akiyama, 2010).
Table 2.
ARCI missense variant carrier patients described to date and bioinformatic prediction of variant outcomes
| Patient | Missense mutation | Location in the protein | Predicted splicing defect | Missense prediction scores | Status | Second mutation | Phenotype | Ethnicity | Sex | Observations | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Align GV‐GDa | SIFT | Mutation taster | |||||||||||
| 31 | c.130C>G; p.(Arg44Gly) | – | None | C0 | D | P | het | c.2033A>G; p.(Asn678Ser) | HI | unknown ethnicity | Not reported | Mild HI phenotype. This patient also carries the TGM1 c.401A>G mutation | Scott et al. (2013) |
| 32 | c.179G>C; p.(Arg60Pro) | – | None | C0 | D | DC | het | c.1300C>T; p.(Arg434*) | HI | unknown ethnicity | Female | – | Scott et al. (2013) |
| 33 | c.1033A>C; p.(Thr345Pro) | – | None | C0 | T | P | hom | – | CIE | Japanese | Female | A 37‐year‐old woman with CIE accompanied by malignant melanoma | Natsuga et al. (2007) |
| 34 | c.1160G>A; p.(Ser387Asn) | – | None | C0 | T | P | het | c.4158_4160del; p.(Thr1387del) | HI | Japanese | Male | Moderate clinical severity | Akiyama et al. (2006 |
| 35 | c.1446A>C; p.(Glu482Asp) | – | None | C0 | T | P | het | c.7444C>T; p.(Arg2482*) | HI | unknown ethnicity | Not reported | – | Scott et al., 2013 |
| 31 | c.2033A>G; p.(Asn678Ser) | – | None | C0 | T | P | het | c.130C>G; p.(Arg44Gly) | HI | unknown ethnicity | Not reported | Mild HI phenotype. This patient also carries the TGM1 c.401A>G mutation | Scott et al., 2013 |
| 36 | c.2634C>G; p.(Phe878Leu) | – | None | C0 | T | DC | het | c.4139A>G; p.(Asn1380Ser) | CIE | Czech | Not reported | Fine, whitish scales, and generalized erythema | Bučková et al., 2016 |
| 37 | c.2638G>C; p.(Val880Leu) | – | None | C0 | D | DC | het | c.3673C>T; p.(Arg1225*) | ARCI | Caucasian | Female | 69 years old at the moment of study | Sitek et al. (2018) |
| 38 | c.2956C>T; p.(Arg986Trp) | – | None | C65 | D | DC | het | c.5940–1G>C | CIE | Japanese | Female | 9‐year‐old girl with generalized scales on an erythrodermic skin, mild ectropion, alopecia, and mild auricular malformation |
Fukuda et al. (2012);
Numata et al. (2015) |
| 39 | c.2956C>T; p.(Arg986Trp) | – | None | C45 | D | DC | hom | – | CIE | Japanese | Not reported | – | Numata et al. (2015) |
| 40 | c.2956C>T, p.(Arg986Trp) | – | None | C65 | D | DC | het | c.5778 + 2 T>C | ARCI | Palestinian Armenian and Palestinian Catholic | Male | CEE, hypotrichosis, severe nail dystrophy, FTT | Bochner et al. (2017) |
| 41 | c.3085G>A; p.(Glu1029Lys) | – | None | C55 | D | DC | het | c.859C>T; p.(Arg287*) | HI | Chinese | Not reported | – | Numata et al. (2015) |
| 42 | c.3265G>T; p.(Val1089Phe) | – | None | C45 | D | DC | het | c.5128 + 3A>G | HI | Scandinavian | Not reported | Patient presented with collodion membrane at birth, ectropion, anhidrosis, and PPK | Hellström‐Pigg et al. (2016) |
| 43 | c.3299 T>G; p.(Met1100Arg) | – | Predicted change at acceptor site 5 bps upstream: +0.5% | C0 | D | DC | het | c.7164dup; p.(Met2389Tyrfs*27) | CIE/HI | unknown ethnicity | Female | Intermediate phenotype between HI and CIE | Peterson et al. (2013) |
| 44 | c.3407G>A; p. p.(Gly1136Asp) | – | None | C0 | D | DC | het | c.5005C>T; p.(Gln1669*) | CIE | Japanese | Male | Fine, whitish scales on hyperkeratotic, erythrodermic skin, mild ectropion, and eclabium | Akiyama et al. (2008) |
| 45 | c.3470C>T; p.(Ser1157Leu) | TNM | None | C15 | D | DC | hom | – | LI | Saudi | – | Four affected members in the same family. They all showed PPK | Wakil et al. (2016) |
| 46 | c.3470C>T; p.(Ser1157Leu) | TNM | None | C15 | D | DC | het | unknown | CIE | Japanese | Not reported | – | Numata et al. (2015) |
| 47 | c.3535G>A; p.(Gly1179Arg) | TNM | None | C65 | D | DC | hom | – | HI | Hmong/Laotian | Not reported | Sepsis, FTT, corneal perforation, respiratory failure, developmental delay | Thomas et al. (2006) |
| 48 | c.3704G>C; p.(Trp1235Ser) | – | None | C65 | D | DC | het | c.5848C>T; p.(Arg1950*) | CIE | Japanese | Male | 6 years at the moment of the study | Sakai et al. (2009) |
| 36 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C0 | D | DC | het | c.2634C>G; p.(Phe878Leu) | CIE | Czech | Not reported | Fine, whitish scales, and generalized erythema | Bučková et al., 2016 |
| 49 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.5128 + 3A>G | CIE | Japanese | Male | Male born as a collodion baby, with whitish scales and generalized erythrodermic skin | Fukuda et al. (2012); Numata et al. (2015) |
| 50 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.4554G>A; p.(Trp1518*) | HI | Scandinavian | Not reported | Collodion membrane, ectropion, anhidrosis, and PPK | Hellström‐Pigg et al. (2016) |
| 51 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | hom | – | LI | Moroccan | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 52 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.4951G>A; p.(Gly1651Ser) | LI | Algeria | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 53 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | hom | – | LI | Algeria | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 54 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.4070C>A; p.(Ser1357*) | CIE/LI | unknown ethnicity | Female | – | Scott et al. (2013) |
| 55 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.1287 + 2 T>G | CIE | Spanish | Male | Small, whitish scales with eythroderma, PPK, PH, and altered sweating | Esperón‐Moldes et al. (2018) |
| 56 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | hom | – | CIE | Spanish | Female | Small, dark scales with alopecia and PPK | Esperón‐Moldes et al. (2018) |
| 57 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.3837_3838del; p.(Tyr1279*) | LI | Spanish | Female | Small, whitish scales with ectropion, alopecia, and PPK | Esperón‐Moldes et al. (2018) |
| 58 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | hom | – | CIE | Spanish | Female | Small, whitish scales with eythroderma, collodion membrane, PPK, PH, and altered sweating | Esperón‐Moldes et al. (2018) |
| 59 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.1287 + 2 T>G | Not specified | Spanish | Not reported | – | Esperón‐Moldes et al. (2018) |
| 60 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.178C>T; p.(Arg60*) | CIE | Spanish | Female | Big, whitish scales with eythroderma, collodion membrane, alopecia, ectropion, PPK, PH, and altered sweating | Esperón‐Moldes et al. (2018) |
| 61 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.5381 + 5G>A | CIE | Spanish | Male | Small, whitish scales with eythroderma, collodion membrane, PPK, PH, and altered sweating | Esperón‐Moldes et al. (2018) |
| 62 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.5641C>T; p.(Arg1881*) | CIE | Spanish | Male | Small, whitish scales with alopecia, ectropion, eythroderma, PPK, PH, and altered sweating | Esperón‐Moldes et al. (2018) |
| 63 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.6031del; p.(Glu2011Asnfs*17) | CIE | Japanese | Female | At birth, entire body surface covered with thick, gray scales on a background of erythrodermic skin | Murase et al. (2018) |
| 64 | c.4139A>G; p.(Asn1380Ser) | NBF1 | None | C45 | D | DC | het | c.4491_4493del3ins22 | LI | unknown ethnicity | Female | Large, brown plte‐like hyperkeratotic scales, PPK, and hyperlinearity of the trunk | Chao et al. (2018) |
| 65 | c.4142G>A; p.(Gly1381Glu) | NBF1 | None | C65 | D | DC | hom | – | LI | Morocco | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 66 | c.4481 T>C; p.(Ile1494Thr) | NBF1 | None | C25 | D | DC | het | Unknown | CIE | Japanese | Male | 42‐year‐old man with CIE and cutaneous squamous cell carcinoma | Natsuga et al. (2007) |
| 67 | c.4541G>A; p.(Arg1514His) | NBF1 | None | C0 | D | DC | het | c.4896del; p.(Ser1633Hisfs*30) | HI‐like | Scandinavian | Not reported | Collodion membrane, ectropion, anhidrosis, and PPK | Hellström‐Pigg et al. (2016) |
| 68 | c.4541G>A; p.(Arg1514His) | NBF1 | None | C0 | D | DC | hom | – | CIE | Japanese | Male | 52 years at the moment of the study | Sakai et al. (2009) |
| 69 | c.4541G>A; p.(Arg1514His) | NBF1 | None | C0 | D | DC | hom | – | LI | Mali | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 70 | c.4544G>A; p.(Arg1515Gln) | NBF1 | None | C0 | D | DC | het | c.4553G>A; p.(Trp1518*) | CIE | Jewish | Not reported | – | Israeli et al. (2013) |
| 71 | c.4615G>A; p.(Glu1539Lys) | NBF1 | None | C55 | D | DC | hom | – | LI | Algeria | Not reported | Milder form of ichthyosis with smaller and whitish scales | Lefèvre et al. (2003) |
| 72 | c.4676G>T; p.(Gly1559Val) | – | None | C65 | D | DC | hom | – | CIE | Pakistani | Not reported | Five affected members with small, fine scales, erythroderma, PPK, and mild ectropion. Legs showed brownish scales similar to LI | Nawaz et al. (2012) |
| 73 | c.4676G>T; p.Gly1559Val | – | None | C65 | D | DC | hom | – | ARCI | Pakistani | Female | 26 years old at the moment of study | Sitek et al. (2018) |
| 74 | c.4723A>C; p.(Thr1575Pro) | – | None | C0 | D | DC | het | c.6031del; p.(Glu2011Asnfs*17) | CIE | Japanese | Female | 3‐year‐old girl with generalised scales, erythroderma, ectropion, eclabium, severely deformed ears, and alopecia | Fukuda et al. (2012); Numata et al. (2015) |
| 75 | c.4723A>C; p.(Thr1575Pro) | – | None | C0 | D | DC | het | c.4951G>A; p.(Gly1651Ser) | CIE | Japanese | Male | 3‐month‐old boy born as a collodion baby, with generalized whitish scales on a erythrodermic skin | Fukuda et al. (2012); Numata et al. (2015) |
| 75 | c.4951G>A; p.(Gly1651Ser) | – | None | C55 | D | DC | het | c.4723A>C; p.(Thr1575Pro) | CIE | Japanese | Male | 3‐month‐old boy born as a collodion baby, with generalized whitish scales on a erythrodermic skin | Fukuda et al. (2012); Numata et al. (2015) |
| 76 | c.4951G>A; p.(Gly1651Ser) | – | None | C55 | D | DC | hom | – | LI | Algeria | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 52 | c.4951G>A; p.(Gly1651Ser) | – | None | C55 | D | DC | het | c.4139A>G; p.(Asn1380Ser) | LI | Algeria | Not reported | Collodion membrane, large dark scales, ectropion, and PPK | Lefèvre et al. (2003) |
| 77 | c.5393C>T; p.(Pro1798Leu) | – | None | C0 | D | DC | het | unknown | CIE | Japanese | Female | Less than one year at the moment of the study | Sakai et al. (2009) |
| 78 | c.5690G>C; p.(Arg1897Thr) | – | Exonic substitution exon 37 (change at donor site:−85.3%) | C65 | D | DC | het | unknown | HI | Eritrean/Jamaican | Not reported | – | Thomas et al. (2008) |
| 79 | c.5779G>T; p.(Val1927Leu) | – | Exonic substitution exon 39 (change at acceptor site: −59.0%) | C25 | D | DC | het | c.6610C>T, p.(Arg2204*) | HI | Ecuadorian | Male | Alive (4‐year‐old) at publication | This report |
| 80 | c.5884G>A; p.(Gly1962Ser) | – | Exonic substitution exon 39(change at donor site: −99%) | C55 | D | DC | het | c.6858del; p.(Phe2286Leufs*6) | HI | Chinese | Female | 5 months at publication. Severe HI phenotype. | Loo et al. (2018) |
| 81 | c.5936C>G; p.(Ala1979Gly) | – | None | C0 | D | DC | het | c.6858del; p.(Phe2286Leufs*6) | HI atypical | unknown ethnicity | Male | HI atypical, chrysalis | Scott et al. (2013) |
| 82 | c.5939C>A; p.(Thr1980Lys) | – | None | C0 | D | DC | het | unknown | CIE | Japanese | Female | One year at the moment of the study | Sakai et al. (2009) |
| 83 | c.5985G>A; p.(Met1995Ile) | TNM | Novel acceptor splice site with similar scores as native site | C0 | T | DC | het | c.1194_1221del; p.(Gln400Phefs*18) | HI | Japanese | Female | 2.5 years old at publication, clinical features typical of HI | Tanahashi et al. (2016) |
| 84 | c.6263 T>C; p.(Leu2088Pro) | TNM | None | C65 | D | DC | het | c.1002_1004delinsT; p.(Thr335Alafs*5) | CIE | Scandinavian | Not reported | – | Hellström‐Pigg et al. (2016) |
| 85 | c.6431 T>C; p.(Phe2144Ser) | – | None | C65 | D | DC | het | c.4139A>G; p.(Asn1380Ser) | CIE | Japanese | Female | A 5‐year‐old girl born as a collodion baby. Clinical features typical of CIE | Shimizu et al. (2013) |
| 86 | c.6443C>A; p.(Pro2148Gln) | – | None | C65 | D | DC | het | c.5232G>A; p.(Trp1744*) | HI | Chinese | Female | Typical HI fetus terminated with two more cases in the family | Xie et al. (2016) |
| 87 | c.6551A>T; p.(Asn2184Ile) | – | None | C55 | D | P | het | c.6696_6699dup; p.(Asp2234*) | CIE | Japanese | Female | Mild CIE with periodic exacerbation | Wada et al. (2017) |
| 88 | c.6900C>A; p.(Phe2300Leu) | NBF2 | None | C15 | D | DC | hom | – | LI | Saudi | Not reported | Large scales with erythroderma and keratoderma. | Wakil et al. (2016) |
| 89 | c.7093G>A; p.(Asp2365Asn) | NBF2 | None | C0 | D | P | het | c.5229del; p.(Trp1744Glyfs*24) | HI | Italian | Not reported | 6 years old at publication, nystagmus, PDA, neonatal sepsis | Kelsell et al. (2005) |
| 90 | c.7187G>C; p.(Arg2396Thr) | NBF2 | None | C65 | D | DC | het | c.986–719_1061+1902del; p.(Asp330Serfs*2) | ARCI | Caucasian | Male | Less than one year old at the moment of study. Osteopenia | Sitek et al. (2018) |
| 91 | c.7412G>A; p.(Gly2471Glu) | – | None | C65 | D | DC | het | c.7137del; p.(Met2380Cysfs*25) | HI‐like | Scandinavian | Not reported | Collodion membrane, ectropion, anhidrosis, and PPK | Hellström‐Pigg et al. (2016) |
| 92 | c.7436G>A; p.(Arg2479Lys) | – | Exonic substitution exon 50 (change at donor site: −99.5%) | C25 | D | DC | het | c.3746C>A, p.(Ser1249*) | HI | French | Male | Died soon after birth | Akiyama et al. (2007) |
| 93 | c.7436G>A; p.(Arg2479Lys) | – | Exonic substitution exon 50 (change at donor site: −99.5%) | C25 | D | DC | het | c.6394–2A>G | HI | Spanish | Female | Alive (9 years old) at examination. The patient now shows a CIE phenotype. | Esperón‐Moldes et al. (2018) |
GenBank reference sequence (NM 173,076.2, NP_775099)
ARCI: autosomal recessive congenital ichthyosis; CEE: congenital exfoliative erythroderma; CIE: congenital ichthyosiform erythroderma; D: deleterious; DC: disease‐causing; FTT: failure to thrive; het: heterozygous; HI: harlequin ichthyosis; HI‐like: CIE patients with ultrastructural findings resembling those detected in previous HI cases; hom: homozygous; LI: lamellar ichthyosis; P: polymorphism; PDA: patent ductus arteriosus; PH: palmar hiperlinearity; PPK: palmoplantar keratoderma; T: tolerated.
Align GV‐GD prediction classes form a spectrum (C0, C15, C25, C35, C45, C55, C65) with C65 most likely to interfere with function and C0 least likely.
Given the current available data, further characterization of missense variants, including the confirmation of the zigosity in putative homozygous patients and the assessment of their impact in the splicing process, would be needed to better elucidate this genotype‐phenotype correlation.
In brief, our case expands the spectrum of ABCA12 reported disease‐causing variants. Additionally the literature review of splice‐site and missense ABCA12 mutations performed in this study contributes to further understanding of the complex genotype‐phenotype correlations in the different subtypes of ARCI.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGMENTS
We would like to thank the patient and his family. U.Esperón Moldes was supported by a predoctoral fellowship from Xunta de Galicia.
Montalván‐Suárez M, Esperón‐Moldes US, Rodríguez‐Pazos L, et al. A novel ABCA12 pathologic variant identified in an Ecuadorian harlequin ichthyosis patient: A step forward in genotype‐phenotype correlations. Mol Genet Genomic Med. 2019;7:e608 10.1002/mgg3.608
Funding information
This work was partially supported by Ramón Areces Foundation project awarded to A.Vega, by Spanish Instituto de Salud Carlos III (ISCIII) (INT15/00,070, INT16/00,154, INT17/00,133) and Xunta de Galicia (IN607B) (to A. Vega) and by Universidad Espíritu Santo‐Ecuador given to M. Montalván‐Suárez.
Contributor Information
Manuel Ginarte, Email: mginartev@aedv.es.
Ana Vega, Email: ana.vega@usc.es.
REFERENCES
- Akiyama, M. (2010). ABCA12 mutations and autosomal recessive congenital ichthyosis: A review of genotype/phenotype correlations and of pathogenetic conceptsa. Human Mutation, 31(10), 1090–1096. 10.1002/humu.21326 [DOI] [PubMed] [Google Scholar]
- Akiyama, M. , Sakai, K. , Hatamochi, A. , Yamazaki, S. , McMillan, J. R. , & Shimizu, H. (2008). Novel compound heterozygous nonsense and missense ABCA12 mutations lead to nonbullous congenital ichthyosiform erythroderma. British Journal of Dermatology, 158(4), 864–867. 10.1111/j.1365-2133.2008.08439.x [DOI] [PubMed] [Google Scholar]
- Akiyama, M. , Sakai, K. , Sugiyama‐Nakagiri, Y. , Yamanaka, Y. , McMillan, J. R. , Sawamura, D. , … Shimizu, H. (2006). Compound heterozygous mutations including a de novo missense mutation in ABCA12 led to a case of harlequin ichthyosis with moderate clinical severity. Journal of Investigative Dermatology, 126(7), 1518–1523. 10.1038/sj.jid.5700295 [DOI] [PubMed] [Google Scholar]
- Akiyama, M. , Sugiyama‐Nakagiri, Y. , Sakai, K. , McMillan, J. R. , Goto, M. , Arita, K. , … Shimizu, H. (2005). Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. The Journal of Clinical Investigation, 115(7), 1777–1784. 10.1172/JCI24834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama, M. , Titeux, M. , Sakai, K. , McMillan, J. R. , Tonasso, L. , Calvas, P. , … Shimizu, H. (2007). DNA‐based prenatal diagnosis of harlequin ichthyosis and characterization of ABCA12 mutation consequences. Journal of Investigative Dermatology, 127(3), 568–573. 10.1038/sj.jid.5700617 [DOI] [PubMed] [Google Scholar]
- Bochner, R. , Samuelov, L. , Sarig, O. , Li, Q. , Adase, C. A. , Isakov, O. , … Sprecher, E. (2017). Calpain 12 function revealed through the study of an atypical case of autosomal recessive congenital ichthyosis. Journal of Investigative Dermatology, 137(2), 385–393. 10.1016/j.jid.2016.07.043 [DOI] [PubMed] [Google Scholar]
- Bučková, H. , Nosková, H. , Borská, R. , Réblová, K. , Pinková, B. , Zapletalová, E. , … Fajkusová, L. (2016). Autosomal recessive congenital ichthyoses in the Czech Republic. British Journal of Dermatology, 174(2), 405–407. 10.1111/bjd.13918 [DOI] [PubMed] [Google Scholar]
- Chao, K. , Aleshin, M. , Goldstein, Z. , Worswick, S. , & Hogeling, M. (2018). Lamellar ichthyosis in a female neonate without a collodion membrane. Dermatology Online Journal, 24(2), https://13030/qt24g7w9t8. [PubMed] [Google Scholar]
- Diociaiuti, A. , El Hachem, M. , Pisaneschi, E. , Giancristoforo, S. , Genovese, S. , Sirleto, P. , … Angioni, A. (2016). Role of molecular testing in the multidisciplinary diagnostic approach of ichthyosis Rare skin diseases. Orphanet Journal of Rare Diseases, 11(1), 1–12. 10.1186/s13023-016-0384-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esperón‐Moldes, U. , Ginarte, M. , Rodríguez‐Pazos, L. , Fachal, L. , Pozo, T. , Aguilar, J. L. , … Vega, A. (2018). ABCA12 mutations in patients with autosomal recessive congenital ichthyosis: Evidence of a founder effect in the Spanish population and phenotype‐genotype implications. Journal of Dermatological Science, 91(3), 328–331. 10.1016/j.jdermsci.2018.05.012 [DOI] [PubMed] [Google Scholar]
- Fischer, J. (2009). Autosomal recessive congenital ichthyosis. Journal of Investigative Dermatology, 129(6), 1319–1321. 10.1038/jid.2009.57 [DOI] [PubMed] [Google Scholar]
- Fukuda, S. , Hamada, T. , Ishii, N. , Sakaguchi, S. , Sakai, K. , Akiyama, M. , … Hashimoto, T. (2012). Novel adenosine triphosphate (ATP)‐binding cassette, subfamily A, member 12 (ABCA12) mutations associated with congenital ichthyosiform erythroderma. British Journal of Dermatology, 166(1), 218–221. 10.1111/j.1365-2133.2011.10516.x [DOI] [PubMed] [Google Scholar]
- Goldsmith, T. , Fuchs‐Telem, D. , Israeli, S. , Sarig, O. , Padalon‐Brauch, G. , Bergman, R. , … Nousbeck, J. (2013). The sound of silence: Autosomal recessive congenital ichthyosis caused by a synonymous mutation in ABCA12. Experimental Dermatology, 22(4), 251–254. 10.1111/exd.12110 [DOI] [PubMed] [Google Scholar]
- Grall, A. , Guaguère, E. , Planchais, S. , Grond, S. , Bourrat, E. , Hausser, I. , … Fischer, J. (2012). PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nature Genetics, 44(2), 140–147. 10.1038/ng.1056 [DOI] [PubMed] [Google Scholar]
- Heinz, L. , Kim, G. J. , Marrakchi, S. , Christiansen, J. , Turki, H. , Rauschendorf, M. A. , … Fischer, J. (2017). Mutations in SULT2B1 cause autosomal‐recessive congenital ichthyosis in humans. American Journal of Human Genetics, 100(6), 926–939. 10.1016/j.ajhg.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström‐Pigg, M. , Bygum, A. , Gånemo, A. , Virtanen, M. , Brandrup, F. , Zimmer, A. D. , … Fischer, J. (2016). Spectrum of autosomal recessive congenital ichthyosis in scandinavia: Clinical characteristics and novel and recurrent mutations in 132 patients. Acta Dermato‐Venereologica, 96(7), 932–937. 10.2340/00015555-2418 [DOI] [PubMed] [Google Scholar]
- Israeli, S. , Goldberg, I. , Fuchs‐Telem, D. , Bergman, R. , Indelman, M. , Bitterman‐Deutsch, O. , … Sprecher, E. (2013). Non‐syndromic autosomal recessive congenital ichthyosis in the Israeli population. Clinical and Experimental Dermatology, 38(8), 911–916. 10.1111/ced.12148 [DOI] [PubMed] [Google Scholar]
- Kelsell, D. P. , Norgett, E. E. , Unsworth, H. , Teh, M.‐T. , Cullup, T. , Mein, C. A. , … O’Toole, E. A. (2005). Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. American Journal of Human Genetics, 76(5), 794–803. 10.1086/429844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchmeier, P. , Zimmer, A. , Bouadjar, B. , Rösler, B. , & Fischer, J. (2017). Whole‐exome‐sequencing reveals small deletions in CASP14 in patients with autosomal recessive inherited ichthyosis. Acta Dermato‐Venereologica, 97(1), 102–104. 10.2340/00015555-2510 [DOI] [PubMed] [Google Scholar]
- Lefèvre, C. , Audebert, S. , Jobard, F. , Bouadjar, B. , Lakhdar, H. , Boughdene‐Stambouli, O. , … Fischer, J. (2003). Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Human Molecular Genetics, 12(18), 2369–2378. 10.1093/hmg/ddg235 [DOI] [PubMed] [Google Scholar]
- Lefèvre, C. , Bouadjar, B. , Ferrand, V. , Tadini, G. , Mégarbané, A. , Lathrop, M. , … Fischer, J. (2006). Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Human Molecular Genetics, 15(5), 767–776. 10.1093/hmg/ddi491 [DOI] [PubMed] [Google Scholar]
- Loo, B. K. G. , Batilando, M. J. , Tan, E. C. , & Koh, M. J. A. (2018). Compound heterozygous mutations with novel missense ABCA12 mutation in harlequin ichthyosis. BMJ Case Reports, bcr-2017-222025.10.1136/bcr-2017-222025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase, C. , Takeichi, T. , Sugiura, K. , Kobayashi, M. , Shiomi, K. , Ikebuchi, K. , … Akiyama, M. (2018). Hearing impairment: A secondary symptom in a congenital ichthyosiform erythroderma patient with ABCA12 mutations. Journal of Dermatology, 45, e303–e304. 10.1111/1346-8138.14350 [DOI] [PubMed] [Google Scholar]
- Natsuga, K. , Akiyama, M. , Kato, N. , Sakai, K. , Sugiyama‐Nakagiri, Y. , Nishimura, M. , … Shimizu, H. (2007). Novel ABCA12 mutations identified in two cases of non‐bullous congenital ichthyosiform erythroderma associated with multiple skin malignant neoplasia [3]. Journal of Investigative Dermatology, 127(11), 2669–2673. 10.1038/sj.jid.5700885 [DOI] [PubMed] [Google Scholar]
- Nawaz, S. , Tariq, M. , Ahmad, I. , Malik, N. A. , Baig, S. M. , Dahl, N. , & Klar, J. (2012). Non‐bullous congentital ichthyosiform erythroderma associated with homozygosity for a novel missense mutation in an ATP binding domain of ABCA12. European Journal of Dermatology, 22(2), 178–181. 10.1684/ejd.2011.1638 [DOI] [PubMed] [Google Scholar]
- Numata, S. , Teye, K. , Krol, R. P. , Karashima, T. , Fukuda, S. , Matsuda, M. , … Hashimoto, T. (2015). Mutation study for 9 genes in 23 unrelated patients with autosomal recessive congenital ichthyosis in Japan and Malaysia. Journal of Dermatological Science, 78(1), 82–85. 10.1016/j.jdermsci.2015.02.006 [DOI] [PubMed] [Google Scholar]
- Oji, V. , Tadini, G. , Akiyama, M. , Blanchet Bardon, C. , Bodemer, C. , Bourrat, E. , … Traupe, H. (2010). Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorze 2009. Journal of the American Academy of Dermatology, 63(4), 607–641. 10.1016/j.jaad.2009.11.020 [DOI] [PubMed] [Google Scholar]
- Peterson, H. , Lofgren, S. , Bremmer, S. , & Krol, A. (2013). Novel ABCA‐12 mutations leading to recessive congenital ichthyosis. Pediatric Dermatology, 30(6), 236–237. 10.1111/j.1525-1470.2011.01695.x [DOI] [PubMed] [Google Scholar]
- Radner, F. P. W. , Marrakchi, S. , Kirchmeier, P. , Kim, G. J. , Ribierre, F. , Kamoun, B. , … Fischer, J. . (2013). Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genetics, 9(6), e1003536– 10.1371/journal.pgen.1003536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, K. , Akiyama, M. , Yanagi, T. , McMillan, J. R. , Suzuki, T. , Tsukamoto, K. , … Shimizu, H. (2009). ABCA12 is a major causative gene for non‐bullous congenital ichthyosiform erythroderma. Journal of Investigative Dermatology, 129(9), 2306–2309. 10.1038/jid.2009.23 [DOI] [PubMed] [Google Scholar]
- Scott, C. A. , Plagnol, V. , Nitoiu, D. , Bland, P. J. , Blaydon, D. C. , Chronnell, C. M. , … Kelsell, D. P. (2013). Targeted sequence capture and high‐throughput sequencing in the molecular diagnosis of ichthyosis and other skin diseases. Journal of Investigative Dermatology, 133(2), 573–576. 10.1038/jid.2012.332 [DOI] [PubMed] [Google Scholar]
- Sheth, J. J. , Bhavsar, R. , Patel, D. , Joshi, A. , & Sheth, F. J. (2018). Harlequin ichthyosis due to novel splice site mutation in the ABCA12 gene: Postnatal to prenatal diagnosis. International Journal of Dermatology, 57(4), 428–433. 10.1111/ijd.13923 [DOI] [PubMed] [Google Scholar]
- Shigehara, Y. , Okuda, S. , Nemer, G. , Chedraoui, A. , Hayashi, R. , Bitar, F. , … Shimomura, Y. (2016). Mutations in SDR9C7 gene encoding an enzyme for vitamin A metabolism underlie autosomal recessive congenital ichthyosis. Human Molecular Genetics, 25(20), ddw277 10.1093/hmg/ddw277 [DOI] [PubMed] [Google Scholar]
- Shimizu, Y. , Sugiura, K. , Aoyama, Y. , Ogawa, Y. , Hitomi, K. , Iwatsuki, K. , & Akiyama, M. (2013). Novel ABCA12 missense mutation p.Phe2144Ser underlies congenital ichthyosiform erythroderma. The Journal of Dermatology, 40(7), 581–582. 10.1111/1346-8138.12169 [DOI] [PubMed] [Google Scholar]
- Sitek, J. C. , Kulseth, M. A. , Rypdal, K. B. , Skodje, T. , Sheng, Y. , & Retterstøl, L. (2018). Whole‐exome sequencing for diagnosis of hereditary ichthyosis. Journal of the European Academy of Dermatology and Venereology, 32(6), 1022–1027. 10.1111/jdv.14870 [DOI] [PubMed] [Google Scholar]
- Takeichi, T. , Sugiura, K. , Matsuda, K. , Kono, M. , & Akiyama, M. (2013). Novel ABCA12 splice site deletion mutation and ABCA12 mRNA analysis of pulled hair samples in harlequin ichthyosis. Journal of Dermatological Science, 69(3), 259–261. 10.1016/j.jdermsci.2012.11.004 [DOI] [PubMed] [Google Scholar]
- Tanahashi, K. , Sugiura, K. , Sato, T. , & Akiyama, M. (2016). Noteworthy clinical findings of harlequin ichthyosis: Digital autoamputation caused by cutaneous constriction bands in a case with novel ABCA12 mutations. British Journal of Dermatology, 174(3), 689–691. 10.1111/bjd.14228 [DOI] [PubMed] [Google Scholar]
- Thomas, A. C. , Cullup, T. , Norgett, E. E. , Hill, T. , Barton, S. , Dale, B. A. , … Kelsell, D. P. (2006). ABCA12 is the major harlequin ichthyosis gene. Journal of Investigative Dermatology, 126(11), 2408–2413. 10.1038/sj.jid.5700455 [DOI] [PubMed] [Google Scholar]
- Thomas, A. C. , Sinclair, C. , Mahmud, N. , Cullup, T. , Mellerio, J. E. , Harper, J. , … Kelsell, D. P. (2008). Novel and recurring ABCA12 mutations associated with harlequin ichthyosis: Implications for prenatal diagnosis. British Journal of Dermatology, 158(3), 611–613. 10.1111/j.1365-2133.2007.08277.x [DOI] [PubMed] [Google Scholar]
- Tourette, C. , Tron, E. , Mallet, S. , Levy‐Mozziconacci, A. , Bonnefont, J. P. , D’Ercole, C. , … Bretelle, F. (2012). Three‐dimensional ultrasound prenatal diagnosis of congenital ichthyosis: Contribution of molecular biology. Prenatal Diagnosis, 32(5), 498–500. 10.1002/pd.3839 [DOI] [PubMed] [Google Scholar]
- Umemoto, H. , Akiyama, M. , Yanagi, T. , Sakai, K. , Aoyama, Y. , Oizumi, A. , & Suga, Y. (2011). New insight into genotype/phenotype correlations in ABCA12 mutations in harlequin ichthyosis. Journal of Dermatological Science, 61(2), 136–138. 10.1016/j.jdermsci.2010.11.010 [DOI] [PubMed] [Google Scholar]
- Wada, Y. , Kusakabe, M. , Nagai, M. , Yamamoto, M. , Imai, Y. , Ide, Y. H. , … Yamanishi, K. (2017). Mild case of congenital ichthyosiform erythroderma with periodic exacerbation: Novel mutations in ABCA12 and upregulation of calprotectin in the epidermis. Journal of Dermatology, 44(11), e282–e283. 10.1111/1346-8138.13976 [DOI] [PubMed] [Google Scholar]
- Wakil, S. M. , Binamer, Y. , Al‐Dossari, H. , Al‐Humaidy, R. , Thuraya, R. A. , Khalifa, O. , … Al Owain, M. (2016). Novel mutations in TGM1 and ABCA12 cause autosomal recessive congenital ichthyosis in five Saudi families. International Journal of Dermatology, 55(6), 673–679. 10.1111/ijd.13279 [DOI] [PubMed] [Google Scholar]
- Washio, K. , Sumi, M. , Nakata, K. , Fukunaga, A. , Yamana, K. , Koda, T. , … Yamanishi, K. (2017). Case of harlequin ichthyosis with a favorable outcome: Early treatment and novel, differentially expressed, alternatively spliced transcripts of the ATP‐binding cassette subfamily A member 12 gene. Journal of Dermatology, 44(8), 950–953. 10.1111/1346-8138.13823 [DOI] [PubMed] [Google Scholar]
- Xie, H. , Xie, Y. , Peng, R. , Li, L. , Zhu, Y. , & Guo, J. (2016). Harlequin ichthyosis: A novel compound mutation of ABCA12 with prenatal diagnosis. Clinical and Experimental Dermatology, 41(6), 636–639. 10.1111/ced.12861 [DOI] [PubMed] [Google Scholar]