

Abstract
Background
Neurofibromatosis type 1 (NF1) is an autosomal dominant condition caused by inactivating mutations of the NF1 gene. The wide allelic heterogeneity of this condition, with more than 3,000 pathogenic variants reported so far, is paralleled by its high clinical variability, which is observed even within the same family. The definition of genotype–phenotype correlations has been hampered by the complexity of the NF1 gene and, although a few exceptions have been recognized, the clinical course remains unpredictable in most patients.
Methods
Sequencing of NF1 in patients with cafè‐au‐lait spots identified the c.3112A>G variant. RNA analysis and a minigene assay were employed to investigate splicing.
Results
Here we report a novel genotype–phenotype correlation in NF1: the identification of the missense variant NM_000267.3:c.3112A>G p.(Arg1038Gly) in seven individuals from two unrelated families with a mild phenotype. All the patients manifest cafè‐au‐lait spots without neurofibromas or other NF1–associated complications, and Noonan syndrome features in most cases. The missense variant was not previously reported in available databases, segregates with the phenotype and involves a highly conserved residue. Both a minigene assay and patient's RNA analysis excluded an effect on splicing.
Conclusion
Our data support the correlation of the p.Arg1038Gly missense substitution with the cutaneous phenotype without neurofibromas or other complications. This finding may have relevant implications for patients and genetic counseling, but also to get insights into the function of neurofibromin.
Keywords: Arg1038Gly, Genotype–phenotype correlation, missense mutation, neurofibromatosis type 1, NF1
1. INTRODUCTION
Neurofibromatosis type 1 (NF1) (MIM#162200) is an autosomal dominant disease caused by haploinsufficiency of the NF1 gene (MIM #613113) (Gutmann et al., 2017). The prominent feature of this condition is its extremely variable phenotype, even within the same family (Ferner & Gutmann, 2013).
The NF1 gene encodes for neurofibromin, a 2,838 amino acid long protein, which is involved in multiple cellular processes. The only functionally well–characterized domain is the GAP–related‐domain (GRD), which negatively regulates the RAS pathway. Other regions of the proteins are nevertheless relevant since they harbor pathogenic mutations (Gutmann, Parada, Silva, & Ratner, 2012).
The NF1 gene is characterized by a wide mutational spectrum, with more than 3,000 genomic variants reported so far (Wallis et al., 2018). Approximately 10%–15% of patients harbor missense or inframe mutations: in this case validation may be problematic since less functional data are available for this protein. Importantly, a relevant fraction of exonic point mutations affects splicing, behaving like classical inactivating alleles (Pros et al., 2008).
The loss of heterozygosity of NF1, which was demonstrated in different affected tissues (Garcia‐Linares et al., 2011; De Schepper et al., 2008), is considered the driving pathogenetic mechanism in this condition, making impossible to predict the timing and nature of the somatic NF1 mutations. However, there are some examples of relatively recurrent mutations associated with a partially concordant phenotype: (i) a more severe phenotype with a higher number of neurofibromas and a doubled relative risk to develop MPSNT in patients with a large deletion including NF1 (Kehrer‐Sawatzki, Mautner, & Cooper, 2017); (ii) a milder phenotype without neurofibromas or other complications in patients with the inframe deletion delMet991 (Upadhyaya et al., 2007) a missense substitution involving the codon 1809 associated with cafe‐au‐lait spots (CALs) and Noonan–like features (Pinna et al., 2015; Rojnueangnit et al., 2015); (iii) a more severe phenotype with a higher risk of deep neurofibromas in patients with a missense mutation in one of the codons 844‐848 (Koczkowska et al., 2018).
In this work we report the missense mutation p.(Arg1038Gly) in the NF1 gene segregating in seven patients from two unrelated families who manifest a mild NF1 and propose a novel genotype–phenotype correlation. Pedigrees of both families are displayed in Figure 1a, whereas the clinical features of the available family members are reported in Table 1.
Figure 1.
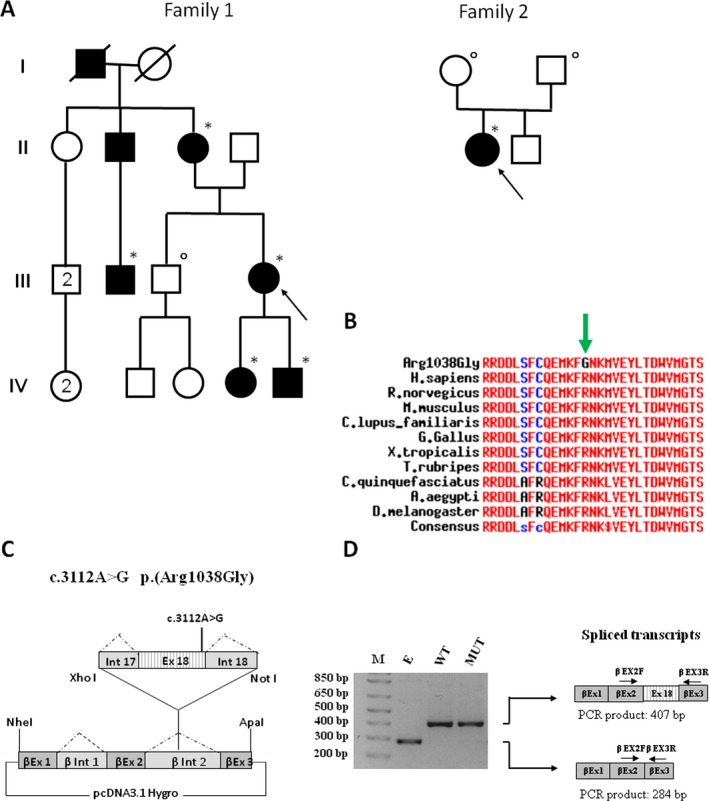
(a) Pedigree of the two families. Black symbols indicate individuals with cafè‐au‐lait spots. Subjects with * harbor the NM_000267.3:c.3112A>G variant of NF1, whereas individuals who were analyzed and do not show the mutation are marked with °. (b) The Arg1038Gly variant affects a highly conserved residue throughout evolution. (c) Schematic representation of the β‐globin minigene construct employed in this study. βEx1, βEx2, βEx3 refer to β‐globin exon 1, 2, and 3. βInt1, βInt2 refer to β‐globin intron 1 and 2. A fragment of 566 nucleotides including exon 18 with at least 100 bp of the flanking introns (Int 17 and Int 18) was amplified from patient III, 5 DNA with primers inserting the XhoI and NotI restrictions sites for the subsequent cloning into the pCDNA3.1‐Beta‐globin vector. One mutant and one wild‐type clone were retrieved for subsequent experiments. (d) Total RNA extracted from HeLa cells transfected with the wild‐type (WT), the mutant construct (MUT) and the empty vector (e) was retro‐transcribed and PCR‐amplified using primers specific for β‐globin exon 2 (β EX2F) and 3 (β EX3R) and separated by agarose gel electrophoresis. The mutant construct (MUT) showed the same splicing pattern as the control (WT).
Table 1.
Clinical features of the seven patients from the two families harboring the p.Arg1038Gly variant
| Clinical features | Family 1 | Family 2 | |||||
|---|---|---|---|---|---|---|---|
| IV, 5 | IV,6 | III,5 | III,3 | II,3 | II,2 | III,1 | |
| Sex | F | M | F | M | F | M | F |
| Age (years) | 10 | 2 | 41 | 38 | 67 | 72 | 30 |
| >5 CALs >1.5 cma | + (>0.5 cm) | + (>0.5 cm) | + | + | + | + | + |
| Axillary/inguinal freckling | − | − | + (groin only) | − | − | − | + |
| Lisch nodules | − | − | − | n.a. | n.a. | n.a. | − |
| Cutaneous neurofibromas | − | − | − | − | − | − | − |
| Symptomatic neurofibromas | − | − | − | − | − | − | − |
| Diffuse plexiform neurofibromas | − | − | − | − | − | − | − |
| Optic pathway glioma | − | − | − | − | − | − | − (UBOs at brain MRI) |
| Short stature | − | − | − | − | − | − | + |
| Macrocephaly | − | + | − | + | − | − | + |
| Pulmonary valve stenosis | −c | −c | −c | − | − | − | −c |
| Facial NS featuresb | + | + | + | − | + | − | + |
| Learning disabilities | − | n.a. | − | − | − | − | − |
For each specific sign: "–" means absent, "+" present, "n.a." = not available.
In all patients CALs were diffuse.
Facial Noonan Syndrome (NS) features include hypertelorism with down–slanting palpebral fissures.
Normal cardiac ultrasound.
Ethical compliance: informed consent was obtained for all the patients.
Family 1 was referred to our outpatient clinic for multiple CALs. The proband (III,5) was a 41‐year old woman whose clinical history was unremarkable. Our physical examination revealed a normal head circumference (+0.4 SD) and showed the presence of more than 10 CALs with a diameter >1.5 cm, mostly at the trunk with rare freckling at the groins and some cherry spots. No cutaneous or subcutaneous neurofibromas could be detected and a detailed ophthalmological evaluation excluded the presence of NF1–related signs. Both her children, a 10‐year old girl (IV,5) and a 2‐year old boy (IV,6), who had normal growth and psychomotor development, showed more than 5 CALs with a diameter >0.5 cm without atypical freckling. Ophthalmological evaluations did not show any sign of NF1 and a cardiac US was normal. Their clinical evaluations revealed hypertelorism with mildly down–slanting palpebral fissures, and the boy also showed macrocephaly (+2.7 SD) with frontal bossing, posteriorly rotated ears and mild retrognathia.
CALs were present also in the proband's mother, her grandfather, the maternal uncle and the first–degree cousin (Figure 1a), neither of whom manifest neurofibromas or other signs of NF1 except for CALs.
Patient II,1 from family 2 is a 30–year old woman who was referred to our center at age 2 for the presence of multiple CALs. The clinical examination at that time revealed normal auxological parameters, hypertelorism, multiple CALs and rare axillary and inguinal freckling, without congenital plexiform neurofibromas, or skeletal anomalies. The subsequent follow‐up revealed a normal psychomotor development, failure to thrive (weight and height at the lower centiles) and a pubertal delay, but no other complications related to NF1 and normal ophthalmological evaluations. An asymptomatic 5‐cm subcutaneous nodule in the back was removed and resulted a lipoma. A brain MRI performed at 19 years was normal, except for the presence of UBOs at the level of left lenticular nucleus and the periventricular white matter. Her family history was unremarkable and her parents did not show cutaneous or ocular signs of NF1.
NF1 and the other genes of the RAS pathway (BRAF, CBL, CDC42, HRAS, KRAS, LZTR1, MAP2K1, MAP2K2, MAP3K8, MYST4, NRAS, PTPN11, RAF1, RASA2, RIT1, RRAS, SHOC2, SOS1, SOS2, SPRED1, SPRY1) were sequenced on an Illumina MiSeq platform with an Agilent Haloplex kit (Santa Clara CA) with coverage >20× in >99% of the target regions.
Molecular analysis identified the heterozygous missense variant NM_000267.3:c.3112A>G p.(Arg1038Gly) in the NF1 gene in patient (III,5) from Family 1, which segregated in all the affected members (Figure 1a). Sequencing of SPRED1 and of the other genes mutated in RASopathies did not detect any pathogenetic variant and MLPA for NF1, which was performed with a commercial kit (MRC Holland, The Netherlands), excluded deletions or duplications. Phenotype was concordant in all family members and characterized by CALs in all patients with Noonan–like features in most cases.
The NM_000267.3:c.3112A>G allele was absent in the HGMD, LOVD, ExAC, GnomAD, or dbSNP databases and has been previously reported in a single unrelated patient fulfilling the diagnostic criteria for NF1 (Corsello et al., 2018) (unfortunately no clinical data are available for this patient). We therefore checked our in–house database of 732 NF1 patients and found the same variant in an unrelated individual (patient II,1 from family 2), who also displays a CALs–only phenotype. Her parents do not show clinical signs of NF1 and, accordingly, do not harbor the missense allele; parental identity was confirmed.
Since the c.3112A>G variant involves the penultimate nucleotide of exon 23 (exon 18 according to the NF1 Consortium Nomenclature) and in silico analyses with different software (Human Splicing Finder, Mutation Taster) predict a possible effect on splicing with the creation of an exonic ESS site, we analyzed cDNA from fresh blood in patient III, 5 as detailed elsewhere (Heim et al., 1995). Direct sequencing of the RT‐PCR amplicon containing exon 13‐27 did not detect any aberrant splicing. Since mutant mRNA may undergo NMRD, we also employed a minigene assay (Figure 1c) to exclude an effect on transcript maturation using a β‐globin hybrid construct as previously reported (Cassina et al., 2017). As showed in Figure 1d, cells transfected with the mutant minigene construct show the same splicing pattern as those expressing the WT plasmid, suggesting that the nucleotide change does not affect splice site recognition. Direct sequencing of the PCR fragment confirmed this finding. The novel NF1 variant has been submitted to the LOVD database [www.lovd.nl/NF1 (DB‐ID NF1_002517)].
There are different pieces of evidence supporting the pathogenicity of this variant: it is extremely rare and it is predicted to cause a nonconservative amino acid change, resulting in the replacement of a positively charged arginine with a neutral glycine. This residue lays within a highly conserved region: arginine 1038 is conserved up to arthropods (Figure 1b) and a different missense substitution affecting the same codon (p.Arg1038Ser) was previously reported in a NF1 patient (Lee et al., 2006), underlying its relevant role for protein structure and/or function. Most of the prediction software tools (PolyPhen2, SIFT, Mutation Taster) we examined, including those integrating multiple tools such as CADD and REVEL (with score of 30 and 0.661, respectively) are in favor of a pathogenetic role of this substitution. Based on our studies on the transcript, the functional consequences of this mutation rely on the presence of the amino acid substitution on a normally spliced protein product. Population, in silico and clinical data (segregation with the cutaneous phenotype in multiple family members with cosegregation analysis of 1/32 according to Jarvik & Browning, 2016 and de novo appearance in a sporadic case) allow to classify this variant as pathogenic (class 5) according to the American College of Medical Genetics and Genomics Standards and Guidelines (Richards et al., 2015).
The effects on protein function are difficult to establish considering the lack of information about this domain of neurofibromin. It is possible that it exhibits a hypomorphic activity, similarly to other alleles that have been associated with a mild phenotype (Pinna et al., 2015; Upadhyaya et al., 2007). Arginine 1038 is located 160 amino acids upstream to the GRD domain of neurofibromin. Nevertheless, it may have functional consequences on the RAS pathway, since it might affect the interaction with downstream Ras‐effectors, as in the case of the R1809C mutation (Wallis et al., 2018). This hypothesis is intriguing since some of our patients display Noonan–like features, but needs functional confirmation.
Unlike the striking clinical variability usually observed within NF1 individuals, the phenotype of the patients harboring the c.3112A>G mutations is quite homogeneous (Table 1): all individuals display CALs, with freckling being present in 28%; none of them shows cutaneous neurofibromas (that are present in most NF1 patients >20 years) (Friedman & Birch, 1997) (Upadhyaya et al., 2007), symptomatic spinal or plexiform neurofibromas, ocular hamartomas or symptomatic OPG (brain MRI was performed only in one patient and showed UBOs). Similarly, learning disabilities were not reported in these patients. It should be noted that some clinical signs, such as deep asymptomatic neurofibromas, may be missed during clinical evaluations. Moreover, the absence of NF1–associated complications, particularly those with lower prevalence, must be confirmed in larger cohorts to draw definitive conclusions.
An increased prevalence of nontruncating mutations in NF1 patients with Noonan–like features was previously observed (De Luca et al., 2005), and another study hypothesized the role of nontruncating mutations on the NF1 cardiac phenotype (Ben‐Shachar et al., 2013). None of the patients harboring the p.Arg1038Gly mutations displays pulmonary stenosis (or other cardiac manifestations), but at least five individuals show facial features resembling the Noonan phenotype. This clinical association may be underestimated in our series, since most patients have been examined in adulthood, where Noonan–associated facial features are harder to detect.
Interestingly, some of the mutations reported in exon 17 and exon 18 have been associated with a phenotype overlapping with other Ras–MAPK disorders: the inframe deletion in exon 17 (Upadhyaya et al., 2007) was related to a higher incidence of pulmonary stenosis without neurofibromas. The p.(Met1035Arg) allele was described in a patient with a Leopard–like phenotype (Wu et al., 1996). LOVD and HGMD report thirteen missense substitutions in exon 18 (including one benign variant): except for four cases (Fauth et al., 2009) (Kluwe, Friedrich, Peiper, Friedman, & Mautner, 2003; Xu, Yang, Hu, & Li, 2014), most of them are classified as variants of unknown significance and unfortunately there are no clinical data, nor it is clear whether RNA maturation is affected, as for the c.3114G>T p.(Arg1038Ser) (Lee et al., 2006) allele. Two missense variants in the initial part of exon 18, p.(Leu1015Pro) (Kluwe et al., 2003) and p.(Cys1016Arg) (Fauth et al., 2009), have been associated with a severe phenotype with the development of MPSNT.
Our findings demonstrated the pathogenicity of the c.3112A>G p.Arg1038Gly variant and support its association with a cutaneous phenotype with lower frequency of NF1–associated complications and Noonan–like features in some individuals. Further studies on larger NF1 cohorts for which clinical data are available are required to corroborate this novel genotype–phenotype correlation. The confirmation of this hypothesis will not only facilitate clinicians in genetic counseling and surveillance programs, but will also help in defining the role of neurofibromin within cells.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank the patients and their families for their collaboration.
Trevisson E, Morbidoni V, Forzan M, et al. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol Genet Genomic Med. 2019;7:e616 10.1002/mgg3.616
Funding information
This work was supported by the University of Padova [CPDA140508/14 to E.T.].
REFERENCES
- Ben‐Shachar, S. , Constantini, S. , Hallevi, H. , Sach, E. K. , Upadhyaya, M. , Evans, G. D. , & Huson, S. M. (2013). Increased rate of missense/in‐frame mutations in individuals with NF1‐related pulmonary stenosis: A novel genotype‐phenotype correlation. European Journal of Human Genetics, 21(5), 535–539. 10.1038/ejhg.2012.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina, M. , Cerqua, C. , Rossi, S. , Salviati, L. , Martini, A. , Clementi, M. , & Trevisson, E. (2017). A synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome. European Journal of Human Genetics, 25(3), 371–375. 10.1038/ejhg.2016.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello, G. , Antona, V. , Serra, G. , Zara, F. , Giambrone, C. , Lagalla, L. , … Piro, E. (2018). Clinical and molecular characterization of 112 single‐center patients with Neurofibromatosis type 1. Italian Journal of Pediatrics, 44, 10.1186/s13052-018-0483-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca, A. , Bottillo, I. , Sarkozy, A. , Carta, C. , Neri, C. , Bellacchio, E. , … Dallapiccola, B. (2005). NF1 gene mutations represent the major molecular event underlying neurofibromatosis‐Noonan syndrome. American Journal of Human Genetics, 77(6), 1092–1101. 10.1086/498454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schepper, S. , Maertens, O. , Callens, T. , Naeyaert, J. M. , Lambert, J. , & Messiaen, L. (2008). Somatic mutation analysis in NF1 cafe au lait spots reveals two NF1 hits in the melanocytes. The Journal of Investigative Dermatology, 128(4), 1050–1053. 10.1038/sj.jid.5701095 [DOI] [PubMed] [Google Scholar]
- Fauth, C. , Kehrer‐Sawatzki, H. , Zatkova, A. , Machherndl‐Spandl, S. , Messiaen, L. , Amann, G. , … Wimmer, K. (2009). Two sporadic spinal neurofibromatosis patients with malignant peripheral nerve sheath tumour. European Journal of Medical Genetics, 52(6), 409–414. 10.1016/j.ejmg.2009.08.001 [DOI] [PubMed] [Google Scholar]
- Ferner, R. E. , & Gutmann, D. H. (2013). Neurofibromatosis type 1 (NF1): Diagnosis and management. Handbook of Clinical Neurology, 115, 939–955. 10.1016/B978-0-444-52902-2.00053-9 [DOI] [PubMed] [Google Scholar]
- Friedman, J. M. , & Birch, P. H. (1997). Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1,728 patients. American Journal of Medical Genetics, 70(2), 138–143. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9128932. 10.1002/(SICI)1096-8628(19970516)70:2lt;138:AID-AJMG7gt;3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- Garcia‐Linares, C. , Fernández‐Rodríguez, J. , Terribas, E. , Mercadé, J. , Pros, E. , Benito, L. , … Serra, E. (2011). Dissecting Loss of Heterozygosity (LOH) in neurofibromatosis type 1‐associated neurofibromas: Importance of copy neutral LOH. Human Mutation, 32(1), 78–90. 10.1002/humu.21387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann, D. H. , Ferner, R. E. , Listernick, R. H. , Korf, B. R. , Wolters, P. L. , & Johnson, K. J. (2017). Neurofibromatosis type 1. Nature Reviews Disease Primers, 3, 17004 10.1038/nrdp.2017.4 [DOI] [PubMed] [Google Scholar]
- Gutmann, D. H. , Parada, L. F. , Silva, A. J. , & Ratner, N. (2012). Neurofibromatosis type 1: Modeling CNS dysfunction. Journal of Neuroscience, 32(41), 14087–14093. 10.1523/JNEUROSCI.3242-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim, R. A. , Kam‐Morgan, L. N. W. , Binnie, C. G. , Corns, D. D. , Cayouette, M. C. , Farber, R. A. , … Luce, M. C. (1995). Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Human Molecular Genetics, 4(6), 975–981. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7655472. 10.1093/hmg/4.6.975 [DOI] [PubMed] [Google Scholar]
- Jarvik, G. P. , & Browning, B. L. (2016). Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. American Journal of Human Genetics, 98(6), 1077–1081. 10.1016/j.ajhg.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer‐Sawatzki, H. , Mautner, V. F. , & Cooper, D. N. (2017). Emerging genotype‐phenotype relationships in patients with large NF1 deletions. Human Genetics, 136(4), 349–376. 10.1007/s00439-017-1766-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluwe, L. , Friedrich, R. E. , Peiper, M. , Friedman, J. , & Mautner, V. F. (2003). Constitutional NF1 mutations in neurofibromatosis 1 patients with malignant peripheral nerve sheath tumors. Human Mutation, 22(5), 420 10.1002/humu.9193 [DOI] [PubMed] [Google Scholar]
- Koczkowska, M. , Chen, Y. , Callens, T. , Gomes, A. , Sharp, A. , Johnson, S. , … Messiaen, L. M. (2018). Genotype‐phenotype correlation in NF1: Evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. American Journal of Human Genetics, 102(1), 69–87. 10.1016/j.ajhg.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.‐J. , Su, Y.‐N. , You, H.‐L. , Chiou, S.‐C. , Lin, L.‐C. , Yang, C.‐C. , … Yu, C.‐L. (2006). Identification of forty‐five novel and twenty‐three known NF1 mutations in Chinese patients with neurofibromatosis type 1. Human Mutation, 27(8), 832 10.1002/humu.9446 [DOI] [PubMed] [Google Scholar]
- Pinna, V. , Lanari, V. , Daniele, P. , Consoli, F. , Agolini, E. , Margiotti, K. , … De Luca, A. (2015). p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. European Journal of Human Genetics, 23(8), 1068–1071. 10.1038/ejhg.2014.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pros, E. , Gomez, C. , Martin, T. , Fabregas, P. , Serra, E. , & Lazaro, C. (2008). Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Human Mutation, 29(9), E173–193. 10.1002/humu.20826 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojnueangnit, K. , Xie, J. , Gomes, A. , Sharp, A. , Callens, T. , Chen, Y. , … Messiaen, L. (2015). High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: Genotype‐phenotype correlation. Human Mutation, 36(11), 1052–1063. 10.1002/humu.22832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya, M. , Huson, S. m. , Davies, M. , Thomas, N. , Chuzhanova, N. , Giovannini, S. , … Messiaen, L. (2007). An absence of cutaneous neurofibromas associated with a 3‐bp inframe deletion in exon 17 of the NF1 gene (c.2970‐2972 delAAT): Evidence of a clinically significant NF1 genotype‐phenotype correlation. American Journal of Human Genetics, 80(1), 140–151. 10.1086/510781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis, D. , Li, K. , Lui, H. , Hu, K. , Chen, M. J. , Li, J. , … Kesterson, R. A. (2018). Neurofibromin (NF1) genetic variant structure‐function analyses using a full‐length mouse cDNA. Human Mutation, 39(6), 816–821. 10.1002/humu.23421 [DOI] [PubMed] [Google Scholar]
- Wu, R. , Legius, E. , Robberecht, W. , Dumoulin, M. , Cassiman, J. J. , & Fryns, J. P. (1996). Neurofibromatosis type I gene mutation in a patient with features of LEOPARD syndrome. Human Mutation, 8(1), 51–56. 10.1002/(SICI)1098-1004(1996)8:1<51:AID-HUMU7>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- Xu, W. H. , Yang, X. , Hu, X. X. , & Li, S. B. (2014). Fifty‐four novel mutations in the NF1 gene and integrated analyses of the mutations that modulate splicing. International Journal of Molecular Medicine, 34(1), 53–60. 10.3892/ijmm.2014.1756 [DOI] [PMC free article] [PubMed] [Google Scholar]