

Abstract
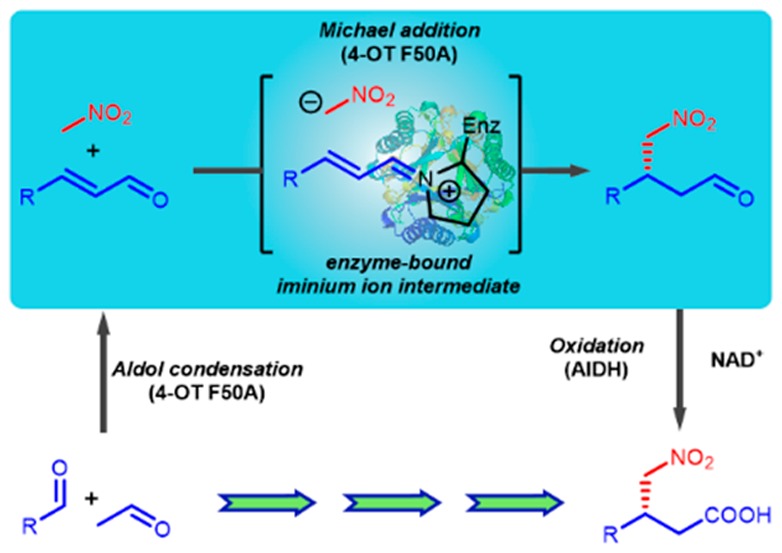
The enzyme 4-oxalocrotonate tautomerase (4-OT) exploits an N-terminal proline as main catalytic residue to facilitate several promiscuous C–C bond-forming reactions via enzyme-bound enamine intermediates. Here we show that the active site of this enzyme can give rise to further synthetically useful catalytic promiscuity. Specifically, the F50A mutant of 4-OT was found to efficiently promote asymmetric Michael additions of nitromethane to various α,β-unsaturated aldehydes to give γ-nitroaldehydes, important precursors to biologically active γ-aminobutyric acids. High conversions, high enantiocontrol, and good isolated product yields were achieved. The reactions likely proceed via iminium ion intermediates formed between the catalytic Pro-1 residue and the α,β-unsaturated aldehydes. In addition, a cascade of three 4-OT(F50A)-catalyzed reactions followed by an enzymatic oxidation step enables assembly of γ-nitrocarboxylic acids from three simple building blocks in one pot. Our results bridge organo- and biocatalysis, and they emphasize the potential of enzyme promiscuity for the preparation of important chiral synthons.
Keywords: biocatalysis, Michael addition, asymmetric synthesis, enzyme catalysis, protein engineering
The γ-nitroaldehydes are important chiral building blocks for the preparation of biologically active γ-aminobutyric acids. The asymmetric synthesis of γ-nitroaldehydes from simple starting materials has become feasible because of outstanding developments within the organocatalysis field, particularly fueled by aminocatalysis.1 This is nicely illustrated by the work of Hayashi and co-workers, who reported that diphenylprolinol silyl ether can promote the asymmetric synthesis of γ-nitroaldehydes through alternative Michael-type reactions: enamine-mediated addition of aldehydes to nitroalkenes, and nitroalkane addition to α,β-unsaturated aldehydes activated as iminium ions.1d−1f
Inspired by these developments in the organocatalysis field, work from our laboratory focused on the development of a biocatalytic procedure for asymmetric synthesis of γ-nitroaldehydes. We reported that 4-oxalocrotonate tautomerase (4-OT), which utilizes a unique N-terminal proline as key catalytic residue, can promiscuously catalyze the Michael addition of acetaldehyde (as well as various other aldehydes) to nitroalkenes yielding enantioenriched γ-nitroaldehydes (Scheme 1A).2 The catalytic mechanism involves the formation of an enamine species between acetaldehyde and the Pro-1 residue (pKa ∼6.4).3,4 Hilvert and co-workers have reported a highly engineered computationally designed artificial aldolase, RA95.5–8, which can catalyze the asymmetric synthesis of γ-nitroketones (but not γ-nitroaldehydes) via acetone addition to nitrostyrenes and nitroalkane addition to conjugated ketones.5 However, a biocatalytic methodology for nitroalkane addition to α,β-unsaturated aldehydes to yield enantioenriched γ-nitroaldehydes is an as yet unmet challenge.
Scheme 1. Proposed Mechanisms for the 4-OT-Catalyzed Michael Additions of Acetaldehyde to Nitroalkenes (A) and Nitromethane to α,β-Unsaturated Aldehydes (B) To Yield γ-Nitroaldehydes.

Here, we report that the F50A mutant of the 4-OT enzyme can efficiently promote asymmetric Michael additions of nitromethane to α,β-unsaturated aldehydes, yielding various γ-nitroaldehydes with high enantiopurity (e.r. up to >99:1) and in high isolated yield (61–96%). The catalytic mechanism appears to involve formation of enzyme-bound iminium ion intermediates in a manner reminiscent of organocatalysis (Scheme 1B).
We previously reported that 4-OT catalyzes the aldol condensation of acetaldehyde with benzaldehyde to yield cinnamaldehyde.3,6 Considering that the active site of 4-OT can accommodate cinnamaldehyde, this aromatic α,β-unsaturated aldehyde was tested as potential Michael acceptor substrate. Cinnamaldehyde was expected to react with Pro-1, the catalytic amine, to form a covalently bound iminium ion intermediate, which could be attacked by nitromethane (Scheme 1B). This Michael reaction was performed in the presence of wild-type 4-OT in HEPES buffer (pH 7.3), containing 5% (v/v) EtOH, 200 mM of nitromethane (1, Scheme 2), and 3 mM of cinnamaldehyde (2a), and reaction progress was monitored by following the depletion of 2a by UV–vis spectrophotometry. Under these conditions, 50% of substrate 2a was consumed in 24 h, and the corresponding product 3a was obtained in 35% isolated yield (as confirmed by 1H NMR spectroscopy). Analysis of product 3a by chiral HPLC revealed high enantiocontrol at the site of addition with formation of the (R)-configured product (e.r. of 86:14). Interestingly, we earlier reported that wild-type 4-OT catalyzes the Michael addition of acetaldehyde to trans-β-nitrostyrene to yield (S)-3a with an e.r. of 95:5.2a Hence, 4-OT catalyzes the synthesis of γ-nitroaldehyde 3a via two enantiocomplementary Michael reactions: enamine-mediated addition of acetaldehyde to trans-β-nitrostyrene and nitromethane addition to cinnamaldehyde likely activated as iminium ion.
Scheme 2. Wild-Type 4-OT-Catalyzed Michael Addition of Nitromethane (1) to Cinnamaldehyde (2a) To Yield γ-Nitroaldehyde (R)-3a.

Encouraged by these initial findings, a systematic mutagenesis approach was applied to enhance this promiscuous Michael addition activity of 4-OT. For this, an earlier constructed collection of 4-OT genes coding for almost all possible single-mutant variants of 4-OT was used.7 Improved variants (>2-fold increase in activity) were identified by monitoring the depletion of 2a in a spectrophotometric kinetic assay in multiwell plates. Given that several mutations at positions Met-45 and Ala-46 (M45G, M45H, M45S, A46H, and A46S) result in a slight improvement in activity (∼3-fold), three mutations at position Phe-50 (F50I, F50V, and F50A) significantly enhanced the Michael addition activity. Assays with the purified mutant enzymes showed a 6-fold, 8-fold and 15-fold increase in activity for F50I, F50V, and F50A, respectively (Figure S1). Further characterization of the Michael reaction between 1 and 2a catalyzed by the best 4-OT variant (F50A) showed that besides increased activity, this mutant enzyme also has enhanced stereoselectivity, allowing the production of optically pure (R)-3a (e.r. 99:1) in high isolated yield of 92% (Table 1, entry 1; Figures S2–S4). These results underscore the potential of the highly promiscuous 4-OT enzyme for evolutionary optimization. At semipreparative scale, the 4-OT(F50A)-catalyzed Michael addition of 1 (50 mM, 152 mg in 50 mL) to 2a (25 mM, 157 mg in 50 mL) gave product (R)-3a (96% conversion in 11 h) in good isolated yield (204.7 mg, 85% yield) and with a high e.r. of 98:2 (Figures S34, S35).
Table 1. 4-OT(F50A)-Catalyzed Nitromethane Addition to α,β-Unstaturated Aldehydes 2a–2k Using Optimized Reaction Conditionsa.


All the reactions were performed in buffer [20 mM HEPES/5% (v/v) ethanol] at pH 6.5 with 4-OT F50A (72 μM, except for 2g and 2i for which 36 μM enzyme was used), 1 (25 mM) and 2a–k (3 mM, except for 2g which was used at 2 mM).
Determined by 1H NMR analysis.
Isolated yield (%).
Determined by chiral HPLC or GC.
The absolute configuration was determined by comparison of chiral HPLC or GC data with those previously reported (see Supporting Information for details).
Apparent kinetic parameters determined with this substrate at a fixed nitromethane concentration of 25 mM: kcat = 0.05 (±0.002) s–1; Km = 367 (±37) μM.
Further purified using flash column chromatography.
Notably, the mutation F50A was previously found to improve the aldol condensation activity of 4-OT.6a This mutation makes the active site pocket of 4-OT more accessible to the outside aqueous environment, without changing the pKa of Pro-1 too much, and likely enhances the aldol condensation activity of 4-OT by promoting the final hydrolysis step in which product is released from Pro-1, which has been suggested to be rate-limiting.6a Similarly, the F50A mutation may increase the Michael addition activity of 4-OT by making the active site more amenable for hydrolytic cleavage of the covalent enzyme–product intermediate.
Having established that 4-OT(F50A) can efficiently promote the asymmetric Michael addition of 1 to 2a, a set of α,β-unsaturated aldehydes was prepared (see Support Information for details) and tested as Michael acceptor substrates. The results demonstrate that the 4-OT(F50A) enzyme has a broad substrate scope, accepting both aromatic and aliphatic Michael acceptor substrates, and catalyzes the addition of 1 to the α,β-unsaturated aldehydes 2b–k to yield the corresponding γ-nitroaldehydes 3b–k with excellent enantiopurity (e.r. up to >99:1) and in good isolated yield (61–96%) (Table 1, Figures S5–S33). Interestingly, the enzymatic Michael reactions with meta- and para-substituted cinnamaldehydes (2c,d and 2f–j) provided the corresponding products as the (R)-configured enantiomers, while those with the ortho-substituted cinnamaldehydes (2b and 2e) yielded the (S)-configured product enantiomers (Table 1, entries 2 and 5). This suggests that positioning substituents on the ortho position of the substrate promoted steric effects, which caused either substrate relocation in the enzyme active site or a stereofacial shielding effect. Notably, the consequence of ortho-substituents on the stereochemical outcome of organo- and biocatalytic reactions with aromatic aldol and Michael acceptor substrates has been observed before.2d,8
We next investigated whether the mechanism of the 4-OT(F50A)-catalyzed Michael reaction proceeds via iminium ion formation between Pro-1 and the α,β-unsaturated aldehyde substrate. A 4-OT(F50A) sample modified by 2a in the presence of NaBH4 and an unmodified 4-OT(F50A) sample were digested with Glu-C (an endoproteinase from Staphylococcus aureus V8), and the generated peptides were characterized by LC-MS (Figures S43–S45). Comparing the peaks of the modified 4-OT(F50A) sample to those of the nonmodified 4-OT(F50A) sample revealed a modification of the fragment PIAQIHILE by a species with a mass of 116 Da. This corresponds to labeling by one cinnamaldehyde molecule. Characterization of the remaining peaks revealed no labeling of other fragments (Figures S44, S45). Within the N-terminal fragment Pro-1 to Glu-9, the most probable positions for alkylation are Pro-1 and His-6. To identify the labeled residue, the modified and unmodified peptides were analyzed by LC-MS/MS (Figure S46). The spectrum of the ion corresponding to the unlabled PIAQIHILE peptide showed the characteristic b5 ion resulting from the peptide fragment PIAQI. MS/MS analysis of the modified PIAQIHILE peptide revealed a mass increase of 116 Da for this b5 ion. Therefore, it can be concluded that Pro-1 is the sole site of modification by 2a. This result supports the hypothesis that Pro-1 functions as an amine catalyst in the enzymatic addition reaction, increasing the electrophilicity of the Michael acceptor through Schiff base formation (Scheme 1B). Replacement of Pro-1 with an alanine in wild-type 4-OT led to a 32-fold decrease in activity for the addition of 1 to 2a (Figure S1), providing further support for this mechanism. Work is in progress to determine the structure of 4-OT(F50A) covalently modified by 2a.
We have previously reported that 4-OT(F50A) can catalyze the aldol condensation of acetaldehyde with benzaldehyde to give cinnamaldehyde.3,6 Here we show that the three different activities observed for the 4-OT(F50A) enzyme can be used to prepare γ-nitroaldehydes in a biocatalytic cascade involving sequential aldol addition of acetaldehyde to a suitable aromatic aldehyde, dehydration, and Michael addition of nitromethane. Inclusion of a suitable aldehyde dehydrogenase and cofactor-recycling NADH oxidase9 in the reaction mixture enabled efficient one-pot synthesis of γ-nitrocarboxylic acids (Scheme 3). Using acetaldehyde (4), benzaldehyde (5a), and nitromethane (1) as starting substrates, product (R)-7a was obtained in 53% isolated yield (65% overall conversion) and with an excellent e.r. of 99:1 (Figures S36–S38). Replacing substrate 5a with 5b, yielded product (S)-7b in 80% isolated yield (>99% overall conversion) and with an excellent e.r. of 99:1 (Figures S39–S42). This simple and effective cascade further demonstrates the tremendous potential of combining different enzymes to construct simple synthetic routes for preparation of important chemical products.
Scheme 3. Four-Step Biocatalytic Cascade Synthesis of γ-Nitrobutyric Acids 7a and 7b in One Pot.

The cascade reactions were performed with 1 (50 mM), 4 (150 mM), and either 5a or 5b (3 mM).
In summary, our results indicate that the active site of 4-OT can give rise to synthetically useful promiscuous activities. Like proline-based organocatalysts, 4-OT utilizes a prolyl amine to attack diverse aldehydes forming reactive enamine and iminium ion intermediates. Hence, this natural enzyme with its unique catalytic amino-terminal proline could possibly accelerate many of the bond-forming reactions promoted by organocatalysts. We have therefore initiated studies aimed at exploring alternative nucleophiles for addition to α,β-unsaturated aldehydes, which would allow for the enzymatic synthesis of additional products.
In contrast to difficult to prepare proline- and peptide-based organocatalysts, the enzyme 4-OT can be produced in large amounts by simple bacterial fermentation. Moreover, the enzymatic reaction proceeds in eco-friendly aqueous buffer rather than in organic solvent. In previous work, we have demonstrated that 4-OT can be engineered into a more efficient biocatalyst for the aldol condensation of acetaldehyde with benzaldehyde to yield cinnamaldehyde, with a >5000-fold enhancement in catalytic efficiency (kcat/Km) and a >107-fold change in reaction specificity.6b It is therefore conceivable that the promiscuous activity of 4-OT(F50A) for the Michael addition of nitromethane to α,β-unsaturated aldehydes can be optimized by directed evolution to generate novel biocatalysts for practical synthesis of chiral precursors for important pharmaceuticals.
Acknowledgments
This research was financially supported by the European Union’s Horizon 2020 Research and Innovation Programme (grant 635595), the European Research Council (grant 713483) and The Netherlands Organization of Scientific Research (grant 724.016.002). We thank M.H. de Vries for support in acquiring mass spectrometry data, and Lieuwe Biewenga for insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b00780.
Additional experimental procedures and compound characterization (PDF)
Author Contributions
† C.G. and M.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Wiesner M.; Revell J. D.; Wennemers H. Tripeptides as Efficient Asymmetric Catalysts for 1,4-Addition Reactions of Aldehydes to Nitroolefins--A Rational Approach. Angew. Chem., Int. Ed. 2008, 47, 1871–1874. 10.1002/anie.200704972. [DOI] [PubMed] [Google Scholar]; b Wiesner M.; Neuburger M.; Wennemers H. Tripeptides of the Type H-D-Pro-Pro-Xaa-NH2 as Catalysts for Asymmetric 1,4-Addition Reactions: Structural Requirements for High Catalytic Efficiency. Chem. - Eur. J. 2009, 15, 10103–10109. 10.1002/chem.200901021. [DOI] [PubMed] [Google Scholar]; c García-García P.; Ladépêche A.; Halder R.; List B. Catalytic Asymmetric Michael Reactions of Acetaldehyde. Angew. Chem., Int. Ed. 2008, 47, 4719–4721. 10.1002/anie.200800847. [DOI] [PubMed] [Google Scholar]; d Gotoh H.; Ishikawa H.; Hayashi Y. Diphenylprolinol Silyl Ether as Catalyst of an Asymmetric, Catalytic, and Direct Michael Reaction of Nitroalkanes with α,β-Unsaturated Aldehydes. Org. Lett. 2007, 9, 5307–5309. 10.1021/ol702545z. [DOI] [PubMed] [Google Scholar]; e Hayashi Y.; Gotoh H.; Hayashi T.; Shoji M. Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]; f Hayashi Y.; Itoh T.; Ohkubo M.; Ishikawa H. Asymmetric Michael Reaction of Acetaldehyde Catalyzed by Diphenylprolinol Silyl Ether. Angew. Chem., Int. Ed. 2008, 47, 4722–4724. 10.1002/anie.200801130. [DOI] [PubMed] [Google Scholar]; g Jentzsch K. I.; Min T.; Etcheson J. I.; Fettinger J. C.; Franz A. K. Silyl Fluoride Electrophiles for the Enantioselective Synthesis of Silylated Pyrrolidine Catalysts. J. Org. Chem. 2011, 76, 7065–7075. 10.1021/jo200991q. [DOI] [PubMed] [Google Scholar]; h Qiao Y.; He J.; Ni B.; Headley A. D. Asymmetric Michael Reaction of Acetaldehyde with Nitroolefins Catalyzed by Highly Water-Compatible Organocatalysts in Aqueous Media. Adv. Synth. Catal. 2012, 354, 2849–2853. 10.1002/adsc.201200215. [DOI] [Google Scholar]; i Alza E.; Pericàs M. A. A Highly Selective, Polymer-Supported Organocatalyst for Michael Additions with Enzyme-Like Behavior. Adv. Synth. Catal. 2009, 351, 3051–3056. 10.1002/adsc.200900817. [DOI] [Google Scholar]; j Riente P.; Mendoza C.; Pericás M. A. Functionalization of Fe3O4 Magnetic Nanoparticles for Organocatalytic Michael Reactions. J. Mater. Chem. 2011, 21, 7350–7355. 10.1039/c1jm10535c. [DOI] [Google Scholar]; k Donslund B. S.; Johansen T. K.; Poulsen P. H.; Halskov K. S.; Jørgensen K. A. The Diarylprolinol Silyl Ethers: Ten Years After. Angew. Chem., Int. Ed. 2015, 54, 13860–13874. 10.1002/anie.201503920. [DOI] [PubMed] [Google Scholar]; l Hojabri L.; Hartikka A.; Moghaddam F. M.; Arvidsson P. I. A. New Imidazole-Containing Imidazolidinone Catalyst for Organocatalyzed Asymmetric Conjugate Addition of Nitroalkanes to Aldehydes. Adv. Synth. Catal. 2007, 349, 740–748. 10.1002/adsc.200600316. [DOI] [Google Scholar]; m Palomo C.; Landa A.; Mielgo A.; Oiarbide M.; Puente Á; Vera S. Water-Compatible Iminium Activation: Organocatalytic Michael Reactions of Carbon-Centered Nucleophiles with Enals. Angew. Chem., Int. Ed. 2007, 46, 8431–8435. 10.1002/anie.200703261. [DOI] [PubMed] [Google Scholar]; n Tsogoeva S. B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 11, 1701–1716. 10.1002/ejoc.200600653. [DOI] [Google Scholar]; o Almaşi D.; Alonso D. A.; Nájera C. Organocatalytic Asymmetric Conjugate Additions. Tetrahedron: Asymmetry 2007, 18, 299–365. 10.1016/j.tetasy.2007.01.023. [DOI] [Google Scholar]; p Mase N.Enamine Catalysis of Michael Reactions. In Science of Synthesis: Asymmetric Organocatalysis; List B., Maruoka K., Eds.; Thieme: Stuttgart, Germany, 2012; pp 135–216. [Google Scholar]; q Nödling A. R.; Świderek K.; Castillo R.; Hall J. W.; Angelastro A.; Morrill L. C.; Jin Y.; Tsai Y. H.; Moliner V.; Luk L. Y. P. Reactivity and Selectivity of Iminium Organocatalysis Improved by a Protein Host. Angew. Chem., Int. Ed. 2018, 57, 12478–12482. 10.1002/anie.201806850. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Betancort J. M.; Barbas C. F. III Catalytic Direct Asymmetric Michael Reactions: Taming Naked Aldehyde Donors. Org. Lett. 2001, 3, 3737–3740. 10.1021/ol0167006. [DOI] [PubMed] [Google Scholar]
- a Zandvoort E.; Geertsema E. M.; Baas B. J.; Quax W. J.; Poelarends G. J. Bridging Between Organocatalysis and Biocatalysis: Asymmetric Addition of Acetaldehyde to β-Nitrostyrenes Catalyzed by a Promiscuous Proline-Based Tautomerase. Angew. Chem., Int. Ed. 2012, 51, 1240–1243. 10.1002/anie.201107404. [DOI] [PubMed] [Google Scholar]; b Miao Y.; Geertsema E. M.; Tepper P. G.; Zandvoort E.; Poelarends G. J. Promiscuous Catalysis of Asymmetric Michael-Type Additions of Linear Aldehydes to β-Nitrostyrene by the Proline-Based Enzyme 4-Oxalocrotonate Tautomerase. ChemBioChem 2013, 14, 191–194. 10.1002/cbic.201200676. [DOI] [PubMed] [Google Scholar]; c Geertsema E. M.; Miao Y.; Tepper P. G.; de Haan P.; Zandvoort E.; Poelarends G. J. Biocatalytic Michael-Type Additions of Acetaldehyde to Nitroolefins with the Proline-Based Enzyme 4-Oxalocrotonate Tautomerase Yielding Enantioenriched γ-Nitroaldehydes. Chem. - Eur. J. 2013, 19, 14407–14410. 10.1002/chem.201302351. [DOI] [PubMed] [Google Scholar]; d Miao Y.; Tepper P. G.; Geertsema E. M.; Poelarends G. J. Stereochemical Control of Enzymatic Carbon–Carbon Bond-Forming Michael-Type Additions by “Substrate Engineering”. Eur. J. Org. Chem. 2016, 32, 5350–5354. 10.1002/ejoc.201601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandvoort E.; Baas B. J.; Quax W. J.; Poelarends G. J. Systematic Screening for Catalytic Promiscuity in 4-Oxalocrotonate Tautomerase: Enamine Formation and Aldolase Activity. ChemBioChem 2011, 12, 602–609. 10.1002/cbic.201000633. [DOI] [PubMed] [Google Scholar]
- Poddar H.; Rahimi M.; Geertsema E. M.; Thunnissen A. M. W. H.; Poelarends G. J. Evidence for the Formation of an Enamine Species During Aldol and Michael-type Addition Reactions Promiscuously Catalyzed by 4-Oxalocrotonate Tautomerase. ChemBioChem 2015, 16, 738–741. 10.1002/cbic.201402687. [DOI] [PubMed] [Google Scholar]
- Garrabou X.; Verez R.; Hilvert D. Enantiocomplementary Synthesis of γ-Nitroketones Using Designed and Evolved Carboligases. J. Am. Chem. Soc. 2017, 139, 103–106. 10.1021/jacs.6b11928. [DOI] [PubMed] [Google Scholar]
- a Zandvoort E.; Geertsema E. M.; Quax W. J.; Poelarends G. J. Enhancement of the Promiscuous Aldolase and Dehydration Activities of 4-Oxalocrotonate Tautomerase by Protein Engineering. ChemBioChem 2012, 13, 1274–1277. 10.1002/cbic.201200225. [DOI] [PubMed] [Google Scholar]; b Rahimi M.; van der Meer J. Y.; Geertsema E. M.; Poddar H.; Baas B. J.; Poelarends G. J. Mutations Closer to the Active Site Improve the Promiscuous Aldolase Activity of 4-Oxalocrotonate Tautomerase More Effectively than Distant Mutations. ChemBioChem 2016, 17, 1225–1228. 10.1002/cbic.201600149. [DOI] [PubMed] [Google Scholar]
- van der Meer J. Y.; Poddar H.; Baas J. B.; Miao Y.; Rahimi M.; Kunzendorf M. A.; van Merkerk R.; Tepper P. G.; Geertsema E. M.; Thunnissen A. M. W. H.; Quax W. J.; Poelarends G. J. Using Mutability Landscapes of a Promiscuous Tautomerase to Guide the Engineering of Enantioselective Michaelases. Nat. Commun. 2016, 7, 10911. 10.1038/ncomms10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez A.; van Gemmeren M.; List B. Unexpected Beneficial Effect of ortho-Substituents on the (S)-Proline-Catalyzed Asymmetric Aldol Reaction of Acetone with Aromatic Aldehydes. Synlett 2014, 25, 961–964. 10.1055/s-0033-1340920. [DOI] [Google Scholar]
- Biewenga L.; Saravanan T.; Kunzendorf A.; van der Meer J. Y.; Pijning T.; Tepper P. G.; van Merkerk R.; Charnock S. J.; Thunnissen A. M. W. H.; Poelarends G. J. Enantioselective Synthesis of Pharmaceutically Active γ-Aminobutyric Acids Using a Tailor-Made Artificial Michaelase in One-Pot Cascade Reactions. ACS Catal. 2019, 9, 1503–1513. 10.1021/acscatal.8b04299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.