

Abstract
Transcriptional repressor EthR from Mycobacterium tuberculosis is a valuable target for antibiotic booster drugs. We previously reported a virtual screening campaign to identify EthR inhibitors for development. Two ligand binding orientations were often proposed, though only the top scoring pose was utilized for filtering of the large data set. We obtained biophysically validated hits, some of which yielded complex crystal structures. In some cases, the crystallized binding mode and top scoring mode agree, while for others an alternate ligand binding orientation was found. In this contribution, we combine rigid docking, molecular dynamics simulations, and the linear interaction energy method to calculate binding free energies and derive relative binding energies for a number of EthR inhibitors in both modes. This strategy allowed us to correctly predict the most favorable orientation. Therefore, this widely applicable approach will be suitable to triage multiple binding modes within EthR and other potential drug targets with similar characteristics.
EthR (Figure 1a) is a well-studied transcriptional repressor from Mycobacterium tuberculosis that represses the expression of the Bayer–Villager mono-oxygenase protein EthA, the in vivo activator of antibiotic pro-drug ethionamide. The homodimeric protein belongs to the family of TetR repressors comprising an N-terminal DNA-binding domain and a C-terminal ligand binding domain. Importantly, EthR ligands inhibiting DNA-binding have been shown to increase ethA expression and significantly boost ethionamide efficacy in in vivo models.1,−4,17 Consequently, EthR inhibitors are under intense investigation as booster codrugs.5−9,12−15 Several approaches have been used to explore novel chemotypes for binding the lipophilic EthR ligand site (Figure 1b), including traditional high-throughput library screening,10 fragment-based drug discovery,12,16 and our previously reported virtual screening (VS) program based on the GOLD software.18 We reported our screen of over 400 000 ligands against EthR, a cohort filtered from an initial starting library in excess of six million compounds derived from the Drugs Now subset of the ZINC database.18 Our predocking filtering strategy included restricting compound volume to 200–700 Å and the requirement of at least one H-bond acceptor, compatible with the size and nature of the EthR binding channel, as well as tailoring physiochemical properties to largely lipophilic binding site.18 In most cases, rigid receptor docking (RRD) yielded two distinct orientations of binding to EthR; however, only the top scoring pose was used for further filtering. Subsequent in vitro assays yielded several biophysically and structurally confirmed hits, though the in crystallo binding mode in some cases was different in orientation to the top scoring virtual screening pose. For one compound we observed both orientations within the crystallized complex, each modeled with 50% occupancy. Importantly, because of the design of the VS, each compound represented a different scaffold, as confirmed in the structural analysis of EthR complexes performed by Tanina and colleagues in 2018.19
Figure 1.
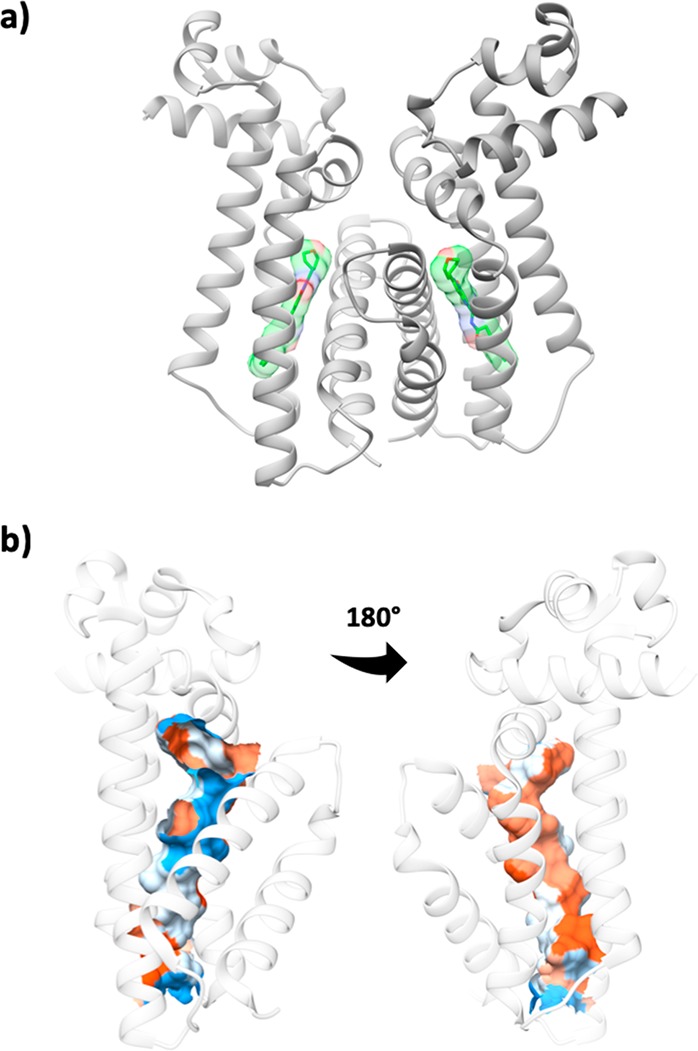
(a) Ribbon diagram of EthR-85, with compound 85 in stick depiction with molecular surface. Carbon atoms of the ligand are shown in green. (b) The binding site of EthR is largely lipophilic (colored orange/red) with few heteroatoms available to act as hydrogen bond donors or acceptors (blue). Image prepared in Chimera using pocket analysis by CASTp20 for EthR (PDB 1U9N).
In recent years, both RRD and induced-fit docking (IFD) protocols have become reasonably proficient at reproducing crystallographic binding modes, as evaluated by the D3R Grand Challenges (Drug Design Data Resource).21−23 RRD and IFD have been applied in virtual screening and structure-based drug design to discover and develop novel ligands for a wide variety of medically relevant targets.24 EthR poses a challenge to RRD due to the composition of the binding site: it is highly lipophilic with two asparagine residues (Asn 176 and Asn 179, respectively) located in the center of the pocket. The side chains of these two residues are typically involved in hydrogen bonding and “anchoring” the compound, allowing for the proposed flipping of binding modes.
While automated ligand docking approaches can be combined with molecular dynamics (MD) simulations, explicit MD simulations themselves are not yet feasible for screening libraries containing millions of compounds.25,26 The combined RDD–MD approach presented here refines the binding poses in explicit solvent (often absent in RRD), and following RDD–MD with the calculation of relative ligand binding energies quantifies the binding event in solution to prioritize favorably binding ligands.27,28 In this contribution we combine RRD, MD simulations, and the linear interaction energy (LIE) method27,29 to calculate the relative free energies of binding for four inhibitors in their two proposed orientations.
From our previous virtual screening,18 we identified compounds 3, 10, and 85 as promising EthR ligands, and successfully cocrystallized EthR with 10. The structures of these compounds are given in the supplementary material in Chart S1. Here we report in addition the cocrystallized structures of EthR with 3 and 85, both at a resolution of 1.8 Å.
Compound 3 has two similar poses where the methyl-oxopyrazine moiety is exposed to solvent (Figure 2a). Key interactions between compound 3 and the EthR pocket include a hydrogen bond between the R159 side chain and the oxopyrazine carbonyl oxygen (2.6 Å, only present in one pose), an OH···N hydrogen bond from the hydroxyl oxygen of Y148 to a pyrazine nitrogen (2.9–3.3 Å), and a CH···O interaction to L87 (2.8 Å). A close-up of these interaction patterns can be seen in Figure S1. The B-factors of the ligand itself are approximately twice the magnitude of the average B-factor for the surrounding protein, an indication of increased ligand flexibility.
Figure 2.

Ligand binding pocket of (a) EthR-compound 3, ligand in plum; (b) EthR-compound 85, ligand in green; (c) EthR-compound 10,18 ligand A in yellow and ligand B in blue; and (d) EthR-BDM31343, ligand in dark red.7 Hydrogen bonds are highlighted as dotted magenta lines, and close contacts are in blue. Only key residues are shown for clarity.
No hydrogen bond interactions are observed in the crystal structure of EthR cocrystallized with compound 85 (Figure 2b). In this case binding appears to be driven by van der Waals interactions (Figure S2). A close contact is formed between the central carbonyl on the pyrazolopyrimidine core to the sulfur atom of M142 (2.5 Å). Further details on data collection and refinement statistics are presented in Table S1.
These three crystal structures highlight the differences between the virtual screening pose used for ligand selection and the experimentally observed binding mode. Specifically, the VS provided two orientations for compound 3, the top scoring pose of which matches the crystal structure. Conversely, for compound 85, the VS protocol yielded only one orientation, which does not match the crystal structure. Finally, two orientations were suggested for compound 10 (Figure 2c) and both are observed in crystallo. We used these cases and the crystal structure of the EthR-BDM31343 complex (PDB 3TP0,7 1.9 Å resolution, Figure 2d) to study alternate binding conformations. BDM31343 presents an unambiguous single binding mode, and docking studies undertaken in 201311 demonstrated that docking of this compound (under the VS protocol by which we identified compounds 10, 3, and 85(11,18)) produced five poses of one orientation, in agreement with the crystal structure. We hypothesized that had we utilized relative binding energies in selecting which orientation from virtual screening was energetically favorable, we would have correctly predicted the experimental binding mode. This suggests that a more quantitative approach is required to evaluate the likelihood of observing different poses. Here we used the calculated binding energies in explicit solvent as the criteria to determine the most favorable pose and compared these results to our crystallographic data. It was found that the MD–LIE approach can confidently identify and rank correct binding modes.
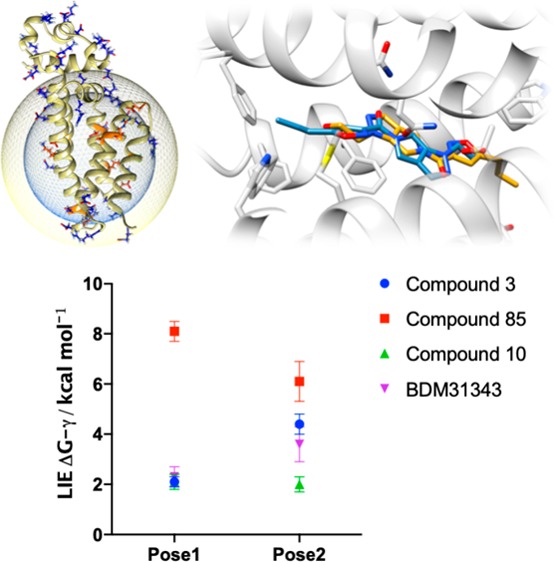
For each EthR–ligand complex, MD–LIE calculations in explicit solvent (24 Å sphere radius) were carried out employing the highest scoring VS pose (“pose 1”) and the highest-scoring alternate pose for the opposite binding orientation (“pose 2”). For compounds 85 and BDM31343 where no second orientation was proposed by docking, the alternate binding mode within the linear tube-like EthR binding channel was created manually through an approximately 180° rotation, maintaining the hydrogen bonding pattern and the hydrophobic interactions on either side of the amide bond. Each simulation was performed in quadruplicate employing different starting velocities (see the Supporting Information for details).30 Following equilibration, the complexes were unrestrained for an initial 10 ns production MD run, extended to 20 ns when required, leading to a total of 40–80 ns of unrestrained MD per system. Each compound was also independently simulated in explicit solvent. Further details are provided in the Supporting Information. All four replicates were used to calculate a relative binding energy, given in Table 1. Ligand positions per pose can be seen in Figure S4.
Table 1. Summary of Results of LIE Calculations Per Compound, Per Pose Where the Poses Are Related by a 180° Inversion within the Binding Sitea.
| ligand
in water (kcal mol–1) |
ligand
in protein (kcal mol–1) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | virtual screening pose | binding mode in crystallo | ⟨Ul-svdw⟩ | ⟨Ul-sel⟩ | ⟨Ul-svdw⟩ | ⟨Ul-sel⟩ | β | averageΔGbind–γ (kcal mol–1 ± SE) | |
| 3 | 1 | √ | √ | –28.0 | –58.9 | –52.4 | –43.9 | 0.43 | 2.1 ± 0.2 |
| 2 | √ | × | –28.0 | –58.9 | –55.3 | –37.2 | 0.43 | 4.4 ± 0.4 | |
| 85 | 1 | √ | × | –32.7 | –73.9 | –63.0 | –42.3 | 0.43 | 8.1 ± 0.4 |
| 2 | × | √ | –32.7 | –73.9 | –62.0 | –47.5 | 0.43 | 6.1 ± 0.8 | |
| 10 | 1 | √ | √ | –25.9 | –40.1 | –52.2 | –21.6 | 0.37 | 2.1 ± 0.3 |
| 2 | √ | √ | –25.9 | –40.1 | –50.1 | –22.9 | 0.37 | 2.0 ± 0.3 | |
| BDM31343 | 1 | √ | √ | –27.6 | –42.7 | –48.7 | –28.4 | 0.43 | 2.3 ± 0.4 |
| 2 | × | × | –27.6 | –42.7 | –48.5 | –25.6 | 0.43 | 3.6 ± 0.7 | |
Ticks denote where (a) the pose could be observed in the five solutions from virtual screening and (b) the pose corresponds to the binding mode in the crystal. For BDM31343, virtual screening poses refer to docking output presented previously.11 Averages were derived from the final 10 ns of converged simulation time. Relative binding energies are shown unadjusted for the γ factor.
Per-residue contributions to binding energy (Figure 3 and Table S2) were calculated for each complex. These calculations allowed us to confirm the relevance of the interactions observed in the crystal structure and identify any exploitable interactions for ligand development that were not captured by the crystal structure. This was particularly relevant for compounds 3 and 85 which were indicated to be more flexible and of lower occupancy, respectively, in the EthR binding site in crystallo. While BDM31343 has been reported to have an EC50 of 3.3 × 10–6 mol, compounds 3, 10, and 85 displayed no antitubercular booster activity.18 Although the compounds appear quite similar in terms of molecular weight and size, our lead compounds display significantly more polar surfaces compared to BDM31343, which may be responsible for a reduced uptake through the very hydrophobic M. tuberculosis cell wall. The detailed energetic characterization of protein–ligand interactions reported here will be used to further optimize our lead compounds.
Figure 3.
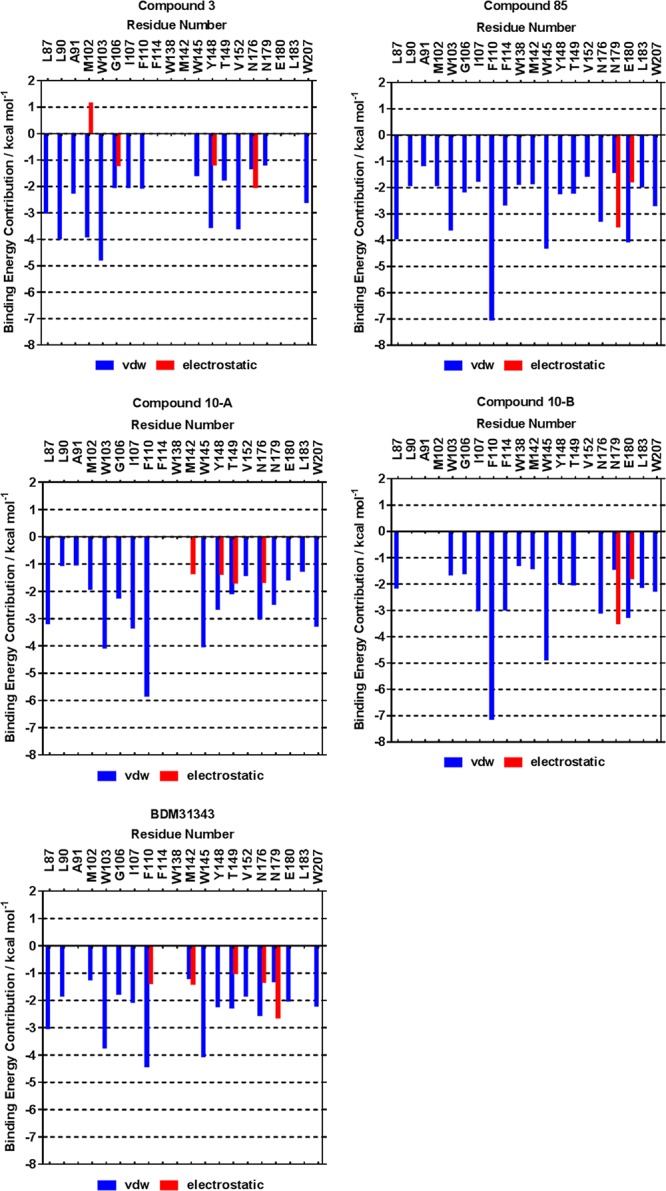
Mean contribution of energy to the overall binding, per-residue of the binding site, for both (a) electrostatic and (b) van der Waals energies for compounds 3, 10, and 85 and BDM31343. Energy contributions over the course of the simulation are given in Figure S3. Only values exceeding ±1 kcal mol–1 are shown. Graphs prepared in Graphpad Prism 7.
Figure 3 shows the electrostatic and van der Waals contribution of the relevant binding site residues for each ligand (contributions for compound 10 are deconvoluted as 10-A and 10-B). Negative binding energy contributions denote a favorable interaction. As anticipated, given the highly lipophilic nature of the binding channel, binding is mostly driven by van der Waals interactions (shown in blue). The common largest contributions arise from residues F110, W103, W145, and W207. Electrostatic interactions are mostly due to residues N176 and N179, which are the two key hydrogen bonding residues of the EthR binding channel. Compound 3 adopts a different binding position than the other presented ligands; it interacts more strongly with L90, M102, W103, and V152 and weakly with F110, W145, N176, and N179. Binding by compound 85 is dominated by van der Waals interactions; compound 85 makes electrostatic interactions with only N179 and E180. Clear differences can be seen between poses A and B of compound 10; the alternate positions of the carbonyl oxygen forms an HB interaction with N176 in pose A, while in pose B this interaction occurs preferentially with N179. Similarly, only pose A of compound 10 interacts with Y148 and T149 and the binding energy includes a contribution from M142; pose B interacts favorably with E180. Compound 3 also benefits from the strong electrostatic contributions by G106 due to Cα-H···O and Cα-H···N hydrogen bonds. A Cα-H···S weak hydrogen bond from the sulfur of M142 to compound 10-A is responsible for a M142 contribution to binding energy, which is more pronounced for compound 85 where a short Cα-H···S contact by the M142 sulfur can be observed in the crystal structure and the van der Waals energy contribution ranges from −1.60 to −2.49 kcal mol–1. M142 is also involved in BDM31343 binding: the M142 sulfur interacts with the cyano-group of the ligand. BDM31343 makes more favorable electrostatic interactions than the other ligands, and van der Waals interactions are weaker in energy contribution. Overall, analysis of the energy contributions to ligand binding corroborate the interactions inferred from the crystal structures, emphasizing the lipophilicity-driven nature of the binding to EthR. This data can be used to quantitatively evaluate the binding event and help inform design decisions—at which positions to try to strengthen hydrogen bond acceptors, for example, or to exploit “weak” hydrogen bonding opportunities such as those seen via G106 with compound 3 or M142 with BDM31343 and compound 85. Given the evidence of weak binding by 85, both compounds 3 and 10-A provide the best starting points for further development.
Figure 4a shows the overlay of the crystal structure of compound 3, with each of the two simulated poses (taken as an average structure at the 10 ns time point). Pose 1 (the highest-scoring VS mode) is a match for the crystallographically observed binding mode and is also the most energetically favorable binder as compared to pose 2 (Table 1, ΔΔG = 2.3 ± 0.6 kcal mol–1). For compound 85, the results of the virtual screening suggested only one pose (pose 1). However, it is pose 2 which is energetically more favorable (ΔΔG = 2.0 ± 1.2 kcal mol–1, Table 1). In the case of compound 10, the dual occupancy found in the crystal structure is in line with our MD–LIE free energy calculations, which demonstrates that the binding free energy for these poses is almost identical (ΔΔG = 0.1 ± 0.6 kcal mol–1, Figure 4c). Finally, for BDM31343 (Figure 4d) pose 1 is energetically favorable (ΔΔG = 1.3 ± 1.1 kcal mol–1), and it corresponds to the pose found in the crystal structure. Therefore, we can conclude that both virtual screening and the MD–LIE estimate correctly predict the binding mode of compounds 3 and 10 and BDM31343. However, it is only the MD–LIE approach that correctly identified the experimentally observed binding mode of compound 85. Therefore, we have demonstrated that the MD–LIE approach allows one to accurately determine and rank potential poses from the in silico screening output.
Figure 4.
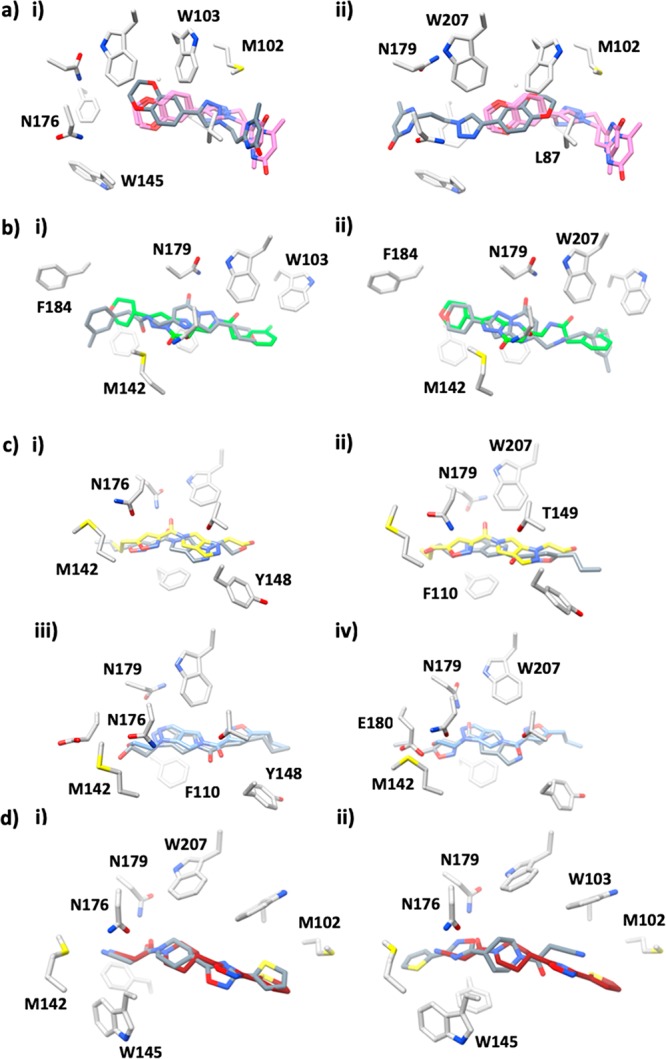
Comparison of the average structure at 10 ns for each molecular dynamics simulation with the crystal structure. For matching modes, the closest replica is used; replicate 1 is given as representative for alternative mode. (a) Compound 3 (in pink) vs (i) pose 1 and (ii) pose 2. (b) Compound 85 (in green) vs (i) pose 1 and (ii) pose 2. (c) Compound 10-A (in yellow) vs (i) pose 1 and (ii) pose 2; compound 10-B (in blue) vs (iii) pose 1 and (iv) pose 2. (d) BDM31343 (in dark red) vs (i) pose 1 and (ii) pose 2.
In summary, this contribution has addressed a key issue posed to computer-aided drug discovery on the EthR target. In this system, the cylindrical, hydrophobic binding site may give rise to multiple likely binding modes which may be difficult to infer only by docking score rank. Determination of the most favorable pose(s) requires crystallographic confirmation, which may be difficult to achieve. The inclusion of the MD–LIE approach to calculate the relative binding energy calculations is an established practice for ranking small cohorts of screening outputs. Though still not viable for routine screening at scale,31,32 its application on the EthR systems has the potential to accelerate the identification and evaluation of novel ligands. Further studies would be required to determine if MD–LIE is sensitive enough for smaller fragment-sized molecules with weak binding affinities, or if indeed a one-simulation end-point method such as MM/GBSA or MM/PBSA could be a higher-throughput method with sufficient accuracy. MD–LIE or an equivalent method could be used to prioritize outputs from crystallographic fragment screening,33 where multiple, millimolar-affinity binding events across a protein surface are frequently observed.34
Recent literature has demonstrated that multiple binding modes within series can be commonplace35 and often are unapparent unless crystal structures are obtained. This is also true of EthR, for example with BDM31343 and related compound BDM14500.5 In the absence of a crystal structure (or indeed a complete set of crystal structures to understand structure–activity relationships or SAR), MD–LIE can offer an alternative stop-gap measure where assays confirm binding but cocrystal structures remain elusive. However, further study is required to determine if our approach is applicable to targets beyond EthR. Furthermore, per-residue energy contributions to the binding event will enable medicinal chemistry design strategies by characterizing the protein–ligand interaction and highlighting where gains could be made (or unfavorable interactions are occurring).36
Ultimately, the pilot experiment presented here suggests a role for relative binding energies and per-residue contributions in not just ranking ligand binders but also confirming ligand binding modes. Furthermore, we have contributed two more crystal structures to the growing data on EthR complexes, which are a valuable resource for further study.19 Moving forward, because of the hydrophobicity of the EthR binding channel it will be all the more important to exploit unconventional binding modes and chemistries to optimize hits toward active cell-penetrant lead compounds, and inclusion of MD–LIE in this pipeline will ensure the best candidates are prioritized.
Acknowledgments
This work was conducted with the resources of the Swedish National Infrastructure for Computing (SNIC). N.J.T. gratefully acknowledges support from the Cambridge Crystallographic Data Centre and the EPSRC (EP/J500215/1) for a predoctoral fellowship and an EMBO Short Term Fellowship (ASTF 123-2015) which funded travel and subsistence for this work to be carried out. This work was also supported by the Swedish Foundation for International Co-operation in Research and Higher Education (STINT). We are grateful to Ian Edwards and Erin Dickinson for their excellent technical support in protein production and crystallization and the Diamond Light Source for beamtime and their excellent assistance on beamlines I03 and I04-1, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.9b00741.
Experimental Methods and supplementary chart and tables (PDF)
The authors declare no competing financial interest.
Notes
The crystal structure data for EthR with compound 3 and compound 85 are deposited in the PDB under respective accession codes 6R1P and 6R1S.
Supplementary Material
References
- Dover L. G.; Corsino P. E.; Daniels I. R.; Cocklin S. L.; Tatituri V.; Besra G. S.; Fütterer K. Crystal Structure of the TetR/CamR Family Repressor Mycobacterium Tuberculosis EthR Implicated in Ethionamide Resistance. J. Mol. Biol. 2004, 340 (5), 1095–1105. 10.1016/j.jmb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Engohang-Ndong J.; Baillat D.; Aumercier M.; Bellefontaine F.; Besra G. S.; Locht C.; Baulard A. R. EthR, a Repressor of the TetR/CamR Family Implicated in Ethionamide Resistance in Mycobacteria, Octamerizes Cooperatively on Its Operator: Regulation of Ethionamide Resistance in Mycobacteria. Mol. Microbiol. 2004, 51 (1), 175–188. 10.1046/j.1365-2958.2003.03809.x. [DOI] [PubMed] [Google Scholar]
- Frénois F.; Engohang-Ndong J.; Locht C.; Baulard A. R.; Villeret V. Structure of EthR in a Ligand Bound Conformation Reveals Therapeutic Perspectives against Tuberculosis. Mol. Cell 2004, 16 (2), 301–307. 10.1016/j.molcel.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Frénois F.; Baulard A. R.; Villeret V. Insights into Mechanisms of Induction and Ligands Recognition in the Transcriptional Repressor EthR from Mycobacterium Tuberculosis. Tuberculosis 2006, 86 (2), 110–114. 10.1016/j.tube.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Willand N.; Dirié B.; Carette X.; Bifani P.; Singhal A.; Desroses M.; Leroux F.; Willery E.; Mathys V.; Déprez-Poulain R.; et al. Synthetic EthR Inhibitors Boost Antituberculous Activity of Ethionamide. Nat. Med. 2009, 15 (5), 537–544. 10.1038/nm.1950. [DOI] [PubMed] [Google Scholar]
- Willand N.; Desroses M.; Toto P.; Dirié B.; Lens Z.; Villeret V.; Rucktooa P.; Locht C.; Baulard A.; Deprez B. Exploring Drug Target Flexibility Using in Situ Click Chemistry: Application to a Mycobacterial Transcriptional Regulator. ACS Chem. Biol. 2010, 5 (11), 1007–1013. 10.1021/cb100177g. [DOI] [PubMed] [Google Scholar]
- Carette X.; Blondiaux N.; Willery E.; Hoos S.; Lecat-Guillet N.; Lens Z.; Wohlkonig A.; Wintjens R.; Soror S. H.; Frenois F.; et al. Structural Activation of the Transcriptional Repressor EthR from Mycobacterium Tuberculosis by Single Amino Acid Change Mimicking Natural and Synthetic Ligands. Nucleic Acids Res. 2012, 40 (7), 3018–3030. 10.1093/nar/gkr1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flipo M.; Desroses M.; Lecat-Guillet N.; Dirié B.; Carette X.; Leroux F.; Piveteau C.; Demirkaya F.; Lens Z.; Rucktooa P.; et al. Ethionamide Boosters: Synthesis, Biological Activity, and Structure-Activity Relationships of a Series of 1, 2, 4-Oxadiazole EthR Inhibitors. J. Med. Chem. 2011, 54 (8), 2994–3010. 10.1021/jm200076a. [DOI] [PubMed] [Google Scholar]
- Flipo M.; Desroses M.; Lecat-Guillet N.; Villemagne B.; Blondiaux N.; Leroux F.; Piveteau C.; Mathys V.; Flament M.-P.; Siepmann J.; et al. Ethionamide Boosters. 2. Combining Bioisosteric Replacement and Structure-Based Drug Design To Solve Pharmacokinetic Issues in a Series of Potent 1, 2, 4-Oxadiazole EthR Inhibitors. J. Med. Chem. 2012, 55 (1), 68–83. 10.1021/jm200825u. [DOI] [PubMed] [Google Scholar]
- Flipo M.; Willand N.; Lecat-Guillet N.; Hounsou C.; Desroses M.; Leroux F.; Lens Z.; Villeret V.; Wohlkönig A.; Wintjens R.; et al. Discovery of Novel N -Phenylphenoxyacetamide Derivatives as EthR Inhibitors and Ethionamide Boosters by Combining High-Throughput Screening and Synthesis. J. Med. Chem. 2012, 55 (14), 6391–6402. 10.1021/jm300377g. [DOI] [PubMed] [Google Scholar]
- Tatum N. J.; Villemagne B.; Willand N.; Deprez B.; Liebeschuetz J. W.; Baulard A. R.; Pohl E. Structural and Docking Studies of Potent Ethionamide Boosters. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2013, C69, 1243–1250. 10.1107/S0108270113028126. [DOI] [PubMed] [Google Scholar]
- Villemagne B.; Flipo M.; Blondiaux N.; Crauste C.; Malaquin S.; Leroux F.; Piveteau C.; Villeret V.; Brodin P.; Villoutreix B. O.; et al. Ligand Efficiency Driven Design of New Inhibitors of Mycobacterium Tuberculosis Transcriptional Repressor EthR Using Fragment Growing, Merging, and Linking Approaches. J. Med. Chem. 2014, 57 (11), 4876–4888. 10.1021/jm500422b. [DOI] [PubMed] [Google Scholar]
- Crauste C.; Willand N.; Villemagne B.; Flipo M.; Willery E.; Carette X.; Dimala M. M.; Drucbert A.-S.; Danze P.-M.; Deprez B.; et al. Unconventional Surface Plasmon Resonance Signals Reveal Quantitative Inhibition of Transcriptional Repressor EthR by Synthetic Ligands. Anal. Biochem. 2014, 452, 54–66. 10.1016/j.ab.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Nikiforov P. O.; Surade S.; Blaszczyk M.; Delorme V.; Brodin P.; Baulard A. R.; Blundell T. L.; Abell C. A Fragment Merging Approach towards the Development of Small Molecule Inhibitors of Mycobacterium Tuberculosis EthR for Use as Ethionamide Boosters. Org. Biomol. Chem. 2016, 14 (7), 2318–2326. 10.1039/C5OB02630J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov P. O.; Blaszczyk M.; Surade S.; Boshoff H. I.; Sajid A.; Delorme V.; Deboosere N.; Brodin P.; Baulard A. R.; Barry C. E.; et al. Fragment-Sized EthR Inhibitors Exhibit Exceptionally Strong Ethionamide Boosting Effect in Whole-Cell Mycobacterium Tuberculosis Assays. ACS Chem. Biol. 2017, 12 (5), 1390–1396. 10.1021/acschembio.7b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surade S.; Ty N.; Hengrung N.; Lechartier B.; Cole S.; Abell C.; Blundell T. A Structure-Guided Fragment-Based Approach for the Discovery of Allosteric Inhibitors Targeting the Lipophilic Binding Site of Transcription Factor EthR. Biochem. J. 2014, 458, 387–394. 10.1042/BJ20131127. [DOI] [PubMed] [Google Scholar]
- Dover L. G.; Alahari A.; Gratraud P.; Gomes J. M.; Bhowruth V.; Reynolds R. C.; Besra G. S.; Kremer L. EthA, a Common Activator of Thiocarbamide-Containing Drugs Acting on Different Mycobacterial Targets. Antimicrob. Agents Chemother. 2007, 51 (3), 1055–1063. 10.1128/AAC.01063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatum N. J.; Liebeschuetz J.; Cole J. C.; Frita R.; Herledan A.; Baulard A.; Willand N.; Pohl E. New Active Leads for Tuberculosis Booster Drugs by Structure-Based Drug Discovery. Org. Biomol. Chem. 2017, 15, 10245–10255. 10.1039/C7OB00910K. [DOI] [PubMed] [Google Scholar]
- Tanina A.; Wohlkönig A.; Soror S. H.; Flipo M.; Villemagne B.; Prevet H.; Déprez B.; Moune M.; Perée H.; Meyer F.; et al. A Comprehensive Analysis of the Protein-Ligand Interactions in Crystal Structures of Mycobacterium Tuberculosis EthR. Biochim. Biophys. Acta, Proteins Proteomics 2019, 1867 (3), 248–258. 10.1016/j.bbapap.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Tian W.; Chen C.; Lei X.; Zhao J.; Liang J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46 (W1), W363–W367. 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plewczynski D.; Łaźniewski M.; Augustyniak R.; Ginalski K. Can We Trust Docking Results? Evaluation of Seven Commonly Used Programs on PDBbind Database. J. Comput. Chem. 2011, 32 (4), 742–755. 10.1002/jcc.21643. [DOI] [PubMed] [Google Scholar]
- Wang T.; Wu M.-B.; Zhang R.-H.; Chen Z.-J.; Hua C.; Lin J.-P.; Yang L.-R. Advances in Computational Structure-Based Drug Design and Application in Drug Discovery. Curr. Top. Med. Chem. 2015, 16, 901–916. 10.2174/1568026615666150825142002. [DOI] [PubMed] [Google Scholar]
- Gaieb Z.; Liu S.; Gathiaka S.; Chiu M.; Yang H.; Shao C.; Feher V. A.; Walters W. P.; Kuhn B.; Rudolph M. G.; et al. D3R Grand Challenge 2: Blind Prediction of Protein-Ligand Poses, Affinity Rankings, and Relative Binding Free Energies. J. Comput.-Aided Mol. Des. 2018, 32 (1), 1–20. 10.1007/s10822-017-0088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59 (9), 4103–4120. 10.1021/acs.jmedchem.5b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastelli G.; Degliesposti G.; Del Rio A.; Sgobba M. Binding Estimation after Refinement, a New Automated Procedure for the Refinement and Rescoring of Docked Ligands in Virtual Screening. Chem. Biol. Drug Des. 2009, 73 (3), 283–286. 10.1111/j.1747-0285.2009.00780.x. [DOI] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59 (9), 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- Åqvist J.; Marelius J. The Linear Interaction Energy Method for Predicting Ligand Binding Free Energies. Comb. Chem. High Throughput Screening 2001, 4 (8), 613–626. 10.2174/1386207013330661. [DOI] [PubMed] [Google Scholar]
- Almlöf M.; Carlsson J.; Åqvist J. Improving the Accuracy of the Linear Interaction Energy Method for Solvation Free Energies. J. Chem. Theory Comput. 2007, 3 (6), 2162–2175. 10.1021/ct700106b. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-de-Terán H.; Åqvist J. Linear Interaction Energy: Method and Applications in Drug Design. Methods Mol. Biol. 2012, 819, 305–323. 10.1007/978-1-61779-465-0_20. [DOI] [PubMed] [Google Scholar]
- King G.; Warshel A. A Surface Constrained All-atom Solvent Model for Effective Simulations of Polar Solutions. J. Chem. Phys. 1989, 91 (6), 3647–3661. 10.1063/1.456845. [DOI] [Google Scholar]
- Ganesan A.; Coote M. L.; Barakat K. Molecular Dynamics-Driven Drug Discovery: Leaping Forward with Confidence. Drug Discovery Today 2017, 22 (2), 249–269. 10.1016/j.drudis.2016.11.001. [DOI] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59 (9), 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- Patel D.; Bauman J. D.; Arnold E. Advantages of Crystallographic Fragment Screening: Functional and Mechanistic Insights from a Powerful Platform for Efficient Drug Discovery. Prog. Biophys. Mol. Biol. 2014, 116 (2), 92–100. 10.1016/j.pbiomolbio.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. The Rise of Fragment-Based Drug Discovery. Nat. Chem. 2009, 1 (3), 187–192. 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- Drwal M. N.; Bret G.; Perez C.; Jacquemard C.; Desaphy J.; Kellenberger E. Structural Insights on Fragment Binding Mode Conservation. J. Med. Chem. 2018, 61 (14), 5963–5973. 10.1021/acs.jmedchem.8b00256. [DOI] [PubMed] [Google Scholar]
- Hudson L.; Mui J.; Vázquez S.; Carvalho D. M.; Williams E.; Jones C.; Bullock A. N.; Hoelder S. Novel Quinazolinone Inhibitors of ALK2 Flip between Alternate Binding Modes: Structure-Activity Relationship, Structural Characterization, Kinase Profiling, and Cellular Proof of Concept. J. Med. Chem. 2018, 61 (16), 7261–7272. 10.1021/acs.jmedchem.8b00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.