

Abstract
A targeted ultrahigh-performance liquid chromatography tandem mass spectrometry with electrospray ionization (UHPLC-ESI-MS/MS) method has been developed for the quantification of tryptophan and its downstream metabolites from the kynurenine and serotonin pathways. The assay coverage also includes markers of gut health and inflammation, including citrulline and neopterin. The method was designed in 96-well plate format for application in multiday, multiplate clinical and epidemiology population studies. A chromatographic cycle time of 7 min enables the analysis of two 96-well plates in 24 h. To protect chromatographic column lifespan, samples underwent a two-step extraction, using solvent protein precipitation followed by delipidation via solid-phase extraction (SPE). Analytical validation reported accuracy of each analyte <20% for the lowest limit of quantification and <15% for all other quality control (QC) levels. The analytical precision for each analyte was 2.1–12.9%. To test the applicability of the method to multiplate and multiday preparations, a serum pool underwent periodic repeat analysis during a run consisting of 18 plates. The % CV (coefficient of variation) values obtained for each analyte were <15%. Additional biological testing applied the assay to samples collected from healthy control participants and two groups diagnosed with inflammatory bowel disease (IBD) (one group treated with the anti-inflammatory 5-aminosalicylic acid (5-ASA) and one group untreated), with results showing significant differences in the concentrations of picolinic acid, kynurenine, and xanthurenic acid. The short analysis time and 96-well plate format of the assay makes it suitable for high-throughput targeted UHPLC-ESI-MS/MS metabolomic analysis in large-scale clinical and epidemiological population studies.
Population wide metabolic phenotyping allows for the untargeted discovery of metabolic biomarkers of health and disease. The use of untargeted mass spectrometry (MS)-based metabolite profiling in large epidemiological cohorts is now widespread.1−5 However, the relative lack of sensitivity of full-scan instrumentation (QTOF, Orbitrap) compared with tandem MS and the fact that stable isotope-labeled standards to compensate for matrix effects are seldom used in screening mode mean that the study design often relies on the observation of relative fold changes between analytes.
Therefore, a prudent study design employs untargeted metabolic profiling to identify potential pathways of interest, followed by confirmation using a fully quantified approach.6 The greater sensitivity of tandem MS allows for the measurement of pathway intermediates that are present at low concentrations and that may not have been detected in the untargeted assays, thereby enabling greater detail in the reporting of mechanistic changes within specific pathways.
The biochemical fate of the essential amino acid tryptophan is one such pathway. While tryptophan is important for protein synthesis,7 it is also metabolized to a number of bioactive compounds that function in physiological processes including immunoregulation,8 inflammation,9,10 and neurotransmission.11 Tryptophan has also been identified as a key metabolite in host-gut microbiome signaling,12 and its bioavailability (and therefore that of its downstream metabolites) is influenced by the microbial balance within the gut.12
The majority of available tryptophan is metabolized in mammalian systems through the kynurenine pathway via the tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO) enzymes (Figure S1). Downstream, this pathway contains many neurologically active compounds including kynurenic acid,13 quinolinic acid,14 and 3-hydroxykynurenine (3-HK),13 with quinolinic acid reported as having neurotoxic properties in the central nervous system (CNS).14,15 A secondary metabolic pathway leads to the production of the neurotransmitter serotonin.11 Dysregulation of the kynurenine metabolic route has been reported in multiple neurological conditions including dementia,16−18 depression,19−21 schizophrenia,22,23 and anorexia.24
Conditions of gut dysfunction including irritable bowel syndrome (IBS),25,26 inflammatory bowel disease (IBD),27,28 gut dysfunction linked acute pancreatitis,29,30 and environmental enteric dysfunction and enteric infections31−33 have also been reported to be associated with changes in concentrations of metabolites in the kynurenine pathway. As such, it is also important to measure additional biomarkers of gut health and systemic inflammation; for example, citrulline is indicative of enterocyte mass reduction and is observed to decrease with various villus atrophy syndromes.34 Additionally, neopterin is an inflammatory mediator with circulating concentrations elevated in many conditions when the cellular immune system is activated.35,36 A comprehensive review of the metabolism and role of kynurenines in health and disease has been published by Cervenka et al.9
Tryptophan and its metabolites have previously been quantified using LC-UV37 and LC-MS/MS.28,38−44 However, existing methodology either has been applied to relatively small studies (<20 individuals) for biological validation28,40,41,43 or has only focused on the major metabolites such as kynurenine and tryptophan.37,39,42 Because of the rise in molecular phenomics in epidemiology, metabolite analysis is increasingly being applied to large population cohorts,1,45 and therefore the methodology presented here was designed to be sufficiently stable to allow for multiday and multibatch preparations. A 96-well plate format was advantageous for this purpose, whereas previous methods have typically performed sample preparation in individual plastic sample tubes.38,39,43 The resulting method was demonstrated to be stable over a multiday run consisting of 18 plates.
As a proof of concept, the method was subsequently applied to a sample set consisting of healthy controls and two groups of participants diagnosed with ulcerative colitis (UC), a subtype of IBD. One IBD group was untreated, while the second was treated with the anti-inflammatory 5-aminosalicylic acid (5-ASA). The sample set was considered as appropriate for method testing as variations in the concentration of serum tryptophan have been previously associated with subtypes of IBD including both UC and Crohn’s disease.27,28
Experimental Section
Chemicals and Reagents
The analyte standards (listed in Table 1) were purchased from Sigma-Aldrich (Gillingham, U.K.) except for neopterin, NAD+, and quinolinic acid, which were purchased from Cayman Chemicals (Ann Arbor, MI, U.S.A.), and nicotinic riboside, which was purchased from Toronto Research Chemicals (Toronto, Canada). Stable isotope-labeled (SIL) standards were used as internal standards (listed in Table 1) and were purchased from Toronto Research Chemicals (Toronto, Canada) except for citrulline-D4, which was purchased from Cambridge Isotopes (Cambridge, MA, U.S.A.). LC-MS-grade acetonitrile and formic acid were purchased from Sigma-Aldrich (Gillingham, U.K.). LC-MS-grade water was purchased from Fisher Scientific (Loughborough, U.K.). Phenomenex PHREE solid-phase extraction plates were purchased directly from Phenomenex (Macclesfield, U.K.).
Table 1. MS Conditions and Chromatographic Retention Time for Each Metabolite and Labeled Internal Standard (Ordered by Retention Time).
| metabolite name | SIL internal standard | parent m/z | quantifier m/z (Q ion) | qualifier m/z (q ion) | retention time (min) | Q ion dwell time (s) | MS polarity | Q ion cone voltage (V) | Q ion collision energy (V) |
|---|---|---|---|---|---|---|---|---|---|
| citrulline-D4 | 180.1 | 74.1 | 117.1 | 0.55 | 0.003 | + | 5 | 20 | |
| citrulline | citrulline-D4 | 176.1 | 113.1 | 70.1 | 0.55 | 0.003 | + | 5 | 9 |
| βNM | neopterin-13C5 | 335.1 | 123.1 | 97.1 | 0.67 | 0.010 | + | 5 | 15 |
| nicotinamide riboside-D3 | 258.1 | 126.1 | 109.1 | 0.73 | 0.004 | + | 5 | 20 | |
| nicotinamide riboside | nicotinamide riboside-D3 | 255.1 | 106.1 | 123.1 | 0.74 | 0.010 | + | 5 | 28 |
| picolinic acid-D3 | 127.1 | 81.1 | 53.1 | 0.86 | 0.005 | + | 36 | 16 | |
| picolinic acid | picolinic acid-D3 | 124.1 | 78.1 | 51.1 | 0.88 | 0.020 | + | 34 | 14 |
| neopterin-13C5 | 259.1 | 210.1 | 197.1 | 0.89 | 0.020 | + | 5 | 18 | |
| neopterin | neopterin-13C5 | 254.1 | 206.1 | 190.1 | 0.89 | 0.020 | + | 5 | 18 |
| nicotinic acid-D4 | 128.1 | 81.1 | 56.1 | 0.94 | 0.005 | + | 36 | 10 | |
| nicotinic acid | nicotinic acid-D4 | 124.1 | 78.1 | 53.1 | 0.95 | 0.020 | + | 34 | 14 |
| quinolinic acid-D3 | 171.1 | 81.1 | 109.1 | 0.99 | 0.006 | + | 5 | 20 | |
| quinolinic acid | quinolinic acid-D3 | 168.1 | 78.1 | 106.1 | 1.01 | 0.006 | + | 5 | 20 |
| dopamine-D4 | 158.1 | 95.1 | 123.1 | 1.10 | 0.010 | + | 26 | 22 | |
| dopamine | dopamine-D4 | 154.1 | 91.1 | 119.1 | 1.11 | 0.010 | + | 16 | 20 |
| NAD+ | N/A(monitored compound only) | 664.1 | 136.1 | 428.1 | 1.16 | 0.004 | + | 44 | 42 |
| 3-HK-13C2-15N | 228.1 | 110.1 | NA | 1.31 | 0.015 | + | 10 | 16 | |
| 3-HK | 3-HK-13C2-15N | 225.1 | 162.1 | 110.1 | 1.31 | 0.020 | + | 10 | 18 |
| serotonin | dopamine-D4 | 160.1 | 132.1 | 105.1 | 1.94 | 0.025 | + | 30 | 18 |
| kynurenine-D4 | 213.1 | 98.1 | 150.1 | 2.11 | 0.026 | + | 18 | 12 | |
| kynurenine | kynurenine-D4 | 209.1 | 94.1 | 146.1 | 2.15 | 0.026 | + | 30 | 12 |
| 3-HAA-D3 | 157.1 | 83.1 | 111.1 | 2.72 | 0.050 | + | 5 | 22 | |
| 3-HAA | 3-HAA-D3 | 154.0 | 80.0 | 108.0 | 2.76 | 0.080 | + | 5 | 22 |
| tryptophan-D5 | 208.3 | 120.3 | 164.3 | 3.23 | 0.015 | - | 30 | 15 | |
| tryptophan | tryptophan-D5 | 203.1 | 116.1 | 142.1 | 3.26 | 0.020 | - | 30 | 18 |
| xanthurenic acid-D4 | 210.1 | 164.1 | 136.1 | 3.39 | 0.017 | + | 8 | 26 | |
| xanthurenic acid | xanthurenic acid-D4 | 206.1 | 132.1 | 136.1 | 3.41 | 0.017 | + | 30 | 26 |
| kynurenic acid-D5 | 195.1 | 149.1 | 94.1 | 3.78 | 0.017 | + | 8 | 18 | |
| kynurenic acid | kynurenic acid-D5 | 190.1 | 144.1 | 116.1 | 3.80 | 0.017 | + | 44 | 18 |
| 5-HIAA D5 | 197.1 | 150.1 | 122.1 | 3.87 | 0.017 | + | 6 | 14 | |
| 5-HIAA | 5-HIAA-D5 | 192.1 | 146.1 | 118.1 | 3.88 | 0.017 | + | 40 | 18 |
| I-3-AA-D4 | 180.1 | 133.1 | 106.1 | 4.16 | 0.017 | + | 6 | 18 | |
| I-3-AA | I-3-AA-D4 | 176.1 | 103.1 | 77.1 | 4.17 | 0.017 | + | 4 | 28 |
Analytical Protocol
Preparation of Standard and Quality Control Diluent
Two diluents were prepared for use in the preparation of calibration and quality control (QC) stocks. Diluent-D1 consisted of water with 1 mg/mL (0.1%) citric acid, and diluent-D2 consisted of water with 0.1 mg/mL (0.01%) citric acid.
Standard Parent Stock Solution Preparation
Stock solutions (1 mg/mL) for 5-hydroxyindole-3-acetic acid (5-HIAA), nicotinamide adenine dinucleotide (NAD+), citrulline, dopamine, picolinic acid, serotonin, quinolinic acid, 3-hydroxykynurenine (3-HK), nicotinic acid, kynurenine, β-nicotinamide mononucleotide (βNM), tryptophan, and nicotinamide riboside were dissolved in 100% diluent-D1. Stock solutions (1 mg/mL) for xanthurenic acid, 3-hydroxyanthranilic acid (3-HAA), indole-3-acetic acid (I-3-AA), and kynurenic acid were dissolved in 60% 0.1 M sodium hydroxide (NaOH) and 40% diluent-D1. A 1 mg/mL stock solution of neopterin was prepared using 100% dimethyl sulfoxide (DMSO).
Dilution of Calibration Standards and Quality Control Standards
Parent 1 mg/mL stock solutions were diluted into two duplicate solutions using diluent-D2 as presented in Tables S1 and S2 to produce suitable calibration and QC ranges for each target analyte. The final concentration ranges for each target analyte are presented in Table S3.
Preparation of Stable Isotope-Labeled Stock Solutions
Stock solutions (1 mg/mL) for 5-hydroxyindole-3-acetic acid-D5 (5-HIAA-D5), citrulline-D4, dopamine-D4, picolinic acid-D3, quinolinic acid-D3, 3-hydroxykynurenine-13C2-15N (3-HK-13C2-15N), nicotinic acid-D4, kynurenine-D4, tryptophan-D5, and nicotinamide riboside-D3 were dissolved in 100% diluent-D1. Stock solutions (1 mg/mL) for xanthurenic acid-D4, 3-hydroxyanthranilic acid-D3 (3-HAA-D3), indole-3-acetic acid-D4 (I-3-AA-D4), and kynurenic acid-D5 were dissolved in 60% 0.1 M sodium hydroxide (NaOH) and 40% diluent-D1.
A 1 mg/mL stock solution of neopterin-13C5 was prepared using 100% dimethyl sulfoxide (DMSO). The internal standard (IS) working solution was prepared using the dilution sequence presented in Table S4.
Plasma and Serum Sample Extraction and Preparation
For the assay validation, human plasma and serum samples were purchased from Seralabs (now BioIVT, West Sussex, U.K.). Six samples (3 male, 3 female) were purchased for both plasma and serum. The plasma and serum samples were obtained from different groups of individual donors. Pooled human plasma and serum samples (60 individuals: 30 male, 30 female) used in the validation were also purchased from Seralabs (BioIVT). Again, plasma and serum donors were from different groups.
Human plasma and serum samples were left to thaw at 4 °C and then vortex-mixed. An aliquot (30 μL) of each sample, calibration standard, quality control, and blank was transferred to a 600 μL Eppendorf 96-well plate (Figure S2), and 10 μL of internal standard working solution was added to each well containing sample, calibration standard, quality control, and single blank wells but not to double blank wells.
Protein and phospholipid removal was performed using solvent precipitation in combination with a pass through a 96-well Phenomenex PHREE SPE plate (Phenomenex, Macclesfield, U.K.). Prior to use, the PHREE SPE plates were prewashed using 250 μL of methanol containing 10 mM ammonium formate added to each well, with elution to waste by centrifugation at 500g for 5 min.
Methanol (250 μL) containing 10 mM ammonium formate was added to each well in the analytical plate. The analytical plates were then foil-capped, vortex-mixed, and briefly centrifuged (500g for 30 s at 20 °C) to ensure that the precipitated samples were at the bottom of the wells. Following this, the total content of the analytical plate wells were pipet-mixed using a multichannel pipet and transferred to the prewashed PHREE SPE plate.
Samples were drawn through the PHREE plate into a fresh collection plate (700 μL high-recovery plate, Waters, Wilmslow, U.K.) via centrifugation at 500g for 5 min. The PHREE plate was then washed with a further 250 μL of methanol containing 10 mM ammonium formate and eluted into the same high-recovery collection plate by centrifugation at 500g for 5 min.
The collection plate was then taken to dryness under a low-flow stream of nitrogen overnight. Dry extracts were resuspended in 40 μL of water containing 10 mM ammonium formate and 0.5% formic acid. Five μL of each well was injected onto the UHPLC-ESI-MS/MS system for analysis.
UHPLC-ESI-MS/MS Analysis
The LC instrument setup consisted of a Waters Acquity UHPLC solvent management system and a Waters 2777C external autosampler (Waters, Wilmslow, U.K.). Chromatographic separation was performed with a Waters HSS T3 2.1 × 150 mm, 1.8 μm column (Waters, Wilmslow, U.K.). The mobile phase was composed of 0.1% formic acid in water (v/v) (A) and 0.1% formic acid in acetonitrile (v/v) (B). The column temperature was maintained at 45 °C, and linear gradient elution was performed at 0.6 mL/min starting at 1% B increasing to 10% B over 3 min, then increasing to 90% B at 4 min, and finally returning to 1% B at 4.1 min for column reequilibration, which was completed at 5 min. The weak and the strong washes were 95:5 water/acetonitrile (0.2% formic acid) (v/v) and 100% isopropanol (0.5% formic acid), respectively. During method development, significant carryover was observed, so an extensive needle wash cycle was employed, increasing the overall run time to 7 min per injection.
MS detection was performed with a Waters Xevo TQ-S tandem quadrupole instrument (Waters, Wilmslow, U.K.) using electrospray ionization (ESI) in positive ion mode, except for the analysis of tryptophan, which was collected using negative ESI. Multiple reaction monitoring (MRM) was used for the quantification of each compound; the specific metabolite transitions are presented in Table 1. The MS conditions for each analyte were determined via direct infusion of individual standard solutions.
Nitrogen was used as the desolvation gas, and argon was used as the collision gas. The following generic source conditions were used in positive ionization mode: capillary voltage, 2.5 kV; source offset, 30 V; desolvation temperature, 600 °C; source temperature, 150 °C, desolvation gas flow, 1000 L/h; cone gas flow, 150 L/h; nebulizer gas, 7.0 bar; collision gas, 0.15 mL/min. For the analysis of tryptophan in negative ionization mode, the capillary voltage was changed to 0.25 kV, while the remaining parameters were the same as those detailed for positive ionization mode. Compound-specific parameters are detailed in Table 1.
Data Processing
Raw UHPLC-ESI-MS/MS spectral data were processed using TargetLynx application package within MassLynx (v4.1) software (Waters Corporation). Further statistical analysis was performed within R (v3.5.1) run in RStudio (v1.1.456).
Method Validation
As far as practical, the method validation was based on “Bioanalytical method validation—Guidance for industry” by the U.S. Food and Drug Administration (FDA).46
Linear Range
Suitable calibration ranges were selected based upon analysis of a pooled sample of plasma and adjusted accordingly. For each analyte that used a SIL internal standard, or surrogate SIL internal standard, peak area response ratios were calculated and plotted against the nominal concentration. A linear fit was employed, and a 1/X2 weighting factor was applied. This was repeated for three separate days of the validation to assess linearity.
Lower Limit of Quantification
Lower limit of quantification (LLOQ) values were accepted based on an analytical precision cutoff of a 20% relative standard deviation rather than signal-to-noise (as discussed in ref (47)). The LLOQ also had to pass the carryover criteria described below.
Intra- and Interday Accuracy and Precision
For intraday assay precision, calibration standards were prepared alongside six replicate QC samples. Different QC concentrations were prepared for each analyte (Table S6), ranging from the LLOQ to the upper limit of quantification (ULOQ). For acceptance, 67% of QCs (50% at each level) were required to be within 15% (LLOQ 20%) of their nominal concentration. For interday precision, this was repeated on three separate days.
Carryover
Carryover was assessed by analyzing diluent that had been through the extraction procedure and did not contain either standards or SIL internal standards (termed “double blank”) directly after an ULOQ calibration standard. Carryover acceptance criteria required a MS MRM transition response in the double blank of ≤20% of the MRM transition response from the LLOQ standards. Carryover for the SIL internal standards was accepted if the MRM transition response in the double blank was ≤5% of the response seen from an injection of the SIL internal standards mixture (see Table S4).
Stability
Stability was assessed using QC standards prepared in the diluent described earlier. Quantification of the samples and subsequent stability assessment was made following sample storage at 24 h at 4 °C in the autosampler, in addition to being stored at −20 °C for 1 and 2 weeks. This was repeated with a low and high QC (QC 2 and QC 6).
Analytical Recovery
Because the method is for endogenous metabolites, a true analyte-free matrix is unavailable; therefore, recovery from analytical samples was estimated using the SIL internal standard compounds. To assess the extraction recovery, SIL internal standards were spiked into matrix both before and after extraction via the protein precipitation/SPE approach. This was repeated six times, with six different sources of biofluid.
Analyte/Blank Matrix Interferences
Interferences between target analytes and the blank matrix were assessed by analyzing six double blanks and inspecting the responses within the spectral windows of the analytes and comparing these with mean analyte responses in LLOQ calibration standards. For acceptance, a minimum of five double blanks had to be <20% of the LLOQ signal.
Matrix Signal Effects
Because a true analyte-free matrix is unavailable, matrix effects (ion suppression/enhancement) were evaluated using SIL internal standards spiked into both double blank samples and pooled development plasma samples after extraction through the SPE plate.
Biological Sample Testing
Long-Term Multiday Assay Performance Testing
To assess multiplate assay performance and reproducibility, a pool of serum was analyzed after every 10th injection during an analytical run that consisted of 18 plates of serum samples prepared according to the plate layout shown in Figure S2. Data were acquired over 9 days, with a total of 92 injections of the serum pool analyzed in this way. For large multiplate, multiday studies such as this, two sample plates were prepared (Figure S2) and analyzed in each 24 h time period.
Application of the Assay to Clinical Samples
Sample Collection Information
The study was approved by the London Northwest NHS trust and National Research Ethics Service (REC no. 14/EM/1290). Plasma samples were obtained from 29 study participants. Ten participants were controls and had no clinical diagnosis of UC (8 male, 2 female, age 29–35, mean age = 33.0), 9 participants had a clinical diagnosis of UC and were not undergoing treatment (5 male, 4 female, age 21–71, mean age = 40.6), and 10 participants with a clinical diagnosis of UC were undergoing treatment with the anti-inflammatory 5-ASA (8 male, 2 female, age 20–71, mean age = 46.2).
Blood was taken from fasting subjects in heparin bottles (Vacutainer Plus Venous Blood Collection Heparin Tube) and centrifuged at 1 000g and 4 °C for 10 min immediately after collection. Plasma was then aliquoted into labeled tubes (Eppendorf 2 mL), ensuring that no red blood cells or clots were carried over. All samples were then immediately frozen and stored in −20 °C freezers for a maximum of 24 h before transfer to a −80 °C freezer.
Samples were randomized, aliquoted, extracted, and analyzed using the protocol described above. Biological samples were analyzed as a set of 40. Each sample set was preceded by a calibration standard set and a full set of analytical QC standards, at LLOQ, low, mid, high, and ULOQ concentrations for each of the respective analytes (QC concentrations are listed in Table S3). The calibration and analytical QC set was repeated at the end of each plate (Figure S2). An additional repeat of each analytical QC concentration (Table S3) was included and analyzed after every 10th injection throughout the acquisition sequence to ensure calibration and quantification performance across the run.
A biological QC was prepared by mixing equal volumes (10 μL) from each study sample into a homogeneous pool; this approach was adapted from previous traditional use in nontargeted metabolomics48 and was included in the protocol to demonstrate and monitor the precision of quantification of each analyte in a repeat analysis of a biological sample. The biological QC was analyzed at intervals of 10 samples throughout the run to further monitor assay performance for biological samples.
Results and Discussion
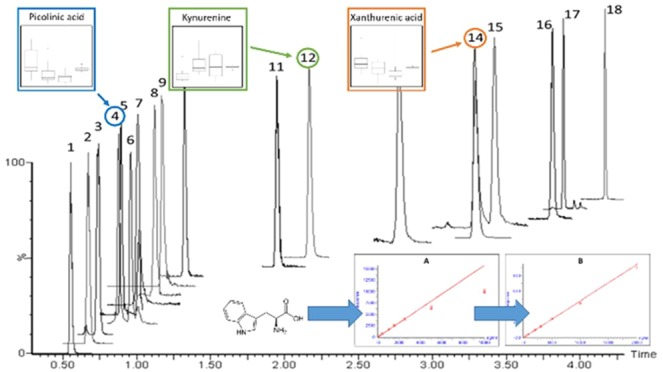
The described assay enables the rapid and reproducible quantification of a panel of metabolites associated with gut health and tryptophan metabolism via the kynurenine and serotonin pathways. The limitations of LC-MS-based assays for the quantification of analytes in complex matrixes such as plasma/serum resulting from so-called matrix effects are well-known.49−51 To some extent such effects can be compensated for by the use of an appropriate SIL internal standard; while this approach is not perfect, it is preferable to the use of surrogate or external standards. We have therefore attempted to match each of the analytes in this method with a suitable SIL with tryptophan and 14 of its metabolites covered in this way. Two further targeted metabolites, βNM and serotonin used neopterin-13C5 and dopamine-D4 as surrogate internal standards to enable at least semiquantification. The remaining targeted analyte (NAD+) was included in the assay for nonquantitative “fold change” monitoring without internal standard correction. A 96-well plate approach, incorporating an extensive sample cleanup, with a two-step extraction using solvent protein precipitation followed by delipidation via SPE (developed to protect chromatographic column lifespan) combined with a 4.2 min chromatographic separation (Figure 1) was designed for application in high-throughput analysis of the target analytes in clinical population cohorts.
Figure 1.

UHPLC chromatogram of the 18 standards of analysis: 1, citrulline; 2, beta-nicotinic mononucleotide; 3, nicotinamide riboside; 4, picolinic acid; 5, neopterin; 6, nicotinic acid; 7, quinolinic acid; 8, dopamine; 9, NAD+*; 10, 3-OH-kynurenine; 11, serotonin; 12, kynurenine; 13, 3-HAA; 14, tryptophan; 15, xanthurenic acid; 16, kynurenic acid; 17, 5-HIAA; 18, indole-3-acetic acid. *NAD+ was not considered for quantification; it was monitored only.
The separation was stable and reproducible, as evidenced by intrabatch retention time CVs of <0.7% across six replicate injections of ULOQ analytical QC samples. Comparison of retention times between two different columns and LC solvent batches showed differences of <0.1 min for all analytes.
For selective detection, MRM transitions were developed for each metabolite and SIL internal standard as listed in Table 1. Where possible, the losses of 18 and 44 (water and CO2) were excluded to obtain unique transitions; this was to avoid cross-contamination from these common losses. In the absence of an analyte-free plasma or serum, water was used as the blank matrix for the preparation of calibration and analytical QC samples. Water was selected as the diluent because it makes minimal assumptions when used as a blank matrix. It is an accepted approach in such situations (see review by Thakare et al.52) that has previously been used in endogenous metabolite quantification.53,54 The method underwent validation and application to a human clinical study containing cases of inflammatory bowel disease to ensure compatibility with the physiological and pathological variation of tryptophan metabolite concentrations.
Method Validation
Analytical Specificity
The MRM transition window for each metabolite and the corresponding SIL internal standard was assessed for analyte MRM transition specificity. Biological samples (both serum and plasma) were extracted and analyzed using the LC-MS conditions described earlier. For 14 of the analytes, there was no chromatographic coelution of interferences in the MRM transition window. While nicotinic and picolinic acids shared the same quantification transition (124.1 → 78.1), they were fully resolved chromatographically. Qualifier transition ions for every metabolite in the assay were also included in the method to help ensure that the correct chromatographic peak was assigned to each metabolite in the biological samples. Nicotinic acid has a qualification transition (124.1 → 53.1) that was not shared by picolinic acid. Additionally, the MRM transition window for xanthurenic acid (206.1 → 132.1) contained a peak for the naturally occurring 13C second isotope of tryptophan; again, these were chromatographically resolved with no interference between these analytes, as can be observed in Figure 1.
Analytical Range and Linearity
Analytical ranges were determined either by the LLOQ achievable by the extraction and analytical setup or by analysis of biological samples and subsequent adjustment of the ranges. Calibration curves were found to be linear (>0.990) over the selected ranges described in Table S5.
A challenge for the method development was that the quantified metabolites are present at very different concentrations to one another and therefore require appropriate linear ranges of the standards to achieve quantification, challenging the linear dynamic range of the instrument. For example, serum neopterin has previously been reported at concentrations 10 000 times lower than that of serum tryptophan.55 Therefore, in this assay neopterin was validated at a range of 0.2–20 ng/mL, while tryptophan was validated over a range of 200–20 000 ng/mL. However, the high ULOQ concentration required for the analysis of tryptophan initially resulted in a nonlinear response, caused by in-source ionization saturation. This was overcome using fast polarity switching and determining the analyte using negative ESI. All metabolites, except for tryptophan, were analyzed using positive ESI and were tuned for maximum sensitivity across the linear range. However, tryptophan and its SIL internal standard were analyzed with a low capillary voltage (0.25 kV). This had an effect of detuning the tryptophan transition for sensitivity with the benefit of negating the effect of in-source saturation, resulting in linearity of the calibration over the desired range (Figure S3).
The results of the intraday and interday precision and accuracy of the assay are presented in Table S6. All metabolites demonstrated an analytical accuracy <15% (LLOQ < 20%). The intra- and interday precision of the assay ranged from 1.3% to 15.4% for the intraday determinations and 2.1–12.9% for the interday comparison.
Carryover
All target metabolites demonstrated minimal carryover (<1.1%). Carryover for internal standards was 0.1–2.8%.
Stability
During method development, poor stability of some metabolites in aqueous solutions was observed. Previously the literature has reported that metabolites in the kynurenine pathway undergo rapid oxidation in aqueous solutions.56−59 To overcome this instability, stock calibration standards and quality control standards were prepared using 0.1% citric acid in water, and subsequent dilutions were prepared with 0.01% citric acid in water. Citric acid, in excess, acts as an antioxidant and a preservative in solution.
Standards prepared in citric acid were assessed for stability after 24 h at 4 °C in an autosampler and at both 1 and 2 weeks at −20 °C. This was repeated with a low and a high QC (QC 2 and QC 6). The QC samples that underwent quantification after storage reported acceptable stability of >97% for the high QC for all metabolites and >90% in the low QC. The exception was picolinic acid, where stability was 90.74% after 1 week and 83.33% after 2 weeks. The results obtained for this metabolite must therefore be interpreted with caution if the analysis of samples is significantly delayed following collection. Full results are presented in Table S7. Twenty-four h autosampler stability was chosen as a key time period for stability assessment because in large multiplate, multiday studies that the assay was designed for two sample plates underwent extraction and analysis in each 24 h time period.
Analytical Recovery
Plasma metabolite recoveries were typically >90%, with only dopamine-D4 (83%), quinolinic acid-D3 (80%), and nicotinamide riboside-D3 (53%) being the exceptions. Serum metabolite recoveries were typically >85%, with quinolinic acid-D3 (79%) and nicotinamide riboside-D3 (52%) being the exceptions (Tables S8 and S9).
Matrix Effects
Observed matrix effects were minimal for most metabolites in both serum and plasma, with results suggesting a matrix area response of 77–112% when comparing SIL standards spiked into either plasma or serum with the response of SIL standards spiked into a blank diluent (Table S10). The exception in both biofluids was citrulline. The MRM transition responses for the SIL internal standard of citrulline spiked into, and extracted from, both plasma and serum were 23.5% and 18.4%, respectively, when compared with the values for the spiked diluent blank. It is likely that the observed matrix effects were due to the short retention time of citrulline and its SIL internal standard. However, the inclusion of the SIL analogue as an internal standard in the assay protocol should compensate for these matrix effects when quantifying citrulline in these biofluids.
Biological Sample Testing
Long-Term Multiday Assay Performance Testing
Following multiday analysis of serum, metabolite concentrations from pooled biological QCs were calculated. Three analytes (3-HAA, βNM, and dopamine) were found to be below the LLOQ in the pooled serum. However, for the remaining detectable analytes, the % CV values ranged from 2.8% to 20.0%. All analytes in the range had CV values of <15% except for nicotinamide riboside, which had a % CV of 20%. This result shows the methods reproducibility across multiplate analysis, therefore highlighting its applicability to the large-scale profiling and quantified analysis of the analyte panel in both clinical research and epidemiology studies. Data for each analyte are presented in Table S11.
It should be noted that the quality acceptance criteria for each metabolite in an analysis is based only upon the calibration samples and analytical QCs. The biological QCs are used to demonstrate the precision of quantification for individual analytes over the duration of a study. In a clinical study, the overall distribution of one or more metabolites may be below the LLOQ, with only a few study samples having values above the LLOQ. Therefore, when samples are pooled to create the biological QC, metabolite concentrations may result that are below the assay LLOQ for these analytes. In this instance, biological QCs will not provide useful information on the precision of measurement of these analytes in biological samples.
Application of the Assay to Clinical Samples
Following analytical validation, the method was then tested in clinical samples obtained from two groups of participants diagnosed with UC (treated and untreated) and a control group in order to show clinical application in a “proof of concept” study. IBD is a broad clinical classification that includes both Crohn’s disease and ulcerative colitis (UC). IBD is characterized by chronic inflammation within the colon and the small intestine.60 The pathogenesis of the condition has not been fully elucidated and is thought to be a combination of genetic and environmental factors.60 In addition, evidence suggests that the equilibrium between gut microbial composition and host immune response at the mucosal layer plays a role in the disease.60−62
The concentration of tryptophan in the circulatory system is regulated by both bioavailability in diet and subsequent metabolism by microbial colonies that are present in the gut.12 The assay was applied to plasma samples from a subset of study participants diagnosed with UC, to investigate metabolic differences reflective of their gut health. The samples were obtained from subjects from three groups including controls with no diagnosis of UC or IBD, participants with a clinical diagnosis of UC who are not undergoing treatment, and participants with a clinical diagnosis of UC who are being treated with 5-ASA, prescribed to help reduce inflammation and alleviate symptoms.
A biological QC was created by pooling equal volumes of all samples from the study. This biological QC was then analyzed at intervals of 10 samples across the analytical run to determine the variation in the extraction. The CVs for the calculated concentration values of analytes that fell within the range of quantification in the biological QC were <10% (Table S12).
Following data acquisition and postacquisition processing, the groups were assessed for significant differences using a one-way analysis of variance (ANOVA) test. ANOVA was performed on those metabolites, which were detected and quantified within the calibration range, as well as the kynurenine/tryptophan ratio. The ANOVA analysis demonstrated significant differences between the three patient classes for the metabolites picolinic acid (p = 0.009), xanthurenic acid (p = 0.017), and kynurenine (p = 0.043). However, when controlling for the family-wise error rate using the Bonferroni method to adjust for multiple testing, adjusted p values for each of the metabolites were as follows: picolinic acid (adjusted p = 0.130), xanthurenic acid (adjusted p = 0.250), and kynurenine (adjusted p = 0.643). Metabolite concentrations for picolinic acid, xanthurenic acid, and kynurenine underwent Tukey HSD testing within each ANOVA, with kynurenine, xanthurenic acid, and picolinic acid demonstrating statistically significant intergroup differences (discussed below).
A statistically significant increase in kynurenine concentration was observed in UC treated (p = 0.042) and untreated (p = 0.16) patient groups compared with the control group (Figure 2). This supports previous literature where kynurenine was reported at increased concentrations in the serum of clinical cases of IBD patients (UC = 7, Crohn’s disease = 5) compared to a control group (n = 12).63 However, it should be noted that this previous study also reported a statistically significant increase in kynurenic acid concentrations in the IBD groups,63 which was not replicated in the present study, where only a nonsignificant decrease for kynurenic acid was observed in the clinical UC cases (both treated and untreated) compared with the control group (ANOVA, p = 0.674).
Figure 2.

Box plots presenting three analytes that significantly differ in concentrations between the study groups. The upper box plots show the full analytical range with the lower limit of quantification in blue and the upper limit of concentration in red. The lower box plots present the same data, but the y-axis has been shrunk for clarity. ANOVA analysis revealed significant differences between the control and patient groups for kynurenine (p = 0.043), picolinic acid (p = 0.009), and xanthurenic acid (p = 0.017). Data for the biological QC have been included to show the measurement precision of each metabolite in a repeat biological sample, compared with the variation within the clinical samples.
The same paper also reported a nonsignificant decrease of xanthurenic acid in the IBD groups compared to the control group.63 This previous finding is supported by our study, with both untreated UC and treated UC groups showing a statistically significant decrease in the concentration compared to the control group (control → UC treated p = 0.161, control → IBD untreated p = 0.013).
For picolinic acid, untreated UC and treated UC groups showed a decrease in concentration compared with controls (control → UC treated p = 0.060, control → UC untreated p = 0.009). Previously, a reduction in serum picolinic acid in Crohn’s disease had been reported; however, there was no significant change in UC.28
It should be noted that the picolinic acid analytical standards showed signs of long-term instability in solution (83.3% after 2 weeks at −20 °C). However, the data from pooled biological QC samples show stable and reproducible biological measurements of picolinic acid for the duration of a 9-day, 18-plate analysis (Table S11). This demonstrates that the method is fit for its purpose in identifying concentration changes between groups in a study that is discovery in design. Despite this, if increased confidence in the concentration of endogenous picolinic acid quantification is required, we would recommend further investigation and optimization of all stages of sample collection, treatment, and long-term storage for the analyte.
The limitation of the proof of concept UC data set analyzed here is the relatively small number of samples without adjustment for clinical confounders. However, despite these limitations, the results do show a graded difference in pairwise comparisons between untreated, treated, and control groups.
Conclusions
A UHPLC-ESI-MS/MS method for the analysis of 18 metabolites linked to gut health and tryptophan metabolism has been developed for application to human plasma and serum. The method was designed to use a 96-well plate format to facilitate multiple-day, large-batch analysis of samples enabling its application to large-scale clinical and epidemiology-based population studies. The method employed a two-step extraction, using solvent protein precipitation followed by delipidation via SPE. Extracted analytes then underwent chromatographic separation using a 4.2 min reversed-phase gradient (7 min total cycle time). The assay was validated and applied to a clinical study of UC using plasma samples obtained from controls and both untreated and treated patients. Reduced amounts of picolinic acid and xanthurenic acid and increased quantities of kynurenine were observed in the plasma of patients with UC using this approach.
Acknowledgments
The MRC-NIHR National Phenome Centre is supported by the U.K. Medical Research Council in association with National Institute of Health Research (England) (Grant MC_PC_12025). Infrastructure support for this work was provided by the NIHR Imperial Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, NIHR, or the Department of Health. The UK Dementia Research Institute (DRI) is an initiative funded by the Medical Council, Alzheimer’s Society, and Alzheimer’s Research UK.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b05884.
Tryptophan metabolism via the serotonin and kynurenine pathways (adapted from ref (64)); 96-well plate map layout used in the assay; effect of detuning the tryptophan transition in negative ionization MS detection mode; overview describing the preparation of an upper limit of quantification stock; preparation of working calibration and QC dilutions; final concentrations of each level of the calibration series and QC series used in the assay; preparation of the internal standard working solution used in the assay; metabolite inter-run calibration linearity; intraday and interday accuracy and precision; results from stability testing of the analytes; results of recovery study for plasma; results of recovery study for serum; results of studies into matrix effects during analysis; mean concentrations, standard deviation, and % coefficient of variation values derived from the quantification of analytes from repeat injections of a pool of serum; and mean concentration values for repeat biological replicates (PDF)
Author Contributions
∇ L.W. and L.C.N. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Lewis M. R.; Pearce J. T. M.; Spagou K.; Green M.; Dona A. C.; Yuen A. H. Y.; David M.; Berry D. J.; Chappell K.; Horneffer-van der Sluis V.; Shaw R.; Lovestone S.; Elliott P.; Shockcor J.; Lindon J. C.; Cloarec O.; Takats Z.; Holmes E.; Nicholson J. K. Development and application of ultra-performance liquid chromatography-TOF MS for precision large scale urinary metabolic phenotyping. Anal. Chem. 2016, 88 (18), 9004–9013. 10.1021/acs.analchem.6b01481. [DOI] [PubMed] [Google Scholar]
- Want E. J.; Wilson I. D.; Gika H.; Theodoridis G.; Plumb R. S.; Shockcor J.; Holmes E.; Nicholson J. K. Global metabolic profiling procedures for urine using UPLC–MS. Nat. Protoc. 2010, 5 (6), 1005–1018. 10.1038/nprot.2010.50. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Broadhurst D.; Begley P.; Zelena E.; Francis-McIntyre S.; Anderson N.; Brown M.; Knowles J. D.; Halsall A.; Haselden J. N.; Nicholls A. W.; Wilson I. D.; Kell D. B.; Goodacre R. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Lin W.; Broadhurst D.; Begley P.; Brown M.; Zelena E.; Vaughan A. A.; Halsall A.; Harding N.; Knowles J. D.; Francis-McIntyre S.; Tseng A.; Ellis D. I.; O’Hagan S.; Aarons G.; Benjamin B.; Chew-Graham S.; Moseley C.; Potter P.; Winder C. L.; Potts C.; Thornton P.; McWhirter C.; Zubair M.; Pan M.; Burns A.; Cruickshank J. K.; Jayson G. C.; Purandare N.; Wu F. C. W.; Finn J. D.; Haselden J. N.; Nicholls A. W.; Wilson I. D.; Goodacre R.; Kell D. B. Molecular phenotyping of a UK population: defining the human serum metabolome. Metabolomics 2015, 11, 9–26. 10.1007/s11306-014-0707-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. A.; Moore S.; Playdon M.; Kritchevsky S.; Newman A. B.; Satterfield S.; Ayonayon H.; Clish C.; Gerszten R.; Harris T. B. Metabolites associated with risk of developing mobility disability in the health, aging and body composition study. J. Gerontol., Ser. A 2019, 74 (1), 73–80. 10.1093/gerona/glx233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzoulaki I.; Ebbels T. M. D.; Valdes A.; Elliott P.; Ioannidis J. P. A. Design and analysis of metabolomics studies in epidemiologic research: A primer on -omic technologies. Am. J. Epidemiol. 2014, 180 (2), 129–139. 10.1093/aje/kwu143. [DOI] [PubMed] [Google Scholar]
- Richard D. M.; Dawes M. A.; Mathias C. W.; Acheson A.; Hill-Kapturczak N.; Dougherty D. M. L-Tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int. J. Tryptophan Res. 2009, 2, 45–60. 10.4137/IJTR.S2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mándi Y.; Vécsei L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119 (2), 197–209. 10.1007/s00702-011-0681-y. [DOI] [PubMed] [Google Scholar]
- Cervenka I.; Agudelo L. Z.; Ruas J. L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. 10.1126/science.aaf9794. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Guillemin G. J. Kynurenine pathway metabolites in humans: disease and healthy states. Int. J. Tryptophan Res. 2009, 2, IJTR.S2097. 10.4137/IJTR.S2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Hu N.; Yang D.; Oxenkrug G.; Yang Q. Regulating the balance between the kynurenine and serotonin pathways of tryptophan metabolism. FEBS J. 2017, 284 (6), 948–966. 10.1111/febs.14026. [DOI] [PubMed] [Google Scholar]
- Kennedy P. J.; Cryan J. F.; Dinan T. G.; Clarke G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. 10.1016/j.neuropharm.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Wang X.-D.; Notarangelo F. M.; Wang J.-Z.; Schwarcz R. Kynurenic acid and 3-hydroxykynurenine production from D-kynurenine in mice. Brain Res. 2012, 1455, 1–9. 10.1016/j.brainres.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierozan P.; Biasibetti H.; Schmitz F.; Ávila H.; Parisi M. M.; Barbe-Tuana F.; Wyse A. T. S.; Pessoa-Pureur R. Quinolinic acid neurotoxicity: Differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863 (12), 3001–3014. 10.1016/j.bbamcr.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Guillemin G. J. Quinolinic acid: neurotoxicity. FEBS J. 2012, 279 (8), 1355. 10.1111/j.1742-4658.2012.08493.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee P.; Goozee K.; Lim C. K.; James I.; Shen K.; Jacobs K. R.; Sohrabi H. R.; Shah T.; Asih P. R.; Dave P.; ManYan C.; Taddei K.; Lovejoy D. B.; Chung R.; Guillemin G. J.; Martins R. N. Alterations in serum kynurenine pathway metabolites in individuals with high neocortical amyloid-β load: A pilot study. Sci. Rep. 2018, 8 (1), 8008. 10.1038/s41598-018-25968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin G. J.; Rahman A.; Ting K. K.; Cullen K.; Braidy N.; Chung R.; Wu W.; Brew B. J. Involvement of the kynurenine pathway in Alzheimer’s disease. Alzheimer's Dementia 2010, 6 (4), e21. 10.1016/j.jalz.2010.08.063. [DOI] [Google Scholar]
- Gulaj E.; Pawlak K.; Bien B.; Pawlak D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55 (2), 204–211. 10.2478/v10039-010-0023-6. [DOI] [PubMed] [Google Scholar]
- Myint A.-M.; Kim Y. K.; Verkerk R.; Scharpé S.; Steinbusch H.; Leonard B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J. Affective Disord. 2007, 98 (1), 143–151. 10.1016/j.jad.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Réus G. Z.; Jansen K.; Titus S.; Carvalho A. F.; Gabbay V.; Quevedo J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: evidences from animal and human studies. J. Psychiatr. Res. 2015, 68, 316–328. 10.1016/j.jpsychires.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogyu K.; Kubo K.; Noda Y.; Iwata Y.; Tsugawa S.; Omura Y.; Wada M.; Tarumi R.; Plitman E.; Moriguchi S.; Miyazaki T.; Uchida H.; Graff-Guerrero A.; Mimura M.; Nakajima S. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. 10.1016/j.neubiorev.2018.03.023. [DOI] [PubMed] [Google Scholar]
- Erhardt S.; Schwieler L.; Imbeault S.; Engberg G. The kynurenine pathway in schizophrenia and bipolar disorder. Neuropharmacology 2017, 112, 297–306. 10.1016/j.neuropharm.2016.05.020. [DOI] [PubMed] [Google Scholar]
- Kegel M. E.; Bhat M.; Skogh E.; Samuelsson M.; Lundberg K.; Dahl M.-L.; Sellgren C.; Schwieler L.; Engberg G.; Schuppe-Koistinen I.; Erhardt S. Imbalanced Kynurenine Pathway in Schizophrenia. Int. J. Tryptophan Res. 2014, 7, IJTR.S16800. 10.4137/IJTR.S16800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demitrack M. A.; Heyes M. P.; Altemus M.; Pigott T. A.; Gold P. W. Cerebrospinal fluid levels of kynurenine pathway metabolites in patients with eating disorders: Relation to clinical and biochemical variable. Biol. Psychiatry 1995, 37 (8), 512–520. 10.1016/0006-3223(94)00173-Z. [DOI] [PubMed] [Google Scholar]
- Berstad A.; Raa J.; Valeur J. Tryptophan: ‘essential’ for the pathogenesis of irritable bowel syndrome?. Scand. J. Gastroenterol. 2014, 49 (12), 1493–1498. 10.3109/00365521.2014.936034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G.; McKernan D. P.; Gaszner G.; Quigley E. M.; Cryan J. F.; Dinan T. G. A distinct profile of tryptophan metabolism along the kynurenine pathway downstream of toll-like receptor activation in irritable bowel syndrome. Front. Pharmacol. 2012, 3, 90. 10.3389/fphar.2012.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M. A.; Ciorba M. A.; Meckel K.; Lim C. K.; Guillemin G. J.; Weber C. R.; Bissonnette M.; Pekow J. R. Tryptophan metabolism through the kynurenine pathway is associated with endoscopic inflammation in ulcerative colitis. Inflammatory Bowel Dis. 2018, 24 (7), 1471–1480. 10.1093/ibd/izy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaus S.; Schulte B.; Al-Massad N.; Thieme F.; Schulte D. M.; Bethge J.; Rehman A.; Tran F.; Aden K.; Häsler R.; Moll N.; Schütze G.; Schwarz M. J.; Waetzig G. H.; Rosenstiel P.; Krawczak M.; Szymczak S.; Schreiber S. Increased tryptophan metabolism Is associated with activity of inflammatory bowel diseases. Gastroenterology 2017, 153 (6), 1504–1516. 10.1053/j.gastro.2017.08.028. [DOI] [PubMed] [Google Scholar]
- Mole D. J.; Webster S. P.; Uings I.; Zheng X.; Binnie M.; Wilson K.; Hutchinson J. P.; Mirguet O.; Walker A.; Beaufils B.; Ancellin N.; Trottet L.; Bénéton V.; Mowat C. G.; Wilkinson M.; Rowland P.; Haslam C.; McBride A.; Homer N. Z. M.; Baily J. E.; Sharp M. G. F.; Garden O. J.; Hughes J.; Howie S. E. M.; Holmes D. S.; Liddle J.; Iredale J. P. Kynurenine–3–monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 2016, 22 (2), 202–209. 10.1038/nm.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworek J.; Szklarczyk J.; Jaworek A. K.; Nawrot-Pora̧bka K.; Leja-Szpak A.; Bonior J.; Kot M. Protective effect of melatonin on acute pancreatitis. Int. J. Inflammation 2012, 2012, 173675. 10.1155/2012/173675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosek M. N.; Mduma E.; Kosek P. S.; Lee G. O.; Svensen E.; Pan W. K. Y.; Olortegui M. P.; Bream J. H.; Patil C.; Asayag C. R.; Sanchez G. M.; Caulfield L. E.; Gratz J.; Yori P. P. Plasma tryptophan and the kynurenine-tryptophan ratio are associated with the acquisition of statural growth deficits and oral vaccine underperformance in populations with environmental enteropathy. Am. J. Trop. Med. Hyg. 2016, 95 (4), 928–937. 10.4269/ajtmh.16-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayneris-Perxachs J.; Swann J. R. Metabolic phenotyping of malnutrition during the first 1000 days of life. Eur. J. Nutr. 2018, 10.1007/s00394-018-1679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayneris-Perxachs J.; Lima A. A. M.; Guerrant R. L.; Leite Á. M.; Moura A. F.; Lima N. L.; Soares A. M.; Havt A.; Moore S. R.; Pinkerton R.; Swann J. R. Urinary N-methylnicotinamide and β-aminoisobutyric acid predict catch-up growth in undernourished Brazilian children. Sci. Rep. 2016, 6, 19780. 10.1038/srep19780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crenn P.; Messing B.; Cynober L. Citrulline as a biomarker of intestinal failure due to enterocyte mass reduction. Clin. Nutr. 2008, 27 (3), 328–339. 10.1016/j.clnu.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Baydar T.; Yuksel O.; Sahin T. T.; Dikmen K.; Girgin G.; Sipahi H.; Kurukahvecioglu O.; Bostanci H.; Sare M. Neopterin as a prognostic biomarker in intensive care unit patients. J. Crit. Care 2009, 24 (3), 318–321. 10.1016/j.jcrc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Melichar B.; Spisarová M.; Bartoušková M.; Krčmová L. K.; Javorská L.; Študentová H. Neopterin as a biomarker of immune response in cancer patients. Ann. Transl. Med. 2017, 5 (13), 280. 10.21037/atm.2017.06.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Tang A. Simultaneous determination of kynurenine and tryptophan in serum by high performance liquid chromatography. Chin. J. Chromatogr. 2006, 24 (2), 140–143. 10.1016/S1872-2059(06)60009-6. [DOI] [PubMed] [Google Scholar]
- Hu L.-J.; Li X.-F.; Hu J.-Q.; Ni X.-J.; Lu H.-Y.; Wang J.-J.; Huang X.-N.; Lin C.-X.; Shang D.-W.; Wen Y.-G. A simple HPLC–MS/MS method for determination of tryptophan, kynurenine and kynurenic acid in human serum and its potential for monitoring antidepressant therapy. J. Anal. Toxicol. 2017, 41 (1), 37–44. 10.1093/jat/bkw071. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Louie A.; Yang Q.; Massenkoff N.; Xu C.; Hunt P. W.; Gee W. A simple LC–MS/MS method for determination of kynurenine and tryptophan concentrations in human plasma from HIV-infected patients. Bioanalysis 2013, 5 (11), 1397–1407. 10.4155/bio.13.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos J.; Renau N.; Valverde O.; Aznar-Laín G.; Gracia-Rubio I.; Gonzalez-Sepulveda M.; Pérez-Jurado L. A.; Ventura R.; Segura J.; Pozo O. J. Targeting tryptophan and tyrosine metabolism by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1434, 91–101. 10.1016/j.chroma.2016.01.023. [DOI] [PubMed] [Google Scholar]
- Midttun Ø; Hustad S.; Ueland P. M. Quantitative profiling of biomarkers related to B-vitamin status, tryptophan metabolism and inflammation in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23 (9), 1371–1379. 10.1002/rcm.4013. [DOI] [PubMed] [Google Scholar]
- Wang W.; Zhuang X.; Liu W.; Dong L.; Sun H.; Du G.; Ye L. Determination of kynurnine and tryptophan, biomarkers of indoleamine 2,3-dioxygenase by LC–MS/MS in plasma and tumor. Bioanalysis 2018, 10 (16), 1335–1344. 10.4155/bio-2018-0041. [DOI] [PubMed] [Google Scholar]
- Hényková E.; Vránová H. P.; Amakorová P.; Pospíšil T.; Žukauskaitė A.; Vlčková M.; Urbánek L.; Novák O.; Mareš J.; Kaňovský P.; Strnad M. Stable isotope dilution ultra-high performance liquid chromatography–tandem mass spectrometry quantitative profiling of tryptophan-related neuroactive substances in human serum and cerebrospinal fluid. J. Chromatogr. A 2016, 1437, 145–157. 10.1016/j.chroma.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Chen G.-y.; Zhong W.; Zhou Z.; Zhang Q. Simultaneous determination of tryptophan and its 31 catabolites in mouse tissues by polarity switching UHPLC-SRM-MS. Anal. Chim. Acta 2018, 1037, 200–210. 10.1016/j.aca.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekmeneva E.; dos Santos Correia G.; Gómez-Romero M.; Stamler J.; Chan Q.; Elliott P.; Nicholson J. K.; Holmes E. Ultra-Performance liquid chromatography–high-resolution mass spectrometry and direct infusion–high-resolution mass spectrometry for combined exploratory and targeted metabolic profiling of human urine. J. Proteome Res. 2018, 17 (10), 3492–3502. 10.1021/acs.jproteome.8b00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA . Guidance for Industry Bioanalytical Method Validation. Biopharmaceutics; FDA: 2018; https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (accessed February 14, 2019).
- Tiwari G.; Tiwari R. Bioanalytical method validation: An updated review. Pharm. Methods 2010, 1 (1), 25–38. 10.4103/2229-4708.72226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gika H. G.; Theodoridis G. A.; Wingate J. E.; Wilson I. D. Within-day reproducibility of an HPLC–MS-based method for metabonomic analysis: Application to human urine. J. Proteome Res. 2007, 6 (8), 3291–3303. 10.1021/pr070183p. [DOI] [PubMed] [Google Scholar]
- Guo X.; Lankmayr E. Phospholipid-based matrix effects in LC–MS bioanalysis. Bioanalysis 2011, 3 (4), 349–352. 10.4155/bio.10.213. [DOI] [PubMed] [Google Scholar]
- Chiu M. L.; Lawi W.; Snyder S. T.; Wong P. K.; Liao J. C.; Gau V. Matrix effects - a challenge toward automation of molecular analysis. JALA 2010, 15 (3), 233–242. 10.1016/j.jala.2010.02.001. [DOI] [Google Scholar]
- Trufelli H.; Palma P.; Famiglini G.; Cappiello A. An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrom. Rev. 2011, 30 (3), 491–509. 10.1002/mas.20298. [DOI] [PubMed] [Google Scholar]
- Thakare R.; Chhonker Y. S.; Gautam N.; Alamoudi J. A.; Alnouti Y. Quantitative analysis of endogenous compounds. J. Pharm. Biomed. Anal. 2016, 128, 426–437. 10.1016/j.jpba.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Gray N.; Zia R.; King A.; Patel V. C.; Wendon J.; McPhail M. J. W.; Coen M.; Plumb R. S.; Wilson I. D.; Nicholson J. K. High-speed quantitative UPLC-MS analysis of multiple amines in human plasma and serum via precolumn derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate: Application to acetaminophen-induced liver failure. Anal. Chem. 2017, 89 (4), 2478–2487. 10.1021/acs.analchem.6b04623. [DOI] [PubMed] [Google Scholar]
- Zhao X.-E.; Zhu S.; Yang H.; You J.; Song F.; Liu Z.; Liu S. Simultaneous determination of amino acid and monoamine neurotransmitters in PC12 cells and rats models of Parkinson’s disease using a sensitizing derivatization reagent by UHPLC–MS/MS. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015, 995–996, 15–23. 10.1016/j.jchromb.2015.05.017. [DOI] [PubMed] [Google Scholar]
- Geisler S.; Mayersbach P.; Becker K.; Schennach H.; Fuchs D.; Gostner J. M. Serum tryptophan, kynurenine, phenylalanine, tyrosine and neopterin concentrations in 100 healthy blood donors. Pteridines 2015, 26, 31–36. 10.1515/pterid-2014-0015. [DOI] [Google Scholar]
- Rescigno A.; Sanjust E.; Soddu G.; Rinaldi A. C.; Sollai F.; Curreli N.; Rinaldi A. Effect of 3-hydroxyanthranilic acid on mushroom tyrosinase activity. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1998, 1384 (2), 268–276. 10.1016/S0167-4838(98)00018-1. [DOI] [PubMed] [Google Scholar]
- Yen G.-C.; Hsieh C.-L. Antioxidant effects of dopamine and related compounds. Biosci., Biotechnol., Biochem. 1997, 61 (10), 1646–1649. 10.1271/bbb.61.1646. [DOI] [PubMed] [Google Scholar]
- Dykens J. A.; Sullivan S. G.; Stern A. Oxidative reactivity of the tryptophan metabolites 3-hydroxyanthranilate, cinnabarinate, quinolinate and picolinate. Biochem. Pharmacol. 1987, 36 (2), 211–217. 10.1016/0006-2952(87)90691-5. [DOI] [PubMed] [Google Scholar]
- Darlington L. G.; Forrest C. M.; Mackay G. M.; Smith R. A.; Smith A. J.; Stoy N.; Stone T. W. On the biological importance of the 3-hydroxyanthranilic acid: anthranilic acid ratio. Int. J. Tryptophan Res. 2010, 3, IJTR.S4282. 10.4137/IJTR.S4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier R. J.; Podolsky D. K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448 (7152), 427–434. 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Manichanh C.; Borruel N.; Casellas F.; Guarner F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9 (10), 599–608. 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- Round J. L.; Mazmanian S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest C. M.; Youd P.; Kennedy A.; Gould S. R.; Darlington L. G.; Stone T. W. Purine, kynurenine, neopterin and lipid peroxidation levels in inflammatory bowel disease. J. Biomed. Sci. 2002, 9 (5), 436–442. 10.1159/000064554. [DOI] [PubMed] [Google Scholar]
- Badawy A. A. B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan Res. 2017, 10, 1–20. 10.1177/1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.