

Abstract
Spinal muscular atrophy (SMA) is a progressive motor neuron disease caused by loss or mutation of the survival motor neuron 1 (SMN1) gene and retention of SMN2. We performed targeted capture and sequencing of the SMN2, CFTR, and PLS3 genes in 217 SMA patients. We identified a 6.3 kilobase deletion that occurred in both SMN1 and SMN2 (SMN½) and removed exons 7 and 8. The deletion junction was flanked by a 21 base pair repeat that occurred 15 times in the SMN½ gene. We screened for its presence in 466 individuals with known SMN1 and SMN2 copy number. In individuals with 1 SMN1 and 0 SMN2 copies the deletion occurred in 63% of cases. We modeled the deletion junction frequency and determined that the deletion occurred in both SMN1 and SMN2. We have identified the first deletion junction where the deletion removes exons 7 and 8 of SMN½. As it occurred in SMN1 it is a pathogenic mutation. We called variants in the PLS3 and SMN2 genes and tested for association with mild or severe exception patients. The variants A-44G, A-549G and C-1897T in intron 6 of SMN2 were significantly associated with mild exception patients but no PLS3 variants correlated with severity. The variants occurred in 14 out of 58 of our mild exception patients indicating that mild exception patients with an intact SMN2 gene and without modifying variants occur. This sample set can be used in association analysis of candidate genes outside of SMN2 that modify the SMA phenotype.
Keywords: SMA, SMN1, SMN2, deletion, modifier
Introduction
Spinal muscular atrophy (SMA) is one of the most common inherited neuromuscular disorders, with an incidence of approximately 1 in 10,000 live births. The disease causes destruction of the alpha motor neurons and remains a leading genetic cause of infant death (Roberts et al. 1970; Pearn 1973, 1978; Crawford and Pardo 1996; Sugarman et al. 2012). SMA is an autosomal recessive disorder caused by loss of function of the survival motor neuron 1 gene (SMN1) and retention of the SMN2 gene (Lefebvre et al. 1995). The human SMN1 and SMN2 genes both encode SMN mRNA and make SMN protein. However, the SMN2 gene produces lower levels of SMN protein. In the absence of functional SMN1, SMA results due to low SMN levels (Lefebvre et al. 1995, 1997; Coovert et al. 1997). The SMN1 and SMN2 genes differ by a single nucleotide: a C to T base change in exon 7 of SMN2 (Lefebvre et al. 1995). Although this is a synonymous mutation, it disrupts the binding of splicing modifiers leading to inefficient incorporation of exon 7 in the mRNA (Lorson et al. 1999; Monani et al. 1999; Cartegni and Krainer 2002; Kashima and Manley 2003). This results in a truncated protein that cannot oligomerize efficiently and is rapidly degraded (Coovert et al. 1997; Lorson et al. 1998; Le et al. 2005; Burnett et al. 2009). 95% of SMA patients have loss of SMN1 by either deletion or gene conversion of SMN1 to SMN2, while 5% of patients have a mutation in the SMN1 gene that typically disrupts the production of fully-functional SMN protein (Lefebvre et al. 1995; Burghes 1997; Burghes and Beattie 2009; Burghes and McGovern 2017).
The phenotypic severity of SMA, which is classified into clinical subtypes based on the age of the initial onset of symptoms and maximum achieved motor milestones, ranges from the most severe infantile form, SMA type 0, to the least severe adult onset form, SMA type 4 (Munsat 1991; Arnold and Burghes 2013; Arnold et al. 2015). SMA type 0 manifests with congenital weakness, joint contractures, and ventilator dependence from birth and is relatively rare. SMA type 1 infants present with symptoms between birth and 6 months of age and a majority die, or become ventilator dependent, before 2 years of age. SMA type 2 patients present with symptoms between 6 months and 12 months of age and achieve the ability to sit independently but are never able to walk. SMA type 3 patients present with symptoms between the ages of 12 months and 21 years and achieve independent ambulation for at least part of their life. SMA type 4 patients manifest with symptoms that first appear in adulthood and like SMA type 0 patients are rare. In some cases, the clinical subtypes have been further subdivided. For example, SMA type 3 can be divided into type 3a and 3b, depending on whether onset occurs prior to or after 3 years of age, respectively (Zerres and Rudnik-Schoneborn 1995). These clinical subtypes have been defined in untreated SMA patients. However, in the current era of treatment, the administration of therapies now results in an alteration of both the phenotypic severity and progression of SMA (Mendell et al. 2017; Finkel et al. 2017; Mercuri et al. 2018). The effect is highly dependent on timing of all treatments where earlier interventions are most effective. Later treatment, however, still results in increased survival and a preservation of function (Gidaro and Servais 2018).
The SMN2 copy number correlates with phenotypic severity, where higher copy numbers result in a less severe SMA phenotype (McAndrew et al. 1997; Burghes 1997; Feldkötter et al. 2002; Mailman et al. 2002; Jedrzejowska et al. 2009; Calucho et al. 2018). However, exceptions to this rule are known to occur (Cobben et al. 1995; Prior et al. 2004). For example, siblings with identical SMN2 copies inherited from the same 5q13 region yet discordant SMA phenotypes have been reported (Burghes et al. 1994; Cobben et al. 1995; Hahnen et al. 1995; McAndrew et al. 1997; Prior et al. 2004; Oprea et al. 2008; Jedrzejowska et al. 2008; Bernal et al. 2011). These sib pairs are referred to as “discordant siblings” and they can occur in all phenotypic forms of SMA. For example, there are SMA type 1 and type 2 siblings reported with the same SMN2 copy number (DiDonato et al. 1994, 1997; Pane et al. 2017), as well as siblings with SMA type 2 and type 3b, and cases of SMA type 3a with a phenotypically normal sibling (Cobben et al. 1995; Hahnen et al. 1995). Furthermore, 3 copies of SMN2 is the most common genotype for both SMA type 2 and type 3 (Calucho et al. 2018), indicating that SMN2 copy number alone poorly predicts severity of the disease in individuals with 3 or more copies of SMN2.
Discrepancy between SMN2 copy number and the expected SMA phenotype may result from partial deletions of SMN2 and modifying variants inside or outside of the SMA region. In the first case, the SMA genomic region is known to be unstable and prone to rearrangements, as evidenced by multiple banding patterns in pulsed-field gel electrophoresis experiments performed using probes for SMN1 and the adjacent gene neuronal anti-inhibitory protein (NAIP) (Campbell et al. 1997). The region is often depicted as an inverted repeat of 500 kilobases (kb) (Lefebvre et al. 1995). However, the region appears considerably more complex with multiple arrangements in different individuals (Campbell et al. 1997; Burghes 1997). In humans the NAIP gene and the Small EDRK-Rich Factor (SERF1A) gene flank the SMN1 gene (Chen et al. 1998), while a NAIP pseudogene and a copy of the SERF1B flank the SMN2 gene. Due to the high variation and difficulty in assembling this large repeat region, no consensus maps of the SMA region exists. The loss of the SMN1 gene can occur via deletion or by gene conversion to SMN2 (Campbell et al. 1997; DiDonato et al. 1997; Burghes 1997). The larger deletions are also marked by loss of the intact NAIP or the probe XS2G3 (Roy et al. 1995; Thompson et al. 1995). To date, there is limited knowledge about the extent of deletions and no deletion junction has been defined which removes exons 7 and 8 of either SMN1 or SMN2 (SMN½). However, at least one internal Alu-mediated deletion has been reported that eliminates exons 5 and 6 (Wirth et al. 1999).
Several genetic modifiers of the SMA phenotype have been reported. The c.859G>C variant in exon 7 of SMN2 acts as a positive modifier which results in an approximate 20% increase in full-length SMN RNA and a milder SMA phenotype (Prior et al. 2009; Vezain et al. 2010). Indeed, this variant has never been reported in a severe SMA type 1 patient but is found in SMA type 2 patients with 2 copies of SMN2 in a heterozygous state, and in SMA type 3 patients with 2 copies of SMN2 in a homozygous state (Bernal et al. 2010). This clearly indicates that the amount of SMN produced by a particular SMN2 gene is critical in determining phenotypic severity. Additional variants in SMN2 introns 6 and 7 have been shown to alter the incorporation of the critical exon 7 (Wu et al. 2017). One proposed modifier that lies outside the SMA region is the plastin 3 (PLS3) gene located on the X chromosome whose expression level has been reported to alter the severity of the SMA phenotype. This is based on higher expression levels of PLS3 in lymphoblasts from mild exception patients but the effect is sex-dependent and only partially penetrant. (Oprea et al. 2008; Bernal et al. 2011). The degree to which PLS3 expression modifies the SMA phenotype remains controversial, in part due to experiments in SMA mice showing no survival or electrophysiological benefit of PLS3 over-expression (McGovern et al. 2015; Burghes and McGovern 2017). No DNA variant that accounts for the altered PLS3 expression has been reported to date.
We have utilized an adaption of the targeted sequencing technique Multiplexed Direct Genomic Selection (MDiGS) (Alvarado et al. 2014) to define all the variants that occur in the SMN1, SMN2, and PLS3 genes of 217 SMA patients. We defined, for the first time, a deletion junction that removed SMN½ exons 7 and 8 but retained exons 1–6. We tested for this deletion junction in a separate group of 466 individuals who have various copy numbers of SMN1 and SMN2. This data, as well as pedigree analysis, showed that the deletion can occur in SMN1 or SMN2. By using the read depth across the SMN2 gene we determined the copy number of SMN½ and the copy number was consistent with that determined by droplet digital PCR (ddPCR). Variants and indels in SMN2 were analyzed for their association in SMA patients with exceptionally mild or severe phenotypes not predicted by their copy number of SMN2. The intron 6 variants A-44G, A-549G, and C-1897T of SMN2 showed a statistically significant correlation with milder than expected exception patients. These variants were found in SMN2 but are typically associated with the SMN1 gene (Monani et al. 1999; Wu et al. 2017). No variant or indel in PLS3 was found to significantly associate with mild or severe SMA patients. Thus, a majority of patients had fully intact SMN2 genes with no evidence of modifying mutations within the SMN2 gene implying that modifying variants in a majority of cases lie outside of the SMA region. The patient sample set presented here is an ideal dataset for the further testing of candidate modifiers of SMA.
Methods
DNA samples
This study used multiple sources of DNA including samples previously collected for linkage analysis or molecular studies of SMA (Burghes et al. 1994; McAndrew et al. 1997; Miller et al. 2001) under the Institutional Review Board (IRB) of The Ohio State University OSU1988H0371 that were de-identified and determined to be exempt by the IRB. This consisted of 80 OSU SMA samples as well as 10 parents or siblings. Thirteen new SMA samples were collected under IRB No. 2015H0115, giving a total of 93 SMA patients. From Massachusetts General Hospital (MGH), we included 127 samples from the Project Cure SMA Longitudinal Pediatric Data Repository, University of Utah IRB No. 8751 and Partners IRB No. 2016-P000469. The MGH samples were deidentified to the OSU investigators. A total of 217 SMA samples were sequenced and analyzed by MDiGS.
A set of 466 de-identified independent samples with known and varying copy number of SMN1 and SMN2 from The Ohio State University Molecular Pathology Laboratory were used to screen for deletion junction frequency. These samples were determined exempt by the OSU IRB.
Classification of patients
A total of 217 SMA patients were sequenced. Phenotypic information was known for all but 29 samples. Phenotype was reported when samples were collected by the physician and was based on the approved criteria of age of symptom onset and maximum achieved motor function (non-sitters, sitters, and walkers). Based on the phenotype and MDiGS determined copy number, we classified each patient as concordant, mild, or severe. We defined concordant patients as those who had an expected SMN2 copy number for their given level of disease severity. For testing purposes, we used the following model to define expected copy number: SMA type 1 has 2 copies of SMN2, type 2 has 3 copies of SMN2, and type 3 has 4 copies of SMN2. This model allows us to identify exception patients (discordant) that have either a milder or more severe phenotype compared to SMA patients that are concordant with SMN2 copy number and severity. This model allows for testing the association of SMN2 and PLS3 variants with severity of SMA while accounting for SMN2 copy number. In the cohort we studied (n = 217), we identified 77 exception patients, 58 with a milder than expected phenotype and 19 with a more severe phenotype.
MDiGS sequencing
Genes of interest were captured and sequenced using an adaptation of the Multiplexed Direct Genomic Selection (MDiGS) assay (Alvarado et al. 2014). Using the same method as described in Alvarado et al, we pooled 48 indexed DNA samples. For BAC capture we used the clones RP11–652K3 (CFTR), RP11–1056O6 (CFTR), and RP11–268A15 (PLS3) obtained from the BACPAC Resource Center at Children’s Hospital Oakland Research Institute in Oakland, California. RP11–652K3 and RP11–1056O6 have approximately 88 kb of overlap and together cover the entire CFTR gene (Osoegawa et al. 2001). For the capture of SMN2 we used a 35.5 kb portion of the clone RP1–215P15. This portion contains the entire SMN2 gene flanked by BamHI that had previously been cloned into the BAC pIndigoBac5 (Epicentre) SMN26.6 (BAC5 SMN2) (Hao et al. 2011). Four cosmids (108F4, 121C9, 22A5, 30C9) flanking the SMN2 gene were used to block non-specific capture. We confirmed that the cosmids do not contain SMN2 by PCR (DiDonato et al. 1994; DiDonato 1995; Thompson et al. 1995). The biotinylated captured BACs were then hybridized with the pooled indexed DNA library for more than 70 hours. The DNA library then contained only sequences hybridized to SMN, PLS3, or CFTR. The 48 samples were run on a single MiSeq lane and then decoded using the indices.
Bioinformatics
Reads were aligned using the program STAR (Dobin et al. 2012) to a custom made genome based on human reference hg19. This genome masked the SMN1, SMN2, CFTR, and PLS3 genes. In addition, the sequence of the clones RP1–215P15, RP11–652K3, RP11–1056O6, and RP11–268A15 were appended to the genome as separate contigs. Only the 35.5 kb sequence of RP1–215P15 that was captured was included in the genome.
Copy number was determined by calculating the ratio of reads aligned to RP1–215P15 (SMN2) compared to RP11–652K3 (CFTR) and RP11–1056O6 (CFTR). This is the ratio of reads aligned to SMN2 (and SMN1 if present) compared to CFTR (i.e. SMN½ / CFTR). We used 3 samples with known SMN2 copy number to normalize the read counts as the clones have different capture efficiencies and lengths. Since the copy number of CFTR should be 2 for all individuals, as it is on an autosomal chromosome, this normalized ratio gave the number of copies of SMN1 and SMN2. Additionally, RP1–215P15 was divided into equally-sized bins 1,815 bp in length and SMN½ copy number was found for each bin, again by calculating the SMN½/ CFTR ratio of reads in that region.
Variants and indels were called using the Genome Analysis Toolkit (DePristo et al. 2011). Duplicate reads were marked and removed using Picard Tools MarkDuplicates. Reads were realigned along known insertions and deletions (indels) in the SMN2, CFTR, and PLS3 genes. Base quality scores were then recalibrated for each read. HaplotypeCaller was used to call the variants and the variants were then hard filtered. The presence and copy number of SMN1 is determined by analyzing variant calls for the C to T change at the +6 position of exon 7. Variants were tested for association with mild or severe exception patients by performing a Fisher’s exact test using a 2 × 2 contingency table and comparing allele counts in exception patients to concordant patients. We defined concordant patients as those who had an expected SMN2 copy number for their given level of disease severity. P-values were corrected for multiple testing using a False Discovery Rate calculation in R (R Core Team 2013). To reduce false positives, variants were only tested if more than 15% of reads at that locus contained the variant.
We analyzed the captured region for possible long deletions in our samples using two methods. First, we designed a Python script that extracted all aligned reads with gaps greater than 20 base pairs based on CIGAR strings containing the ‘N’ value (Li et al. 2009). The script then printed out the length of every gap and the number of occurrences per sample. Second, we plotted the CFTR vs. SMN½ read count ratio over bins (1815 bp per bin) for each sample. Any sudden decrease in copy number in a particular bin suggested a possible deletion at that location. Individual reads at sites of possible deletions were analyzed for gaps, partial alignments, and mismatches to determine the break point. Break points were then confirmed via PCR. Similarly, a mismatch between the copy number when determined over the whole gene and copy number when determined at exon 7 was evidence of a possible duplication or deletion.
All scripts have been deposited to GitHub and are available at https://github.com/BurghesLab/SMN2Analysis.
Determination of SMN1 and SMN2 copy number by ddPCR
Copy number as determined by MDiGS was validated using droplet digital PCR on the QX200 Droplet Digital PCR system (BioRad). Locked nucleic acid (LNA) probes with a competitive non-extending oligo were used for to determine SMN1 and SMN2 copy number using the primer set (5’-AATGCTTTTTAACATCCATATAAAGCT-3’ and 5’-CCTTAATTTAAGGAATGTGAGCACC-3’) (Anhuf et al. 2003), SMN1 5’-FAM-CAGG+GTT+T+C+AGACAAA-3’ with competitive oligo 5`-ATTTTCCTTACAGGGTTTtAGACAAAATCAAAAGA-PHO-3’, and SMN2 5’-FAM-TGATTTTGT+C+T+A+A+AA+CCCT-3’, with competitive oligo 5`-ATTTTCCTTACAGGGTTTcAGACAAAATCAAAAGA-PHO-3’ (Pyatt and Prior 2006). In every multiplex reaction exon 14 of CFTR was amplified as a two-copy control and used to determine copy number using (FP 5`-AGAGAGAAGGCTGTCCTTAGT-3’, RP 5`-GAGTGTGTCATCAGGTTCAGG-3’, probe 5`-HEX-TTCTGAGCAGGGAGAGGCGATACT-3’).
PCR detection of the SMN½ intron 6 deletion junction
Once possible deletion junctions were identified bioinformatically they were confirmed using conventional PCR. Primers were designed that flank the junction and did not contain repeats as identified by using Repeat Masker (Smit et al. 2013). The forward primer used was 5’-CAGTTATCTGACTGTAACACTGTAGGC-3’ and the reverse primer used was 5’-GTTGTTGCTTATGCTGGTCTTG-3’ to generate a 650 bp product. For 3 individuals the PCR product was subcloned into pCR 2.1-TOPO TA vector (Thermo Fisher) and Sanger sequenced for confirmation.
Determination of the inheritance of alleles in family with deletion junction
ddPCR: To determine inheritance of alleles primers and probes were made to SMN exon 1 (FP 5’-TGTTCCGCTCCCAGAAG-3’ and RP 5’-CTCATCGCCATAGCAAACC-3’, 5’-FAM-TTAAGAGTGACGACTTCCGCCGC-3’), exon 2a (FP 5’-TTATTTCTTACCCTTTCCAGAGC-3’ and RP5’-AAATGAAGCCACAGCTTTATCA-3’, 5’-FAM-TCTGACATTTGGGATGATACAGCACTGA-3’), and exon 6 (FP 5’-CACCTCCCATATGTCCAGATTC-3’ and RP 5’-CCAGTATGATAGCCACTCATGT-3’, 5’-FAM-TCTTGATGATGCTGATGCTTTGGGAAGT-3’). To determine the number of junction fragments created by the deletion we designed a primer probe set where the forward primer (FP 5`-ATACAAGTTGGCTGGGCACAA-3’) was located in intron 6 and the reverse primer (RP 5`-TTTTACTATGTTGGCCAGGCTG-3’) and probe (5`FAM-TGGATCACCTGAGATCAGGAGTTCC-3’) were located in exon 8. 10–20ng of genomic DNA was used in each ddPCR reaction. All ddPCR reactions were multiplexed with the same CFTR primer/probe set used to determine SMN½ copy number. Copy number of SMN1 and SMN2 at exon 7 was determined via ddPCR using primers as described above.
Markers flanking SMN: Two flanking markers were followed to determine inheritance of the junction fragment in the family. Inheritance of marker D5S39, which is linked to (Mfd27), was as described previously (Weber et al. 1991). A second marker D5S832 (A31) was amplified from individuals using primers as described previously (Wirth et al. 1995). The PCR products were subcloned into pCR 2.1-TOPO TA vector (Thermo Fisher) and Sanger sequenced to determine the number of CA repeats. A minimum of 6 clones were analyzed for each individual in the pedigree. The alleles are given by the number of CA detected. The third marker AG1-CA was analyzed as described previously (DiDonato et al. 1994).
Results
Identification of SMN½ deletion junction
To analyze the genotype/phenotype relationship in SMA, we captured and sequenced the CFTR, SMN2, and PLS3 genes of 217 SMA patients using the MDiGS procedure (Alvarado et al. 2014). We used the clones BAC5SMN2 (SMN2), RP11–652K3 (CFTR), RP11–1056O6 (CFTR), and RP11–268A15 (PLS3) as bait for the capture procedure. The average number of total reads per sample was approximately 940,000, with 19.2% of reads aligning to the clone sequences which is similar to previously reported results (Alvarado et al. 2014). Copy number of SMN2 was determined by calculating the ratio of reads aligned to the CFTR contigs compared to the SMN2 contig.
In order to detect partial deletions of SMN½ we divided the SMN2 contig into bins of 1,815 bp and copy number was determined at each bin. Out of 217 samples sequenced 17 samples displayed a decrease in the copy number over 3 consecutive bins in the 3’ region of the SMN½ genes (Fig. 1A). Upon analysis of the read alignments in this region we found reads that contained a 6310 bp deletion. The deletion junction occurred within a 21 bp repeat that had an exact match a total of 15 times in the 35.5 kb segment containing the SMN2 gene that we captured (Fig. 1B). Primers were made to unique sequences flanking the repeats and the deletion junction was amplified by PCR. The resulting fragment was Sanger sequenced to confirm the deletion in 3 samples (Fig. 1C). From this assay alone it is impossible to determine if the deletion occurs in SMN1 or SMN2. Nonetheless, this is the first deletion junction identified that eliminates the critical exons 7 and 8 of SMN½. The deletion was detected in 17 out of 217 SMA patients sequenced. The full-length copy numbers of SMN1 and SMN2 of these patients is listed in Supplementary Table 2. Of these 17 patients, 2 had SMN1: OSU284 and MGH300. OSU284 had 1 copy of SMN1 and 1 copy of SMN2, while MGH300 had 1 copy of SMN1 and 2 copies of SMN2. Of the 15 remaining patients, 2 had 1 copy of SMN2, 6 had 2 copies of SMN2, 6 had 3 copies of SMN2, and 1 had 4 copies of SMN2. However, this sample is not completely random as the MGH samples where partially selected to include patients that either had a milder or more severe phenotype than expected. In the case of the OSU samples the deletion was found in 9 out of 80 samples, which were all collected in an unbiased manner. The frequency of deletion was then determined by PCR in a separate set of samples with known SMN1 and SMN2 copy number to develop a model of deletion described below.
Fig. 1A-C.
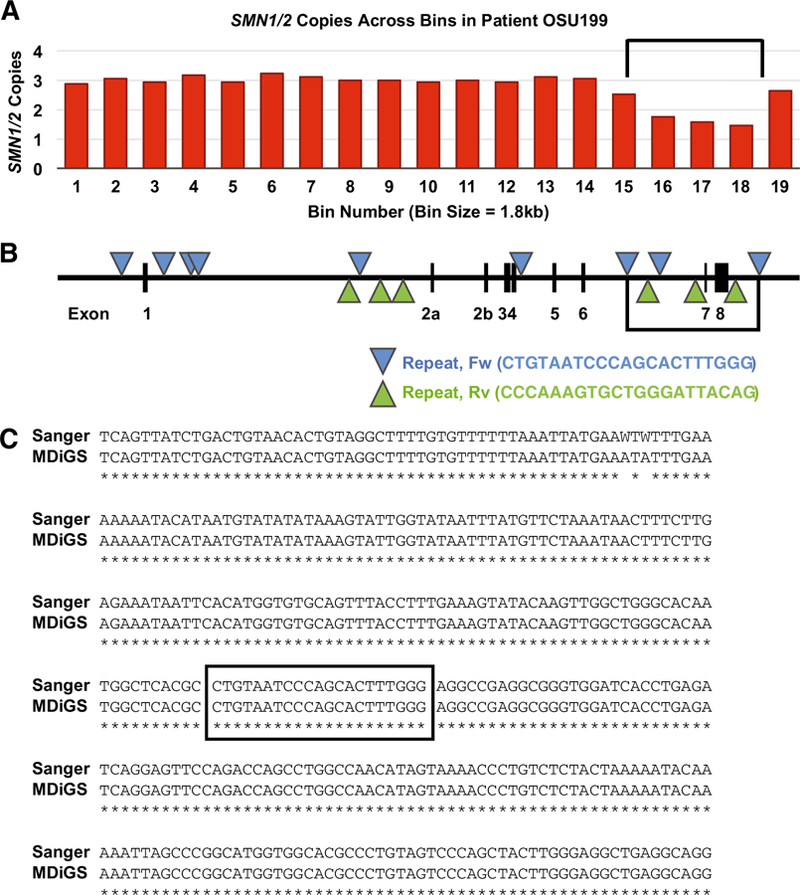
Detection of 6.3 kb deletion junction bioinformatically, location of repeats in SMN2, and verification of the deletion junction using Sanger sequencing after amplification using PCR. A The ratio of reads aligned to CFTR compared to SMN½ was calculated and plotted for 19 bins, each with a size of 1.8 kb. A decrease in the ratio over 3 bins in the 3’ end of SMN½ is shown here for sample 199. Analysis of the sequencing reads resulted in the detection of a 6.3 kb deletion. The location of the deletion is indicated by black bars. B. A diagram illustrating where each bin aligns on the SMN2 gene. Also shown are the locations of the 21 bp repeats in SMN2, depicted as triangles. These 21 bp repeats match the Alu-repeat sequence and are known to be involved in deletions and rearrangements (Rüdiger et al. 1995). The black bars indicate location of the deletion. C. The deletion junction was amplified using PCR, subcloned, and Sanger sequenced for verification. Shown is a sequence alignment between the Sanger sequencing and the MiSeq MDiGS reads. An asterisk (*) represents identical nucleotides. A “W” base call can be either an “A” or a “T”. The boxed nucleotides indicate the repeat that contains the junction.
Inheritance of the junction in an SMA family
To determine if the deletion occurred in SMN1 or SMN2, we analyzed the inheritance of the deletion junction using the marker AG1-CA, the flanking single copy polymorphic markers D5S823 and D5S107, and copy number analysis as measured by ddPCR. The pedigree of Family 32 with proband OSU199 is shown in Fig. 2. The proband had a severe SMA type 1/0 phenotype which was consistent with 2 copies of an SMN2 partial deletion detected using MDiGS. Copy number of SMN½ was determined at exons 1, 2a, 6, and 7. Only exon 7 primers and probe were able to distinguish between SMN1 and SMN2 exon 7. The parents (II:OSU201 and II:OSU200) and paternal grandparents (I:OSU201.1 and I:OSU201.2) of the proband were also analyzed.
Fig. 2.
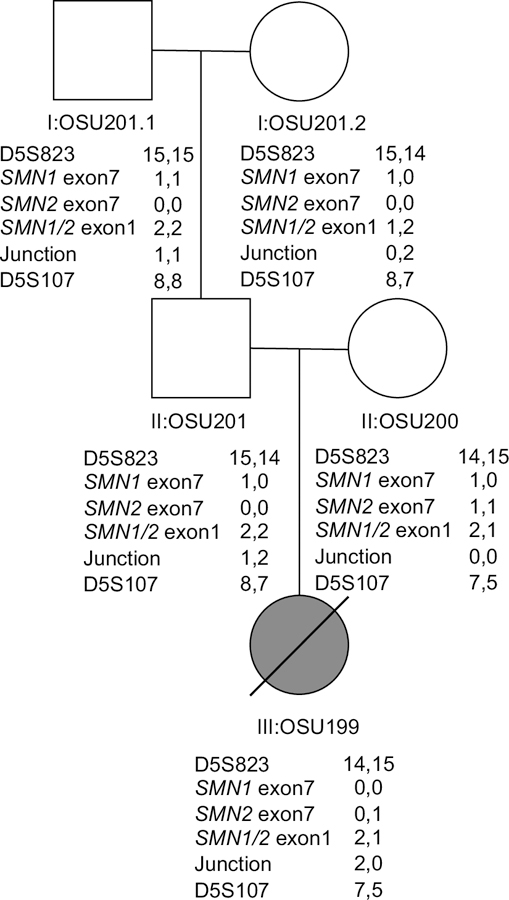
A 6.3 kb deletion was detected in the proband (III:OSU199). The copy number of SMN1 exon 7, SMN2 exon 7, SMN½ exon 1, and the deletion junction were measured using ddPCR in patient III:OSU199, her parents (II:OSU201 and II:OSU200), as well as paternal grandparents (I:OSU201.1 and I:OSU201.2). Shown are the measured copy numbers, from which we have inferred the number of copies per chromosome. We also show the genotype for the markers D5S823 and D5S107 which flank the region containing the SMN½ genes. The proband III:OSU199 had 1 copy of SMN2 exon 7, 3 copies of SMN½ exon 1, and 2 copies of the deletion junction. The father (II:OSU201) had 1 copy of SMN1, 4 copies of SMN½ exon 1, and 3 copies of the deletion junction. The mother had 0 copies of the deletion junction, meaning the proband inherited 2 copies of the deletion junction from the father on the same chromosome. The grandfather (I:OSU201.1) had 2 copies of SMN1 exon 7, 4 copies of SMN½ exon 1, and 2 copies of the deletion junction. The grandmother had 1 copy of SMN1, 3 copies of SMN½ exon 1, and 2 copies of the deletion junction. Analysis of polymorphic markers D5S823 and D5S107 was consistent with the deletion being inherited from the grandmother.
The proband (III:OSU199) was determined to have 1 copy of SMN2 at exon 7, 3 copies of SMN2 at exon 1, and 2 copies of the deletion junction. The mother (II:OSU200) had 3 copies of SMN½ exon 7 (1 copy of SMN1, 2 copies of SMN2), 3 copies of SMN½ at exon 1, and 0 copies of the deletion junction, meaning all SMN½ copies are intact. The father (II:OSU201) had 1 copy of SMN1 exon 7, 4 copies of SMN½ exon 1 and 3 copies of the deletion junction. These data indicate the proband inherited 2 deletion junctions from the father and 1 SMN2 from the mother and the 2 deletion junctions lie on the same chromosome. The paternal grandmother (I:OSU201.2) had 1 copy of SMN1 exon 7, but 3 copies of SMN½ exon 1, and 2 copies of the deletion junction. The grandfather (I:OSU201.1) had 2 copies of SMN1 exon 7, 4 copies of SMN½ exon 1, and 2 copies of the deletion junction.
In order to determine the origin of the deletion alleles, we examined the polymorphic markers AG1-CA, D5S823 and D5S107 (Weber et al. 1991; DiDonato et al. 1994; Wirth et al. 1995). The marker D5S107 lies near D5S39 and the markers are closely associated with each other (Weber et al. 1991). The marker AG1-CA was not informative on the origin of the deleted chromosome. The flanking markers D5S823 and DS107 (also D5S78 not shown) where consistent with the inheritance of the deletion from the grandmother with no recombination between the markers. The dosage of the deletion junction as well as SMN1 and SMN2 and SMN½ probes between SMN½ exons 1–6 were also consistent with the deletion being inherited from the grandmother (see Fig.2).
Thus, the grandmother had a deletion in both SMN1 and SMN2 which was inherited by the proband. These markers provide strong evidence that the deletion junction occurs in both SMN1 and SMN2 in this family.
Model of deletion junction frequency in individuals with varying SMN1 and SMN2 copy number
To determine the frequency of the deletion junction, we screened a separate panel of 466 samples with varying copy numbers of SMN1 and SMN2 using PCR. The results are shown in Table 1. The group with the highest frequency of deletion were individuals who had 1 copy of SMN1 and 0 copies of SMN2 with a frequency of 0.63. The genotype with the next highest deletion frequency was 2 SMN1, 0 SMN2 with a frequency of 0.46. For genotypes with the same copy number of SMN1 deletion frequency increased as copy number of SMN2 decreased. For example, the deletion frequency was 0, 0.02, 0.06, and 0.35 for individuals with 0 SMN1 and 4, 3, 2, and 1 copies of SMN2 respectively. Similarly, for genotypes with the same copy number of SMN2, deletion frequency increased as copy number of SMN1 decreased. The deletion frequency in patients with (3 SMN1; 0 SMN2), (2 SMN1; 0 SMN2), and (1 SMN1; 0 SMN2) was 0.10, 0.46, and 0.63, respectively.
Table 1.
Prevalence of deletion junction amongst a panel of 466 individuals with different copy numbers of SMN1 and SMN2.
| Known Copy Number |
||||
|---|---|---|---|---|
| SMN1 | SMN2 | Total Positive | Total Samples Screened | Frequency |
| 0 | 1 | 6 | 17 | 0.35 |
| 0 | 2 | 3 | 50 | 0.060 |
| 0 | 3 | 1 | 50 | 0.020 |
| 0 | 4 | 0 | 50 | 0.0 |
| 1 | 0 | 12 | 19 | 0.63 |
| 2 | 0 | 23 | 50 | 0.46 |
| 3 | 0 | 3 | 30 | 0.10 |
| 2 | 1 | 17 | 50 | 0.34 |
| 2 | 2 | 0 | 50 | 0.0 |
| 1 | 1 | 17 | 50 | 0.34 |
| 1 | 2 | 2 | 50 | 0.04 |
To determine if the deletion junction occurred in both SMN1 and SMN2 we compared two probability models that calculated the log odds ratio of having the deletion based on the copy numbers of SMN1 and SMN2. Model 1 assumed the deletion junction frequency is dependent only on SMN2. Model 2 assumed the frequency is dependent upon the cross-classification of SMN1 and SMN2.
Model 1:
Logit { P(Y=1) } = β0 + β1I(X2 = 1) +β2I(X2 = 2) +β3I(X2 = 3)+ β4I(X2 = 4)
Model 2:
Logit
Where X1 is SMN1 copy number, X2 is SMN2 copy number, and I is the indicator function, which is 1 if the event occurs and 0 if the event does not occur.
By using the frequencies in Table 1 and comparing Model 1 to Model 2 using a likelihood ratio test, a p-value of 0.005 is obtained in favor of Model 2. As a result, the model that allows different probabilities of deletion by the cross-classified SMN1 and SMN2 fit the data better than the model that assumes the probability of deletion depends only on SMN2. This supports the hypothesis that the deletion junction occurred in both SMN1 and SMN2. This model however does not make any assumptions about the frequency in which the deletion occurs in each gene. It is notable that all groups with 1 copy of SMN2 and varying copy numbers of SMN1 had a relatively similar rate of deletion at frequencies of 0.34, 0.34 and 0.35. This indicates a higher deletion rate in SMN2 than SMN1 however SMN1 deletions do occur as evidenced by the increased rate in the 1 SMN1; 0 SMN2 individuals compared to 2 SMN1; 0 SMN2 individuals.
Correlation curve of SMN2 as determined by MDiGS compared to ddPCR
SMN2 copy number as determined by MDiGS was validated in a subset of the sequenced samples (n = 180) using ddPCR. Of the 180 samples, 172 were confirmed to be correct (95.5%). The correlation curve (Fig. 3) had an R2 value of 0.9108. Of the 8 discrepant samples, 3 samples had low read counts, which can complicate bioinformatic analysis of copy number. In these samples, MDiGS indicated 3 copies of SMN2 in 2 cases whereas ddPCR indicated 2 copies of SMN2. In 1 case, MDiGS indicated 4 copies of SMN2 whereas ddPCR indicated 3 copies of SMN2. Thus, when read depth is low MDiGS over-estimated SMN2 copy number by 1. Of the remaining 5 discrepant copy numbers, 3 were underestimates with MDiGS. In 2 cases, MDiGS indicated 3 copies of SMN2 while ddPCR indicated 4 copies of SMN2. In 1 case, MDiGS indicated 4 copies of SMN2 and ddPCR 5 copies of SMN2. In the other 2 samples, overestimates occurred with MDiGS indicating 4 and 3 copies of SMN2 and ddPCR indicating 3 and 2 copies of SMN2. In all cases the difference between the two methods was a single copy. The exact reason for these discrepancies is unclear but could result from sample quality affecting the sequencing measures. We note that this rate of discrepancy is similar to previously reported rates when comparing sequencing and ddPCR copy number determination (Eisfeldt et al. 2018).
Fig. 3.
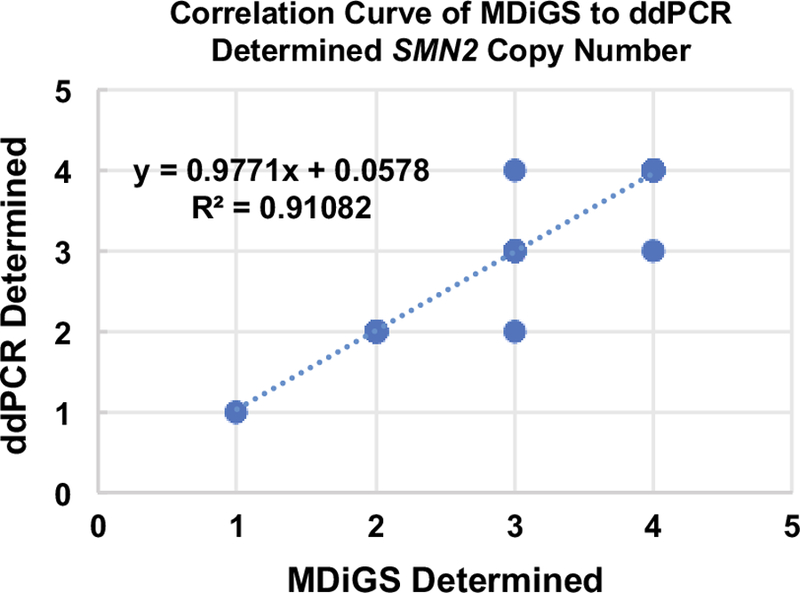
Correlation curve of SMN2 copy number as determined by MDiGS compared to ddPCR. A total of 180 samples were analyzed, 172 of which were confirmed to be correct (95.5%).
Analysis of SMN2 variants that can modify the SMA phenotype
Variants, insertions, and deletions were identified for all samples. There were 157 SNPs in SMN½ amongst all patients examined. A total of 177 samples were examined from the 217 sequenced. Of the samples eliminated from analysis, 5 had SMN1, 25 had either no phenotypic data or the exact phenotype was unclear, 8 had low read counts, and 2 had discrepant copy number of SMN2. As previously described, we classified each patient as concordant (Type 1 = 2 copies of SMN2, Type 2 = 3 copies of SMN2 and Type 3 = 4 copies of SMN2), mild, or severe. We analyzed each variant for segregation by comparing allele counts in exception cases (mild or severe) to allele counts in concordant cases using a Fisher’s exact test. P-values were corrected for multiple testing using False Discovery Rate. Samples were included for this analysis if there was phenotypic information available and if the MDiGS determined SMN2 copy number matched the SMN2 copy number determined by ddPCR or an alternative quantitative dosage assay (Mailman et al. 2002).
The SNPs with the highest significance are shown in Table 2. A total of 3 SNPs in intron 6, namely A-44G, A-549G, and C-1897T, were found to significantly associate with mild exception patients after correcting for multiple testing. Variant A-549G (NC_000005.9:g.69371799A>G; rs564142907; GnomAD frequency of 0.0018) was found to be most significant (adjusted p-value of 0.00294) with identification in 9 mild exception patients and never in concordant or severe exception patients. Similarly, A-44G (NC_000005.9:g.69372304A>G; rs212216; GnomAD frequency of 0.0001) was found in 6 mild exception patients but never in severe exception patients or concordant patients (adjusted p-value of 0.04982). C-1897T (NC_000005.9:g.69370451C>T; rs1381625877; GnomAD frequency of 0.0025) was found in 8 mild exception patients and 1 concordant (adjusted p-value of 0.03704). In all cases, only 1 allele of the variant was present in each patient. All 3 of these variants are known to associate more commonly with SMN1 but can also be present in SMN2 due to gene conversion events (Burghes 1997; Monani et al. 1999; Wu et al. 2017). Two other variants that are also usually in SMN1, T-478C (NC_000005.9:g.69371870C>T; rs1457707829), and C-255T (NC_000005.9:g.69372088C>T; rs1317747440; GnomAD frequency of 0.0005), were detected in 5 mild exception patients however they did not reach statistical significance with an adjusted p-values of 0.1066 and 0.1126, respectively. Interestingly, A-44G has recently been shown to increase the level of full-length transcript when it is present in SMN2. C-255T and A-549G were also studied in the same experiment but were not found to have an effect (Wu et al. 2017). All of these variants had an extremely low frequency of less than 1% as reported by GnomAD. However, this frequency may be incorrect as it is not clear if the variants were properly designated as being in SMN1 or SMN2. Moreover, some of these variants have been reported previously with significantly higher allele frequencies (Monani et al. 1999). These data suggest 3 variants in intron 6 of SMN2 result in a milder SMA phenotype than expected.
Table 2.
Association between SNPs and exception SMA phenotypes.
| Concordant | Mild | Severe | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Location | Variant | Refa | Altb | Ref | Alt | Ref | Alt | Ref | Alt | P-Value | Adjusted P-Value |
| Intron 6 | A-549G | A | G | 309 | 0 | 143 | 9 | 39 | 0 | 1.872E-5 | 0.00294 |
| Intron 6 | A-44G | A | G | 310 | 0 | 156 | 6 | 39 | 0 | 0.000952 | 0.04982 |
| Intron 6 | C-1897T | C | T | 308 | 1 | 146 | 8 | 39 | 0 | 0.000471 | 0.03704 |
| Intron 6 | C-478T | C | T | 309 | 0 | 151 | 5 | 39 | 0 | 0.002716 | 0.10662 |
| Intron 6 | C-255T | C | T | 311 | 0 | 165 | 5 | 39 | 0 | 0.003588 | 0.11265 |
Ref = Reference Allele
Alt = Alternate Allele
We also identified and tested 80 SMN½ indel alleles for association with either the mild or severe exception patients. There were no significant differences in the frequencies of these alleles after multiple testing correction. Similarly, we tested for segregation of PLS3 variants with exception patients after separating all patients by sex as PLS3 is on the X chromosome. Including indels, we tested 399 PLS3 variants in males and 636 in females. We found no SNPs or indels in PLS3 that segregated with mild or severe exception patients after multiple testing correction. This includes the rs871773 variant which is known to increase PLS3 expression in colon cancer (Szkandera et al. 2013). We found rs871773 in 6 female mild exception patients, but also 3 female severe exception patients with a corrected p-value of 0.6749. These data suggest that there is no evidence of a modifying variant in PLS3. Data for all SMN½ and PLS3 variants can be found in Supplemental Table 1.
Mutations in SMN2 or SMN1 in SMA patients
We found 5 exonic variants in our patient samples (n = 217). One was the novel mutation p.A75T (c.223G>A; NC_000005.9:g.69361861G> A) in SMN2 exon 2b of one SMA patient. To our knowledge, this variant has never been reported in either SMN1 or SMN2. This variant was present in a SMA type 3 patient with 4 copies of SMN2. As the phenotype is concordant with SMN2 copy number, there is no evidence this mutation affects SMA phenotype. Indeed, the variant was predicted by PolyPhen to be benign with a score of 0.005.
We also identified the variant p.Y130C (c.389A>G; rs397514517; NC_000005.9:g.70238300A>G) in exon 3 of one patient. This patient had 1 copy of SMN1, 2 copies of SMN2, and was diagnosed as SMA type 3b. The SMN p.Y130C variant was previously reported as an SMA-causing mutation (Prior 2007), though the phenotype is milder compared to a 3 copy SMN2 individual. Therefore, the variant appears to result in a milder SMA phenotype.
Two patients had the c.859G>C (rs121909192; NC_000005.9:g.69372372G>C, GnomAD frequency of 0.0017) mutation in exon 7 which has previously been shown to modify SMN production by modulating the binding of splicing factors to exon 7 (Prior et al. 2009).The first patient with this variant was determined to have 2 copies of SMN2 using the MDiGS sequencing data and phenotypically was a SMA type 3a SMA patient. The second patient was determined to have 2 copies of SMN2 and was a SMA type 3b patient. Additionally, this patient was determined to have 2 copies of the c.859G>C allele. These phenotypes are milder than expected, as the vast majority of 2 copy SMN2 patients have a severe type 1 phenotype (Feldkötter et al. 2002; Calucho et al. 2018). This milder phenotype is consistent with previous reports of SMA patients with the c.859G>C variant (Prior et al. 2009).
The c.84C>T (rs1554066599; NC_000005.9:g.69359244C>T) variant in exon 2a was detected in 11 patients. This is a synonymous variant located 3 bp from the beginning of exon 2a. This variant was found in 9 concordant, 1 mild, and 2 severe exception patients and was not significantly associated with either a milder (adjusted p-value 0.881) or more severe (adjusted p-value 1.000) phenotype. This is in contrast with previous data reporting it as a possible pathogenic variant, as it was found in an SMA patient with 1 copy of SMN1 (Wang et al. 2010).
Finally, we detected the c.462A>G (rs1450194682; NC_000005.9:g.69362949A>G) variant in exon 3 of numerous patients. This allele was detected 268 times out of a total of 403 alleles. It was not found to segregate with either the mild (adjusted p-value 1.000) or severe exception patients (adjusted p-value 1.000).
Alignment and map of the SMA region
The complexity of the SMA region has caused notable difficulty in constructing a correct map of the region. There are multiple different arrangements of the region, as evidenced by the multiplicity of banding patterns obtained on pulsed field gel electrophoresis experiments when using SMN2 and NAIP probes. To determine one arrangement of the SMA region, we assembled a map using overlapping PACs that originated from a single library (Fig. 4A) (Osoegawa et al. 2001). We used a total of 10 clones, with an average overlap of 71850 bp between adjacent clones and the smallest overlap being 32833 bp. There was only a single base pair mismatch among all the overlaps. It has been reported that heterozygous SNPs occur on average every 1.1 kb making it very likely this assembly is one chromosome (Ceballos et al. 2018). The clones we used were all from the RP11 library (Osoegawa et al. 1998, 2001), specifically RP11–619K7, RP11–1012N14, RP11–1415C14, RP11–1414O21, RP11–497H16, RP11–1005E12 , RP11–1432L1 , RP11–974F13 , RP11–195E2 , and RP11–1280N14 (Fig. 4B). The total length of the assembly was 1.358 Mbp.
Fig. 4A, B.
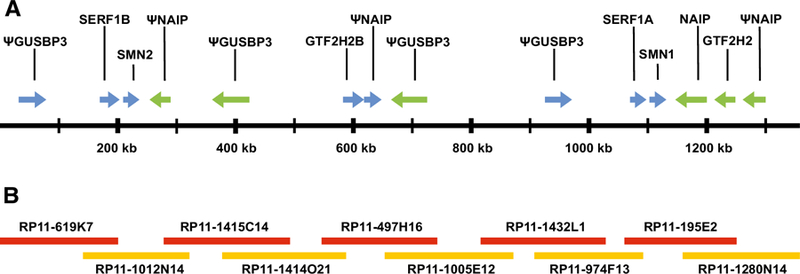
Map of the SMA region that was assembled using overlapping clones that originate from the same chromosome. A Map of the region showing all genes and their orientation. SMN1 and SMN2 are in the same orientation and are approximately 848 kb away from each other. Lying between SMN1 and SMN2 are 2 NAIP pseudogenes (one containing NAIP exons 6–17 and the other contained NAIP exons 3 and 6–9) and 2 GUSBP3 pseudogenes, as well as SERF1A and GTF2H2B. Pseudogenes are indicated with a (Ψ). B Overlapping clones used to construct the region. The smallest overlap between clones was 32,833 bp while the average overlap was 71,850 bp. There was only a single mismatched base pair out of all the overlapping regions.
We identified and labeled genes and pseudogenes on the assembled map that can be seen in Fig 4. SMN1 and SMN2 were located approximately 848 kb away from each other and were in the same orientation. Approximately 6.5 kb upstream of SMN2 was SERF1B and 16.4 kb downstream of SMN2 was a NAIP pseudogene containing NAIP exons 6–17 (Ensembl exons ENSE00003489009 through ENSE00003505062). The gene GTF2H2B was 338 kb downstream of SMN2, followed by a second NAIP pseudogene containing NAIP exon 3 (Ensembl exon ENSE00003668305) and exons 6–9 (Ensembl exons ENSE00003489009 through ENSE00002219419). Approximately 6.5 kb upstream of SMN1 was SERF1A and 16.4 kb downstream was NAIP. Approximately 80 kb downstream of SMN1 was GTF2H2, followed by a NAIP pseudogene that contains NAIP exons 6–13 (Ensembl exons ENSE00003489009 through ENSE00003590701). There were also 4 copies of GUSBP3 pseudogene containing various combinations of exons and in different orientations.
Discussion
The SMA region was originally described as an inverted repeat which was based on mapping and pulsed field analysis of YAC clones obtained from the region (Melki et al. 1994; Lefebvre et al. 1995). However, YAC clones from this repeated region often contain rearrangements with loss of markers (Carpten et al. 1994). Here, we have assembled a map of the SMA region on one chromosome using overlapping clones (Fig 4). It is notable that in this arrangement there is not an inverted duplication. Instead, SMN1 and SMN2 are in the same orientation as is NAIP and a NAIP pseudogene. The arrangement of the SMA region we present is consistent with the occurrence of unequal crossover events leading to deletion of the SMN1 gene as has been previously reported (Wirth et al. 1997). This arrangement is also consistent with the studies of CA-dinucleotide markers in the region that show multiple copies but of varying number. The studies of these markers would also be consistent with multiple possible arrangements and additional copies of certain markers, including AG1-CA and CAAT1 (DiDonato et al. 1994; Burghes et al. 1994). Further experiments will be needed to determine which orientation is most common in the population. This could be approached by long read sequencing technologies such as Nanopore MinION whole genome sequencing or 10X Genomics sequencing, which barcodes the small DNA fragments to allow for direct assembly. Performing such an analysis would give insight into the process of gene conversion. If SMN1 is converted to SMN2 by mutation, the SMN2 gene will then lie adjacent to the NAIP gene instead of the NAIP pseudogene. Similarly, if SMN2 is converted to SMN1 then the 2 SMN1 genes can be on the same chromosome, one adjacent to NAIP and the other adjacent the NAIP pseudogene.
It is known that SMN1 and SMN2 can be lost by deletion but deletion junctions have not been reported that remove SMN½ exons 7 and 8 (Burghes 1997). Using a ligation mediated PCR assay for SMN½ copy number, exon 1 of SMN½ has been previously examined in SMA patients, carriers, and normal individuals. In these studies, SMA cases were presented where there was a loss of SMN1 exons 1–6, but with the presence of SMN1 exon 7 (Arkblad et al. 2006). Additionally, there were cases with excess exons 1–6 but missing exons 7–8 (Arkblad et al. 2006). The loss of SMN exons 7 and 8 was described as a polymorphism as it was present in normal individuals with 2 copies of SMN1 (Arkblad et al. 2006; Calucho et al. 2018). However, here we report a 6.3 kb deletion that eliminates exons 7 and 8 and can occur in either the SMN1 or SMN2 gene (Fig 1). As the deletion occurs in SMN1 and can cause SMA, it is a disease-causing variant and it cannot be referred to as a polymorphism. Interestingly, we did not detect loss of SMN1 exons1–6 but these alleles clearly exist and are SMA alleles as they occur in SMA patients (Arkblad et al. 2006). Evidence of the 6.3 kb deletion occurring in SMN1 comes from dosage of the 3’ and 5’ end of SMN½ genes examined across 3 generations in one family (Fig. 2), as well as polymorphic markers D5S823 and D5S107 that trace the origin of the mutation. This analysis indicates that the mutant allele originates in the grandmother, who is deleted for both SMN1 and SMN2 on the same chromosome.
Additionally, we developed models based on the deletion frequency in patients with varying copy numbers of SMN1 and SMN2. In comparing the models, the model where the deletion occurs in both SMN1 and SMN2 is most consistent with the observed deletion frequency data. Together, the data show strong evidence of the deletion occurring in both SMN1 and SMN2. The frequency of this deletion appears to be higher in SMN2 than SMN1 as indicated by the similar frequency of the deletion in the groups containing a single copy of SMN2 but different SMN1 copy number. In general, there is correlation of the 5’ end markers and the copy number of SMN½, which would not be the case if this was a highly frequent deletion in SMN1. A puzzling feature of the SMA region is the frequency of SMN1 loss versus the frequency of SMN2 loss. In the case of SMN1 loss, the frequency is 1/10,000 (0.01%), which is also the frequency of SMA (Pearn 1978). Whereas with SMN2 loss, the frequency in the general population is 10–15%, which is 1000 times higher than SMN1 (Mailman et al. 2002). Perhaps the reason for this is selection against loss of SMN1 but not SMN2 as the loss of SMN1 in the population gives rise to SMA and the loss of SMN2 is not detrimental.
We identified 15 repeats in the SMN½ gene that have an exact 21 bp match to the 3’ end of the deletion junction. The sequence of this 21 bp repeat matches perfectly the first 21 bp of the Alu core element (Rüdiger et al. 1995). Interestingly, a deletion junction of SMN exons 5 and 6 has been found near a similar repeat, though with a slightly different sequence (Wirth et al. 1999). It is possible other less frequent deletion junctions are occurring at these other repeats. The repeats also lie in the adjacent NAIP gene, which had a total of 24 repeats with an exact 21 bp match. With NAIP known to be deleted in approximately 43% of type 1 SMA patients (Roy et al. 1995; Thompson et al. 1995; Burlet et al. 1996), it is possible a deletion junction exists spanning NAIP and SMN1 that is flanked by these repeats. Our attempts to find such a deletion junction were unsuccessful, though future experiments which expand the captured region to include NAIP would greatly increase the chances of detection. NAIP has previously been suggested as a possible modifier of SMA as it is more commonly deleted in SMA type 1 patients than in SMA type 2 and 3 patients (Roy et al. 1995; Velasco et al. 1996). However, this seems unlikely as NAIP deletions are still detected in some SMA type 2 and 3 patients and NAIP is still present in some SMA type 1 patients. Finally there are severe cases with mutations within SMN1 that do not remove NAIP (Roy et al. 1995; Thompson et al. 1995; Burlet et al. 1996; Burghes 1997). More likely, there is a large deletion which eliminates SMN1 and a portion of NAIP and gives rise to SMA type 1 individuals. Since SMA type 2 and 3 individuals have a higher SMN2 copy number it is likely that gene conversion of SMN1 to SMN2 and not gene deletion causes the disease (Burghes 1997). As a result, deletions of NAIP would be less common in SMA type 2 and 3 individuals and the implied correlation between NAIP deletions and SMA type 1 is not unexpected.
In this study, we sequenced the SMN2 genes of SMA patients in order to find modifiers of SMA. The sequencing data was analyzed for variants as well as SMN2 copy number which was then verified using ddPCR (Fig. 3). From this analysis, we found 3 variants, A-44G, A-549G, and C-1897T in intron 6 of SMN2, that are significantly associated with mild exception patients. All 3 are variants more prevalent in the SMN1 gene (Monani et al. 1999; Wu et al. 2017) but may be present in SMN2 due to gene conversion events (Campbell et al. 1997; DiDonato et al. 1997; Burghes 1997). The variants C-1897T and A-549G have not been previously implicated as SMA modifiers before. The third variant A-44G has previously been suspected of being a modifier as it has been shown to increase the amount of full-length SMN (Wu et al. 2017). Here we confirm that prediction, as A-44G was associated with 6 mild exception patients and never with concordant or severe exception patients, strongly suggesting the presence of this variant resulted in a milder than expected SMA phenotype. Five patients were identified as having 2 or more of these variants and there was some evidence of an additive effect. For example, patient MGH157 had 3 copies SMN2 and 1 copy of the modifying variant (C-1897T) and was a SMA type 3a individual. Patients MGH335, OSU01–001, and OSU01–002 also had 3 copies of SMN2 but in addition these individuals each had all 3 of the variants and displayed milder type 3b or type 4 SMA. This data indicates that patients with multiple variants may manifest a milder than anticipated phenotype. However not all discordant sibling pairs contained variants in SMN2. For example, OSU01–001 and OSU01–002 had identical SMN2 genotypes yet OSU01–001 had a SMA type 3b phenotype while OSU01–002 had SMA type 4. Thus, in this case it is likely that individual OSU01–002 has additional modifiers outside of the SMA region that could account for the milder phenotype.
Aside from variants in SMN2, genes outside of the SMA region as well as epigenetic changes have been implicated in modifying SMA. Epigenetic effects have been proposed to affect SMA phenotype, both within and outside of the SMN2 gene, though evidence for this is limited (Hauke et al. 2009; Zheleznyakova et al. 2013, 2015). One study found differences in CpG methylation at certain CpG dinucleotides, two of which were near an alternative transcription start site of SMN2 (Hauke et al. 2009). Although these 2 CpG dinucleotides were shown to modulate expression of a particular isoform of SMN2, this isoform makes up less than 5% of total SMN2 transcripts and hence does not result in a significant change in total SMN protein (Hauke et al. 2009). Outside of SMN2, methylation differences have been found between lymphoblast samples from SMA patients and healthy controls near the genes CHML and ARHGAP22 which are related to the activity of Rab and Rho GTPases (Zheleznyakova et al. 2013). However, the ability of these methylation changes to alter the SMA phenotype has yet to be demonstrated.
The PLS3 gene was determined to be a sex-specific protective modifier that is over-expressed in lymphoblasts from SMA females (Oprea et al. 2008). However, further studies of female discordant SMA siblings showed higher PLS3 expression in the severe sibling demonstrating that the effect is non-penetrant in certain patients (Bernal et al. 2011). To date it is unclear why elevated PLS3 expression is found in some female patients but not others. Additionally, male exception cases exist which PLS3 cannot explain (Burghes et al. 1994; Cobben et al. 1995; McAndrew et al. 1997; Cuscó et al. 2006). Furthermore, mice experiments remain controversial in that our group showed no improvement in SMA mice upon overexpression of PLS3 while the Wirth group show marginal improvement (Ackermann et al. 2013; McGovern et al. 2015). Studies by Oprea et al. maintain that the modification can only occur in SMA type 2 patients and not in those with SMA type 1 and consequently the severe phenotypes cannot be modified (Oprea et al. 2008). However, cases with discordant SMA type 1 and type 2 siblings have been reported (Pane et al. 2017). Mild SMA animals treated with both PLS3 and a suboptimal dose of corrective antisense oligonucleotide (ASO) have an improved phenotype (Strathmann et al. 2018), but it is not known if it is due to improved uptake of oligonucleotide or a direct effect of PLS3. In sum, the mechanism and degree to which PLS3 is a modifier of SMA remains uncertain.
In this study, we captured PLS3 and analyzed the gene for variants, separated by sex, that alter SMA phenotype. We tested all alleles that were present in the 268A15 clone that contains PLS3 and none of them had a statistically significant segregation with neither mild nor severe exception SMA patients. This includes the variant rs871773 which is known to increase expression of PLS3 in colon cancer (Szkandera et al. 2013). In males, rs871773 was found in 1 concordant and 1 mild exception patient, whereas in females it was found in 6 mild and 3 severe exception patients. In short, our data does not support the hypothesis that PLS3 modifies the SMA phenotype as no variant was found to statistically segregate with exception patients. Finally, SMA patients with a milder phenotype than expected for their copy number are not exclusively females as twenty-four of our 58 mild exception patients were males. Clearly, other modifiers besides PLS3 exist.
The NCALD gene is another proposed modifier of SMA. A family with 5 asymptomatic SMN1 deleted SMA patients were found to have a 17 bp deletion adjacent a possible super enhancer that lies 600 kb upstream of the gene (Riessland et al. 2017). However, this deletion alone is insufficient to explain the protective effect. Indeed, we have tested for this deletion in discordant sibling pairs and found it in both mild and severe exception patients as well as concordant patients (data not shown). Instead, as demonstrated by Riessland et al., the protective effect only occurs when a second CT insertion in intron1 of NCALD is also present. Analysis of this variant using the splicing prediction program Alamut indicated that the CT insertion creates a strong cryptic splice site. This could disrupt the expression of the gene and explain why the samples selected displayed low NCALD expression (Riessland et al. 2017). We examined this variant and found it present in the severe sibling of a discordant pair as well as in one sibling of a concordant pair (data not shown). Thus, the NCALD mutations on their own do not appear to be modifiers. The two mutations would have to occur together in the same individual however the mechanism by which these two changes together alter NCALD expression is unclear (Riessland et al. 2017). The combination of both alleles is a rare event and as such can only explain a small percent of exception cases.
The work we have presented here has several important conclusions. First, not all SMN2 genes are equivalent as evidence by the existence of variants that segregate with milder phenotypes. Second, there must be variants that modify SMA that exist outside of SMN2. The SMN2 variants we found to significantly associate with milder SMA individuals can only explain a small fraction (14 out of 58) of our mild exception patients. Amongst all our patient samples we had 11 cases of discordant siblings and all were confirmed to have identical genotypes in the SMA region. Hence the majority of SMA patients, including exception patients, have fully intact SMN2 genes. This strongly supports the notion that in the majority of discrepant cases genetic modifiers lie outside the SMN region. Third, no variant in PLS3 segregated with the mild exception patients, not even when accounting for sex. Fourth, the data generated by this adaptation of the MDiGS technique had enough statistical power to identify significant variant associations with milder phenotypes, even when the variant was present in only a limited number of individuals. This makes the MDiGS technique a viable option for determining modifiers using targeted sequencing. Finally, the patient samples and data described here are an ideal dataset for identifying modifying variants outside of the SMA region. These fully sequenced samples have a confirmed SMN2 copy number and well-characterized phenotypic information. Any candidate modifiers of SMA can be tested for their presence in our confirmed exception cases, or their absence in our confirmed concordant cases.
Supplementary Material
Acknowledgements
These studies were supported by a grant from the Muscular Dystrophy Association MDA352913. In addition, funding was provided from the National Institutes of Child Health and Development (NICHD) R01HD069045 Award to KJS for development of the Project Cure SMA Longitudinal Pediatric Data Repository. Corey Ruhno was supported by NICHD grant HD060586. We would also like to thank the Ohio Supercomputing Center. Anton J. Blatnik III assisted with sequencing of polymorphic markers. Finally, thank you to Dr. Jennifer Sinnott in The Ohio State University Department of Statistics for assisting us with the development of the models used in this paper.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- Ackermann B, Kröber S, Torres-Benito L, et al. (2013) Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 1–20. doi: 10.1093/hmg/dds540 [DOI] [PubMed] [Google Scholar]
- Alvarado DM, Yang P, Druley TE, et al. (2014) Multiplexed direct genomic selection (MDiGS): A pooled BAC capture approach for highly accurate CNV and SNP/INDEL detection. Nucleic Acids Res 42:1–10. doi: 10.1093/nar/gku218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anhuf D, Eggermann T, Rudnik-Schöneborn S, Zerres K (2003) Determination of SMN1 and SMN2 copy number using TaqManTM technology. Hum Mutat 22:74–78. doi: 10.1002/humu.10221 [DOI] [PubMed] [Google Scholar]
- Arkblad EL, Darin N, Berg K, et al. (2006) Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord 16:830–838. doi: 10.1016/j.nmd.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Arnold WD, Burghes AHM (2013) Spinal muscular atrophy: Development and implementation of potential treatments. Ann Neurol 74:348–62. doi: 10.1002/ana.23995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WD, Kassar D, Kissel JT (2015) Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 51:157–167. doi: 10.1002/mus.24497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal S, Alías L, Barceló MJ, et al. (2010) The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet 47:640–2. doi: 10.1136/jmg.2010.079004 [DOI] [PubMed] [Google Scholar]
- Bernal S, Also-Rallo E, Martínez-Hernández R, et al. (2011) Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord 21:413–9. doi: 10.1016/j.nmd.2011.03.009 [DOI] [PubMed] [Google Scholar]
- Burghes AHM (1997) When is a deletion not a deletion? When it is converted. Am J Hum Genet 61:9–15. doi: 10.1086/513913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AHM, Beattie CE (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10:597–609. doi: 10.1038/nrn2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AHM, Ingraham SE, Kóte-Jarai Z, et al. (1994) Linkage mapping of the spinal muscular atrophy gene. Hum Genet 93:305–12 [DOI] [PubMed] [Google Scholar]
- Burghes AHM, McGovern VL (2017) Genetics of Spinal Muscular Atrophy. Mol Cell Ther Mot Neuron Dis 121–139. doi: 10.1016/B978-0-12-802257-3.00006-7 [DOI] [Google Scholar]
- Burlet P, Bürglen L, Clermont O, et al. (1996) Large scale deletions of the 5q13 region are specific to Werdnig-Hoffmann disease. J Med Genet 33:281–283. doi: 10.1136/jmg.33.4.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett BG, Muñoz E, Tandon A, et al. (2009) Regulation of SMN protein stability. Mol Cell Biol 29:1107–15. doi: 10.1128/MCB.01262-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calucho M, Bernal S, Alías L, et al. (2018) Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord 28:208–215. doi: 10.1016/j.nmd.2018.01.003 [DOI] [PubMed] [Google Scholar]
- Campbell L, Potter A, Ignatius J, et al. (1997) Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am J Hum Genet 61:40–50. doi: S0002–9297(07)64275-X [pii]\r 10.1086/513886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten JD, DiDonato CJ, Ingraham SE, et al. (1994) A YAC contig of the region containing the spinal muscular atrophy gene (SMA): Identification of an unstable region. Genomics 24:351–356. doi: 10.1006/geno.1994.1626 [DOI] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN. Nat Genet 30:377–384. doi: 10.1038/ng854 [DOI] [PubMed] [Google Scholar]
- Ceballos FC, Hazelhurst S, Ramsay M (2018) Assessing runs of Homozygosity: A comparison of SNP Array and whole genome sequence low coverage data. BMC Genomics 19:1–12. doi: 10.1186/s12864-018-4489-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Baird SD, Mahadevan M, et al. (1998) Sequence of a 131-kb region of 5q13.1 containing the spinal muscular atrophy candidate genes SMN and NAIP. Genomics 48:121–127. doi: 10.1006/geno.1997.5141 [DOI] [PubMed] [Google Scholar]
- Cobben JM, Van Der Steege G, Grootscholten P, et al. (1995) Deletions of the Survival Motor Neuron Gene in Unaffected Siblings of Patients with Spinal Muscular Atrophy. Am J Hum Genet 57:805–808 [PMC free article] [PubMed] [Google Scholar]
- Coovert DD, Le TT, McAndrew PE, et al. (1997) The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6:1205–1214. doi: 10.1093/hmg/6.8.1205 [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3:97–110. doi: 10.1006/nbdi.1996.0010 [DOI] [PubMed] [Google Scholar]
- Cuscó I, Barceló MJ, Rojas-García R, et al. (2006) SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol 253:21–5. doi: 10.1007/s00415-005-0912-y [DOI] [PubMed] [Google Scholar]
- DePristo M a, Banks E, Poplin R, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–8. doi: 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato CJ (1995) THE SPINAL MUSCULAR ATROPHY GENE: “ISOLATION AND CHARACTERIZATION OF THE GENETIC AND PHYSICAL REGION SURROUNDING THE GENE LOCUS AND IDENTIFICATION OF CANDIDATE cDNAs.” The Ohio State University [Google Scholar]
- DiDonato CJ, Ingraham SE, Mendell JR, et al. (1997) Deletion and conversion in spinal muscular atrophy patients: is there a relationship to severity? Ann Neurol 41:230–237. doi: 10.1002/ana.410410214 [DOI] [PubMed] [Google Scholar]
- DiDonato CJ, Morgan K, Carpten JD, et al. (1994) Association between Ag1-CA alleles and severity of autosomal recessive proximal spinal muscular atrophy. Am J Hum Genet 55:1218–1229 [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, et al. (2012) STAR : ultrafast universal RNA-seq aligner 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeldt J, Nilsson D, Andersson-Assarsson JC, Lindstrand A (2018) AMYCNE: Confident copy number assessment using whole genome sequencing data. PLoS One 13:1–14. doi: 10.1371/journal.pone.0189710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkötter M, Schwarzer V, Wirth R, et al. (2002) Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 70:358–68. doi: 10.1086/338627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, et al. (2017) Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 377:1723–1732. doi: 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- Gidaro T, Servais L (2018) Nusinersen treatment of spinal muscular atrophy: current knowledge and existing gaps. Dev Med Child Neurol doi: 10.1111/dmcn.14027 [DOI] [PubMed] [Google Scholar]
- Hahnen E, Forkert R, Marke C, et al. (1995) Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet 4:1927–1933. doi: 10.1093/hmg/4.10.1927 [DOI] [PubMed] [Google Scholar]
- Hao LT, Burghes AHM, Beattie CE (2011) Generation and Characterization of a genetic zebrafish model of SMA carrying the human SMN2 gene. Mol Neurodegener 6:1–9. doi: 10.1186/1750-1326-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauke J, Riessland M, Lunke S, et al. (2009) Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet 18:304–317. doi: 10.1093/hmg/ddn357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejowska M, Borkowska J, Zimowski J, et al. (2008) Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet 16:930–4. doi: 10.1038/ejhg.2008.41 [DOI] [PubMed] [Google Scholar]
- Jedrzejowska M, Milewski M, Zimowski J, et al. (2009) Phenotype modifiers of spinal muscular atrophy: the number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease. Acta Biochim Pol 56:103–108. doi: 20091732 [pii] [PubMed] [Google Scholar]
- Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34:460–3. doi: 10.1038/ng1207 [DOI] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach MER, et al. (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet 14:845–57. doi: 10.1093/hmg/ddi078 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–65 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, et al. (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16:265–9. doi: 10.1038/ng0797-265 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 96:6307–11. doi: 10.1073/pnas.96.11.6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, et al. (1998) SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 19:63–66. doi: 10.1038/ng0598-63 [DOI] [PubMed] [Google Scholar]
- Mailman MD, Heinz JW, Papp AC, et al. (2002) Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 4:20–6. doi: 10.1097/00125817-200201000-00004 [DOI] [PubMed] [Google Scholar]
- McAndrew PE, Parsons DW, Simard LR, et al. (1997) Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 60:1411–22. doi: 10.1086/515465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern VL, Massoni-Laporte A, Wang X, et al. (2015) Plastin 3 expression does not modify spinal muscular atrophy severity in the Δ7 SMA mouse. PLoS One 10:1–19. doi: 10.1371/journal.pone.0132364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki J, Lefebvre S, Burglen L, et al. (1994) De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science (80-) 264:1474–1477. doi: 10.1126/science.7910982 [DOI] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, et al. (2017) Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377:1713–1722. doi: 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- Mercuri E, Darras BT, Chiriboga CA, et al. (2018) Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 378:625–635. doi: 10.1056/NEJMoa1710504 [DOI] [PubMed] [Google Scholar]
- Miller RG, Moore DH, Dronsky V, et al. (2001) A placebo-controlled trial of gabapentin in spinal muscular atrophy. J Neurol Sci 191:127–131. doi: 10.1016/S0022-510X(01)00632-3 [DOI] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, et al. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8:1177–1183. doi: 10.1093/hmg/8.7.1177 [DOI] [PubMed] [Google Scholar]
- Munsat TL (1991) International SMA Collaboration. Neuromuscul Disord 1:81. doi: 10.1016/0960-8966(91)90052-T [DOI] [PubMed] [Google Scholar]
- Oprea GE, Kröber S, McWhorter ML, et al. (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320:524–7. doi: 10.1126/science.1155085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, Mammoser AG, Wu C, et al. (2001) A bacterial artificial chromosome library for sequencing the complete human genome. Genome Res 11:483–496. doi: 10.1101/gr.169601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, Woon PY, Zhao B, et al. (1998) An improved approach for construction of bacterial artificial chromosome libraries. Genomics 52:1–8. doi: 10.1006/geno.1998.5423 [DOI] [PubMed] [Google Scholar]
- Pane M, Lapenta L, Abiusi E, et al. (2017) Longitudinal assessments in discordant twins with SMA. Neuromuscul Disord 27:890–893. doi: 10.1016/j.nmd.2017.06.559 [DOI] [PubMed] [Google Scholar]
- Pearn JH (1973) The Gene Frequency of Acute Werdnig-Hoffmann Disease (SMA Type 1). A Total Population Survey in North-East England. J Med Genet 10:260–265. doi: 10.1136/jmg.10.3.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearn JH (1978) Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 15:409–413. doi: 10.1136/jmg.15.6.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW (2007) Spinal Muscular Atrophy Diagnostics. J Child Neurol 22:952–956. doi: 10.1177/0883073807305668 [DOI] [PubMed] [Google Scholar]
- Prior TW, Krainer AR, Hua Y, et al. (2009) A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet 85:408–13. doi: 10.1016/j.ajhg.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ (2004) Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet 130 A:307–310. doi: 10.1002/ajmg.a.30251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyatt RE, Prior TW (2006) A feasibility study for the newborn screening of spinal muscular atrophy. Genet Med 8:428–437. doi: 10.1097/01.gim.0000227970.60450.b2 [DOI] [PubMed] [Google Scholar]
- R Core Team (2013) R: A Language and Environment for Statistical Computing [Google Scholar]
- Riessland M, Kaczmarek A, Schneider S, et al. (2017) Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am J Hum Genet 100:297–315. doi: 10.1016/j.ajhg.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DF, Chavez J, Court SDM (1970) The genetic component in child mortality. Arch Dis Child 45:33–38. doi: 10.1136/adc.45.239.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N, Mahadevan MS, McLean M, et al. (1995) The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 80:167–178. doi: 10.1016/0092-8674(95)90461-1 [DOI] [PubMed] [Google Scholar]
- Rüdiger NS, Gregersen N, Kielland-brandt MC (1995) One short well conserved region of Alu-sequences is involved in human gene rearrangements and has homology with prokaryotic chi. Nucleic Acids Res 23:256–260. doi: 10.1093/nar/23.2.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathmann EA, Peters M, Hosseinibarkooie S, et al. (2018) Evaluation of potential effects of Plastin 3 overexpression and low-dose SMN-antisense oligonucleotides on putative biomarkers in spinal muscular atrophy mice 4:1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman EA, Nagan N, Zhu H, et al. (2012) Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of 472400 specimens. Eur J Hum Genet 20:27–32. doi: 10.1038/ejhg.2011.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkandera J, Winder T, Stotz M, et al. (2013) A common gene variant in PLS3 predicts colon cancer recurrence in women. Tumor Biol 34:2183–2188. doi: 10.1007/s13277-013-0754-7 [DOI] [PubMed] [Google Scholar]
- Thompson TG, Didonato CJ, Simard LR, et al. (1995) A novel cDNA detects homozygous microdeletions in greater than 50% of type I spinal muscular atrophy patients. Nat Genet 9:56–62. doi: 10.1038/ng0195-56 [DOI] [PubMed] [Google Scholar]
- Velasco E, Valero C, Valero A, et al. (1996) Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies ofcBCD541 and SMA phenotype. Hum Mol Genet 5:257–263. doi: 10.1093/hmg/5.2.257 [DOI] [PubMed] [Google Scholar]
- Vezain M, Saugier-Veber P, Goina E, et al. (2010) A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat 31:1110–1125. doi: 10.1002/humu.21173 [DOI] [PubMed] [Google Scholar]
- Wang CC, Chang JG, Chen YL, et al. (2010) Multi-exon genotyping of SMN gene in spinal muscular atrophy by universal fluorescent PCR and capillary electrophoresis. Electrophoresis 31:2396–2404. doi: 10.1002/elps.201000124 [DOI] [PubMed] [Google Scholar]
- Weber JL, Polymeropoulos MH, May PE, et al. (1991) Mapping of human chromosome 5 microsatellite DNA polymorphisms. Genomics 11:695–700. doi: 10.1016/0888-7543(91)90077-R [DOI] [PubMed] [Google Scholar]
- Wirth B, El-Agwany A, Baasner A, et al. (1995) Mapping of the spinal muscular atrophy (SMA) gene to a 750-kb interval flanked by two new microsatellites. Eur J Hum Genet 3:56–60. doi: 10.1159/000472274 [DOI] [PubMed] [Google Scholar]
- Wirth B, Herz M, Wetter A, et al. (1999) Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 64:1340–1356. doi: S0002–9297(07)62279–4 [pii]\n 10.1086/302369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, Schmidt T, Hahnen E, et al. (1997) De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 61:1102–11. doi: 10.1086/301608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wang S-H, Sun J, et al. (2017) A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet 26:2768–2780. doi: 10.1093/hmg/ddx166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schoneborn S (1995) Natural history in proximal spinal muscular atrophy: clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 52: [DOI] [PubMed] [Google Scholar]
- Zheleznyakova GY, Nilsson EK, Kiselev AV., et al. (2015) Methylation levels of SLC23A2 and NCOR2 genes correlate with spinal muscular atrophy severity. PLoS One 10:1–14. doi: 10.1371/journal.pone.0121964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheleznyakova GY, Voisin S, Kiselev AV, et al. (2013) Genome-wide analysis shows association of epigenetic changes in regulators of Rab and Rho GTPases with spinal muscular atrophy severity. Eur J Hum Genet 21:988–93. doi: 10.1038/ejhg.2012.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.