

Abstract
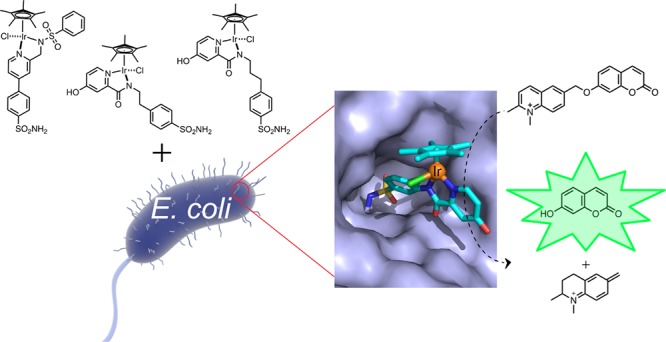
Artificial metalloenzymes combine a synthetic metallocofactor with a protein scaffold and can catalyze abiotic reactions in vivo. Herein, we report on our efforts to valorize human carbonic anhydrase II as a scaffold for whole-cell transfer hydrogenation. Two platforms were tested: periplasmic compartmentalization and surface display in Escherichia coli. A chemical optimization of an IrCp* cofactor was performed. This led to 90 turnovers in the cell, affording a 69-fold increase in periplasmic product formation over the previously reported, sulfonamide-bearing IrCp* cofactor. These findings highlight the versatility of carbonic anhydrase as a promising scaffold for whole-cell catalysis with artificial metalloenzymes.
Keywords: artificial metalloenzymes, biocatalysis, organometallic chemistry, protein engineering, synthetic biology
Artificial metalloenzymes (ArMs) result from anchoring a synthetic metallocofactor within a protein scaffold, thereby combining attractive features of enzymatic and transition metal catalysis.1 Enzymes are known for high reaction rates, turnover numbers, and selectivity,2,3 whereas synthetic transition metal catalysts offer a broad range of reactivities, some of which have no equivalent in nature.4 Merging both features promises new catalytic, diagnostic, and therapeutic approaches and opportunities for biotechnology, synthetic biology, and medicine.1,5A variety of ArM-catalyzed transformations have been reported, mostly relying on purified protein samples.1,5−9 With the long-term goal of complementing natural enzymes with ArMs in a cellular environment, we have selected E. coli’s periplasm10,11 and its outer-membrane12,13 to compartmentalize these hybrid catalysts to minimize inhibition of the cofactor by glutathione.7 In this context, two β-barrel host proteins have been reported: nitrobindin12 and streptavidin.10,11,13,14 Both compartmentalization strategies offer advantages and limitations. On the one hand, a higher amount of host protein can be accumulated in the periplasm. On the other hand, the access of the cofactor and its substrate to the periplasm is hampered by the outer-membrane’s permeability.15 Herein, we report on the following: (i) the use of wild-type human carbonic anhydrase II (CAII)16−19 for the assembly of an ArM in a cellular environment, (ii) the chemical optimization of the sulfonamide-bearing IrCp* cofactor for transfer hydrogenation, and (iii) comparison of the artificial transfer hydrogenase (THase) performance for both compartmentalization strategies (i.e., CAIIperiplasm and CAIIsurface display).
Mammalian cell-surface variants of CA are well-established markers for various tumor types including the following: gliomas/ependymomas, mesotheliomas, papillary/follicular carcinomas and carcinomas of the bladder, uterine cervix, kidneys, lungs, neck, breast, brain etc.20−23 Several sulfonamide-based anticancer drugs24−26 are used to inhibit CA by binding to the zinc in the active site, thus halting the carbon dioxide to bicarbonate interconversion. With medical applications in mind,27 we hypothesized it may be possible to rely on wild-type CA to target cells overexpressing this protein to accumulate a sulfonamide-bearing metal cofactor, which displays a bioorthogonal reactivity toward a caged drug. This strategy would allow this prodrug to be site-specifically uncaged in the immediate proximity of the cells overexpressing CA. In the past, genetic optimization strategies have been amply applied to improve the catalytic performance of ArMs.1 For medical applications, however, one is limited to the protein that is overexpressed on the target cells, thus restricting the optimization of the ArM to chemical strategies (i.e., variation of the spacer–cofactor moiety). The performance of the ArM was evaluated in the presence of wild-type CAII both in E. coli’s periplasm and displayed on its outer membrane (CAIIperiplasm and CAIIsurface display, respectively).
For the periplasmic compartmentalization, CAII was N-terminally fused to the signal peptide of the outer membrane protein A (OmpA) to ensure its secretion to the periplasm of E. coli (Figure 1a).11,28 For the surface display, CAII was anchored in the outer membrane by fusing it to a truncated E. coli lipoprotein Lpp (residues 1–9), followed by the first five β-sheets of OmpA (residues 46–159, Figure 1b).13,29 To probe the localization and functionality of CAII, the fluorescent probe 4 consisting of a carboxyfluorescein linked to an arylsulfonamide (Figure 1c) was synthesized (see Supporting Information). Probe 4 and a primary anti-CAII antibody in combination with a fluorescent secondary antibody were used to stain E. coli cells containing (i) an empty vector (negative control 1) or plasmids targeting CAII to (ii) the cytoplasm (CAIIcytoplasm, negative control 2), (iii) the periplasm (CAIIperiplasm), or (iv) the cell surface (CAIIsurface display). The samples were analyzed by fluorescence microscopy and flow cytometry (Figure 2). Cells containing the empty vector or CAIIcytoplasm could not be stained with probe 4 or with the anti-CAII antibody (Figure 2). In contrast, cells containing CAIIperiplasm or CAIIsurface display were stained with probe 4 (Figure 2a, c). The flow cytometry analysis (Figure 2c, d) corroborated the observations of the fluorescence microscopy. Nearly the same fluorescence increase was observed for both the compartmentalization in the periplasm and on the cell surface when using probe 4. Nonetheless, conclusions about the amount of bound-fluorescent probe 4 are difficult to draw since the probe 4 may behave different in the periplasm and on the surface (Figure 2c). Staining with probe 4 confirms that induction of CAIIperiplasm and CAIIsurface display lead to the expression of functional CAII. The anti-CAII antibody is not able to enter the periplasm of E. coli cells13 and, thus, only labels cells expressing CAIIsurface display (Figure 2b, d). The immunostaining, validated with the probe-labeling experiments, suggests that CAII is correctly localized and functional. Furthermore, the labeling with probe 4 suggests that neutral and hydrophobic probes and metallocofactors around 700 Da can passively diffuse into the periplasm. For streptavidin-based ArMs, we had speculated that biotin present on the cofactor may favor the accumulation of biotinylated cofactors in the E. coli periplasm11 by hijacking an active, energy-dependent, and carrier-mediated biotin uptake.30−32
Figure 1.

In cellulo assembly of artificial transfer hydrogenases (THases). (a) Human carbonic anhydrase II (CAII) is secreted to the periplasm by fusion to an N-terminal signal peptide from the outer membrane protein A (OmpA). (b) CAII is displayed on the cell surface of E. coli by fusion to a truncated lipoprotein and the outer membrane protein A (Lpp-OmpA). Sulfonamide-bearing IrCp* piano-stool complexes were evaluated for reducing a self-immolating substrate, releasing the fluorescent umbelliferone 2 (a, b). (c) The localization of CAII in the periplasm and on the outer-membrane was confirmed by fluorescence, relying on the sulfonamide-bearing carboxyfluorescein probe 4.
Figure 2.
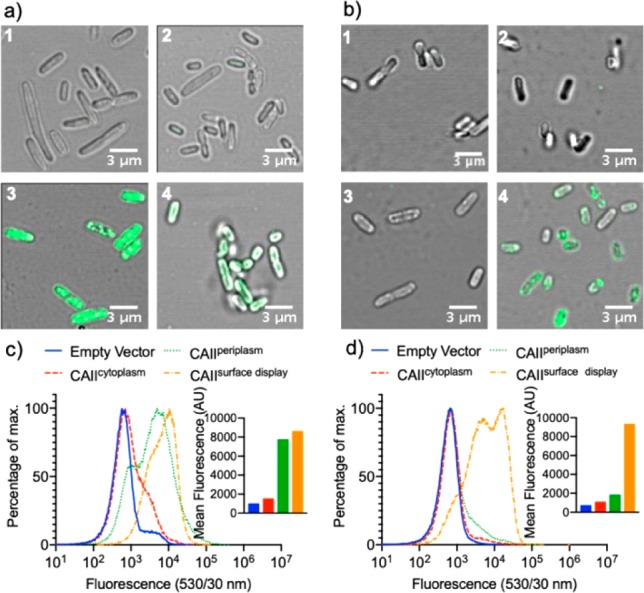
Compartmentalization and expression of CAII in the cytoplasm, in the periplasm, and on the surface of E. coli. Fluorescence microscopy of E. coli cells: (a) stained with the fluorescent probe 4 and (b) immunostained with an anti-CAII antibody. For (a) and (b), E. coli cells containing an empty vector (1), CAIIcytoplasm (2), CAperiplasm (3), and CAIIsurface display (4) were stained. Flow cytometry analysis of (c) cells stained with a fluorescent probe 4 and (d) immuno-stained cells with an anti-CAII antibody. The inset displays the arithmetic mean of 200 000 cells for the different compartmentalization systems.
After establishing the compartmentalization of CAII in the periplasm and on the surface of E. coli cells, we tested the use of these systems for the assembly of THases. The activity of these two systems was evaluated by using a self-immolative fluorogenic substrate, which facilitates high-throughput screening. Upon N=C reduction of the quinolinium substrate 1, the fluorescent umbelliferone 2 and a iminoquinonemethide 3 (Figure 1) are produced.10 The highly electrophilic intermediate 3 spontaneously reacts with nucleophiles present in solution (either water or nucleophilic amino acids).33 To determine the catalytic activity of these systems, we adapted the assay conditions developed for a streptavidin-based THase.10 In short, CAII was expressed at 30 °C for 4 h, cells of an OD600 = 3 were harvested, and the pellet was washed with 0.8 mM [Cu(gly)2] and resuspended in the cofactor buffer (2 μM cofactor, 100 mM MOPS, pH 7, 154 mM NaCl). After incubation with the cofactor 5–10 for 1 h, the cells were pelleted and washed twice to remove unbound cofactor. The cell pellet was resuspended in the reaction buffer (1 M formate, 500 μM substrate 1, 0.4 M MOPS, pH 7) and was incubated for 16 h at 30 °C shaking at 280 rpm (see Supporting Information). The uncaging of umbelliferone 2, resulting from the reduction of quinolinium 1, was monitored by fluorescence (323 nm excitation, 451 nm emission). To exclude the possibility that natural E. coli enzymes uncage substrate 1, E. coli cells were treated as described above, except that the cofactors were omitted. Without cofactor incubation, product 2 formation was not observed, independent of the expression system (i.e., CAIIcytoplasm, CAIIperiplasm, CAIIsurface display, Figure 3b). Next, we tested the previously reported piano-stool cofactor 5.17,18 Using either the periplasm- or the surface-displayed THase, conversions, albeit limited, were 5- and 9-fold higher than the conversion observed for cofactor 5 unspecifically bound to cells expressing CAIIcytoplasm (Figure 3b). Next, we chemically optimized the THase activity by varying the first coordination sphere around the IrCp* moiety. Building on the picolinamide-based ligands reported by Do,34−36 Himeda,37 and Duhme-Klair,38 we anchored an arylsulfonamide moiety via a spacer on 2-picolinamide (Figure 3a; see Supporting Methods in the Supporting Information for the cofactor synthesis).
Figure 3.

Screening cofactors 5–10 for THase whole-cell activity. (a) Structure of the piano-stool complexes 5–10 bearing a sulfonamide anchor. (b) Cellular uncaging of fluorogenic substrate 1 by cofactors 5–10·CAII. (c) Ir-amount, Ir-uptake, and corresponding turnover number (TON) for cofactor 7 determined by ICP-MS. The background was subtracted due to varying degrees of unspecific activity; see Figure S5. Data displayed are the means ± standard deviation of experiments performed in triplicate; the individual data and additional controls are depicted in Figure S5.
Gratifyingly, the THase activity for the optimized cofactors 6–10 were markedly superior to those observed with cofactor 5 either in vitro (Figure S3) or for whole-cell catalysis (Figure 3b, Table 1). The background for the negative controls, cells containing CAIIcytoplasm or an empty vector, remains low (Figure 3b and Figure S5a). These results suggest that the introduced 4-hydroxypicolinamide moiety has a beneficial effect on THase activity. Removal of the electron-donating hydroxy-group (compare cofactors 7 and 8) had a deleterious effect on the THase activity for whole-cell catalysis: cofactor 7 is nearly 3-fold more active than cofactor 8 for CAIIsurface display.
Table 1. Summary of the Dissociation Constant Kd for Cofactors 5–10 and Corresponding Whole-Cell Umbelliferone 2 Formation.
| Cofactor | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|
| Kd (nM) | 15 ± 2 | >1000 | 35 ± 11 | 49 ± 8 | 149 ± 30 | 20 ± 5 |
| Umbelliferone 2 formationa by CAIIperiplasm | 100 ± 33 | 1255 ± 60 | 6863 ± 179 | 4454 ± 662 | 4341 ± 39 | 2072 ± 70 |
| Umbelliferone 2 formationa by CAIIsurface display | 168 ± 33 | 844 ± 103 | 5723 ± 64 | 2018 ± 192 | 2659 ± 287 | 1716 ± 120 |
For comparison, the product formation monitored by fluorescence was normalized and cofactor 5·CAIIperiplasm was set to 100. Data displayed are the means ± standard deviation of experiments performed in triplicate.
Next, we investigated the effect of the spacer on the THase activity and the corresponding dissociation constant Kd with purified CAII (Figure 3a and Table 1). For cofactors consisting of the same first coordination sphere (i.e., 4-hydroxy-2-picolinamide) and arylsulfonamide anchor, the nature and the length of the spacer has a pronounced effect on the dissociation constant. Cofactor 6, bearing the shortest spacer, displayed the highest dissociation constant (Kd > 1000 nM), suggesting it probably dissociates during the washing step. Accordingly, the whole-cell activity is very low. Whereas the Kd for the cofactors bearing two (i.e., cofactors 7 and 8) and three atom spacers (i.e., cofactors 9 and 10) are roughly comparable, thus ensuring quantitative binding to CAII under catalytic conditions, the THase activity significantly differs, emphasizing the critical influence of the second coordination sphere on catalytic performance (Figure 3b and Table 1).
To highlight the subtle differences in the second coordination sphere resulting from the different spacers, we determined the cofactor’s localization within CAII by X-ray crystallography. High-resolution X-ray data for cofactor 7–10·CAII were obtained (Table S6 and Figure 4). Residual electron density in the Fo – Fc and the anomalous difference density map were observed, highlighting the presence of cofactors 7–10 in the funnel-shaped vestibule of CAII. Modeling of cofactor 7–10 into the electron density projected the iridium in the position of the anomalous density peak (Figure S7). As observed for cofactor 5,17 the Ir-atom is not fully occupied. It was modeled in the (S)-configuration with an occupancy of 60% for cofactor 7, 9, and 10 and 90% for cofactor 8 (Table S6).
Figure 4.

Structural characterization of IrCp* piano-stool cofactors bound to CAII. (a) Overlay of the Ir-position from cofactor 5 (gray, PDB: 3ZP9), cofactor 7 (cyan, PDB: 6QFU), cofactor 8 (lilac, PDB: 6QFV), cofactor 9 (dark green, PDB: 6QFW) and cofactor 10 (pink, PDB: 6QFX bound to wild-type CAII). The Ir-atoms are depicted as spheres in the color of the corresponding protein backbone and the ligand surrounding the metal was omitted for clarity. (b) Cofactor 7, (c) cofactor 8, (d) cofactor 9, (e) cofactor 10, bound to wild-type CAII. The protein is depicted as a transparent surface and an orange cartoon representation. Amino acids in the proximity of the cofactor are highlighted as sticks and labeled. The cofactors are represented as sticks, and the Ir and Zn ions, as orange and light-yellow spheres, respectively, and surrounded by the corresponding anomalous electron density (red mesh at 5 σ). The distance from the amide oxygen of the cofactors to the N of Q92 is highlighted in yellow dashes and labeled; chloride, green; nitrogen, blue; oxygen, red; sulfur, yellow.
Comparing the localization of the different cofactors reveals that the Ir-atoms of cofactor 7 and 8 are located deepest inside the hydrophobic funnel of CAII (Figure 4a). The firm positioning of cofactor 7 and 8 within CAII is secured by a hydrogen bond between the oxygen of the cofactors’ amide group and the nitrogen of the side chain of glutamine 92 (Q92, O···N distance 2.8 and 2.9 Å for cofactor 7 and 8, respectively). For cofactors 9 and 10, the O···N distance is 4.3 and 4.5 Å, respectively. The localization of cofactor 10 is most diffuse, as reflected by the smear of the anomalous electron density (Figure 4e).
To determine the uptake of cofactor 7, the iridium concentration for the empty vector, CAIIcytoplasm, CAIIperiplasm, and CAIIsurface display was determined by ICP-MS (Figure 3c and Figure S5b). These data allow determination of the turnover number (TON) per iridium for the whole-cell THase. The ICP-MS sample preparation was identical to that implemented for whole-cell catalysis (see Supporting Information). The following results were obtained: cofactor 7·CAIIperiplasm 348 ± 43 nM Ir thus amounting to 93 ± 11 turnovers (TON); cofactor 7·CAIIsurface display 315 ± 8 nM Ir thus corresponding to 85 ± 3 TON (Figure 3c). These data suggest that similar amounts of THases are compartmentalized on the cell surface and in the periplasm as observed in the flow cytometry analysis with probe 4 (Figure 2c). Although periplasmic-expression levels typically exceed surface-displayed expression levels,39 the Ir concentration and corresponding TONs suggest that the outer membrane limits the access of the cofactor and the substrate to the compartmentalized CAII. Accordingly, the surface-display strategy seems most suitable to develop ArMs for synthetic biology and therapeutic purposes.
With the long-term goal of exploiting overexpressed CA on cancer cells to concentrate organometallic cofactors and to uncage drugs, this study has revealed the following features: (i) picoline-amide containing cofactors lead to significantly higher whole-cell THase activity compared to the previously reported picoline-sulfonamide containing cofactor 5;17 (ii) chemical optimization is a powerful means to improve catalytic performance of a THase using wild-type CAII; (iii) CAII is a promising monomeric globular scaffold for in vivo catalysis, leading to ≥90 TONs; (iv) CAIIperiplasm and CAIIsurface display are excellent platforms for ArM-based whole-cell catalysis. These results confirm that CA is a propitious scaffold for the accumulation of ArMs on the surface of tumor cells.40 Tumor-localized ArMs may be valorized for the development of innovative cancer therapies, including targeted-drug delivery. Current efforts are aimed at adapting this protocol to cancer cell models overexpressing CA on their surface and using NADH as a hydrogen source instead of formate.41
Acknowledgments
T.R.W. thanks the Swiss National Science Foundation (Grant SNF, 200020_182046/1), the NCCR “Molecular Systems Engineering”, and the ERC (the DrEAM) for generous support of this work. J.G.R. thanks the EMBO for a long-term fellowship (ALTF 194-2017). Dr. Roman Jakob is thanked for helpful discussions about X-ray crystallography and structure solution, Dr. Ioana Craciun for help with the microscope, and Dr. Rosario Vanella for the help with the flow cytometer.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b01006.
Kd-measurements, supporting activity data, structural analysis, cofactor characterization and material and methods (PDF)
Author Contributions
† J.G.R. and Y.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Schwizer F.; Okamoto Y.; Heinisch T.; Gu Y.; Pellizzoni M. M.; Lebrun V.; Reuter R.; Kohler V.; Lewis J. C.; Ward T. R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Huisman G. W.; Kazlauskas R. J.; Lutz S.; Moore J. C.; Robins K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T. The fourth wave of biocatalysis is approaching. Philos. Trans. R. Soc., A 2018, 376, 1–7. 10.1098/rsta.2017.0063. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis, 1st ed.; University Science Books: Sausalito, CA, United States, 2010. [Google Scholar]
- Rebelein J. G.; Ward T. R. In vivo catalyzed new-to-nature reactions. Curr. Opin. Biotechnol. 2018, 53, 106–114. 10.1016/j.copbio.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Liang A. D.; Serrano-Plana J.; Peterson R. L.; Ward T. R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res. 2019, 52, 585–595. 10.1021/acs.accounts.8b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markel U.; Sauer D. F.; Schiffels J.; Okuda J.; Schwaneberg U. Towards the Evolution of Artificial Metalloenzymes-A Protein Engineer’s Perspective. Angew. Chem., Int. Ed. 2019, 58, 4454–4464. 10.1002/anie.201811042. [DOI] [PubMed] [Google Scholar]
- Jeschek M.; Panke S.; Ward T. R. Artificial Metalloenzymes on the Verge of New-to-Nature Metabolism. Trends Biotechnol. 2018, 36, 60–72. 10.1016/j.tibtech.2017.10.003. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Ward T. R. Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew. Chem., Int. Ed. 2016, 55, 7344–7357. 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Rebelein J. G.; Mallin H.; Trindler C.; Pellizzoni M. M.; Ward T. R. Genetic Engineering of an Artificial Metalloenzyme for Transfer Hydrogenation of a Self-Immolative Substrate in Escherichia coli’s Periplasm. J. Am. Chem. Soc. 2018, 140, 13171–13175. 10.1021/jacs.8b07189. [DOI] [PubMed] [Google Scholar]
- Jeschek M.; Reuter R.; Heinisch T.; Trindler C.; Klehr J.; Panke S.; Ward T. R. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537, 661–665. 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- Grimm A. R.; Sauer D. F.; Polen T.; Zhu L. L.; Hayashi T.; Okuda J.; Schwaneberg U. A Whole Cell E. coli Display Platform for Artificial Metalloenzymes: Poly(phenylacetylene) Production with a Rhodium-Nitrobindin Metalloprotein. ACS Catal. 2018, 8, 2611–2614. 10.1021/acscatal.7b04369. [DOI] [Google Scholar]
- Heinisch T.; Schwizer F.; Garabedian B.; Csibra E.; Jeschek M.; Vallapurackal J.; Pinheiro V. B.; Marliere P.; Panke S.; Ward T. R. E. coli surface display of streptavidin for directed evolution of an allylic deallylase. Chem. Sci. 2018, 9, 5383–5388. 10.1039/C8SC00484F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y.; Kojima R.; Schwizer F.; Bartolami E.; Heinisch T.; Matile S.; Fussenegger M.; Ward T. R. A cell-penetrating artificial metalloenzyme regulates a gene switch in a designer mammalian cell. Nat. Commun. 2018, 9, 1943. 10.1038/s41467-018-04440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; Horanyi P. S. How hydrophobic molecules traverse the outer membranes of gram-negative bacteria. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10929–10930. 10.1073/pnas.1106927108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnard F. W.; Heinisch T.; Nogueira E. S.; Schirmer T.; Ward T. R. Human carbonic anhydrase II as a host for piano-stool complexes bearing a sulfonamide anchor. Chem. Commun. 2011, 47, 8238–8240. 10.1039/c1cc10345h. [DOI] [PubMed] [Google Scholar]
- Monnard F. W.; Nogueira E. S.; Heinisch T.; Schirmer T.; Ward T. R. Human carbonic anhydrase II as host protein for the creation of artificial metalloenzymes: the asymmetric transfer hydrogenation of imines. Chem. Sci. 2013, 4, 3269–3274. 10.1039/c3sc51065d. [DOI] [Google Scholar]
- Heinisch T.; Pellizzoni M.; Durrenberger M.; Tinberg C. E.; Kohler V.; Klehr J.; Haussinger D.; Baker D.; Ward T. R. Improving the Catalytic Performance of an Artificial Metalloenzyme by Computational Design. J. Am. Chem. Soc. 2015, 137, 10414–10419. 10.1021/jacs.5b06622. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Kajetanowicz A.; Ward T. R. Carbonic anhydrase II as host protein for the creation of a biocompatible artificial metathesase. Org. Biomol. Chem. 2015, 13, 5652–5655. 10.1039/C5OB00428D. [DOI] [PubMed] [Google Scholar]
- Potter C. P.; Harris A. L. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br. J. Cancer 2003, 89, 2–7. 10.1038/sj.bjc.6600936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said J. Biomarker discovery in urogenital cancer. Biomarkers 2005, 10 (Suppl 1), 83–86. 10.1080/13547500500215050. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Kaufman G. K.; Urbach A. R.; Gitlin I.; Gudiksen K. L.; Weibel D. B.; Whitesides G. M. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev. 2008, 108, 946–1051. 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone G.; Supuran C. T. Carbonic anhydrase IX: Biochemical and crystallographic characterization of a novel antitumor target. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 404–409. 10.1016/j.bbapap.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Krall N.; Pretto F.; Decurtins W.; Bernardes G. J.; Supuran C. T.; Neri D. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew. Chem., Int. Ed. 2014, 53, 4231–4235. 10.1002/anie.201310709. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Supuran C. T. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin. Ther. Pat. 2018, 28, 729–740. 10.1080/13543776.2018.1508453. [DOI] [PubMed] [Google Scholar]
- Can D.; Spingler B.; Schmutz P.; Mendes F.; Raposinho P.; Fernandes C.; Carta F.; Innocenti A.; Santos I.; Supuran C. T.; Alberto R. [(Cp-R)M(CO)3] (M = Re or 99mTc) Arylsulfonamide, arylsulfamide, and arylsulfamate conjugates for selective targeting of human carbonic anhydrase IX. Angew. Chem., Int. Ed. 2012, 51, 3354–3357. 10.1002/anie.201107333. [DOI] [PubMed] [Google Scholar]
- Coverdale J. P. C.; Romero-Canelon I.; Sanchez-Cano C.; Clarkson G. J.; Habtemariam A.; Wills M.; Sadler P. J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347–354. 10.1038/nchem.2918. [DOI] [PubMed] [Google Scholar]
- Takahara M.; Hibler D. W.; Barr P. J.; Gerlt J. A.; Inouye M. The ompA signal peptide directed secretion of Staphylococcal nuclease A by Escherichia coli. J. Biol. Chem. 1985, 260, 2670–2674. [PubMed] [Google Scholar]
- Francisco J. A.; Earhart C. F.; Georgiou G. Transport and anchoring of beta-lactamase to the external surface of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 2713–7. 10.1073/pnas.89.7.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piffeteau A.; Gaudry M. Biotin uptake: influx, efflux and countertransport in Escherichia coli K12. Biochim. Biophys. Acta, Biomembr. 1985, 816, 77–82. 10.1016/0005-2736(85)90395-5. [DOI] [PubMed] [Google Scholar]
- Finkenwirth F.; Kirsch F.; Eitinger T. Solitary BioY proteins mediate biotin transport into recombinant Escherichia coli. J. Bacteriol. 2013, 195, 4105–4111. 10.1128/JB.00350-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piffeteau A.; Zamboni M.; Gaudry M. Biotin transport by a biotin-deficient strain of Escherichia coli. Biochim. Biophys. Acta, Biomembr. 1982, 688, 29–36. 10.1016/0005-2736(82)90575-2. [DOI] [PubMed] [Google Scholar]
- Bruins J. J.; Albada B.; van Delft F. ortho-Quinones and Analogues Thereof: Highly Reactive Intermediates for Fast and Selective Biofunctionalization. Chem. - Eur. J. 2018, 24, 4749–4756. 10.1002/chem.201703919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo A. H.; Bose S.; Do L. H. Intracellular Chemistry: Integrating Molecular Inorganic Catalysts with Living Systems. Chem. - Eur. J. 2018, 24, 10584–10594. 10.1002/chem.201800504. [DOI] [PubMed] [Google Scholar]
- Yang L.; Bose S.; Ngo A. H.; Do L. H. Innocent But Deadly: Nontoxic Organoiridium Catalysts Promote Selective Cancer Cell Death. ChemMedChem 2017, 12, 292–299. 10.1002/cmdc.201600638. [DOI] [PubMed] [Google Scholar]
- Ngo A. H.; Ibanez M.; Do L. H. Catalytic Hydrogenation of Cytotoxic Aldehydes Using Nicotinamide Adenine Dinucleotide (NADH) in Cell Growth Media. ACS Catal. 2016, 6, 2637–2641. 10.1021/acscatal.6b00395. [DOI] [Google Scholar]
- Kanega R.; Onishi N.; Wang L.; Murata K.; Muckerman J. T.; Fujita E.; Himeda Y. Picolinamide-Based Iridium Catalysts for Dehydrogenation of Formic Acid in Water: Effect of Amide N Substituent on Activity and Stability. Chem. - Eur. J. 2018, 24, 18389–18392. 10.1002/chem.201800428. [DOI] [PubMed] [Google Scholar]
- Raines D. J.; Clarke J. E.; Blagova E. V.; Dodson E. J.; Wilson K. S.; Duhme-Klair A. K. Redox-switchable siderophore anchor enables reversible artificial metalloenzyme assembly. Nat. Catal. 2018, 1, 680–688. 10.1038/s41929-018-0124-3. [DOI] [Google Scholar]
- Cornelis P. Expressing genes in different Escherichia coli compartments. Curr. Opin. Biotechnol. 2000, 11, 450–4. 10.1016/S0958-1669(00)00131-2. [DOI] [PubMed] [Google Scholar]
- Ghattas W.; Dubosclard V.; Wick A.; Bendelac A.; Guillot R.; Ricoux R.; Mahy J. P. Receptor-Based Artificial Metalloenzymes on Living Human Cells. J. Am. Chem. Soc. 2018, 140, 8756–8762. 10.1021/jacs.8b04326. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Kohler V.; Ward T. R. An NAD(P)H-Dependent Artificial Transfer Hydrogenase for Multienzymatic Cascades. J. Am. Chem. Soc. 2016, 138, 5781–4. 10.1021/jacs.6b02470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.