

Abstract
Brain atrophy, white matter abnormalities, and ventricular enlargement have been described in different neuromuscular diseases (NMDs). We aimed to provide a comprehensive overview of the substantial advancement of brain imaging in neuromuscular diseases by consulting the main libraries (Pubmed, Scopus and Google Scholar) including the more common forms of muscular dystrophies such as dystrophinopathies, dystroglycanopathies, myotonic dystrophies, facioscapulohumeral dystrophy, limb-girdle muscular dystrophy, congenital myotonia, and congenital myopathies. A consistent, widespread cortical and subcortical involvement of grey and white matter was found. Abnormalities in the functional connectivity in brain networks and metabolic alterations were observed with positron emission tomography (PET) and single photon emission computed tomography (SPECT). Pathological brain changes with cognitive dysfunction seemed to be frequently associated in NMDs. In particular, in congenital muscular dystrophies (CMDs), skeletal muscular weakness, severe hypotonia, WM abnormalities, ventricular dilatation and abnormalities in cerebral gyration were observed.
In dystroglycanopathy 2I subtype (LGMD2I), adult patients showed subcortical atrophy and a WM periventricular involvement, moderate ventriculomegaly, and enlargement of subarachnoid spaces. Correlations with clinical features have been observed with brain imaging characteristics and alterations were prominent in congenital or childhood onset cases. In myotonic dystrophy type 2 (DM2) symptoms seem to be less severe than in type 1 (DM1).
In Duchenne and Becker muscular dystrophies (DMD, BMD) cortical atrophy is associated with minimal ventricular dilatation and WM abnormalities.
Late-onset glycogenosis type II (GSD II) or Pompe infantile forms are characterized by delayed myelination. Only in a few cases of oculopharyngeal muscular dystrophy (OPMD) central nervous system involvement has been described and associated with executive functions impairment.
Keywords: brain, imaging, MRI, muscular dystrophy
Introduction
The interest in brain imaging of inherited neuromuscular disorders has increased in recent years. Several brain imaging studies on neuromuscular diseases (NMDs) have been published to consider both muscular impairment and central nervous system (CNS) involvement.1 In the past three decades, researchers have started to focus not only on genetic, biopsy, or muscular imaging biomarkers but also on brain involvement. To date, information on brain involvement in ‘less rare’ and best-known inherited pathologies such as myotonic dystrophies, type 1 (DM1), and type 2 (DM2) are available, while for other NMDs (i.e. facioscapulohumeral dystrophy) CNS involvement needs to be investigated. Nevertheless, the origin and spatiotemporal evolution of CNS involvement, as well as their correlation with neuropsychological and clinical features, still remain unclear. Correlations between brain neuroimaging parameters and clinical symptoms have been reported in different studies but results are often difficult to replicate. This might be related to small sample sizes or heterogeneity in the phenotypes assessed. Moreover, we have to consider that clinical symptomatology might not be linked to specific brain alterations but more probably to dysfunctional networks. In particular, brain imaging studies have been improved by the development of more sophisticated technologies and experimental paradigms. Several imaging methods have been applied to detect brain involvement in patients affected by NMDs, including structural and functional, such as cranial computed tomography (CT), magnetic resonance imaging (MRI), single photon emission computed tomography (SPECT), positron emission tomography (PET), and recently, ultrasound. After the first CT scans, more advanced MRI techniques were applied: morphological to examine the brain structure or functional to investigate brain network activity, cerebral glucose metabolism, or perfusion. The new application of ultrasound (transcranial B-mode sonography) allowed the examination of in vivo brain structural integrity, making easier to evaluate CNS disorders in congenital forms and neonatology patients. The fact that brain examination is expensive and not easy to conduct in young or claustrophobic patients, or in the case of respiratory insufficiency, has to be considered. For this reason, all these limitations have required the application of noninvasive, easily performed investigation techniques (i.e. transcranial B-mode sonography or arterial spin labeling), especially in children.
Brain involvement in NMDs has been well described and proved to be clinically relevant. Several MRI studies revealed a wide spectrum of brain abnormalities, including diffused grey matter (GM) and white matter (WM) abnormalities and ventricular enlargement.
This review is based on papers reporting neuroimaging results on NMDs. Scientific papers were searched from 1980 to May 2018 on the main databases (PubMed, MedLine, Google Scholar). We included only free and full available texts written in English. We excluded papers concerning pharmacological trials. The temporal interval of 1980–2018 was selected in order to include all the information regarding some conditions [i.e. congenital muscular dystrophies (CMDs)]; while the number of studies on muscular dystrophies has increased in recent decades, consistent reports are still missing for other NMDs. We considered the papers on the following NMDs: dystrophinopathies, dystroglycanopathies, myotonic dystrophies, facioscapulohumeral dystrophy, limb-girdle muscular dystrophies, congenital myotonias, congenital myopathies, and related terms or abbreviations. We included the following imaging techniques in the research: ‘computed tomography’ or ‘CT’, ‘magnetic resonance imaging’ or ‘MRI’, ‘single photon emission computed tomography’ or ‘SPECT’, ‘positron emission tomography’ or ‘PET’, ultrasound, ‘transcranial sonography’ or ‘TCS’. We included only those papers containing information about brain involvement.
The total number of papers found in 38 years (1980–2018) on brain imaging was 246.
Considering our inclusion criteria, that excluded single cases, pharmaceutical trials and papers appearing more than one time in the search, we included a final number of 89 articles in this review.
Brain imaging techniques
Brain involvement has been studied in NMDs using both quantitative and nonquantitative techniques that analyzed the morphological structure and functional activity. At the beginning of the 1980s cranial studies, using conventional CT were performed and MRI-descriptive and conventional investigations were conducted to assess WM and GM abnormalities. Conventional morphological brain investigations allowed the description and quantification of morphological features (i.e. WM changes). These were rater-dependent and made results more controversial and less uniform to reproduce. Qualitative scales have been created to grade the severity and location of WM lesions (WMLs) using visual rating scales (i.e. Fazekas or Wahlund scale) on T2-weighted or fluid-attenuated inversion recovery (FLAIR) sequences. In particular, age-related WM changes (ARWMCs) has been proved to be a valid qualitative scale to grade lesions in dystrophies. Ranging from 0 (no lesion) to 3 (diffuse involvement) it allowed the assessment of four different areas separated for each hemisphere.2
The recent technological advances introduced more sophisticated techniques and experimental paradigms of study. Through quantitative analysis results became more reproducible, allowing quantification of more complex and specific brain proprieties. For this reason, the number of nonquantitative conventional morphological brain MRI studies decreased due to the development of observed independent techniques [i.e. voxel-based morphometry (VBM), diffusion tensor imaging (DTI), functional MRI (fMRI)]. At a macro-structural level via brain parenchymal fraction (BPF) methods, global brain atrophy can be quantified. VBM estimates the local amount of a specific tissue. Using the automatic classification of T1-weighted images VBM leads to extract, for each voxel GM, WM, and cerebrospinal fluid and compare tissues probability maps of global density (or regional volume) for each part. VBM investigation can be used to estimate clusters of focal cortical or subcortical alteration (mainly GM) comparing different groups.3
Whole brain volume or regions of interest (ROIs) can be calculated to detect changes in density. Recently, surface-based analysis addressed structural information about the morphological organization of GM volumes. The surface-based morphometry allows the assessment of the cortex surface and extraction of different parameters (i.e. cortical thickness, surface area, cortical folding, and gyrification or any combination of them). Cortical thickness is a measure of the amount of GM estimated at each point on the cortex. It may be employed to detect pathological changes associated with the disease process. Contiguous maps of cortical thickness can be generated and carefully used as a marker of the integrity of the cerebral cortex, related to the size, number, and density of cells.4 The DTI technique quantifies (in vivo) WM microstructures analyzing the magnitude and directionality of diffusivity proprieties of water molecules in tissues. Some pathological conditions can alter the diffusion proprieties of water in the fiber tracts reducing, for example, the anisotropic propriety of water molecules within the WM. DTI is a variant of MRI used to map the diffusion process and identify changes in organization in the WM microstructure, such as fiber density and WM integrity;5 extracting several parameters could be used to investigate different properties. Fractional anisotropy (FA) is an indirect measure of the directionality and coherence of fibers. A lower FA shows a reduction in the density of WM, a loss of structural organization (axonal bundle coherence), or a variation in membrane permeability to water.6 Mean diffusivity measures the overall magnitude of water diffusion without any directional information. Other parameters such as the diffusivity along the principal axis (AD) and the average of the diffusivities along the two minor axes (RD) allow evaluation of the structural damage. The first could reflect axonal pathological changes, the second could be more related to demyelination and axonal membranes alteration.7
Functional brain imaging techniques attempt to measure neuronal activity by detecting changes in metabolism (the metabolic activity of oxygen or glucose in different tissues). fMRI is used to study cerebral activity indirectly.8 It measures the difference in magnetization between oxygen-rich and oxygen-poor blood in different tissue and is based on the linkage between cerebral blood flow (CBF) and neuronal activation. Oxygen consumption is higher during neural activation and, to meet this growth of demand, increased CBF is needed for the active brain cells. The blood-oxygen-level-dependent (BOLD) signal is related to the level of energy used by neurons due to changing O2 in the blood.9
Spontaneous fluctuation of the BOLD signal is present even without any experimental paradigm or any other explicit input. In fact, fMRI examination can be performed during a specific task or by a resting-state fMRI (rs-fMRI) detecting the spontaneous variance of the BOLD signal. Task-based fMRI is used to identify regions that are functionally involved during a specific task performance. The rs-fMRI is used to explore the brain circuits and identify functionally connected neural networks (i.e. default mode network).10
Other functional techniques that are based on brain hemodynamic include SPECT and PET.11 SPECT generates tomographic images of the three-dimensional (3D) distribution of a specific radiopharmaceutical and can directly display measures of regional brain hemodynamics (i.e. 133xenon) affording whole brain coverage. Moreover, regional Cerebral Blood Flow (rCBF) maps can be obtained by SPECT in order to outline the regions with abnormal perfusion.12
PET imaging provides in vivo quantitative measures of CBF with the injection of radioactive tracers, such as fluorodeoxyglucose (FDG)-PET that allow the assessment of neuronal glucose metabolism. Using different tracers, PET describes the metabolic activity of glucose in tissues with a positron and emitting radioisotopes. According to neuronal metabolic activity, it indirectly assesses the mental activity corresponding to glucose uptake. Recently, transcranial sonography (TCS) has been used in childhood NMDs. TCS is a well-established diagnostic method mainly used to assess extrapyramidal movement disorders in neurological disease. It started to be used to study structural CNS changes in NMDs. It is a noninvasive tool that provides reliable additional information about hyperechogenicity of the CNS based on the increasing amounts of iron, bound to proteins other than ferritin.13 All the brain imaging techniques described above have their own advantages in specific clinical settings, leading to improved knowledge about the morphological and functional activity in the brain of patients in different clinical conditions (Table 1).
Table 1.
Comparison of neuroimaging techniques used in various neuromuscular disorders.
| Neuromuscular diseases | Technique |
|||||
|---|---|---|---|---|---|---|
| CT | MRI | fMRI | SPECT | PET | TCS | |
| Dystroglycanopathies | + | ++ | − | − | − | − |
| Congenital muscular dystrophies | + | ++ | − | − | − | − |
| Myotonic dystrophy type 1 | + | ++ | ++ | ++ | ++ | + |
| Myotonic dystrophy type 2 | + | ++ | + | + | + | + |
| Dystrophinopathies | ++ | ++ | + | + | + | − |
| Glycogenosis disease type II | + | + | + | − | − | − |
| Oculopharyngeal muscular dystrophy | − | − | − | − | − | − |
| Facioscapulohumeral dystrophy | ± | + | − | − | − | − |
Changes: −, not described or reported; ±, slight brain changes; +, significant brain changes; ++, marked structural or functional abnormality.
CT, computerized tomography; fMRI, functional MRI; MRI, magnetic resonance imaging; PET, positron emission tomography; SPECT, single photon emission computed tomography; TCS, transcranial sonography.
Neuromuscular disease with brain involvement
Neuromuscular disorders are a group of heterogeneous, hereditary, genetic, or acquired diseases affecting mainly skeletal muscle. Major advances in molecular genetic research led to discovering the pathogenic determinant of the dystrophinopathies in the 1980s. Later, an increased number of genes have been found to be associated with different forms of muscular dystrophy. This led to the classification of them in terms of molecular and genetic features (genes and functions of the respective proteins affected) rather than on clinical symptoms (clinical pattern of weakness, mode of inheritance, and age of onset).14 The main categories of inherited neuromuscular diseases are neuropathies (diseases of a peripheral nerve), myopathies, motor neuron diseases, and muscular dystrophies. Not so long ago, muscular dystrophies were mainly divided into six categories such as Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy, limb-girdle muscular dystrophy (LGMD), distal myopathies, CMDs, facioscapulohumeral muscular dystrophy and myotonic dystrophies. Genetic investigations have expanded our knowledge in muscular dystrophies and now more than 50 forms of NMDs have been identified on molecular bases, each one with different clinical features (e.g. muscle pattern of involvement, variable course different genotype–phenotype correlation).15 In recent years next-generation sequencing technology has led to the discovery of novel causative genes and differentiation of novel myopathies.16 Brain involvement has been reported in different types of NMDs. Recently, neuromuscular conditions have started to be considered even for their multisystemic features and for the implications they have on CNS, such as myotonic dystrophies, dystroglycanopathies and glycogenosis disease type II. The genotype/phenotype correlation remains controversial considering the wide variability of clinical expression. Nevertheless, although rare, correlations between genotype and phenotype have been established but further investigations are needed.17–20
Dystroglycanopathies
Dystroglycanopathies are a group of autosomal recessive conditions characterized by reduced glycosylation of α-dystroglycan (α-DG). Clinical manifestations are extremely variable, and a wide spectrum of phenotypic outcomes has been identified. A different pattern of disability symptomatology has been reported: from the more severe congenital form of Walker Warburg syndrome (WWS) to the milder adult-onset form of LGMD. WWS is characterized by a severe brain involvement (see below congenital forms description). The adult-onset form of LGMD may not present brain or eye involvement.21 There are many LGMD subtypes (i.e. sarcoglycanopathy, calpainopathy, dysferlinopathy). Commonly, type 1 (i.e. LGMD1A, LGMD1B) refers to genetic types showing dominant inheritance (requiring one mutation for symptoms to result). Type 2 refers to recessive inheritance (requiring two mutations from each parent for symptoms to result).22 Unfortunately, only a few studies have analyzed brain involvement in LGMD.
Dystroglycanopathies 2I subtype (LGMD2I) is due to the mutation of the fukutin-related protein (FKRP) gene. It is characterized by a mild to severe phenotype with prominent skeletal muscle involvement. Bourteel and colleagues assessed 11 French patients with LGMD2I (FKRP) mutation.23 They underwent clinical, neuropsychological, and brain MRI investigation. Myocardiopathy and respiratory insufficiency were preponderant. Memory impairment was detected in four patients. Subcortical atrophy and a WM periventricular involvement were associated with changes in α-DG expression in brain.23 A second study, with a smaller sample size of 10 patients with LGMD2I, showed heterogeneous results characterized by nonspecific WM abnormalities, moderate ventriculomegaly, and enlargement of subarachnoid spaces. In one patient, cerebellar atrophy was detected. Neuropsychological evaluation showed a deficit in executive functions and visuospatial abilities without effects on intelligent quotient (IQ) score. In this case, Authors argued that abnormal glycosylation of α-DG in LGMD2I might affect brain development and cognitive performance. Frontal and parietal lobes would be mainly involved even though there was not a specific MRI pattern.
A study performed on five patients affected by LGMD due to LAMA2 mutations by Gavassini and colleagues showed various degrees of CNS involvement with widespread brain WM hyperintensities.24 A mild deficit in executive functions and low IQ scores were noted in three patients. Interestingly, in one patient brain MRI abnormalities preceded the clinical onset of muscle weakness. So, authors argued that WM abnormalities were always present and could be considered a diagnostic criterion for LGMD associated with LAMA2 mutations. Only one study on LGMD2M reported normal brain MRI and cognitive functioning.25
Detailed knowledge about other LGMDs disease forms is lacking (i.e. LGMD type 2N; LGMD2N). LGMD2N is due to mutations in the POMT2 gene. It is characterized by wasting and weakness of the muscles in the shoulder and hip region. Østergaard and colleagues performed a brain MRI study in 10 patients.26 They respectively showed abnormality in 3 of the 10 patients in brain imaging evaluation. They found mild ventricular enlargement due to central and cortical atrophy, periventricular hyperintensities, and frontal atrophy of the left hemisphere in the last cases. MRI abnormalities were associated with lower Mini-Mental State Examination (MMSE) scores, suggesting a link with cognitive impairment. Finally, in LGMD2K (POMT1), intellectual disability with low IQ without structural brain abnormalities Were reported in all patients.27,28
Congenital muscular dystrophies
CMDs are a group of muscular disorders characterized by an early onset of skeletal muscular weakness (within 1 year of age), severe hypotonia, with a variable involvement of organs, such as the brain, and characterized by different outcomes. The prevalence of CMDs is not well established. Several classifications of CMDs are reported in the literature.29,30 Based on the combination of clinical, biochemical, molecular, and genetic findings, Voit and Tomè (2004) distinguished CMDs into four groups: Merosinopathy deficit of lamin α2 or ‘merosin’; dystroglycanopathies caused by glycosylation of α-DG abnormality including WWS, muscle-eye-brain diseases (MEBs), Fukuyama CMD (FCMD), and MDC1B, MDC1C, MDC1D; rigid spine syndrome; Ullrich dystrophy; and primary or secondary α7 integrin deficiency.31
Here, we will mainly focus on congenital dystroglycanopathies (FCMD, MEBs, and WWS diseases) as the literature is more plentiful and the clinical manifestations are more evident.31 Notably, clinical variability is shown among CMDs forms, sometimes through subclinical appearance. Brain involvement is frequent in CMDs and is the main feature in WWS and MEBs, subtypes of CMD, with a prevalent involvement of ocular structures.32
The FCMD type is characterized by a progressive muscular deficit, mental retardation and speech disorders associated with a plain brain involvement. CT examination performed on 22 patients revealed WM abnormalities, ventricular dilatation, cortical sulci, longitudinal cerebral fissure, and Sylvian fissure alterations.33 Abnormalities in cerebral gyration are frequent. Micropolygyria of the cerebrum and cerebellum with loss of cytoarchitecture has been detected using brain MRIs in patients with FCMD.34 In addition, brain MRI shows WM abnormalities, cobblestone occipital cortex, and cerebellar abnormalities.33,35 Nevertheless, the initial WM involvement seems to not progress or change over time.36 MEB disease is characterized by brain malformation, with a cortical defective migration dysfunction, WM abnormalities, and anomalies of the brainstem and cerebellum.37 Recently Yiş and colleagues detected WM abnormalities, ventriculomegaly, and multiple cerebellar cysts in a cohort of 34 patients.38 Others reported cortical gyration abnormalities at the frontal level, microcystic degeneration, and polymicrogyria of the temporal lobe associated with cognitive deficits in 13-year-old girl with long-term survival.15
The brain structural MRI pattern in MEB CMDs is similar to that reported for patients with FCMD, with lissencephaly type II/pachygyria, brain stem, and cerebellar abnormalities. WWS is the most severe form of CMD with an evident involvement of muscles, ocular system, and CNS, and is associated with mental retardation. It is a severe disorder and children do not survive more than 1 year. It is characterized by clinical malformation featuring ‘cobblestone lissencephaly’ cortex, ventricular enlargement, obstructive hydrocephalus, neuronal heterotopias, agyria, polymicrogyria, corpus callosum agenesis, and diffuse WM changes as shown in brain MRI examination.15,39,40
Myotonic dystrophy type 1: Steinert’s disease
DM1 is an autosomal dominant inheritance, multisystemic disorders. It is the most prevalent muscular dystrophy; occurring in 1:8000 people worldwide.41 Muscular, cardiac, endocrine, ocular, hormonal, and neurological systems are involved in the disease on variable levels.42 The Cytosine-Thymine-Guanine (CTG) expansion size seems to be directly correlated to the age of onset (inverse correlation) and to a more severe clinical phenotype (positive correlation).43 DM1 can be divided into four subtypes based on CTG repeat sizes and onset of symptoms, each one associated with specific clinical features: (1) congenital (CDM1); (2) childhood onset, or infantile and juvenile form (JDM1); (3) adult onset; and (4) late onset/asymptomatic.42,44 CNS involvement in DM1 is commonly observed but characterized by between-study heterogeneity.45 Higher impairment has been seen in patients with early onset.46,47 Congenital and juvenile forms are the most severe characterized by a poorer phenotype and prognosis.48 Nevertheless, some congenital patients show normal intellectual functioning and brain structure.15 Cognitive impairment in the adult-onset form is more subtle. CNS involvement has been highlighted in several studies.49,50 Neurocognitive deficits mainly affect memory, executive, verbal, and visuospatial functions. Behavioral changes, apathy, change of personality, anosognosia, and daytime somnolence have been found and are associated to a poorer quality of life.41,51–53 A cognitive progression over time has been confirmed.54–57 Brain changes have been reported but, until now, the pathophysiology and temporal development of brain involvement still remain unclear. Premature aging neurofibrillary tangles (NFTs) and a related degenerative disorder process have been observed in different cerebral structures.47 Widespread neuronal loss (cortical and subcortical), increase in perivascular Virchow–Robinson space, rarefaction in deep WM, mild gliosis, and capillary hyalinization have been detected in postmortem studies.58 Nevertheless, it remains difficult to define a specific lesion pattern since the resulting data are often difficult to replicate. Findings from the first CT scans published in the 1980s, showed a morphological affection in patients with DM1 characterized by ventricular increased and cerebral atrophy.59–62 Microcephaly, hyperostosis internal frontalis, and thickening in the calvarium were detected by Avrahami and colleagues and may be due to brain atrophy.63 A 0.5 T cranial investigation (1988) revealed perivascular hyperintensities as well as a ventricular enlargement in patients compared with controls.64 Early neuroimaging investigations using a structural nonquantitative MRI approach frequently reported WM hyperintense load (WMHL),65–67 subcortical and periventricular WM hyperintense lesions, cerebral atrophy, thickening or atrophy of the corpus callosum, and dilated Virchow–Robin space.48,58,68–71 In particular, WMHLs have been detected using different scales, such as Fazekas or ARWMCs.41,67 The prevalence of WMLs in DM1 patients seems to be related to neuropsychological deficits,47,65 patients’ age, disease progression,47,72 and CTG expansion size.65 The temporopolar WM pathology in DM1 seems to be the most robust and reproducible finding characterizing patients with DM1 rather than DM2, although clinical correlations and evolutions remain unclear (Figure 1).47,65–68,71 A long-term follow-up study suggested that those abnormalities might be progressive over time. WMLs, when absent at baseline, may appear during the course of the disease, proving the existence of a slow demyelinating process in the adult form.73 Progression over time in the number and extent of WMLs and brain atrophy suggests that WM involvement in the adult form is degenerative.73 However, other authors have not found this potential correlation between progression and age,48,51,71,74–76 or neuropsychological testing and the degree of neuromuscular involvement.77
Figure 1.
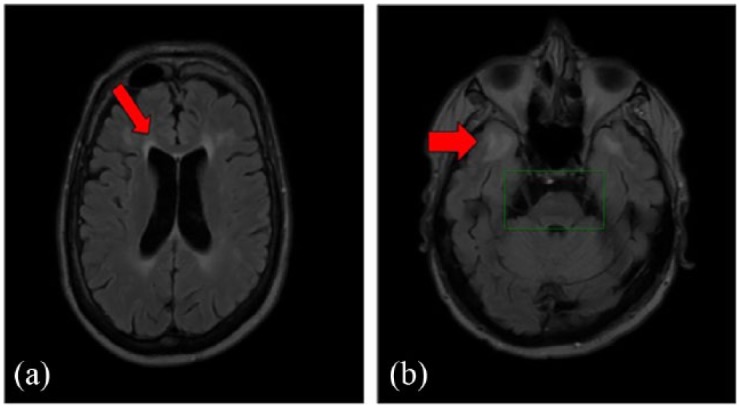
47-year-old male affected by myotonic dystrophy type 1 (with a sister affected by the same pathology) with moderate cognitive involvement, (MIRS) = 3, first symptoms onset at age 26 years, CTG = 413, expansion class = E2.
(a) Marked white matter change abnormality around frontal horn detected in a FLAIR brain MRI scan (red arrow). Left ventricle appears larger than right.
(b) In the temporopolar area at the level of insula marked changes of white matter with major involvement on the left side (red arrow). In brain stem, there is marked atrophy of quadrigeminal bodies and atrophy of pons on the left side (green box). The third ventricle is dilated.
CTG, Cytosine-Thymine-Guanine; FLAIR, fluid-attenuated inversion recovery; MIRS, muscular impairment rating scale; MRI, magnetic resonance imaging.
Figure 2.
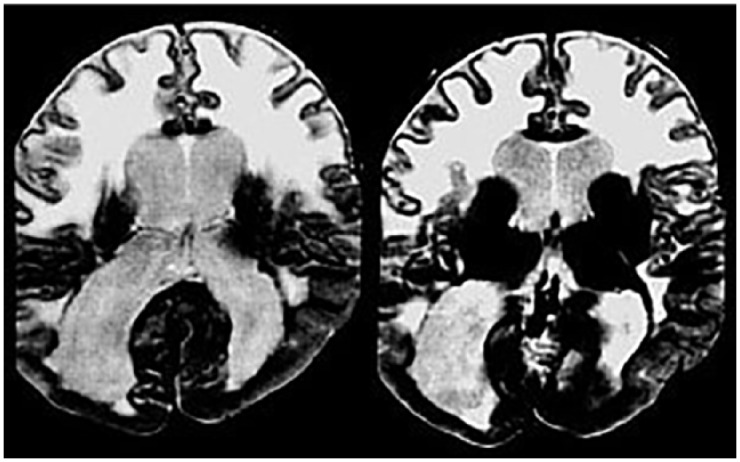
Brain MRI of a patient with CMD.
Axial T2 images show evidence of severe white matter abnormalities, pachygyria in the occipital lobes and ventricular enlargement.
CMD, congenital muscular dystrophy; MRI, magnetic resonance imaging.
Brain atrophy and GM volume reduction in cortical lobes and in deep structures have frequently been reported in literature. It is noted to be mostly symmetric and more pronounced in adult onset but no agreement has been found on the congenital, juvenile, and adult onset form. A recent review published in 2017 revealed a general volume reduction in all cortical lobes, the basal ganglia, and cerebrum.78 The development of observer-independent techniques enabled the estimation of systemic brain atrophy and the relationship with clinical parameters. Comparing GM and WM volumes in noncongenital DM1 patients, Antonini and colleagues found widespread global volume reductions in patients and a negative correlation between age and GM volume.79 No correlation was found between age and WM volume. In light of this, they postulated the existence of a neurodevelopmental GM loss process progressive with age independent from the WMHL degeneration process.79 In contrast, Weber described a correlation between disease duration and WMHL.52 Recently, Baldanzi and colleagues estimated brain atrophy using BPF and found a correlation between atrophy and visuospatial and frontal tasks.41
Several volumetric analysis based on automated methods (e.g. VBM) described widespread GM volume loss both in cortical areas (frontal, temporal, parietal and occipital cortices) and subcortical structures (such as thalami, putamen, and caudate nucleus) and cerebellum with a significant involvement of the corpus callosum (anterior, medioanterior, central, mediodorsal regions) in noncongenital patients.41,47,48,52,72,79–81 Occipital involvement has been detected only in a few cases, which correlates with visuospatial impairment.48
Correlation between sleeplessness and volume loss in the right pallidum and diencephalon has been found.48
Variable reduction of temporal volume has been documented: while described the volume preservation of temporal lobe only few authors have reported volume reduction in the hippocampus.47,52,67,72,82
In patients with different disease onset, GM volume reduction has only been detected in adult onset (not in the congenital form) and an increased volume loss in the cortex is associated with age.48,83,84 Unchanged subcortical GM volume has been found, excluding patients with congenital/childhood onset.72 Correlations between CTG repeat length and GM volumes in motor-related structure and frontal areas, and between pontine WM changes and depression score have been reported.80,81,85
In general, neuroimaging parameters do not correlate with cognitive performance and only an association between delayed recall of verbal memory test and the volume of left postcentral, left middle, and inferior temporal gyri and left supramarginal gyrus has been found.41
Fewer WM changes using a VBM technique have been found. Noncongenital patients showed a diffuse WM volume reduction in every lobe and in the corpus callosum.48,67,80,81
To our knowledge, only two studies have been carried out on cortical thickness. Zanigni and colleagues observed a lateral-occipital thinning in the bilateral cortex, in the right precentral and in the left superior-parietal, fusiform and superior-temporal cortex.72 A more diffused thickness reduction in the frontal, temporal, and occipital cortex has been observed instead.86
The first DTI study was performed by Ota and colleagues and revealed diffusivity abnormalities in the corpus callosum in noncongenital patients with DM1 associated with GM volume reduction.80 Other studies have recently demonstrated diffuse microstructural WM damage with widespread functional anisotropy reduction and increased mean diffusivity in fibers (i.e. corpus callosum, corticospinal tracts)41,47,48,72,85,86 even for child and adolescent onset.84,87,88 In particular, an association between diffusivity values and IQ score was found and between FA and muscular dystrophy values in frontal and temporal lobes and working memory performance.87,88 In noncongenital patients correlations between diffusivity parameters and MMSE,72,85 visuospatial impairment,48 visuomotor coordination and working memory tasks41 or attentional scores47 were detected. CTG repeat length association was found with DTI parameters67,88 even diffused for the entire brain.85,89 Correlations between DTI values and the muscular impairment rating scale (MIRS) score67,85,90 were evidenced and only one study reported correlation with disease duration or age.67 Sleepiness or fatigue were investigated in four studies, but only two found significant correlations.67,90 Some authors, detecting abnormalities in the corpus callosum, suggested that the microstructural abnormalities might be the result of Wallerian degeneration following GM atrophy.80,84 On the other hand, other authors underlined the predominance of WM degeneration and its diffusion when compared with GM effects in DM1.67 Caso and colleagues detected the existence of WM abnormalities in the juvenile form that could lead to GM degeneration. They concluded that microstructural WM change might be due to development change in congenital or child-onset forms while GM degeneration may be more related to a degenerative process considering the aging (premature aging).47 The independence of WM and GM neurodegenerative processes is suggested by the presence of WM T2 hyperintense lesions, whereas there is no correlation with GM loss. This supports the existence of two independent degenerative processes involving WM and cortical degeneration.79
Recently, fMRI studies conducted during a motor task, reported an altered activation in sensory-motor network, including motor control areas, inferior parietal lobes, basal ganglia, and thalami, in patients with DM1 with myotonia compared with controls. The authors hypothesized that a greater motor area activation reflected a compensatory mechanism involved in an accelerated aging process.91 Toth and colleagues investigated the impact of myotonia during a grip task and they found no correlations between clinical characteristic in motor performance and neurofunctional findings, assuming independence between brain functioning and myotonia development.92 Connectivity between brain regions estimated with an rs-fMRI examination revealed an abnormal functional connectivity in the default model network associated with schizotypal-paranoid traits.89
Later, the same research group, investigating social cognition abilities through theory of mind, found a correlation between difficulties in social interactions and personality with connectivity abnormalities.93 Moreover, the dysfunctional network seemed to be correlated with impairment in visuospatial abilities.93 The authors argued that in DM1 patients the decreased frontoparietal connectivity might be linked to cognitive and behavioral disorders and changes in connectivity might represent a compensatory or maladaptive process related to the disease.
Park and colleagues studied functional alterations in the resting state sensorimotor network analyzing the power spectral density (PSD) and noted a widespread PSD alteration in GM and WM. In particular, GM reduction and increased functionality were found in brain regions involved in visual processing, which were associated with motor performance. A subtle increased functional difference was detected in WM associated with motor function. These findings suggest that motor disability in DM1 is not an isolated deterioration of structures involved in motor coordination and regulation but also a multimodal dysfunction that includes the visual system.94
Fiorelli and colleagues conducted the first FDG-PET research in DM1 and found a reduced glucose metabolism in patients compared with healthy controls.95 Later, widespread rising in glucose uptake in the cortical and subcortical structure was found.96 Frontal and temporal lobes are mainly involved.52,95,97,98 Renard and colleagues reported a frontal bilateral reduction in FDG uptake, while no differences were observed in deep GM structures.99
In a recent study Peric and colleagues, excluding congenital and late-onset patients, detected hypometabolism in frontotemporal regions and a correlation between right frontotemporal glucose uptake and executive function.100 SPECT investigation revealed that CBF and perfusion were reduced in DM1, mainly in the frontotemporal region.101 Hypoperfusion, more pronounced in the left hemisphere, was found in frontal and parieto-occipital regions in a large DM1 sample, and this also correlated with the MIRS score.98
Frontal rCBF reduction (frontal mesial and dorsolateral frontal cortex), hypoperfusion in the posterior cortical regions) and in the right parietal lobe correlates with deficits in visuospatial and memory function.102 These results are in agreement with a previous H2O-PET investigation in which authors described a reduction of CBF in the frontotemporal cortex, hypothalamus, and left basal ganglia,77 which was also reported in two other childhood-onset patients.103
Transcranial B-mode sonography is a noninvasive technique used to assess brain structure and integrity, ventricular enlargement, or hydrocephalus. Transcranial ultrasound B-mode examinations have been proved useful to measure ventricular enlargement in adult-onset DM1, as well as to detect changes in the echogenicity of specific brainstem structures. Both studies revealed a brainstem raphe hypoechogenicity and hyperechogenicity of the substantia nigra and by increased width of the third ventricle in patients with DM1.104,105 All these findings contribute to understanding the pathophysiology process of cerebral involvement in DM1. Nevertheless, the conflicting results found in these studies can be explained by the high clinical variability of DM1 and the different age of onset.
Myotonic dystrophy type 2
DM2 shares the RNA pathogenesis with type 1 but fewer studies focusing on DM2 are available. The symptom onset typically occurs in the third decade. Developmental abnormalities and severe childhood disorders are not associated.106 DM2 clinically differs from DM1 in the degree of cognitive impairment. While DM1 congenital forms can exhibit severe mental retardation, this has not been reported in DM2. The first description of brain involvement in patients with DM2, characterized by diffuse and confluent WM hyperintense lesions, was provided by Hund and colleagues, while the early findings of global atrophy were reported by Kassubek and colleagues.107,108 Using a conventional MRI approach diffuse WMHLs have been detected mainly in periventricular space and frontoparieto-occipital structures.52,65,66,77 without any association with cognitive performance.65,77 Some of them demonstrated patterns of hyperintense lesions in DM2 similar to those noted in DM1.66,77 Kornblum and colleagues described atrophy in patients with DM2 using a 1.5 T scanner. They found no significant correlation between BPF and clinical features.66 Later, other studies reported brain atrophy in patients with DM2.52,67,81,109 GM and WM loss along brain midline structures have been identified in patients with DM2.52,67,81 Some VBM analysis found cortical and subcortical (thalamus, hypothalamus, and brainstem) GM reduction but these findings were never replicated. Only diffuse changes in WM and the corpus callosum have been replicated.109 Schenierd-Gold and colleagues observed a GM loss in the cuneus, temporal regions, and amygdala and a prominent WM frontal atrophy without strong correlation between neuroimaging findings and cognitive features.81,110 Interestingly, they found that reductions in mediofrontal GM and cerebellar peduncles and pars of pons WM was associated with excessive day time sleepiness, while brain strain atrophy was correlated with depression.81 A correlation between hippocampal volume and episodic memory was found.52,67
WM alterations in patients with DM2 were carefully detected by DTI techniques and associated with different results. The corpus callosum, internal and external capsules, and limbic system are microstructurally impaired in patients with DM2 compared with controls, nevertheless less than DM1.67 The correlation between abnormal WM and clinical features appears controversial. Found an association between age, disease duration, depression, and motor performance and DTI parameters. No significant correlations were found later between neuropsychological performance and the brain microstructure.67 This discrepancy might be due to small and different sample sizes. No difference in a DTI-ROI-based study was found in FA parameters comparing patients with DM2 with controls.84
The first functional imaging study demonstrated a reduced regional CBF in the orbital and medial frontal structures detected with H2O15-PET in patients with DM2.77 These anatomic changes appear to have some effect on cognition, behavior, and personality, although unlike DM1, DM2 has not been associated with intellectual disability.102
An FDG-PET study found significant widespread hypometabolism of glucose in frontal and temporal lobes and, comparing patterns of GM volumes and hypometabolism (FDG-PET), Weber and collegues colncluded that hypometabolism was an independent phenomenon not resulting from the GM atrophy process.52 Moreover, as previously mentioned, no correlation between functional parameters and neuropsychological features were found.52 On the contrary, Peric and colleagues observed numerous correlations between hypometabolism in prefrontal, insular, and striatal structures and attentive deficit and association between executive dysfunction and frontotemporal hypometabolism.57
According to results obtained in PET studies, Meola and colleagues detected hypometabolism in frontal, parieto-occipital brain regions using SPECT,102 while Romeo found the most significant hypoperfusion in the (left) parietal area.65
Only two studies using TCS have been published.105,111 Significant enlargement of the third ventricle in patients with DM2 compared with controls was found. A correlation between the pathological raphe signal and excessive daytime sleepiness (EDS) was detected.105 Rakocevic-Stojanovic and colleagues observed a higher frequency of brainstem raphe and substantia nigra hypoechogenicity. The diameter of the third ventricle resulted increased and was associated with depression and EDS. Brainstem raphe hypoechogenicity was associated with EDS and diameter of the third ventricle with depression. EDS and fatigue were correlated to brainstem values. No correlation with the substantia nigra with depression or fatigue were found.111 All the above-described neuroimaging evidence suggests a brain involvement in DM2 strengthened by clinical data milder than DM1.
The dystrophinopathies: Duchenne and Becker muscular dystrophies
DMD and BMD are X-linked neuromuscular disorders characterized by a progressive muscular impairment due to a lack of dystrophin. Dystrophin is fundamental for the structural integrity of muscle membranes. DMD is characterized by absent/nonfunctional muscle dystrophin protein. In BMD, dystrophin might be short in size. Progressive weakness of the legs, pelvic and shoulder muscles and heart failure start in early childhood and weakness is generally more severe in patients with DMD than BMD. Striated muscle and heart involvement are mostly described in the literature in patients with DMD. Cardiomyopathy may be the first disease manifestation without evident skeletal myopathy in BMD.112 Striated muscle and heart involvement are deeply described in the literature, in particular in patients with DMD. Male patients with DMD may develop early respiratory muscle failure causing death in the third decades generally due to cardiac and pulmonary complications.113 The occurrence of neurocognitive deficits in DMD are well described and seem to be related to the reduced level of dystrophin isoforms and its location in CNS tissues.114–117 Nevertheless, the role of dystrophin in the brain is not well established. Mutations within the DMD gene result in the absence of specific dystrophin isoforms. The location of dystrophin isoforms has been linked with cognitive impairment in DMD. The lack of dystrophin in the brain is localized in neurons and glial cells in the hippocampal, cortical, and subcortical regions, and Purkinje cells of the cerebellum.118 Information on dystrophin in the brain is based on few studies on human brains.119,120 Isoform Dp427c is predominantly expressed in the neurons of the cortex and hippocampus.121 The Purkinje isoform Dp427p is expressed in cerebellar Purkinje cells, Dp260 and Dp116 isoforms in the retina and the peripheral nerve, Dp71 isoform presents higher levels in the CNS.122,123 Less informtions are available on Dp140.124 Recently DMD expression level in the adult human brain has been found to be higher in the hippocampus, involved in memory and amygdala as well as emotion regulation, rather than in the cerebellum and the pons. The high expression of DMD in subcortical structures might support evidence of memory and emotion deficits in DMD.120 Duchenne himself, described some patients with cognitive deficits.125
It has been estimated that 30% of boys with DMD present cognitive deficits and mental retardation. Children with DMD show lower IQ scores than the normal population (average IQ score in patients with DMD is 85) and this average is one standard deviation below the mean (30% of boys with DMD have an IQ > 70).126 Worse performance in attentional and verbal repetition tasks has been described.127–129 Reading deficits, attention-deficit hyperactivity disorder, autism spectrum disorders, epilepsy, and obsessive–compulsive disorder (OCD) have also been observed.130–134 Spelling, arithmetic, reading difficulties behavioral problems115 and the occurrence of epilepsy135 have been described in BMD but neurodevelopmental disorders have been investigated less than in DMD. Regarding DMD brain imaging, Septien and colleagues used CT scans in 15 patients with DMD. The authors detected cortical atrophy associated with minimal ventricular dilatation probably due to WM abnormalities.136 A decade before Yoshioka and colleagues investigated 30 patients with DMD with the same techniques found subtle cortical atrophy in 67% of them, which was associated with slight ventricular dilation.137 Enlargement of the lateral ventricles of the brain in mdx mice, when compared with controls, has been detected recently.138 Brain MRI evaluation showed that patients with DMD lacking Dp140 isoforms performed worse on neuropsychological evaluation compared with those with preserved Dp140. They found some difference on a structural level where patients showed significantly smaller occipital cortex volumes and a difference in FA parameters in the occipital lobe.139 The authors concluded that the DMD_Dp140(-) mutation may contribute more to GM volume reduction. There was no statistical difference in WM volume. A kind of alteration in WM microstructural integrity (lower WM FA, higher WM mean and radial diffusivity), indicating compromised structural complexity, comparing patients with DMD with healthy controls has been found.139 Using DTI-ROI-based approaches and selecting 16 different structures bilaterally, Fu and colleagues found changes in the corpus callosum and, in particular, in the splenium.113 However, both the small size of ROI used and the lack of multiple correction comparisons are the limits of the study. Lv and colleagues, assessing motor-related brain cortex, found the smaller volume in the left primary sensorimotor cortex in DMD.140 The results on global morphological and microstructural differences in the brain of patients with DMD suggest that these differences arise from altered brain maturation rather than atrophy. Hypometabolism of glucose in boys affected by DMD has been reported in the brain.141 Functional imaging technique showed a reduction of glucose metabolism bilaterally in medial temporal structures and cerebellum and on the right side in sensorimotor and lateral temporal cortex with PET142 and altered metabolite concentrations in the left cerebrum detected by SPECT.143 Using an rs-fMRI paradigm, a decrease of synchronization of spontaneous activity in the motor cortex has been noted,140 underlying the existence of functional abnormalities in boys affected by DMD which might be due to the deficiency of dystrophin in the brain. Unfortunately, less information on brain imaging is available on BMD. This is probably due to a milder phenotype characterizing the disorder that has led researchers to focus on the more severe form of DMDs. Nevertheless, despite various DMDs studies, etiology of the CNS pathology in DMD and BMD remains unclear.
Glycogenosis disease type II
Glycogenosis type II (GSD II), also named Pompe disease, is an autosomal recessive metabolic disorder caused by an accumulation of glycogen. Dysfunctional glycogen storage is due to a deficiency of lysosomal enzyme α-glucosidase.144
Disease onset may occur in childhood or adulthood and infantile or late-onset forms of classification have been proposed.145
The late-onset GSD II form mainly affects limb and respiratory muscles and is more slowly progressive than the infantile-onset form.146
Postmortem evidence of glycogen accumulation in the brain of patients with GSD II has been found,147,148 without evidence of cortical atrophy.149
Infantile-onset Pompe Disease (IOPD) is a characterized by the accumulation of glycogen in skeletal muscle, heart, and central neuronal tissue (cervical spinal cord and the brainstem neurons with minimal build up in the cerebral cortex)150,151 even prenatally.152 Clinically characterized by general muscular weakness and respiratory and cardiac dysfunctions, the symptomatology appears at the age of 6 months and results in death before 1 year of age. Recombinant human α-glucosidase therapy has been introduced since 1999 and has demonstrated an improvement in survival and a reduction of symptoms.
Brain in IOPD disease is characterized by a delayed myelination, cortical atrophy and a widening of the anterior horns of the lateral ventricles.153 Postmortem autopsies revealed glial cell accumulation in the WM in IOPD.150,151
Chien and colleagues described abnormal cranial findings (e.g. dilatation of the lateral ventricles) associated with a delay of CNS myelination, evaluated with repeated cranial MRI after 6 months of Enzyme Replacement Therapy (ERT).154 A slowly progressive neurodegenerative process affecting the motor neurons has been detected in patients with infantile Pompe disease with MRI.155
Moreover, intellectual decline and extensive periventricular subcortical WMLs are associated.156,157 Progressive WM changes in early IOPD childhood have been detected by Bloomfield and colleagues.158
Brain computed tomography (CT) and MRI of one patient case report performed over a 6-year follow up (at 3 and 9 years old) showed progressive global volume loss.159 While ventricle dilatation is a common feature in IOPD, myelination of the cerebral WM has not been detected in T2-weighted images. Interestingly, the effect of glycogen storage in the brain has been studied recently by Ebbink and colleagues in a follow-up study of 11 patients.157 At a brain imaging point, they noted three characteristic stages of WM involvement. The first phase is characterized by periventricular WM pattern at the level of centrum semiovale associated to the widening of lateral ventricles. Growing patients show WM abnormalities spreading to subcortical areas, the internal and external capsule, first in the splenium and later in the genu of the corpus callosum. At 11 years of age, hyperintensities were noted in decussation and corticospinal tracts. This introduces new knowledge about the separate involvement of the brain and motor functioning in Pompe disease, with a slowly progressive WM involvement independent from motor functionality.160
Using VBM and rs-fMRI paradigms in nine patients with late-onset glycogenosis type II, Barroni and colleagues revealed no significant difference in regional GM volumes between patients and changes in brain connectivity in the salience network, cingulate gyrus and medial frontal cortex, implicated in executive functioning. The fMRI findings were congruent with neuropsychological impairment.161
Oculopharyngeal muscular dystrophy
OPMD is an adult-onset autosomal dominant disorder (onset is usually between the fifth or sixth decade of life). It is characterized by progressive eyelid drooping and proximal limb weakness (asymmetric). Pharyngeal muscle involvement produces ptosis and dysphagia.162
Suspicion of OPMD is based on clinical criteria and the diagnosis is confirmed by molecular genetic testing of PABPN1. In a few cases, CNS involvement has been described and is associated with executive functions impairment.
Quality of life is poor mainly because of significant dysphagia.163
Currently, no effective treatment exists for OPMD.
OPMD, described in a French-Canadian family for the first time, has the highest prevalence among Bukhara Jews.164 OPMD is characterized by proximal limb and facial weakness, bilateral ptosis, and dysphagia. It is a myopathy which affects all voluntary muscles and in particular the levator palpebrae, tongue, pharynx, extraocular muscles. A CNS involvement in OPMD has been suggested only in a few cases.165
Nevertheless, although few brain imaging studies are available, cognitive decline and psychological disorders have been described in homozygous patients.166 Blumen and colleagues described 10 homozygous OPMD, detecting lacunar infarcts and leukoaraiosis in a patient with cardiovascular risk factors and brain atrophy.
Behavioral and psychological disturbances seem to correlate with the expansion size in heterozygote OPMD.167
Recently, Chen and colleagues described 11 patients with a probable OPMD diagnosis. Clinical phenotype was characterized by ocular pharyngeal muscle weakness, severe brain involvement (cerebellum and brain stem were mildly atrophied), and intrafamilial variability of muscle impairment. Nevertheless, further molecular investigations are needed to understand the genetic background and pathogenesis.168
Facioscapulohumeral dystrophy
Facioscapulohumeral dystrophy (FSHD) is an autosomal dominant neuromuscular disorder. It is characterized by (asymmetric) involvement of selective muscle groups (face, shoulder, and arms) with a wide spectrum of phenotypic expression and inter- and intrafamilial variability.169 It can be associated with CNS disorders. Previous studies reported extra-muscular involvement and associations with hearing deficits, cardiac complications, and schizophrenia.170 Epilepsy and mental retardation have been observed, even in the absence of neuroradiological abnormalities, in patients with early onset FSHD.171,172 Sporadic association with multiple sclerosis has been reported.173 Fierro and colleagues described a relatively high incidence of WM hyperintensities in the disease.170 In particular, WM hyperintense lesions were mainly located bilaterally in parieto-temporal lobes in 44% of FSHD without a significant hemispheric prevalence. No correlations between severity of WMHL, age, age at onset, duration of the disease, or muscular impairment have been found.170 No cortical atrophy nor ventriculomegaly have been noted. Using Transcranial magnetic stimulation (TMS), Di Lazzaro and colleagues observed a significantly reduced intra-cortical inhibition in patients with FSHD compared with healthy people, which may be due to a compensatory phenomenon of the progressive muscle deficit. VBM analysis revealed clusters of GM volume reduction in the left frontal lobe, in anterior cingulate and right frontopolar cortex in the brain of patients with FSHD.174 The authors hypothesized that the volume loss of the left sensory-motor area could be a consequence or cause of muscular weakness. Quarantelli and colleagues, comparing 30 patients with 39 healthy controls, observed a significant reduction of GM loss mainly in left frontal structures, precentral cortex, the anterior cingulate cortex, and the right frontal region in patients with FSHD and a correlation between GM and clinical severity. In particular, three clusters of significant GM loss were detected: in the left precentral cortex, anterior cingulate, and right frontopolar regions. WM hyperintensities did not correlate with a more prominent GM loss175 and brain tissue volumes did not correlate with disease duration, size of the genetic deletion, or age at onset.
Discussion
Although NMDs refers to a heterogeneous group of disorders with different genetic and phenotypic features many aspects of CNS neuromuscular involvement have been observed. The pathogenic mechanisms involved in NMD brain damage are complex and need further study. Significant progress has been made recently in the brain imaging field. Nevertheless, the resulting data are still discordant and, until now, few longitudinal studies are available on brain imaging changes. Future studies might allow us to understand the pathogenesis of brain changes. We summarized the main brain changes in Table 2 with the principal imaging findings of CNS involvement in NMDs. We have focused on congenital dystrophies, dystrophinopathies, dystroglycanopathies, myotonic dystrophies, facio-scapulo-humeral dystrophy, and LGMD. Each pathology presents typical features, but some common changes can be detected. Conventional MRI reveals a more prominent involvement in the WM. In particular diffused, periventricular and temporopolar, WM hyperintensities, and a thin corpus callosum were detected. Later, the advancement of technology allowed the detection of GM alterations, with cortical and subcortical involvement. In most of the papers assessed, observations on brain involvement are associated with some clinically relevant guidelines for psychological support and special schooling.
Table 2.
Summary of the main results of brain involvement in NMDs.
| Imaging technique |
DM1 | DM2 | LGMD | CMD | DMD | GSD II | OPMD | FSHD |
|---|---|---|---|---|---|---|---|---|
| MRI | Ventricular enlargement | Ventricular enlargement | Ventricular enlargement | Ventricular enlargement | Ventricular enlargement | Ventricular enlargement | ||
| Global atrophy (cortical and subcortical) Thinner corpus callosum Dilated Virchow–Robin spaces Temporopolar WMHLs |
Global atrophy | Global atrophy (fronto-parieto cortex and subcortical) Dilated Virchow–Robin spaces and subarachnoid space |
Abnormalities in brain morphology (gyration, cortical sulci, fissures alteration) Agyria Micropolygyria Polymicrogyria Cobblestone lissencephaly Hydrocephalus Corpus callosum agenesis |
Subtle cortical atrophy | Mild atrophy in cerebellum and brainstem mild cortical atrophy |
Gray Matter reduction Cortical (fronto parietal) Left > right |
||
| WMHL (frontal and temporal lobes); (ATWML) |
Bilateral WMHL (periventricular, frontal parieto-occipital) | WMHL (periventricular and widespread) | Widespread WMHL | Diffused WM abnormalities WMHL (periventricular and subcortical widespread) |
Diffused WMHL (parietal-temporal lobes) | |||
| Gray matter reduction (VBM) Cortical (all lobes) Subcortical (striatum, thalamus, nucleus accumbens, cerebellum) |
Gray matter reduction (VBM) Cortical (frontotemporal) Subcortical (brainstem, thalamus, hypothalamus, mesencephalon, int. pallidum, amygdala) |
Grey matter volume reduction (VBM) |
Gray matter volume reduction (VBM) | |||||
| WM reduction (VBM) Subcortical (all lobes) Corpus callosum Fornix |
WM reduction (VBM) (Subcortical in all lobes, corpus callosum, cerebellum |
|||||||
| DTI | Limbic system tracts Association fibers (fornix, cingulum bundle) Corpus callosum Projection fibers Cerebrum |
Limbic system Association fibers Corpus callosum |
Diffused microstructural impairment, and in occipital lobe Corpus callosum Splenium |
|||||
| PET/SPECT/ fMRI |
<FDG uptake: Cortical (fronto, temporal) Lentiform nucleus < CBF: Cortical (fronto temporal) Hypothalamus |
<FDG uptake: Cortical (fronto, temporal parietal operculum) Subcortical (Thalamus, striatum) <CBF: Cortical (frontoparieto-occipital) |
<FDG uptake: medial temporal structures and cerebellum and on the right side in sensorimotor and lateral temporal fMRI (rstate) decrease of synchronization |
rs-fMRI brain connectivity in salience network, cingulate gyrus and medial frontal cortex |
||||
| Ultrasound | Increased width of the third ventricle Hyperechogenicity: substantia nigra Hypoechogenicity: brainstem raphe |
Increased width of the third ventricle Hyperechogenicity: substantia nigra Hypoechogenicity: brainstem raphe |
ATWML, anterior temporal white matter hyperintense lesion; CBF, cerebral blood flow; CMD, congenital muscular dystrophy; DM1, myotonic dystrophy type 1; DM2, myotonic dystrophy type 2; DMD, Duchenne muscular dystrophy; DTI, diffusion tensor imaging; FDG, F-fluorodeoxyglucose; fMRI, functional magnetic resonance imaging; FSHD, facioscapulohumeral dystrophy; GSD II, glycogenosis type II; LGMD, limb-girdle muscular dystrophy; MRI, magnetic resonance imaging; OPMD, oculopharyngeal muscular dystrophy; PET, positron emission tomography; rs-fMRI, resting-State fMRI; SPECT, single photon emission computed tomography; VBM, voxel-based morphometry; WM, white matter; WMHL, White Matter Hyperintense Load in the table.
Clinical observations revealed some intellectual impairment in a number of NMDs, as well as difficulties in reasoning and memory. Congenital onset cases seem to be more affected than adult-onset forms and exhibit pronounced WM abnormalities and sometimes epilepsy. Cognitive impairment has been found in most patients with dystrophinopathy, who usually have difficulty with abstract reasoning and calculation. Myotonic dystrophy showed cortical atrophy, a mild form of intellectual impairment and behavioral abnormalities. There is evidence of MRI abnormalities of both WM and GM. In a preliminary longitudinal study, cognitive abnormalities appear not to be progressive. We also compared neuroimaging abnormalities in LGMD 2I and in other dystroglycanopathy in relation to the age of disease onset. Nevertheless, until now, the evolution of brain involvement and the correlation with clinical symptomatology is still unclear, and few follow-up studies have been published. Regarding pathomechanisms, there are a number of membrane protein complexes (i.e. dystroglycan complex and sarcoglycan complex) that are signaling at the surface of membrane in both muscle and neuron. Therefore, lack of integrity of the sarcolemmal membrane or neuronal membrane leads both to the dysfunction of neurons and muscle and possible reactive inflammation by microglia and macrophages. An important clinical issue is to advise when imaging techniques are required in muscular patients. In our opinion, these techniques are useful not only for diagnosis but also as an indication in the follow up of patients, especially in children. In adult cases, cognitive behavioral therapy may be used in the follow up of DM1 cases. Presymptomatic behavioral abnormalities such as mood disorders should be screened before open depression or mood disorders might influence negatively quality of life of patients and caregivers.
Future prospective and limitations
Our review suggested that only ventricular enlargement with hydrocephalus are in most cases related to mental retardation. Supra-tentorial and subcortical WM changes might determine a less frequent and milder cognitive abnormality. Accurate histological, immunohistochemical and biochemical evaluations are needed in the brain as well as autoptic studies. An evaluation of 3.0 T MRI and DTI tractography with relation to default connectivity might lead to a better understanding of the pathomechanism of brain abnormalities. Another technique that could evaluate the dysfunctional disease mechanism is the level of oxygen perfusion in the brain by arterial spin labeling (ASL) MRI, an alternative to expensive and invasive PET. ASL is so far been used in the heart and is applicable to the brain to study the energetic state of neurons since this tissue is highly perfused.176,177 A multidisciplinary approach of patients with NMDs with brain involvement or mental retardation is needed. Medical and paramedical personnel to be involved in the team are neurologists, psychologists, neuropsychologists, neuroradiologists, physical therapists, occupational therapists, and social workers. Caregivers can also be supported by such teams. Another important point is that new innovative therapies used in glycogenosis type 2 (ERT in combination with chaperon) or ANTISENSE OLIGONUCLEOTIDES in DMD, possibly available in the future for myotonic dystrophy, need to cross the brain–blood barrier (BBB). In fact, most of the new treatments do not cross the BBB. In Pompe disease, the lack of penetration can cause permanent dysfunction in surviving children with Pompe that present facial abnormalities and respiratory difficulties. Therefore, while motor skills may be improved, intellectual functions might be impaired and not changed with ERT.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
ORCID iD: Corrado Angelini  https://orcid.org/0000-0002-9554-8794
https://orcid.org/0000-0002-9554-8794
Contributor Information
Corrado Angelini, Fondazione Ospedale San Camillo IRCCS, Via Alberoni 70, Venezia, 30126, Italia.
Elena Pinzan, Fondazione Ospedale San Camillo IRCCS, Venezia, Italia.
References
- 1. D’Angelo MG, Bresolin N. Report of the 95th European Neuromuscular Centre (ENMC) sponsored International Workshop Cognitive Impairment in Neuromuscular Disorders, Naarden, The Netherlands, 13–15 July 2001. Neuromuscul Disord 2003; 13: 72–79. [DOI] [PubMed] [Google Scholar]
- 2. Wahlund LO, Barkhof F, Fazekas F, et al. A new rating scale for age-related white matter changes. Stroke 2001; 32: 1318–1322. [DOI] [PubMed] [Google Scholar]
- 3. Ashburner J, Friston KJ. Voxel-based morphometry—the methods. NeuroImage 2000; 11: 805–821. [DOI] [PubMed] [Google Scholar]
- 4. Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 2000; 97: 11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beaulieu C. The basis of anisotropic water diffusion in the nervous system - a technical review. NMR Biomed 2002; 15: 435–455. [DOI] [PubMed] [Google Scholar]
- 6. Mori S, Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron 2006; 51: 527–539. [DOI] [PubMed] [Google Scholar]
- 7. Abhinav K, Yeh FC, Pathak S, et al. Advanced diffusion MRI fiber tracking in neurosurgical and neurodegenerative disorders and neuroanatomical studies: a review. Biochim Biophys Acta 2014; 1842: 2286–2297. [DOI] [PubMed] [Google Scholar]
- 8. Beckmann F, Smith S. Probabilistic independent component analysis for functional magnetic resonance imaging. IEEE Trans Med Imaging 2004; 23: 137–152. [DOI] [PubMed] [Google Scholar]
- 9. Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage 2004; 23: 208–219. [DOI] [PubMed] [Google Scholar]
- 10. Logothetis NK. What we can do and what we cannot do with fMRI. Nature 2008; 453: 869–878. [DOI] [PubMed] [Google Scholar]
- 11. Wintermark M, Sesay M, Barbier E, et al. Comparative overview of brain perfusion imaging techniques. Stroke 2005; 36: e83–e99. [DOI] [PubMed] [Google Scholar]
- 12. Warwick JM. Imaging of brain function using SPECT. Metab Brain Dis 2004; 113–123. [DOI] [PubMed] [Google Scholar]
- 13. Berg D, Roggendorf W, Schroder U, et al. Echogenicity of the substantia Nigra. Arch Neurol 2002; 59: 999–1005. [DOI] [PubMed] [Google Scholar]
- 14. Walter MC, Lochmüller H. Muscular dystrophies. Neurol Clin Neurosci 2007; 85(15): 1142–1167. [Google Scholar]
- 15. Falsaperla R, Praticò AD, Ruggieri M, et al. Congenital muscular dystrophy: from muscle to brain. Ital J Pediatr; 2016; 42(1): 78 Epub ahead of print 31 August 2016. DOI: 10.1186/s13052-016-0289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 2015; 85: 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin F, Wang Z, Lin M, et al. New insights into genotype - phenotype correlations in Chinese facioscapulohumeral muscular dystrophy: a retrospective analysis of 178 patients. Chin Med J; 128(13): 1707–1713. Epub ahead of print 2015. DOI: 10.4103/0366-6999.159336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu J, Hunt SD, Matthias N, et al. Lab Resource: stem cell line generation of an induced pluripotent stem cell line (CSCRMi001-A) from a patient with a new type of limb-girdle muscular dystrophy (LGMD) due to a missense mutation in POGLUT1 (Rumi). Stem Cell Res 2017; 24: 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santoro M, Masciullo M, Silvestri G, et al. Myotonic dystrophy type 1: role of CCG, CTC and CGG interruptions within DMPK alleles in the pathogenesis and molecular diagnosis. Clin Genet 2017; 92: 355–364. [DOI] [PubMed] [Google Scholar]
- 20. Hamilton MJ, Mclean J, Cumming S, et al. Outcome measures for central nervous system evaluation in myotonic dystrophy type 1 may be confounded by deficits in motor function or insight. Front Neurol; 2018; 9: 780 Epub ahead of print 2018. DOI: 10.3389/fneur.2018.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001; 10: 2851–2860. [DOI] [PubMed] [Google Scholar]
- 22. Pegoraro E, Hoffman EP. Limb-girdle muscular dystrophy overview. In: Pagon RA, Bird TD, Dolan CR, et al., (eds). GeneReviews [Internet]. Seattle, WA: University of Washington; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1408/. Updated August 30, 2012. Accessed 2012. [PubMed] [Google Scholar]
- 23. Bourteel H, Vermersch P, Cuisset JM, et al. Clinical and mutational spectrum of limb-girdle muscular dystrophy type 2I in 11 French patients. J Neurol Neurosurg Psychiatry 2009; 80: 1405–1408. [DOI] [PubMed] [Google Scholar]
- 24. Gavassini BF, Carboni N, Nielsen JE, et al. Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve 2011; 44: 703–709. [DOI] [PubMed] [Google Scholar]
- 25. Godfrey C, Escolar D, Brockington M, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol 2006; 60: 603–610. [DOI] [PubMed] [Google Scholar]
- 26. Østergaard ST, Johnson K, Stojkovic T, et al. Limb girdle muscular dystrophy due to mutations in POMT2. J Neurol Neurosurg Psychiatry 2018; 89: 506–512. [DOI] [PubMed] [Google Scholar]
- 27. Godfrey C, Clement ÃE, Mein ÃR, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. Epub ahead of print 18 September 2007;130(Pt 10): 2725–35. DOI: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 28. Lommel M, Hermann R, Uyanik G, et al. Correlation of enzyme activity and clinical phenotype in POMT1- associated dystroglycanopathies. Neurology 2010; 74: 157–164. [DOI] [PubMed] [Google Scholar]
- 29. Parano E, Fiumara A, Falsperla R, et al. Congenital muscular dystrophy: correlation of muscle biopsy and clinical features. Pediatr Neurol 1994; 10: 233–236. [DOI] [PubMed] [Google Scholar]
- 30. Bönnemann CG, Wang CH, Quijano-Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord 2014; 24: 289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Voit T, Tomè F. The congenital muscular dystrophies. In: Engel AG, Franzini-Amstrong C. (eds) Myology-basic and clinical. 3rd ed. New York: McGraw-Hill-Medical Publishing Division, 2004, pp.1203–1238. [Google Scholar]
- 32. Angelini C. Enzyme replacement therapy for the treatment of Pompe disease. Expert Opin Orphan Drugs 2018; 6: 311–318. [Google Scholar]
- 33. Fukuyama Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the fukuyama type - clinical, genetic and pathological considerations. Brain Dev 1981; 3: 1–29. [DOI] [PubMed] [Google Scholar]
- 34. Kamoshita S. Etiology of congenital muscular dystrophy (Fukuyama type). Nihon Rinsho 1977; 35: 3929–3935. [PubMed] [Google Scholar]
- 35. Barkovich AJ. Neuroimaging manifestations and classification of congenital muscular dystrophies. Am J Neuroradiol 1998; 19: 1389–1396. [PMC free article] [PubMed] [Google Scholar]
- 36. Toda T, Segawa M, Nomura Y, et al. Localization of a gene for Fukuyama type congenital muscular dystrophy to chromosome 9q31−33. Nat Genet 1993; 5: 283–286. [DOI] [PubMed] [Google Scholar]
- 37. Hehr U, Uyanik G, Gross C, et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics 2007; 8: 279–288. [DOI] [PubMed] [Google Scholar]
- 38. Yiş U, Uyanik G, Rosendahl DM, et al. Clinical, radiological, and genetic survey of patients with muscle-eye-brain disease caused by mutations in POMGNT1. Pediatr Neurol 2014; 50: 491–497. [DOI] [PubMed] [Google Scholar]
- 39. Devisme L, Bouchet C, Gonzals M, et al. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain 2012; 135: 469–482. [DOI] [PubMed] [Google Scholar]
- 40. Dobyns WB, Patton MA, Stratton RF, et al. Cobblestone lissencephaly with normal eyes and muscle. Neuropediatrics 1996; 27: 70–75. [DOI] [PubMed] [Google Scholar]
- 41. Baldanzi S, Cecchi P, Fabbri S, et al. Relationship between neuropsychological impairment and grey and white matter changes in adult-onset myotonic dystrophy type 1. NeuroImage Clin 2016; 12: 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harper PS. Congenital myotonic dystrophy in Britain: I. Clinical aspects. Arch Dis Child 1975; 50: 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. The International Myotonic Dystrophy Consortium (IDMC). New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1). Neurology 2000; 54: 1218–1221. [DOI] [PubMed] [Google Scholar]
- 44. De Antonio M, Dogan C, Hamroun D, et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol 2016; 172: 572–580. [DOI] [PubMed] [Google Scholar]
- 45. Modoni D, Silvestri G, Pomponi M, et al. Characterization of the pattern of cognitive impairment in myotonic dystrophy type 1. Arch Neuro 2004; 61: 1–5. [DOI] [PubMed] [Google Scholar]
- 46. Ekstrom A, Hakenas-Plate L, Samuelsson L, et al. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet 2008; 926: 918–926. [DOI] [PubMed] [Google Scholar]
- 47. Caso F, Agosta F, Peric S, et al. Cognitive impairment in myotonic dystrophy type 1 is associated with white matter damage. PLoS One; 9 Epub ahead of print 2014; 9(8):e104697. DOI: 10.1371/journal.pone.0104697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cabada T, Iridoy M, Jericó I, et al. Brain involvement in myotonic dystrophy type 1: a morphometric and diffusion tensor imaging study with neuropsychological correlation. Arch Clin Neuropsychol 2017; 32: 401–412. [DOI] [PubMed] [Google Scholar]
- 49. Bosco G, Diamanti S, Meola G, et al. Workshop report: consensus on biomarkers of cerebral involvement in myotonic dystrophy, 2-3 December 2014, Milan, Italy Neuromuscul Disord 2015; 25: 813–823. [DOI] [PubMed] [Google Scholar]
- 50. Gourdon G, Meola G. Myotonic dystrophies: state of the art of new therapeutic developments for the CNS. Front Cell Neurosci; 11 Epub ahead of print 2017; 11:101. DOI: 10.3389/fncel.2017.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve 2007; 36: 294–306. [DOI] [PubMed] [Google Scholar]
- 52. Weber YG, Roebling R, Kassubek J, et al. Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 2010; 74: 1108–1117. [DOI] [PubMed] [Google Scholar]
- 53. Baldanzi S, Bevilacqua F, Lorio R, et al. Disease awareness in myotonic dystrophy type 1: an observational cross-sectional study. Orphanet J Rare Dis; 11 Epub ahead of print 2016;11: 34. DOI: 10.1186/s13023-016-0417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sansone V, Gandossini S, Cotelli M, et al. Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol Sci 2007; 28: 9–15. [DOI] [PubMed] [Google Scholar]
- 55. Modoni A, Silvestri G, Vita MG, et al. Cognitive impairment in myotonic dystrophy type 1 (DM1): a longitudinal follow-up study. J Neurol 2008; 255: 1737–1742. [DOI] [PubMed] [Google Scholar]
- 56. Winblad S, Samuelsson L, Lindberg C, et al. Cognition in myotonic dystrophy type 1: a 5-year follow-up study. Eur J Neurol 2016; 23: 1471–1476. [DOI] [PubMed] [Google Scholar]
- 57. Peric S, Rakocevic V, Gorana S, et al. Clusters of cognitive impairment among different phenotypes of myotonic dystrophy type 1 and type 2. Neurol Sci 2017; 38: 415–423. [DOI] [PubMed] [Google Scholar]
- 58. Di Costanzo A, Di Salle F, Santoro L, et al. Dilated Virchow–Robin spaces in myotonic dystrophy: frequency, extent and significance. Eur Neurol 2001; 46: 131–139. [DOI] [PubMed] [Google Scholar]
- 59. Bird TD, Follett C, Griep E. Cognitive and personality function in myotonic muscular dystrophy. J Neurol Neurosurg Psychiatry 1983; 46: 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pires M, Nunes B, Monteiro L. Computed tomographic findings of brain and skull in myotonic dystrophy. J Neurol Neurosurg Psychiatry 1987; 50: 1387–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cornacchia L, Marina R, Ballarini V, et al. Computed tomographic findings of brain and skull in myotonic dystrophy. J Neurol Neurosurg Psychiatry 1988; 51: 1463–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Walker G, Rosser R, Mastaglia F, et al. Psychometric and cranial CT study in myotonic dystrophy. Clin Exp Neurol 1984; 20: 161–167. [PubMed] [Google Scholar]
- 63. Avrahami E, Katz A, Bornstein N, et al. Computed tomographic findings of brain and skull in myotonic dystrophy. J Neurol Neurosurg Psychiatry 1987; 50: 435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Glantz R, Wright R, Huckman MS, et al. Central nervous system magnetic resonance imaging findings in myotonic dystrophy. Arch Neurol 1988; 45: 36–37. [DOI] [PubMed] [Google Scholar]
- 65. Romeo V, Pegoraro E, Ferrati C, et al. Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol 2010; 257: 1246–1255. [DOI] [PubMed] [Google Scholar]
- 66. Kornblum C, Reul J, Kress W, et al. Cranial magnetic resonance imaging in genetically proven myotonic dystrophy type 1 and 2. J Neurol 2004; 251: 710–714. [DOI] [PubMed] [Google Scholar]
- 67. Minnerop M, Weber B, Schoene-Bake JC, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011; 134: 3527–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abe K, Fujimura H, Soga F. The fluid-attenuated inversion-recovery pulse sequence in assessment of central nervous system involvement in myotonic dystrophy. Neuroradiology 1998; 40: 32–35. [DOI] [PubMed] [Google Scholar]
- 69. Huber S, Kissel J, Shuttleworth E, et al. Magnetic resonance imaging and clinical correlates of intellectual impairment in myotonic dystrophy. Arch Neurol 1989; 46: 536–540. [DOI] [PubMed] [Google Scholar]
- 70. Martinello F, Piazza A, Pastorello E, et al. Clinical and neuroimaging study of central nervous system in congenital myotonic dystrophy. J Neurol 1999; 246: 186–192. [DOI] [PubMed] [Google Scholar]
- 71. Ogata A, Terae S, Fujita M, et al. Anterior temporal white matter lesions in myotonic dystrophy with intellectual impairment: an MRI and neuropathological study. Neuroradiology 1988; 40: 411–415. [DOI] [PubMed] [Google Scholar]
- 72. Zanigni S, Evangelisti S, Giannoccaro MP, et al. Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. NeuroImage Clin 2016; 11: 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Conforti R, Cristofaro M, De, Cristofano A, et al. Brain MRI abnormalities in the adult form of myotonic dystrophy type 1: a longitudinal case series study. Neuroradiol J 2016; 29: 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Di Costanzo A, Di Salle F, Santoro L, et al. Brain MRI features of congenital- and adult-form myotonic dystrophy type 1: case-control study. Neuromuscul Disord 2002; 12: 476–483. [DOI] [PubMed] [Google Scholar]
- 76. Bajrami A, Azman F, Yayla V, et al. MRI findings and cognitive functions in a small cohort of myotonic dystrophy type 1: retrospective analyses. Neuroradiol J 2017; 30: 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Meola G, Sansone V, Perani D, et al. Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology 1999; 53: 1042–1050. [DOI] [PubMed] [Google Scholar]
- 78. Okkersen K, Monckton DG, Le N, et al. Brain imaging in myotonic dystrophy type 1. Neurology 2017; 89: 960–969. [DOI] [PubMed] [Google Scholar]
- 79. Antonini G, Mainero C, Romano A, et al. Cerebral atrophy in myotonic dystrophy: a voxel based morphometric study. J Neurol Neurosurg Psychiatry 2004; 75: 1611–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ota M, Sato N, Ohya Y, et al. Relationship between diffusion tensor imaging and brain morphology in patients with myotonic dystrophy. Neurosci Lett 2006; 407: 234–239. [DOI] [PubMed] [Google Scholar]
- 81. Schneider-Gold C, Bellenberg B, Prehn C, et al. Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS One; 10 Epub ahead of print 2015;10(6):e0130352. DOI: 10.1371/journal.pone.0130352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sugiyama A, Sone D, Sato N, et al. Brain gray matter structural network in myotonic dystrophy type 1. PLoS One 2017; 12: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Perrone D, Valentini F, Servidio S, et al. Analysis of intermittent heating in a multi-component turbulent plasma. Eur Phys J D 2014; 68: 1–7. [Google Scholar]
- 84. Franc DT, Muetzel RL, Robinson PR, et al. Cerebral and muscle MRI abnormalities in myotonic dystrophy. Neuromuscul Disord 2012; 22: 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Serra L, Petrucci A, Spanò B, et al. How genetics affects the brain to produce higher-level dysfunctions in myotonic dystrophy type 1. Funct Neurol 2015; 30: 21–31. [PMC free article] [PubMed] [Google Scholar]
- 86. Yoo WK, Park YG, Choi YC, et al. Cortical thickness and white matter integrity are associated with CTG expansion size in myotonic dystrophy type I. Yonsei Med J 2017; 58: 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wozniak JR, Mueller BA, Ward EE, et al. White matter abnormalities and neurocognitive correlates in children and adolescents with myotonic dystrophy type 1: a diffusion tensor imaging study. Neuromuscul Disord 2011; 21: 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wozniak JR, Mueller BA, Bell CJ, et al. Diffusion tensor imaging reveals widespread white matter abnormalities in children and adolescents with myotonic dystrophy type 1. J Neurol 2013; 260: 1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Serra L, Silvestri G, Petrucci A, et al. Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol 2014; 71: 603–611. [DOI] [PubMed] [Google Scholar]
- 90. Wozniak JR, Mueller BA, Lim KO, et al. Tractography reveals diffuse white matter abnormalities in Myotonic Dystrophy Type 1. J Neurol Sci 2014; 341: 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Caramia F, Mainero C, Gragnani F, et al. Functional MRI changes in the central motor system in myotonic dystrophy type 1. Magn Reson Imaging 2010; 28: 226–234. [DOI] [PubMed] [Google Scholar]
- 92. Toth A, Lovadi E, Komoly S, et al. Cortical involvement during myotonia in myotonic dystrophy: an fMRI study. Acta Neurol Scand 2015; 132: 65–72. [DOI] [PubMed] [Google Scholar]
- 93. Serra L, Mancini M, Silvestri G, et al. Brain connectomics’ modification to clarify motor and nonmotor features of myotonic dystrophy type 1. Neural Plast 2016; 2696085 Epub ahead of print 25 May 2016. DOI: 10.1155/2016/2696085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Park J, Seo J, Cha H, et al. Alterated power spectral density in the resting-state sensorimotor network in patients with myotonic dystrophy type 1. Sci Rep 2018; 8: 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fiorelli M, Duboc D, Mazoyer BM, et al. Decreased cerebral glucose utilization in myotonic dystrophy. Neurology 1992; 42: 91–94. [DOI] [PubMed] [Google Scholar]
- 96. Mielke R, Herholz K, Fink G, et al. Positron emission tomography in myotonic dystrophy. Psychiatry Res 1993; 50: 93–99. [DOI] [PubMed] [Google Scholar]
- 97. Annane D, Fiorelli M, Mazoyer B, et al. Impaired cerebral glucose metabolism in myotonic dystrophy: a triplet-size dependent phenomenon. Neuromuscul Disord 1998; 8: 39–45. [DOI] [PubMed] [Google Scholar]
- 98. Romeo V, Pegoraro E, Squarzanti F, et al. Retrospective study on PET-SPECT imaging in a large cohort of myotonic dystrophy type 1 patients. Neurol Sci 2010; 31: 757–763. [DOI] [PubMed] [Google Scholar]
- 99. Renard D, Collombier L, Castelli C, et al. In myotonic dystrophy type 1 reduced FDG-uptake on FDG-PET is most severe in Brodmann area 8. BMC Neurol 2016; 16: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Peric S, Brajkovic L, Belanovic B, et al. Brain positron emission tomography in patients with myotonic dystrophy type 1 and type 2. J Neurol Sci 2017; 378: 187–192. [DOI] [PubMed] [Google Scholar]
- 101. Chang L, Anderson T, Migneco O, et al. Cerebral abnormalities in myotonic dystrophy. Cerebral blood flow, magnetic resonance imaging, and neuropsychological tests. Arch Neurol 1993; 50: 917–923. [DOI] [PubMed] [Google Scholar]
- 102. Meola G, Sansone V, Perani D, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord 2003; 13: 813–821. [DOI] [PubMed] [Google Scholar]
- 103. Takeda A, Kobayakawa M, Suzuki A, et al. Lowered sensitivity to facial emotions in myotonic dystrophy type 1. J Neurol Sci 2009; 280: 35–39. [DOI] [PubMed] [Google Scholar]
- 104. Peric S, Pavlovic A, Ralic V, et al. Transcranial sonography in patients with myotonic dystrophy type 1. Muscle Nerve 2014; 50: 278–282. [DOI] [PubMed] [Google Scholar]
- 105. Krogias C, Bellenberg B, Prehn C, et al. Evaluation of CNS involvement in myotonic dystrophy type 1 and type 2 by transcranial sonography. J Neurol 2015; 262: 365–374. [DOI] [PubMed] [Google Scholar]
- 106. Udd B, Meola G, Krahe R, et al. Myotonic dystrophy type 2 (DM2) and related disorders. Report of the 180th ENMC Workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. Neuromuscul Disord 2011; 21: 443–450. [DOI] [PubMed] [Google Scholar]
- 107. Hund E, Jansen O, Koch MC, et al. Proximal myotonic myopathy with MRI white matter abnormalities of the brain. Neurology 1997; 48: 33–37. [DOI] [PubMed] [Google Scholar]
- 108. Kassubek J, Juengling FD, Hoffmann S, et al. Quantification of brain atrophy in patients with myotonic dystrophy and proximal myotonic myopathy: a controlled 3-dimensional magnetic resonance imaging study. Neurosci Lett 2003; 348: 73–76. [DOI] [PubMed] [Google Scholar]
- 109. Minnerop M, Luders E, Specht K, et al. Grey and white matter loss along cerebral midline structures in myotonic dystrophy type 2. J Neurol 2008; 255: 1904–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sharma A, Sane H, Badhe P, et al. A clinical study shows safety and efficacy of autologous bone marrow mononuclear cell therapy to improve quality of life in muscular dystrophy patients. Cell Transplant 2013; 22(Suppl. 1): S127–S138. [DOI] [PubMed] [Google Scholar]
- 111. Rakocevic-stojanovic V, Peric S, Savic-pavicevic D, et al. Brain sonography insight into the midbrain in myotonic dystrophy type 2. Muscle Nerve 2016; 53: 700–704. [DOI] [PubMed] [Google Scholar]
- 112. Johnston T, Hor K, Mah M, et al. Description of Becker muscular dystrophy cardiomyopathy natural history by cardiac magnetic resonance imaging from the first to third decades provides insight for cardiac surveillance. Neuromuscul Disord. Epub ahead of print 2018; 28(2018): S29–S146. DOI: 10.1016/j.nmd.2018.06.066. [DOI] [Google Scholar]
- 113. Fu Y, Dong Y, Zhang C, et al. Diffusion tensor imaging study in Duchenne muscular dystrophy. Ann Transl Med 2016; 4: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Haenggi T, Fritschy JM. Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell Mol Life Sci 2006; 63: 1614–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Young HK, Barton BA, Waisbren S, et al. Cognitive and psychological profile of males with becker muscular dystrophy. J Child Neurol 2008; 23: 155–162. [DOI] [PubMed] [Google Scholar]
- 116. Waite A, Brown SC, Blake DJ. The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci 2012; 35: 487–496. [DOI] [PubMed] [Google Scholar]
- 117. Ricotti V, Mandy WPL, Scoto M, et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 2016; 58: 77–84. [DOI] [PubMed] [Google Scholar]
- 118. Uchino M, Teramoto H, Naoe H, et al. Localisation and characterisation of dystrophin in the central nervous system of controls and patients with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry 1994; 57: 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hendriksen RGF, Schipper S, Hoogland G, et al. Dystrophin distribution and expression in human and experimental temporal lobe epilepsy. Front Cell Neurosci 2016; 10: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Doorenweerd N, Mahfouz A, Van Putten M, et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep 2017; 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nudel U, Zuk D, Einat P, et al. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature 1989; 337: 76–78. [DOI] [PubMed] [Google Scholar]
- 122. Lederfein D, Levy Z, Augier N, et al. A 71-kilodalton protein is a major product of the Duchenne muscular dystrophy gene in brain and other nonmuscle tissues. Proc Natl Acad Sci USA 1992; 89: 5346–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Austin R, Howard P, D’souza V, et al. Cloning and characterization of alternatively spliced isoforms of Dp71. Hum Mol Genet 1995; 4: 1475–1483. [DOI] [PubMed] [Google Scholar]
- 124. Morris GE, Simmons C, Man NT. Apo-Dystrophins (DP140 and DP71) and Dystrophin-Splicing Isoforms in Developing Brain. Biochem Biophys Res Commun 1995; 215: 361–367. [DOI] [PubMed] [Google Scholar]
- 125. Duchenne G. Recherches sur la paralysie musculaire pseudohyper trophique, ou paralysie myo-sclerosique. Archives Générales Médecine 1868; 11: 5–25, 179–209, 305–21, 421–43, 552–88. [Google Scholar]
- 126. Cotton S, Nicholas J, Kenneth M. Intelligence and Duchenne muscular dystrophy: full-scale, verbal, and performance intelligence quotients. Dev Med Child Neurol 2001; 43: 497–501. [DOI] [PubMed] [Google Scholar]
- 127. Hinton VJ, Vivo DCDE, Nereo NE. Selective deficits in verbal working memory associated with a known genetic etiology: the neuropsychological profile of duchenne muscular dystrophy NIH Public Access. J Int Neuropsychol Soc. Jan;7(1):45–54. 2001; 7: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hinton VJ, De Vivo DC, Fee R, et al. Investigation of poor academic achievement in children with Duchenne muscular dystrophy. Learn Disabil Res Pract 2004; 19: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hinton VJ, Fee RJ, Goldstein EM, et al. Verbal and memory skills in males with Duchenne muscular dystrophy. Dev Med Child Neurol 2007; 49: 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Billard C, Gillet P, Signoret J, et al. Cognitive functions in Duchenne muscular dystrophy: a reappraisal and comparison with spinal muscular atrophy. Neuromuscul Disord 1992; 2: 371–378. [DOI] [PubMed] [Google Scholar]
- 131. Castles A, Coltheart M. Varieties of developmental dyslexia. Cognition 1993; 47: 149–180. [DOI] [PubMed] [Google Scholar]
- 132. Dorman C, Hurley A, D’Avignon J. Language and learning disorders of older boys with Duchenne muscular dystrophy. Dev Med Child Neurol 1998; 30: 316–327. [DOI] [PubMed] [Google Scholar]
- 133. Pane M, Messina S, Bruno C, et al. Duchenne muscular dystrophy and epilepsy. Neuromuscul Disord 2013; 23: 313–315. [DOI] [PubMed] [Google Scholar]
- 134. Banihani R, Smile S, Yoon G, et al. Cognitive and neurobehavioral profile in boys with duchenne muscular dystrophy. J Child Neurol 2015; 30: 1472–1482. [DOI] [PubMed] [Google Scholar]
- 135. Goodwin F, Muntoni F, Dubowitz V. Epilepsy in Duchenne and Becker muscular dystrophies. Eur J Paediatr Neurol 1997; 1: 115–119. [DOI] [PubMed] [Google Scholar]
- 136. Septien L, Gras P, Borsotti JP, et al. [Mental development in Duchenne muscular dystrophy. Correlation of data of the brain scanner]. Pediatrie 1991; 46: 817–819. [PubMed] [Google Scholar]
- 137. Yoshioka M, Okuno T, Honda Y, et al. Central nervous system involvement in progressive muscular dystrophy. Arch Dis Child 1980; 55: 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Xu S, Shi D, Pratt SJP, et al. Abnormalities in brain structure and biochemistry associated with mdx mice measured by in vivo MRI and high resolution localized 1H MRS. Neuromuscul Disord 2015; 25: 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Doorenweerd N, Straathof CS, Dumas EM, et al. Reduced cerebral gray matter and altered white matter in boys with Duchenne muscular dystrophy. Ann Neurol 2014; 76: 403–411. [DOI] [PubMed] [Google Scholar]
- 140. Lv SY, Zou QH, Cui JL, et al. Decreased gray matter concentration and local synchronization of spontaneous activity in the motor cortex in Duchenne muscular dystrophy. Am J Neuroradiol 2011; 32: 2196–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bresolin N, Castelli E, Comi GP, et al. Cognitive impairment in Duchenne muscular dystrophy. Neuromuscul Disord 1994; 4: 359–369. [DOI] [PubMed] [Google Scholar]
- 142. Lee JS, Pfund Z, Juhász C, et al. Altered regional brain glucose metabolism in Duchenne muscular dystrophy: a pet study. Muscle Nerve 2002; 26: 506–512. [DOI] [PubMed] [Google Scholar]
- 143. Rae C, Scott RB, Thompson CH, et al. Brain biochemistry in Duchenne muscular dystrophy: a 1H magnetic resonance and neuropsychological study. J Neurol Sci 1998; 160: 148–157. [DOI] [PubMed] [Google Scholar]
- 144. van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010; 362: 1396–1406. [DOI] [PubMed] [Google Scholar]
- 145. Winkel LPF, Hagemans MLC, Van Doorn PA, et al. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol 2005; 252: 875–884. [DOI] [PubMed] [Google Scholar]
- 146. American Association of Neuromuscular Medicine Electrodiagnostic Medicine. Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve 2009; 40: 149–160. [DOI] [PubMed] [Google Scholar]
- 147. Raben N, Plotz P, Byrne B. Acid a-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2002; 2: 145–166. [DOI] [PubMed] [Google Scholar]
- 148. Di Rocco M, Buzz D, Tarò M. Glycogen storage disease type II: clinical overview. Acta Myol 2007; 26: 42–44. [PMC free article] [PubMed] [Google Scholar]
- 149. van der Walt J, Swash M, Leake J, et al. The pattern of involvement of adult-onset acid maltase deficiency at autopsy. Muscle Nerve 1987; 10: 272–281. [DOI] [PubMed] [Google Scholar]
- 150. Gambetti P, Dimauro S, Baker L. Nervous system in Pompe’s disease. J Neuropathology Exp Neurol 1971; 30: 412–430. [DOI] [PubMed] [Google Scholar]
- 151. Martin JJ, de Barsy T, Van Hoof F, et al. Pompe’s disease: an inborn lysosomal disorder with storage of glycogen - a study of brain and striated muscle. Acta Neuropathologica 1973; 23: 229–244. [DOI] [PubMed] [Google Scholar]
- 152. Chen CP, Lin SP, Tzen CY, et al. Detection of a homozygous D645E mutation of the acid alpha-glucosidase gene and glycogen deposition in tissues in a second-trimester fetus with infantile glycogen storage disease type II. Prenat Diagn 2004; 24: 231–232. [DOI] [PubMed] [Google Scholar]
- 153. van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003; 112: 332–340. [DOI] [PubMed] [Google Scholar]
- 154. Chien Y-H, Lee N-C, Peng S-F, et al. Brain development in infantile-onset Pompe disease treated by enzyme replacement therapy. Pediatr Res 2006; 60: 349–352. [DOI] [PubMed] [Google Scholar]
- 155. Burrow TA, Bailey LA, Kinnett DG, et al. Acute progression of neuromuscular findings in infantile pompe disease. Pediatr Neurol 2010; 42: 455–458. [DOI] [PubMed] [Google Scholar]
- 156. Ebbink BJ, Aarsen FK, Van Gelder CM, et al. Cognitive outcome of patients with classic infantile Pompe disease receiving enzyme therapy. Neurology 2012; 78: 1512–1518. [DOI] [PubMed] [Google Scholar]
- 157. Ebbink BJ, Poelman E, Plug I, et al. Cognitive decline in classic infantile Pompe disease: an underacknowledged challenge. Neurology 2016; 86: 1260–1261. [DOI] [PubMed] [Google Scholar]
- 158. Bloomfield A, Fletcher J, Hensman P, et al. Rapidly progressive white matter involvement in early childhood: the expanding phenotype of infantile onset Pompe? JIMD Reports 2017; 39: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tan QKG, Stockton DW, Pivnick E, et al. Premature pubarche in children with Pompe disease. J Pediatr 2015; 166: 1075–1078.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ebbink BJ, Poelman E, Aarsen FK, et al. Classic infantile Pompe patients approaching adulthood: a cohort study on consequences for the brain. Dev Med Child Neurol 2018; 60: 579–586. [DOI] [PubMed] [Google Scholar]
- 161. Borroni B, Cotelli MS, Premi E, et al. The brain in late-onset glycogenosis II: a structural and functional MRI study. J Inherit Metab Dis 2013; 36: 989–995. [DOI] [PubMed] [Google Scholar]
- 162. Brais B, Rouleau G, Bouchard J, et al. Oculopharyngeal muscular dystrophy. Semin Neurol 1999; 19: 56–99. [DOI] [PubMed] [Google Scholar]
- 163. Brais B. Oculopharyngeal muscular dystrophy: a polyalanine myopathy. Curr Neurol Neurosci Rep 2009; 9: 76–82. [DOI] [PubMed] [Google Scholar]
- 164. Abu-Baker A, Rouleau GA. Oculopharyngeal muscular dystrophy: recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim Biophys Acta 2007; 1772: 173–185. [DOI] [PubMed] [Google Scholar]
- 165. Linoli G, Tomelleri G, Ghezzi M. Oculopharyngeal muscular dystrophy. Description of a case with involvement of the central nervous system. Pathologica 1991; 83: 325–334. [PubMed] [Google Scholar]
- 166. Blumen SC, Bouchard JP, Brais B, et al. Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotes. Neurology 2009; 73: 596–601. [DOI] [PubMed] [Google Scholar]
- 167. Dubbioso R, Moretta P, Manganelli F, et al. Executive functions are impaired in heterozygote patients with oculopharyngeal muscular dystrophy. J Neurol 2012; 259: 833–837. [DOI] [PubMed] [Google Scholar]
- 168. Chen T, Lu XH, Wang HF, et al. Oculopharyngeal weakness, hypophrenia, deafness, and impaired vision: a novel autosomal dominant myopathy with rimmed vacuoles. Chin Med J 2016; 129: 1805–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Tawil R, Van Der Maarel S, Padberg GW, et al. 171st ENMC International Workshop: Standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2010; 20: 471–475. [DOI] [PubMed] [Google Scholar]
- 170. Fierro B, Daniele O, Aloisio A, et al. Evoked potential study in facio-scapulo-humeral muscular dystrophy. Acta Neurol Scand 1997; 95: 346–350. [DOI] [PubMed] [Google Scholar]
- 171. Funakoshi M, Goto K, Arahata K. Epilepsy and mental retardation in a subset of early onset 4q35-facioscapulohumeral muscular dystrophy. Neurology 1998; 50: 1791–1794. [DOI] [PubMed] [Google Scholar]
- 172. Brouwer OF, Padberg GW, Bakker E, et al. Early onset facioscapulohumeral muscular dystrophy. Muscle Nerve 1995; 18: S67–S72. [PubMed] [Google Scholar]
- 173. Mishra SK, Currier RD, Smith EE, et al. Facioscapulohumeral dystrophy associated with multiple sclerosis. Arch Neurol 1984; 41: 570–571. [DOI] [PubMed] [Google Scholar]
- 174. Di Lazzaro V, Oliviero A, Pilato F, et al. The physiological basis of transcranial motor cortex stimulation in conscious humans. Clin Neurophysiol 2004; 115: 255–266. [DOI] [PubMed] [Google Scholar]
- 175. Quarantelli M, Lanzillo R, Del Vecchio W, et al. Modifications of brain tissue volumes in facioscapulohumeral dystrophy. NeuroImage 2006; 32: 1237–1242. [DOI] [PubMed] [Google Scholar]
- 176. Carlier PG, Bertoldi D, Baligand C, et al. Muscle blood flow and oxygenation measured by NMR imaging and spectroscopy. NMR Biomed 2006; 19: 954–967. [DOI] [PubMed] [Google Scholar]
- 177. Doorenweerd N, Dumas EM, Ghariq E, et al. Decreased cerebral perfusion in Duchenne muscular dystrophy patients. Neuromuscul Disord 2017; 27: 29–37. [DOI] [PubMed] [Google Scholar]
- 178. Angelini C. Challenges and progress in the diagnosis of Congenital Muscular Dystrophies. Expert Opinion on Orphan Drugs 2016; 4:347–358. [Google Scholar]