

Abstract
Ion mobility and mass spectrometry techniques are used to investigate the stabilities of different conformations of bradykinin (BK, Arg1-Pro2-Pro3-Gly4-Phe5-Ser6-Pro7-Phe8-Arg9). At elevated solution temperatures, we observe a slow protonation reaction, i.e., [BK+2H]2++H+ → [BK+3H]3+, that is regulated by trans → cis isomerization of Arg1-Pro2, resulting in the Arg1-Cis-Pro2-cis-Pro3-Gly4-Phe5-Ser6-cis-Pro7-Phe8-Arg9 (all-cis) configuration. Once formed, the all-cis [BK+3H]3+ spontaneously cleaves the bond between Pro2-Pro3 with perfect specificity, a bond that is biologically resistant to cleavage by any human enzyme. Temperature-dependent kinetics studies reveal details about the intrinsic peptide processing mechanism. We propose that nonenzymatic cleavage at Pro2-Pro3 occurs through multiple intermediates and is regulated by trans → cis isomerization of Arg1-Pro2. From this mechanism, we can extract transition state thermochemistry: ΔG‡ = 94.8 ± 0.2 kJ·mol−1, ΔH‡ = 79.8 ± 0.2 kJ·mol−1, and ΔS‡ = −50.4 ± 1.7 J·mol−1·K−1 for the trans → cis protonation event; and, ΔG‡ = 94.1 ± 9.2 kJ·mol−1, →H‡ = 107.3 ± 9.2 kJ·mol−1, and →S‡ = 44.4 ± 5.1 J·mol−1·K−1 for bond cleavage. Biological resistance to the most favored intrinsic processing pathway prevents formation of Pro3-Gly4-Phe5-Ser6-cis-Pro7-Phe8-Arg9 that is approximately an order of magnitude more antigenic than BK.
Graphical Abstract
While investigating the solution structure of bradykinin (BK, Arg1-Pro2-Pro3-Gly4-Phe5-Ser6-Pro7-Phe8-Arg9) by a new hybrid temperature-controlled electrospray ionization (T-ESI),1–3 ion mobility spectrometry (IMS) and mass spectrometry (MS) technique,4 we find evidence for a conformationally regulated proton-transfer event that spontaneously cleaves the Pro2-Pro3 peptide bond with perfect specificity, leading to the smaller, seven-residue BK(3-9) species. This is a remarkable finding. In their comprehensive 1995 review, “Proline motifs in peptides and their biological processing,” Scharpe and co-workers emphasize, “the bond between two Pro residues, when not positioned at the N-terminus, possess a high degree of resistance to any human proteolytic enzyme.”5 Resistance arises because dipeptidyl peptidase IV (DPP-IV) cannot directly cleave Pro-Pro bonds.5 Instead, biological processing of N-terminal residues of BK is initiated by aminopeptidase P elimination of Arg1, and the remaining BK(2-9) sequence is then susceptible to DPP-IV processing.5 Below, we show that the most difficult bond to cleave enzymatically is intrinsically not only most facile, but uniquely specific and cleaved with 100% efficiency in the absence of enzymes. A comparison of our mechanism for spontaneous cleavage of free BK in solution with proposed enzymatic mechanisms suggests that the difference lies in the cis–trans configuration of Arg1-Pro2. The ability to follow detailed configurational changes that lead to peptide processing in the absence of enzymes provides a new means of understanding the role of peptidases and the importance of conformational changes in biological processing.
BK possesses hypotensive activity6 and is involved in numerous physiological conditions, including pain and inflammation.7–9 Structural studies show that conformations of BK depend upon its environment.10 C-terminal Ser6-Arg9 forms a stable turn in polar solvents.11 However, each proline can adopt either cis or trans configurations leading to multiple structures that are difficult to characterize,12 and some to conclude that the N-terminal region of BK is unstructured.13,14
Figure 1 shows typical mass spectra recorded using our T-ESI-IMS-MS4,15,16 technique, acquired upon incubating BK in an ethanol, 0.5% acetic acid solution (for acid concentration dependence, see Supporting Information) at 65 °C. Similar results are found when BK is incubated in water, methanol and propanol (see Supporting Information). Initially, two peaks are observed in the mass spectrum at m/z = 531 and 354, correspond to intact [BK+2H]2+ and [BK+3H]3+, respectively. As time progresses, the abundance of [BK+2H]2+ decreases and [BK+3H]3+ increases. By ~75 min, two new peaks, at m/z = 404 and 254 are observed. These peaks correspond to a complementary pair of fragments formed in solution and are assigned as the seven-residue [BK(3-9)+2H]2+ peptide and a fragment associated with the remaining Arg1-Pro2 dipeptide (discussed in detail below). This pair forms upon cleavage of Pro2-Pro3, which as noted above is enzymatically resistant. By ~150 min, the [BK(3-9)+2H]2+ and Arg1-Pro2 fragments dominate the mass spectrum, and are the only species observed beyond ~300 min. These data indicate that [BK+2H]2+ is converted to [BK+3H]3+ through a slow protonation reaction, and that the triply protonated peptide undergoes a cleavage of the Pro2-Pro3 peptide bond. This transformation is remarkably selective, ultimately forming the seven residue [BK(3-9)+2H]2+ product with ~100% efficiency.
Figure 1.
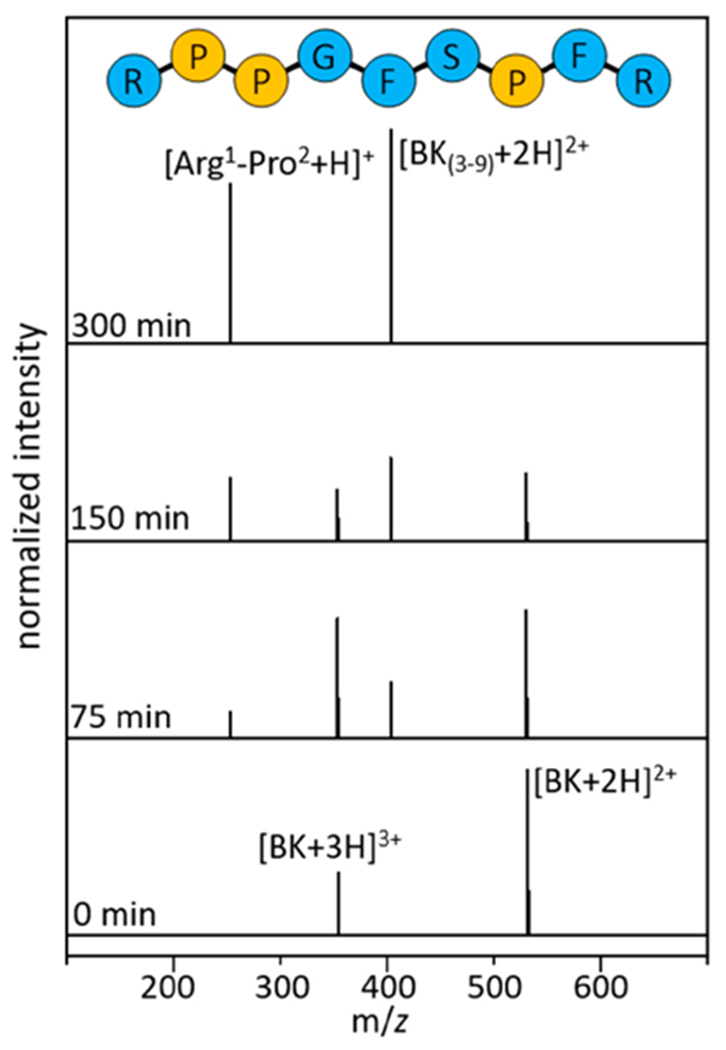
Mass spectral distributions shown at 0, 75, 150, and 300 min.
Of immediate interest is the slow protonation reaction. Such a slow transfer of a proton, the lightest and fastest chemical moiety, suggests that a key barrier must regulate protonation. A clue about this barrier comes from a recent study of polyproline-7 showing that a cooperative cis → trans isomerization of all peptide bonds was responsible for regulating the slowest proton transfer reaction ever measured.17 The slow protonation event observed here suggests a similar configurational change may regulate this system.
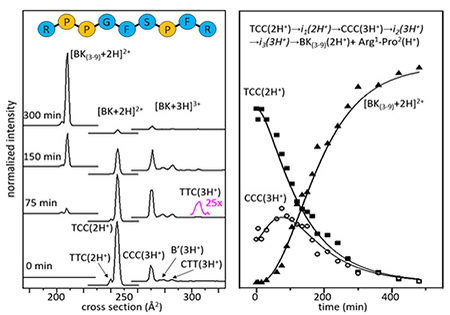
This structural transition can be directly tested for BK by examining IMS cross section distributions shown in Figure 2. The mobility of an ion through a buffer gas, under the influence of a weak field, depends on its collision cross section with the buffer gas.18–21 Structures having different cross sections arise from different combinations of cis- and trans-configurations of Pro2, Pro3, and Pro7 residues, which have been characterized previously by IMS-MS,12 and recently confirmed by cryogenic IR spectroscopy.22 For [BK+2H]2+, there are two experimentally resolved structures: a small peak, representing ~5% of the population at Ω = 240 Å2 and a more abundant species having Ω = 244 Å. The configurations of the three proline residues in these structures have been assigned as trans-Pro2, trans-Pro3, and cis-Pro7 [TTC(2H+)] for Ω = 240 Å2; and, trans-Pro2, cis-Pro3, cis-Pro7 [TCC(2H+)] for Ω = 244 Å2 (see Supporting Information). Four conformations resolved for [BK+3H]3+ are assigned to the following proline configurations: the largest peak at Ω = 269 Å2, corresponds to [CCC(3H+)];12 a peak at Ω = 278 Å2, in which the cis/trans configurations have not been assigned is designated as [B’(3H+)]; a low-abundance species at Ω = 285 Å2 as [CTT(3H+)];12,22 and, the small peak at Ω = 306 Å2 as [TTC(3H+)].12 Figure 2 shows that upon initiation of these cross section measurements, the TCC(2H+) configuration is most abundant. As the reaction progresses (~75 min) the fraction of TCC(2H+) decreases and there is a corresponding increase in the abundance of triply protonated BK in the CCC(3H+) form. This indicates that, as was observed for Pro7,17 the slow-protonation event is regulated by a conformational change; in the case of BK, the trans → cis conversion of the Arg1-Pro2 peptide bond [i.e., the doubly protonated TCC(2H+) conformation] is converted to the triply protonated all-cis CCC(3H+) conformation. In this way, the configuration of Arg1-Pro2 regulates the rate of the proton transfer event. This configurational change must allow access to an additional basic site that is preferentially protonated.
Figure 2.
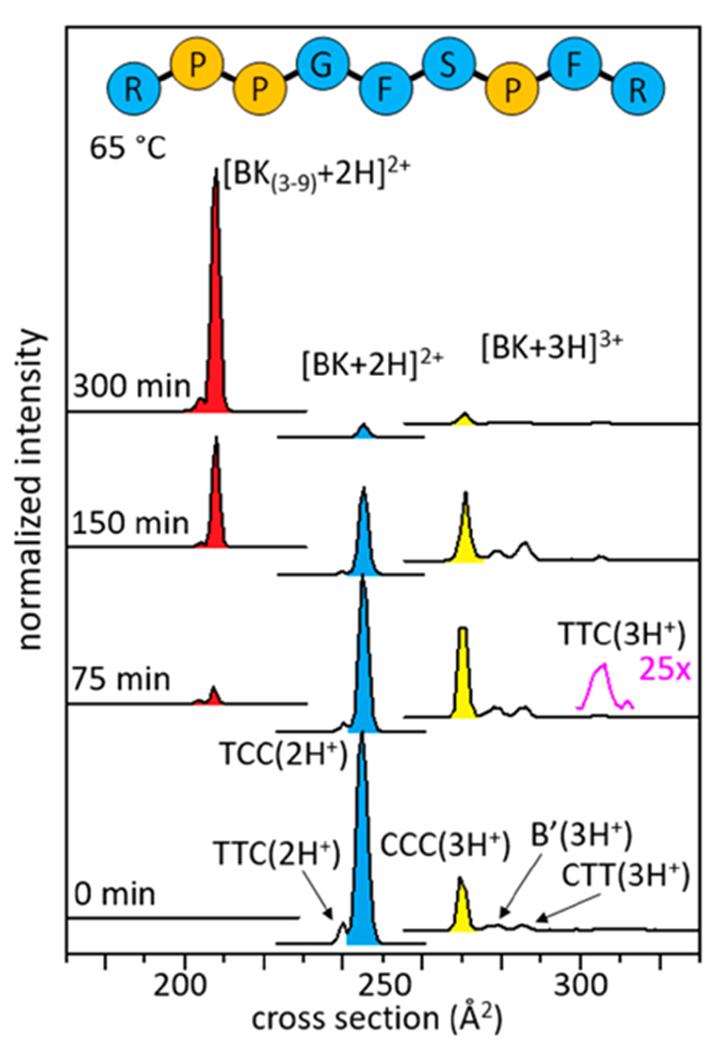
Cross section distributions shown at 0, 75, 150, and 300 min. The blue peak represents the TCC(2H+) configuration, the yellow peak represents CCC(3H+), and the red peak represents [BK(3-9)+2H]2+, which are the most abundant conformations in the IMS-MS distributions. The cross section for Arg1-Pro2 (not shown in the figure) is Ω = 96 Å2.
Two immediate questions emerge. How does Pro2-Pro3 cleavage occur? And, why would the intrinsically most-favored process be avoided biologically? To understand how Pro-Pro bond cleavage occurs in the absence of enzymes we need to understand the reaction mechanism. We begin by noting that the Arg1-Pro2 fragment lacks a C-terminal carboxylic acid moiety and thus likely exists as the well-known cyclized diketopiperazine (DKP) structure of Arg1-Pro2.23 The Arg1-Pro2 DKP species is formed when the N-terminus is in close proximity to the carbonyl carbon between the second and third residues. Scheme 1 illustrates a simple mechanism of formation arising from nucleophilic attack of the amino terminus on the carbonyl carbon atom,23 a process that cleaves the C—N peptide bond, yielding the cyclic Arg1-Pro2 DKP, and the linear seven-residue peptide (Pro3-Gly4-Phe5-Ser6-Pro7-Phe8-Arg9). Previous studies of a C-terminally amidated Ala-Pro-NH2 dipeptide reported an overall reaction rate that is also limited by the trans → cis isomerization of the Ala-Pro peptide bond,23 analogous to results reported here, and further corroborating our cis/trans configurational assignments. It is worth noting that this mechanism does not require the additional protonation event. Thus, the role of the third proton remains somewhat unclear; this change may be off pathway and unassociated with bond cleavage.
Scheme 1.
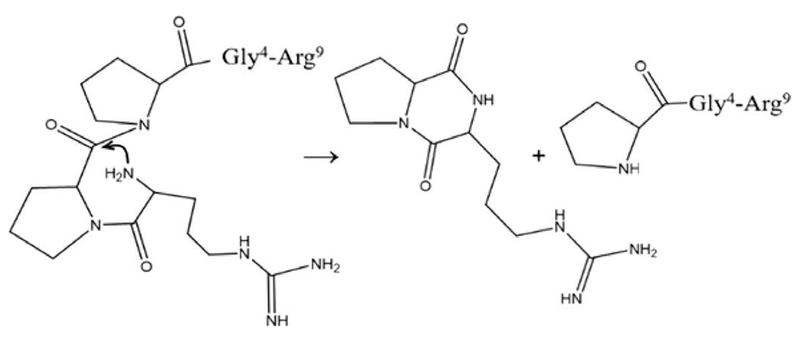
Cleavage mechanism of Pro2-Pro3 by DKP formation with Arg1-Pro2 in the cis configuration
Insight about how this occurs can be gleaned by modeling the experimental kinetics data shown in Figure 3. This approach provides insights about the details of reaction pathways.24,25 We first assume a model reaction pathway and then write differential rate equations for the assumed process. Upon solving the system of differential equations at all times and optimizing this solution with respect to the experiment, we obtain kinetics curves for the model pathway that can be compared with the experiment.
Figure 3.
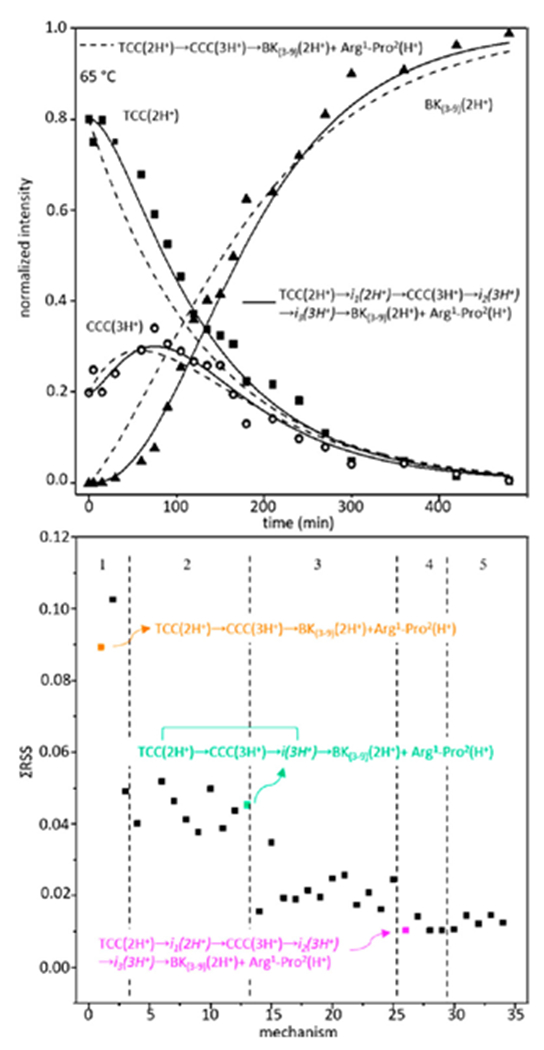
Kinetics data shown at 65 °C (top). Two models are shown with the solid line representing TCC(2H+) → i1(2H+) → CCC(3H+) → i2(3H+) → i3(3H+) →BK(3-9)(2H+)+Arg1-Pro2(H+), among the statistically best models, and the dashed line representing TCC(2H+) → CCC(3H+) → BK(3-9)(2H+)+Arg1-Pro2(H+), the simplest model. Bottom panel shows residual sums of squares values for each model, separated by dashed lines corresponding to 1-5 intermediates. Three example reaction pathways are shown.
Consider the model pathway shown by reaction 1:
| (1) |
this is the simplest model that includes the experimentally resolved intermediate [i.e., formation of CCC(3H+)]. Optimization of this model yields values of k1 = 7.5 × 10−3 s−1 for formation of CCC(3H+) and k2 = 1.4 × 10−2 s−1 for depletion of CCC(3H+) and formation of the product fragments at 65 °C. Dashed lines in Figure 3 represent this model pathway. Although this model captures the general shapes of the experimental kinetics curves, it unfortunately misses some key points. For example, experimental data shows that the TCC(2H+) → CCC(3H+) step is observed after a ~10 min induction period: the resulting shape is not captured by the single-intermediate decay process.
Fortunately, it is possible to calculate many different trial models. We have tested 34 possible model pathways (see Supporting Information). A representation of the quality of each of these processes (that include up to five intermediates and consider sequential and parallel pathways) is shown in Figure 3 as a residual sum of squares plot. From this analysis, we find that the best representation of experiment comes from a model pathway that includes a hidden intermediate24 associated with trans → cis isomerization of Arg1-Pro2 [i.e., TCC(2H+) → i1(2H+) → CCC(3H+)], and two additional intermediates associated with bond cleavage [i.e., CCC(3H+) → i2(3H+) → i3(3H+) → BK(3-9)(2H+)+Arg1-Pro2(H+)], leading to the complete process, reaction 2.
| (2) |
Though the structures of these intermediates remain entirely uncharacterized, evidence for their existence from this analysis is remarkable, as it suggests a minimum number of large barriers (five) associated with spontaneous dissociation of BK. Thus, we note that although this process is spontaneous, it is not easy. Measurements and analysis of these kinetics at different solution temperatures (T = 55, 60, 65, 70, 75, and 78 °C) can be used to obtain Arrhenius plots (see Supporting Information), and from transition state theory we can estimate step-by-step thermochemistry associated with each of these barriers. This analysis for our best pathway (i.e., reaction 2) yields transition state free energies of ΔG‡ = 93.1 ± 12.1, 94.8 ± 0.2, 94.0 ± 9.1, 93.9 ± 9.7, and 94.1 ± 9.2 kJ·mol−1 for each sequential step as reaction 2 progresses. When we partition the free energies into enthalpic (ΔH‡) and entropic (ΔS‡) terms, we find that the contributions of ΔH‡ to the barriers for the two steps TCC(2H+) → i1(2H+) → CCC(3H+) are 97.7 ± 12.1 kJ·mol−1 and 79.8 ± 1.6 kJ·mol−1, respectively. For the three steps after isomerization, which lead to dissociation [i.e., CCC(3H+) → i2(3H+) → i3(3H+) → BK(3-9)(2H+)+Arg1-Pro2(H+)], we obtain ΔH‡ = 105.8 ± 9.1, 106.7 ± 9.7, and 107.3 ± 9.2 kJ⋅mol−1, respectively. The corresponding entropic components of each step shows that a late step associated with trans → cis isomerization of the Arg1-Pro2 bond involves a tight, entropically restricted transition state [ΔS‡ = 15.3 ± 2.5 J⋅K−1⋅mol−1 for the first TCC(2H+) → i1(2H+) step and –50.4 ± 1.7 J⋅mol−1⋅K−1 for the later i1(2H+) → CCC(3H+) transition]. The entropic contributions to the three barriers associated with the CCC(3H+) → i2(3H+) → i3(3H+) → BK(3-9)(2H+)+Arg1-Pro2(H+) rearrangements and bond cleavage shows that each step proceeds through a relatively loose (entropically allowed) transition state (i.e., ΔS‡ = 39.4 ± 4.6, 42.8 ± 5.2, and 44.4 ± 5.1 J⋅mol−1⋅K−1, respectively, for sequential steps leading to bond cleavage).
With this understanding about how spontaneous Pro2-Pro3 dissociation occurs in the absence of enzymes, we turn our attention to the question of why would the most intrinsically favored process be avoided biologically? Our finding, that cleavage between Pro2-Pro3 is the only accessible pathway spontaneously, is remarkable considering that this bond is not processed enzymatically. It turns out that the seven-residue BK(3-9) is nearly an order of magnitude more antigenic than the BK nonapeptide as well as the eight-residue BK(2-9) peptide.26 Rather than form BK(3-9) enzymatically, BK can be processed by aminopeptidase P to form the eight-residue peptide (which lacks typical BK activity), followed by DPP-IV elimination of Pro2-Pro3, thus avoiding the antigenic repercussions associated with BK(3-9) formation.
Finally, it is important that while spontaneous Pro2-Pro3 cleavage is favored intrinsically, this processing is slow. Biologically, the deleterious antigenic impact of spontaneous cleavage is avoided by rapid enzymatic degradation of the Arg1-trans-Pro2 configured BK. Structural studies of penultimate proline containing peptides interacting with the DPP-IV enzyme show that the trans-Pro2-peptide is the bound form which leads to enzyme cleavage,27 consistent with this idea. In this way, the biological processing mechanism naturally regulates antigenicity, as enzymatic degradation of BK leads to inactive species7,28.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported in part by funds from the National Institutes of Health, R01 GM117207-03, the Indiana University Robert and Marjorie Mann endowment fellowship (C.R.C.), and a fellowship from the Indiana University College of Arts and Sciences (T.J.E.-B.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b04751.
Experimental details, sequence determination of [BK(3–9)+2H]2+, cis/trans assignments of [BK+2H]2+ conformations, kinetics data at other temperatures, BK dissociation in other solutions, list of trial models tested, acetic acid dependence plot, and Arrhenius plot (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Benesch JL; Sobott F; Robinson CV Anal. Chem. 2003, 75, 2208–2214. [DOI] [PubMed] [Google Scholar]
- (2).Wang G; Abzalimov RR; Kaltashov IA Anal. Chem. 2011, 83, 2870–2876. [DOI] [PubMed] [Google Scholar]
- (3).Cong X; Liu Y; Liu W; Liang X; Russell DH; Laganowsky AJ Am. Chem. Soc. 2016, 138, 4346–4349. [DOI] [PubMed] [Google Scholar]
- (4).El-Baba TJ; Woodall DW; Raab SA; Fuller DR; Laganowsky A; Russell DH; Clemmer DE J. Am. Chem. Soc. 2017, 139, 6306–6309. [DOI] [PubMed] [Google Scholar]
- (5).Vanhoof G; Goossens F; De Meester I; Hendriks D; Scharpe S FASEB J. 1995, 9, 736–744. [PubMed] [Google Scholar]
- (6).Abelous J; Bardier EC R. Soc. Biol. 1909, 66, 511–512. [Google Scholar]
- (7).Regoli D; Barabe J Pharmacol. Rev. 1980, 32, 1–46. [PubMed] [Google Scholar]
- (8).Dray A; Perkins M Trends Neurosci. 1993, 16, 99–104. [DOI] [PubMed] [Google Scholar]
- (9).Basbaum AI; Bautista DM; Scherrer G; Julius D Cell 2009, 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pierson NA; Chen L; Valentine SJ; Russell DH; Clemmer DE J. Am. Chem. Soc. 2011, 133, 13810–13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Young JK; Hicks RP Biopolymers 1994, 34, 611–623. [DOI] [PubMed] [Google Scholar]
- (12).Pierson NA; Chen L; Russell DH; Clemmer DE J. Am. Chem. Soc. 2013, 135, 3186–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Denys L; Bothner-By A; Fisher G Biochemistry 1982, 21, 6531–6536. [DOI] [PubMed] [Google Scholar]
- (14).Lopez JJ; Shukla AK; Reinhart C; Schwalbe H; Michel H; Glaubitz C Angew. Chem. 2008, 120, 1692–1695. [DOI] [PubMed] [Google Scholar]
- (15).Merenbloom SI; Koeniger SL; Valentine SJ; Plasencia MD; Clemmer DE Anal. Chem. 2006, 78, 2802–2809. [DOI] [PubMed] [Google Scholar]
- (16).Koeniger SL; Merenbloom SI; Valentine SJ; Jarrold MF; Udseth HR; Smith RD; Clemmer DE Anal. Chem. 2006, 78, 4161–4174. [DOI] [PubMed] [Google Scholar]
- (17).Shi L; Holliday AE; Khanal N; Russell DH; Clemmer DE J. Am. Chem. Soc. 2015, 137, 8680–8683. [DOI] [PubMed] [Google Scholar]
- (18).Mason EA; McDaniel EW Transport properties of ions in gases; Wiley: New York, NY, 1988. [Google Scholar]
- (19).Von Helden G; Hsu MT; Kemper PR; Bowers MT J. Chem. Phys. 1991, 95, 3835–3837. [Google Scholar]
- (20).Jarrold MF; Constant VA Phys. Rev. Lett 1991, 67, 2994–2997. [DOI] [PubMed] [Google Scholar]
- (21).Clemmer DE; Jarrold MF J. Mass Spectrom. 1997, 32, 577–592. [Google Scholar]
- (22).Voronina L; Scutelnic V; Masellis C; Rizzo TR J. Am. Chem. Soc. 2018, 140, 2401–2404. [DOI] [PubMed] [Google Scholar]
- (23).Capasso S; Vergara A; Mazzarella LJ Am. Chem. Soc. 1998, 120, 1990–1995. [Google Scholar]
- (24).El-Baba TJ; Kim D; Rogers DB; Khan FA; Hales DA; Russell DH; Clemmer DE J. Phys. Chem. B 2016, 120, 12040–12046. [DOI] [PubMed] [Google Scholar]
- (25).Shi L; Holliday AE; Shi H; Zhu F; Ewing MA; Russell DH; Clemmer DE J. Am. Chem. Soc. 2014, 136, 12702–12711. [DOI] [PubMed] [Google Scholar]
- (26).Redman L; Regoli D; Tustanoff ER Can. J. Biochem. 1979, 57, 529–539. [DOI] [PubMed] [Google Scholar]
- (27).Fischer G; Heins J; Barth A Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1983, 742, 452–462. [DOI] [PubMed] [Google Scholar]
- (28).Kuoppala A; Lindstedt KA; Saarinen J; Kovanen PT; Kokkonen JO Am. J. Physiol.: Heart Circ. Physiol. 2000, 278, H1069–H1074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.