

SUMMARY
FBXW7 is a tumor suppressive E3 ligase, whereas RAS-ERK and mechanistic target of rapamycin kinase (mTORC1) are two major oncogenic pathways. Whether and how FBXW7 regulates these two oncogenic pathways are unknown. Here, we showed that SHOC2, a RAS activator, is a FBXW7 substrate. Growth stimuli trigger SHOC2 phosphorylation on Thr507 by the mitogen-activated protein kinase (MAPK) signal, which facilitates FBXW7 binding for ubiquitylation and degradation. FBXW7-mediated SHOC2 degradation terminates the RAS-MAPK signals and inhibits proliferation. Furthermore, SHOC2 selectively binds to Raptor to competitively inhibit the Raptor-mTOR binding to inactivate mTORC1 and induce autophagy, whereas Raptor binding of SHOC2 inhibits the SHOC2-RAS binding to block the MAPK pathway and proliferation. Finally, SHOC2 is overexpressed in pancreatic cancer, which correlated with poor patient survival. SHOC2 mutations were found in lung cancer tissues with gain-of-function activity. Collectively, the SHOC2-Raptor interaction triggers negative cross-talk between RAS-ERK and mTORC1 pathways, whereas FBXW7 regulates both pathways by targeting SHOC2 for ubiquitylation and degradation.
Graphical Abstract
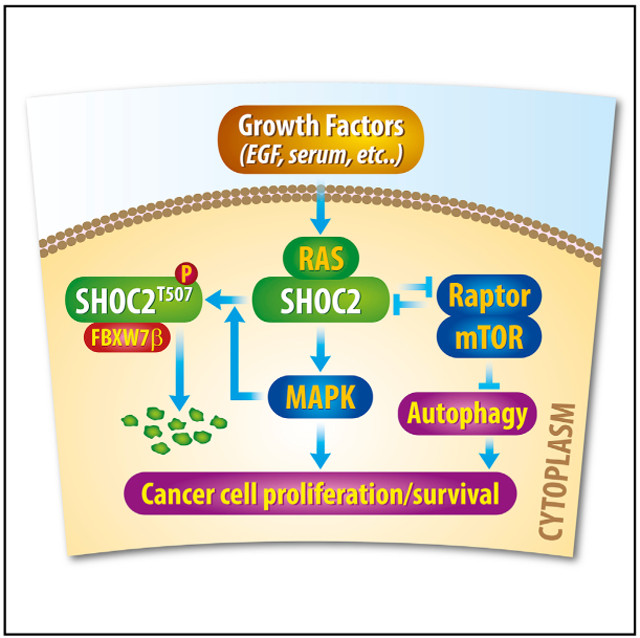
In Brief
In this study, Xie et al. show upon growth stimulation that RAS-MAPK is activated to phosphorylate SHOC2 on T507 to facilitate its binding with FBXW7β for ubiquitylation and degradation, thus establishing a negative feedback loop. Furthermore, the SHOC2-RAPTOR interaction can inactivate either pathway to keep proliferation and autophagy under precise control.
INTRODUCTION
FBXW7, a haploinsufficient tumor suppressor, is the substraterecognizing sub-unit of SCF E3 ubiquitin ligase, which promotes ubiquitylation and degradation of several key molecules governing major signaling pathways, including cellular myelocytomatosis (c-MYC) (Welcker et al., 2004; Yada et al., 2004), nuclear factor κB2 (NFκB2) (p100) (Fukushima et al., 2012), myeloid cell leukemia-1 (MCL-1) (Inuzuka et al., 2011; Wertz et al., 2011), neurofibromatosis type 1 (NF1) (Tan et al., 2011), c-JUN (Gu et al., 2007; Wei et al., 2005), Notch1 (O’Neil et al., 2007), Cyclin E (Koepp et al., 2001), and early meiotic induction protein 1 (EMI1) (Bernis et al., 2007; Margottin-Goguet et al., 2003; Wang et al., 2014). FBXW7 also facilitates non-homologous end joining (NHEJ) repair to maintain genome integrity (Zhang et al., 2016a). FBXW7 interacts with a specific conserved Cdc4 phospho-degron sequence ((L)-X-pT/pS-P-(P)-X-pS/pT) on its substrates. Proper phosphorylation of the substrate is required in most cases for FBXW7 to recognize and target its substrate for ubiquitylation (Welcker and Clurman, 2008). Low levels of FBXW7 expression in cancer tissues correlate with a poor prognosis, higher grade of malignancy, and dedifferentiation of cancer cells in several cancers (Berger et al., 2017; Gao et al., 2014; He et al., 2017; Wang et al., 2016; Wang et al., 2012; Welcker and Clurman, 2008). Interestingly, extracellular signal-regulated kinase (ERK) was reported to phosphorylate FBXW7 and promote its self-ubiquitylation in pancreatic cancer cells (Ji et al., 2015).
SHOC2 was first identified in Caenorhabditis elegans by serving as a scaffold for RAS and RAF and positively regulates the RAS-ERK pathway (Selfors et al., 1998; Sieburth et al., 1998). SHOC2 is an evolutionarily conserved protein, composed of an unstructured N-terminal domain and a long stretch of leucine-rich repeats (LRRS) (Jeoung et al., 2013). The N-terminal domain binds to RAS and RAF to activate ERK1 and ERK2 (Dai et al., 2006; Jeoung et al., 2013; Jeoung et al., 2016). In addition, SHOC2 is upregulated in the majority of human cancers (Young et al., 2013). Interestingly, in cancer cells with constitutive RAS activity, SHOC2 is still active to enhance anchorage-independent growth, clonal survival, and growth in nude mice (Young et al., 2013). In pancreatic cancer cells with RAS mutations, SHOC2 knock down inhibits mitogen-activated protein kinase (MAPK) but not phosphatidylinositol 3-kinase (PI3K) activity (Rodriguez-Viciana et al., 2006), which was also seen in other types of cancer cells with active Ras (Jang et al., 2015). HUWE1 E3 ligase was reported to ubiquitylate SHOC2, not for its degradation, but for facilitating RAF ubiquitylation and degradation (Jang et al., 2014).
In mammalian cells, mechanistic target of rapamycin kinase (mTOR) exists in two multi-protein complexes: mLST8, Raptor, Deptor, and PRAS40 form mTORC1 and mLST8, mSin1, Rictor, Deptor, and Protor-1 and Protor-2 form mTORC2. Although Raptor is necessary for mTORC1 activity, Rictor and mSin1 are needed for mTORC2 activity (Guertin et al., 2006; Sabatini, 2006). mTORC1 is mainly involved in regulation of protein translation, cell size, and cell proliferation by phosphorylating ribo-somal protein S6 kinase (S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (eIF-4E-BP1), whereas mTORC2 regulates cell survival by directly phosphorylating and activating RAC-alpha serine/threonine-protein kinase (AKT) and serum/ glucocorticoid regulated kinase 1 (SGK1) (Guertin and Sabatini, 2006). Furthermore, mTORC is a well-established negative regulator of autophagy (Jung et al., 2010; Shintani and Klionsky, 2004), a process involved in many physiological and pathological processes (Mizushima et al., 2010). Although mTORC1 inhibits autophagosome formation, mTORC2 represses the expression of some autophagy-related genes (ATG) and other autophagy regulators (Cardenas et al., 1999; Levine and Klionsky, 2004; Narita et al., 2009).
Although the RAS-ERK and mTORC1 signals are two common oncogenic pathways, there is no systematic study to investigate whether a cross-talk between two pathways exists to coordinated regulate cell growth and survival. Still unknown is whether and how both pathways are subjected to FBXW7 regulation. In this study, we showed that SHOC2 is a substrate of FBXW7, and subjected to FBXW7-mediated ubiquitylation and degradation upon exposure to growth factors (e.g., epidermal growth factor [EGF]). We further showed that SHOC2 specifically interacts with Raptor. The SHOC2 binding of Raptor inactivates the mTORC1 signal to induce autophagy, whereas the Raptor binding of SHOC2 inactivates the RAS-ERK signal to suppress growth and survival. Finally, SHOC2 was found to be overexpressed in pancreatic cancer, which is positively correlated with poor patient survival, whereas multiple SHOC2 mutations were found in human lung cancer with the gain-of-function activity. Collectively, our study provides, to the best of our knowledge, demonstration that the SHOC2-Raptor interaction triggers negative cross-talk between the RAS-ERK and mTORC1 pathways, which is under tight control by tumor suppressor FBXW7 by targeted degradation of SHOC2.
RESULTS
FBXW7 Binds to SHOC2 and Shortens Its Protein Half-Life
In the process of characterizing Erbin, a negative regulator of RAS/RAF signal, as a substrate of SCFβTrCP E3 ubiquitin ligase (Xie et al., 2015), we unexpectedly found that SHOC2, a positive regulator of RAS/RAF can also be accumulated upon the treatment of MLN4924, a small molecule inhibitor of SCF E3 ligase (Soucy et al., 2009), in a dose -dependent manner in keratinocytes and multiple lung cancer lines (Figures S1A and S1B), suggesting SHOC2 is likely a substrate of SCF E3 ligase. Given that SHOC2 is an oncogenic protein, we reasoned that it would be ubiquitylated and degraded by a tumor suppressor, such as FBXW7. Consensus binding motif search on SHOC2 identified two evolutionarily conserved sites for FBXW7 binding at residues 240–243 (LITL) and 505–508 (LLTH) with one mismatch of P to L or H (Figure S1C) (Kwon et al., 2012; Tan et al., 2011).
We followed up with this lead and found that FBXW7, upon being ectopically expressed in 293 cells, pulled down endogenous SHOC2 (Figures 1A and 1B). Two proteins at endogenous levels also bound to each other in H1299 cells (Figures 1C and 1D). We further found that ectopically expressed FBXW7, but not its F-box-deleted mutant (FBXW7ΔF), reduced the endogenous SHOC2 level, which can be rescued by the treatment of MG132, a proteasome inhibitor (Figure 1E). When co-transfected, FBXW7 reduced the SHOC2 level in a dose-dependent manner (Figure 1F). Furthermore, FBXW7, but not its FBXW7ΔF mutant, shortened the protein half-life of exogenously expressed or endogenous SHOC2 without affecting SHOC2 mRNA (Figures 1G, 1H, S1D, and S1E). On the other hand, Fbxw7 deletion in mouse embryonic fibroblasts caused SHOC2 accumulation (Figure 1I), and FBXW7 knock down extended the protein half-life of SHOC2 (Figure 1J). Finally, we made two SHOC2 mutants, SHOC2-T242A and SHOC2-T507A, within putative FBXW7-binding sites (Figure S1C) to determine which consensus motif is required for FBXW7 binding. We found that FBXW7 bound to wild-type SHOC2 and its T242A mutant but not the T507A mutant (Figure S1F). Although the T242A mutant was still sensitive to FBXW7-induced degradation, as evidenced by the shortened protein half-life, T507A became resistant (Figure 1K), indicating that the Thr507 residue within the second FBXW7-binding motif of the SHOC2 protein is responsible for mediating the SHOC2-FBXW7 binding. Taken together, our results strongly suggest that SHOC2 is a substrate of FBXW7.
Figure 1. FBXW7 Binds to SHOC2 and Shortens Its Protein Half-Life.

(A and B) 293 cellsweretransfected with the indicated plasmidsfor 48 h, followed by immunoprecipitation and immunoblotting (IB) with anti-FLAG antibody (Ab) (A) or anti-mouse lgG antibody (B). WCE, whole-cell extract.
(C and D) H1299 cells were lysed and incubated with normal immunoglobulin G (IgG), anti-SHOC2 (C), or anti-FBXW7 Ab (D), and then pulled down with protein A/G beads, followed by IB.
(E and F) H1299 (E) or 293 (F) cells were transfected with indicated plasmids for 48 h; one indicated sample was treated with MG132 4 h before harvesting, followed by IB.
(G, H, J, and K) 293 (G and K) and H1299 (H and J) cells were transfected with indicated plasmids(G, H, and K) or siRNA targeting FBXW7 (J) for 48 h, followed by cycloheximide (CHX) treatment for different time periods and harvested for IB. The band density was quantified by AlphaEaseFC software. The data shown are from a single representative experiment out of two repeats.
(I) MEF cells derived from two independent Fbxw7-fl/fl mouse embryos (Zhang et al., 2016b) were infected with adenovirus encoding Cre-recombinase (Ad-Cre) to remove floxed exon of Fbxw7 or Ad-GFP control, followed by IB.
Growth Factor Triggers SHOC2 Phosphorylation on Thr507 to Facilitate FBXW7 Binding and Ubiquitylation
Having established that FBXW7 shortened the protein half-life of SHOC2 without affecting its mRNA, we next used both in vivo and in vitro ubiquitylation assays to confirm that SHOC2 is a substrate of FBXW7 for targeted ubiquitylation and degradation. Indeed, FBXW7, but not mutant FBXW7ΔF, promoted polyubiquitylation of wild-type SHOC2, as well as its T242A mutant, but not the T507A mutant (Figures 2A–2C).
Figure 2. FBXW7 Promotes SHOC2 Ubiquitylation, Triggered by MAPK-Mediated Phosphorylation on Thr507.

(A) 293 cells were transfected with the indicated plasmids, lysed with 6 M guanidine solution, pulled down with Ni-bead, and followed by IB for GST-SHOC2, or direct IB.
(B and C) 293 cells were transfected with WT SHOC 2 (B) or SHOC2 mutants (C), followed by IP purification and in vitro ubiquitylation. The blot was probed with anti-GST Ab.
(D and E) A549 cells (D) or Miapaca-2 and H1299 cells (E) were treated with EGF or serum for indicated time periods, followed by IB for p-SHOC2 (T507), SHOC2, p-ERK, and ERK.
(F and G) H1299 cells were transfected with MEK1 siRNA (siMEK1) in the presence (G) or absence (F) of EGF or treated with MEK1 inhibitor (PD98059, MEKi) for 48 h, followed by IB.
(H) A549 cells were transfected with Flag-FBXW7 for 48 h in the presence or absence of EGF (50 ng/ml) for 5 min, followed by immunoprecipitation and IB with indicated Abs. WCE, whole-cell extract.
(I and J) HCT116 FBXW7−/− cells were transfected with indicated plasmids (I) or MEK1 siRNA in the presence or absence of MEKi (PD98059) (J), followed by indicated serum treatment. Cells were then lysed with 6 M guanidine solution, pulled down with Ni-bead, and followed by IB. WCE, whole-cell extract.
It has been established that phosphorylation of a substrate at its F-box binding motif is prerequisite in most cases for its binding to the F-box protein for subsequent ubiquitylation and degradation (Deshaies and Joazeiro, 2009). Because FBXW7 bound to and promoted ubiquitylation of both wild-type SHOC2 and its T242A mutant, but not the T507A mutant, we reasoned that the residue Thr507, but not Thr242, is likely being phosphorylated prior to FBXW7 binding. We, therefore, made a phospho-anti-body, specifically recognizing phospho-Thr507 of SHOC2. Using this antibody, we found that pSHOC2-Thr507 and total SHOC2 were accumulated upon the treatment of MLN4924 in a dose-dependent manner in FBXW7 wild-type cells but to a much lesser degree in FBXW7 null cells (Figures S2A and S2B). To identify signal that could trigger SHOC2-Thr507 phosphorylation, we serum-starved cells, followed by addition of EGF or serum. Significantly, the pSHOC2-Thr507 levels were increased within 5 min after EGF addition in EGF dose-dependent manner and remained high at 10 min (Figure 2D). The pSHOC2-Thr507 levels were also increased by serum addition in a time-dependent manner within 2–4 h (Figure 2E).
We next attempted to identify the kinases that are activated upon exposure to growth factors to phosphorylate SHOC2 on Thr507. It is well-known that growth factor EGF and serum activate the MAPK pathway. Indeed, both EGF and serum induced ERK phosphorylation and activation in dose- and time-dependent manners, and importantly, the pSHOC2-Thr507 levels correlated well with phospho-extracellular regulated MAPK (pERK1 and pERK2) levels (Figures 2D and 2E). Inactivation of pERK by mitogen-activated protein kinase kinase 1 (MEK1) knock down or MEK inhibitor treatment significantly decreased the basal as well as EGF-induced levels of p-SHOC2T507 (Figures 2F and 2G), suggesting the MAPK signal is responsible for SHOC2 phosphorylation on T507. Furthermore, EGF treatment enhanced SHOC2-FBXW7 binding (Figure 2H).
In cells, FBXW7 has three isoforms with α form being in the nucleus, β form in the cytoplasm, and γ form in the nucleolus (Welcker and Clurman, 2008). We next determined which isoform is responsible for SHOC2 ubiquitylation, with focus on the α and β forms, and found that the β form played a major role (Figure 2I), consistent with cytoplasmic localization of SHOC2. Strikingly, FBXW7β-induced SHOC2 polyubiquitylation was near completely abrogated by either MEK1 knock down or MEK inhibitor treatment (Figure 2J). Finally, lentivirus-based FBXW7 knock down significantly extended the protein half-life of pSHOC2-Thr507 (Figure S2C). Collectively, we conclude that the MAPK signal is responsible for SHOC2 phosphorylation on Thr507 upon exposure to growth factors, which facilitates FBXW7 binding for subsequent ubiquitylation.
SHOC2 Promotes Cell Proliferation that Is Blocked by FBXW7 and ERK Silencing
Given that SHOC2 is an activator of the RAS-ERK signal, we measured its effect on cell proliferation in lung cancer cells and found that ectopic expression of SHOC2 indeed promotes growth. This growth-promoting activity was greater in the degradation-resistant T507A mutant but lesser than FBXW7 knock down, as expected (Figures 3A and 3B). When transfected in combination of expressing plasmids (Figure 3C), SHOC2 promoted growth and survival, which were completely abrogated by FBXW7 (Figure 3D; Figures S3A and S3B). On the other hand, apoptotic cell death induced by FBXW7 can be partially blocked by SHOC2 (Figure 3E). Reciprocally, SHOC2 knock down inhibited cell growth and colony formation, which were blocked by simultaneous FBXW7 knockdown (Figures 3F, 3G, S3C, and S3D), whereas SHOC2 knock down promoted apoptosis, which was blocked by FBXW7 knock down (Figure 3H). Furthermore, SHOC2 induced ERK phosphorylation, as a result of RAS-ERK activation, and largely blocked ERK inactivation by FBXW7 or MEK inhibitor PD98059 (Figures 3I, S3E, and S3F). On the other hand, the promotion of growth or survival induced by SHOC2 overexpression or FBXW7 knock down could be largely abrogated by the MEK inhibitor or ERK knock down (Figures 3J–3L and S3G–S3J). Taken together, it appears that SHOC2-mediated cell proliferation by activation of the RAS-ERK signal is negatively regulated by FBXW7. Thus, the growth suppression activity of FBXW7 can be achieved, at least in part, by promoting SHOC2 ubiquitylation and degradation, leading to inactivation of RAS-ERK signaling pathway.
Figure 3. SHOC2 Promotes Cell Growth, Blocked by FBXW7 and ERK Silencing.

(A–H) A549 (A, B, E, and H) or H1299 (C, D, F, and G) cells were transfected with the indicated plasmids or siRNAs. Forty-eight h later, a portion of cells were harvested for RT-PCR or IB analysis (A, C, and F). Another portion was grown for 48 h, cell numbers (left: B, D, and G) were counted, and percentage of apoptotic cells (E and H) were measured. Still other portions were plated into 6-well plates, and colonies with more than 50 cells counted after 9–13 days (right: B, D, and G). The error bars represent SEM from three independent experiments; *p < 0.05, **p < 0.01, and ***p < 0.001.
(I–L) H1299 (I–L) or A549 (I, right) cells were transfected with indicated plasmids or siRNAs and harvested 48 h later for IB (I and J) or counting cell numbers (K) and colonies (L). The error bars represent SEM from three independent experiments; *p < 0.05, **p < 0.01, and ***p < 0.001.
SHOC2 Induces Autophagy by mTORC1 Inactivation
We next pursued the nature of enhanced cell survival and found that cells with ectopic SHOC2 expression (Figure S4A) showed the sign of autophagy with accumulation of autophagic vacuoles, by using two independent methods of Cyto-ID autophagy detection kit (Figures 4A and S4B) and LC3 staining (Figures 4B and S4C). To distinguish induction of autophagy and blockage of degradation, we analyzed autophagic flux in the presence and absence of the lysosomal inhibitor chloroquine (CQ), and found that CQ could further promote LC3-II accumulation, compared to SHOC2 action alone (Figure 4C), suggesting SHOC2 indeed induced autophagy rather than blocked degradation. We next determined the potential contribution of SHOC2-induced autophagy on cell proliferation and survival by ATG5 knock down (Figure 4D) to abrogate autophagy. Although ATG5 knock down alone had no effect on cell growth, it blocked SHOC2-enhanced growth (Figures 4E and S4D). On the other hand, ATG5 knock down significantly inhibited colony formation, and its effect on SHOC2-enhanced survival is less obvious but still statistically different (Figures 4F and S4E).
Figure 4. SHOC2 Promotes Autophagy by mTOR Inactivation for Cell Proliferation.

(A and B) H1299 cells were transfected with GST-SHOC2 for 48 h and then stained with Cyto-ID Autophagy detection kit (A) or with LC3 Ab (B). Cells containing more than 10 autophagic vacuoles (A) or 5 LC3 dots (B) were counted as autophagic cells with at least 200 cells counted in each group. The data shown are from a single representative experiment out of three repeats. ***p < 0.001. Error bar represents SEM. Scale bar, 20 μm.
(C–G) H1299 cells were transfected with GST-SHOC2 or siRNA targeting SHOC2 (siSHOC2) or ATG5 (siATG5) for 24 h, followed by exposure to 10 μM CQ (C) for 24 h or for 48 h (D–G). Cell number was counted (E). A portion of cells were harvested for IB analysis (C, D, and G). The other portion of cells was plated into 6-well plates and colonies counted after 9–13 days (F). The error bars represent SEM from three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001.
(H and I) A549 (H) or Miapaca-2 (I) cells were transfected with indicated plasmids for 48 h, followed by IB with indicated Abs.
Given that mTORCI is a major negative player of autophagy induction, we determined whether SHOC2 promoted autophagy by mTORC1 inactivation. Indeed, ectopically expressed SHOC2 inhibited mTORC1 activity, whereas SHOC2 knock down activated it, as evidenced by decreased or increased phosphorylation of S6K and 4E-BP1, respectively (Figure 4G). Also, ectopic expression of the SHOC2-T507A mutant inhibited mTORC1 activity to the extent similar to wild-type SHOC2 (Figure 4H). We further determined the effect of FBXW7 on SHOC2-induced mTORC1 inactivation. Indeed, ectopic expression of SHOC2 caused a dose-dependent inactivation of mTORC1, which was completely abrogated by simultaneous expression of FBXW7β (Figure 4I). Finally, we found that SHOC2-induced autophagy could be completely blocked by mTORC1 inhibitor rapamycin (Figure S4F). Collectively, our results strongly suggest that autophagy induction indeed plays a role in SHOC2-induced stimulation of growth and survival.
SHOC2 Binds to Raptor and Competitively Inhibits the Raptor-mTOR Binding
To elucidate the mechanism by which SHOC2 inactivates mTORC1, we performed immunoprecipitation assay and found that mTOR and Raptor, but not Rictor, were detected in SHOC2 immunoprecipitates (Figures 5A and S5A), whereas SHOC2 was detected in both Raptor and mTOR immunoprecipitates (Figures 5B, 5C, S5B, and S5C), indicating that SHOC2-Raptor bound together under the physiological condition and that SHOC2 interacted with mTORC1 (by Raptor), but not mTORC2. We next determined whether the SHOC2-Raptor binding would affect the Raptor-mTOR binding and found that SHOC2 competed away Raptor but not Rictor from the mTOR binding in a dose-dependent manner (Figures 5D, 5E, S5D, and S5E). Reciprocally, SHOC2 silencing enhanced the Raptor-mTOR binding without affecting the Rictor-mTOR binding (Figures 5F, 5G, S5F, and S5G). Taken together, these results indicate that SHOC2 binds to Raptor and competitively inhibits Raptor-mTOR binding.
Figure 5. SHOC2 Binds to Raptor and Competitively Inhibits Raptor-mTOR Binding.

(A–C) H1299 (A) or Huh7 (B and C) cell lysates were prepared for IP, followed by IB with indicated Abs.
(D–G) H1299 (D and F) or A549 (E and G) cells were transfected with indicated plasmids or siRNA oligos for 48 h, followed by IP or IB with indicated Abs.
(H and I) 293 cells were transfected with indicated plasmids of SHOC2 (H) or Raptor (I) for 48 h, followed by IP and IB with indicated Abs. The domain structure of Raptor is shown (I, top).
(J and K) H1299 cells were transfected with indicated plasmids for 48 h, followed by IB (J), or staining with Cyto-ID Autophagy detection kit (K). Shown are mean ± SEM from three independent experiments, **p < 0.01, ***p < 0.001.
We then mapped the interaction region(s) between SHOC2 and Raptor. We first did a pull-down assay by using full-length glutathione S-transferase (GST)-SHOC2 as well as three GST-tagged SHOC2 deletion mutants individually with full-length hemagglutinin (HA)-Raptor protein and found all tested constructs bound to Raptor (Figure 5H), indicating the entire region of SHOC2 could bind to Raptor to block its binding with mTOR, although the C terminus (residues 347–582) showed the strongest binding. We then made three deletion mutants of HA-Raptor for binding with endogenous full-length SHOC2 and found the Raptor-binding regions are at the center (residues 351–900) and C terminus (residues 901 to 1335), with the C terminus showing the better binding (Figure 5I). Interestingly, the Raptor-mTOR binding region on Raptor is also mapped at the center (residues 351–900) (Figure 5I), thus providing a structural basis for direct competition between SHOC2 and mTOR for Raptor binding. Finally, we determined whether ectopic expression of these Raptor-binding regions of SHOC2 would trigger autophagy. Indeed, we found a good correlation, as evidenced by the stronger the Raptor binding, the better mTORC1 inactivation, and autophagy induction (Figures 5J, 5K, and S5H).
Raptor Negatively Regulates the RAS-ERK Signal by Disrupting the SHOC2-RAS Binding
We next determined the potential effect of the SHOC2-Raptor binding on the RAS-ERK signal. Ectopic Raptor expression inhibited the SHOC2-RAS binding and ERK phosphorylation in a dose-dependent manner (Figure 6A), whereas Raptor knock down significantly increased both basal and serum-induced SHOC2-RAS binding (Figure 6B) to activate the RAS-ERK signal, as reflected by increased ERK phosphorylation (Figure 6C). Thus, Raptor appears to be a negative regulator of the RAS-ERK signal by competing with RAS for SHOC2 binding.
Figure 6. Raptor Negatively Regulates the RAS-ERK Signal by Disrupting the SHOC2-RAS Binding.

(A–C) MiaPaCa-2 pancreatic cancer cells were transfected with indicated concentrations of HA-Raptorplasmids (A) or siRNA oligos targeting Raptor (B and C) for 48 h, followed by IP or IB with indicated Abs or serum-starved for 24 h, followed by serum addition (C). Cells were harvested at various time points for IB.
(D and E) MiaPaCa-2 cells were transfected with the indicated plasmids (D) or siRNA oligos (E) for 48 h. Transfected cells were plated into 6-well plates. Cell numbers or colonies were counted at 48 h or 9 days later. The error bars represent SEM from three independent experiments; *p < 0.05 and **p < 0.01.
(F and G) H1299 cells were transfected with indicated siRNA oligos or short hairpin RNA (shRNA), followed by IB (F) and clonogenic assay (G).
(H-J) HCT116 cells with or without FBXW7 were transfected with indicated siRNAs for 48h, followed by IB with indicated Abs (H), cell growth analysis using cell number (I), or autophagy analysis using LC3 staining (J). Scale bar, 10 μm. The error bars represent SEM from three independent experiments; *p < 0.05 and **p < 0.01.
We then determined the biological significance of Raptor manipulation on the RAS-ERK signal. Under all tested growth conditions, including low serum (2%), normal serum (10%) or EGF addition, ectopic Raptor expression significantly inhibited cell growth and survival (Figures S6A and S6B), whereas SHOC2-stimulated growth and survival were blocked by Raptor (Figure 6D). On the other hand, SHOC2 knock down-induced suppression of growth or survival was blocked by simultaneous Raptor knock down (Figure 6E). Raptor knock down stimulated cell growth and clonogenic survival, which was abrogated by ERK inactivation by either genetic approach (RNA silencing) (Figures 6F, 6G, and S6C–S6E) or pharmacological approach (MEK inhibitor) (Figures S6F and S6G), indicating that Raptor effect is indeed dependent of the RAS-ERK signal.
Having established a previously unrealized negative cross-talk between MAPK (for proliferation) and mTORC1 (for autophagy) by the SHOC2-Raptor interaction, we next determined the role of FBXW7 in the fine-tuning of this balance, by using paired HCT116 cells with or without FBXW7. As shown in Figures 6H–6J, SHOC2 knock down suppressed cell growth but had no effect on autophagy regardless of FBXW7 status in HCT116 cells with fully activated mTORC1. However, in HCT116 cells harboring wt-FBXW7, Raptor knock down, on one hand, inactivated mTORC1 and induced autophagy and, on the other hand, slightly activated ERK but without affecting overall cell growth. In FBXW7 null HCT116 cells, however, Raptor knock down further inactivated mTORC1 to trigger a much greater degree of autophagy, and at the same time, activated ERK to promote growth. Thus, the FBXW7 effect is largely dependent on cellular levels of SHOC2 and Raptor and their interaction.
The SHOC2 Alterations in Human Cancers
Finally, we searched various genome-wide databases to determine potential alterations of SHOC2 in human cancers. The cBioPortal database (https://www.cbioportal.org/) showed many SHOC2 mutations throughout the coding region in all cancer types (Figure S7A), whereas The Cancer Genome Atlas (TCGA) and the Catalogue of Somatic Mutations (COSMIC) databases revealed multiple missense and truncating SHOC2 mutations in non-small cell lung carcinoma (NSCLC) (Figure S7B). Because there are no available structures for SHOC2 and its homolog, a 3D model was simulated by Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) (Kelley et al., 2015), a structure-prediction software. SHOC2 contains 20 LRRs; however, LRR8 and LRR9 (Q262-S307) are missing in this predicted 3D structure. As a scaffold protein, the concave face and adjacent β-α loops of SHOC2, are likely responsible for binding with its partner proteins. Based on the given model, it is likely that T126S and L199I mutants are the most possible residues that would affect SHOC2 function (Figure S7C). We, therefore, characterized these two mutants and found that they indeed promoted the growth (Figure S7D) and survival of lung cancer cells (Figure S7E) better than wild-type SHOC2, thus having the gain-of-function activity.
Gene expression analysis of two expression databases (Badea et al., 2008; Pei et al., 2009) revealed that SHOC2 is overexpressed in pancreatic ductal adenocarcinoma (PDAC), as compared to normal tissues (Figure 7A). Our immuno-histochemical (IHC) staining of 71 paired PDAC samples (tumor versus adjacent normal tissues from the same patients) revealed that SHOC2 is overexpressed in PDAC, as compared to adjacent tissues from the same patients (Figures 7B and 7C). More significantly, SHOC2 overexpression was found to be correlated with poor survival of PDAC patients (Figure 7D). Thus, it appears that SHOC2 is associated with PDAC tumorigenesis.
Figure 7. Alterations of SHOC2 in Human Pancreatic Cancer.

(A) SHOC2 mRNA was elevated in pancreas tumors. Data were from studies by Badea et al., (2008) and Pei et al., (2009).
(B and C) Human pancreatic tumor tissue microarray was stained with anti-SHOC2 Ab (B). SHOC2 staining indexes in tumors versus adjacent tissues are shown as stacked columns (C). Scale bar, 100 μm.
(D) SHOC2 staining in human pancreatic tumor tissue microarray was scored at the scales of 0–3. Overall survival (OS) was estimated with the Kaplan-Meier method, whereas Log-rank tests were used to compare OS based upon SHOC2 staining (scales 2–3 versus 0–1). All studies were conducted in double-blind manner.
(E and F) Human pancreatic tumor tissue microarray was stained for expression of SHOC2, p-ERK, and p4EBP1 (E) and scored based upon staining intensity. Data were analyzed using SPSS software to obtain correlation coefficient (p < 0.01, p < 0.05, Pearson’s test) (F). Scale bar, 100 μm. (G) Working model. FBXW7 binds to SHOC2 and promotes its ubiquitylation and degradation upon phosphorylation on SHOC2-Thr507 by the MAPK signal. Depletion of SHOC2 inactivates the RAS-ERK signal, thus inhibiting cell proliferation as a mechanism for FBXW7 to suppress growth. Through the SHOC2-Raptor binding, SHOC2 negatively regulates the mTORC1 signal to induce autophagy, whereas Raptor negatively regulates the RAS-ERK signal to block proliferation, establishing the negative cross-talks between two oncogenic pathways.
Correlation between the SHOC2-pERK and SHOC2-p4EBP1 Levels in PDAC Tissues
Finally, we used the IHC to measure the SHOC2 expression in pancreas cancer tissues and correlated it with the staining of pERK (RAS-ERK signal) and p4EBP1 (mTORC1 signal) in the same 99 PDAC tissues. SHOC2 staining is positively correlated with pERK but negatively with mTORC1 signal (Figures 7E and 7F), which is statistically significant, suggesting possible ERK activation and mTORC1 inactivation by SHOC2 in PDAC.
DISCUSSION
In this study, we made several interesting findings, which elucidate how the FBXW7-SHOC2-Raptor axis regulates the RAS-ERK and mTORC1 signaling pathways. First, we found that SHOC2 is a substrate of FBXW7. SHOC2 was first identified in C. elegans by acting as a scaffold for RAS and RAF and positively regulates the RAS-ERK pathway (Selfors et al., 1998; Sieburth et al., 1998). Its mammalian homolog was subsequently identified to bind to RAS/RAF by its N-terminal domain to activate MAPK cascade (Li et al., 2000). However, it is previously unknown how the turn-over of SHOC2 is regulated, although HUWE1 E3 ligase was reported to ubiquitylate SHOC2, which is not for SHOC2 degradation but rather for facilitating RAF ubiquitylation and degradation (Jang et al., 2014). We reported here that upon activation of the MAPK signal by growth factor EGF or serum, SHOC2 is phosphorylated at the Thr507 residue within the FBXW7 consensus binding motif, which facilitates its binding to FBXW7 for subsequent ubiquitylation and degradation (Figure 2). Indeed, ectopic FBXW7 expression shortens the SHOC2 protein half-life, whereas small interfering RNA (siRNA)-based FBXW7 knock down extends it (Figure 1). Thus, SHOC2 joins the long list of FBXW7 substrates (Wang et al., 2012; Wang et al., 2014) as a new oncoprotein member.
Because SHOC2 is an activator of the RAS-ERK signal, whereas FBXW7 is a tumor suppressor that promotes SHOC2 ubiquitylation and degradation, it is not surprising to observe that two proteins counteract with each other in regulation of the growth and survival of lung cancer cells in an ERK-dependent manner, with FBXW7 acting at the upstream (Figure 3). Nevertheless, given SHOC2 is involved in coordination of RAS-ERK pathway activation with tumorigenic growth (Young et al., 2013) and promotes cell motility and metastasis by activation of RAS-PI3K signaling (Kaduwal et al., 2015), our study provides a mechanism for FBXW7 to suppress tumor growth by targeting SHOC2 for degradation.
Quite a surprising observation we made here is that SHOC2 triggers autophagy, and autophagy induction contributes to growth-promotion activity of SHOC2, as it is partially inhibited by ATG5 knock down (Figure 4). Mechanistic investigation led to our second significant finding that SHOC2-induced autophagy is mediated by inactivation of mTORC1 through competitive Raptor binding. Indeed, SHOC2 selectively binds to Raptor, but not Rictor, through the middle and C-terminal regions to deplete Raptor from the mTORC1 complex. Our study showed that the middle region (AA351–AA900) of Raptor interacts with both SHOC2 and mTOR, providing the structural basis for the binding competition. Biologically, the Raptor-binding domains of SHOC2 are sufficient in autophagy induction (Figure 5), suggesting that the SHOC2-Raptor binding indeed has a biological consequence.
Whether and how mTORC1 regulates the RAS-ERK signal by Raptor is completely unknown, although two previous studies from the same lab reported that oncogenic MAPK signaling stimulates mTORC1 activity by promoting Raptor phosphorylation by ribosomal protein S6 kinase (RSK) and ERK1 and ERK2 in HEK293 and HeLa cells (Carrière et al., 2008; Carriere et al., 2011). We have shown here that Raptor negatively regulates the RAS-ERK signal through depletion of SHOC2 from the RAS/RAF complex, leading to suppression of growth and survival of pancreatic, colon, and lung cancer cells (Figure 6). Our study, therefore, establishes a negative cross-talk between two dominant oncogenic pathways with SHOC2 inactivation of Raptor/mTORC1 and Raptor inactivation of RAS/RAF to keep the proliferation and survival signals under the precise control.
It is well-established that both MAPK and mTORC1 are required for growth. Here, we have shown that SHOC2 activates MAPK but inhibits mTORC1. So how is this balanced for overall cell growth? Our data demonstrate that SHOC2 could stimulate cell growth by two approaches: (1) activates MAPK signal, which is known; and (2) inhibits mTORC1 by depleting Raptor (Figures 4 and 5) to trigger autophagy. We proposed that SHOC2-triggered autophagy provides nutrients for cell proliferation because both SHOC2-enhanced growth and clonal survival are significantly reduced if autophagy is blocked (by ATG5 knock down) (Figures 4E and 4F). Furthermore, the balance for overall cell growth is also subjected to FBXW7 regulation. Thus, an accelerated growth is expected in human cancers, particularly with FBXW7 mutations and SHOC2 overexpression, which are frequent events.
We further addressed the potential role of SHOC2 in human cancer. The bioinformatics mining of human cancer databases at both the genomic and expression levels revealed missense and truncating mutations of SHOC2 in human lung cancer. We characterized two mutants with potential alteration on surface structure and found that both of them promote the growth and survival of lung cancer cells (Figure S7), indicating that these mutants are not random but have biological significance. Through the IHC staining of pancreatic cancer tissues and paired adjacent normal tissues from the same patients, we found that SHOC2 is overexpressed in pancreatic cancer tissues, which is importantly correlated with poor patient survival, suggesting that SHOC2 likely acts as an oncoprotein during PDAC development. Finally, we found that in human PDAC tissues, the expression of SHOC2 and pERK is positively correlated, whereas expression of SHOC2 and p4E-BP1 is negatively correlated (Figure 7), implying that differential regulation of SHOC2 on the RAS-ERK and mTORC1 signals might be pathologically relevant.
In summary, our study supports the following model: growth factors activate the SHOC2-RAS-MAPK signal, followed by MAPK phosphorylation of SHOC2 on the Thr507 to trigger FBXW7-mediated SHOC2 degradation. This negative feedback loop is likely a cellular self-protective mechanism to prevent over-activation of the SHOC2-RAS-MAPK signal and is most likely a dynamic process, dependent of FBXW7 availability. Human cancers with FBXW7 inactivation would shut off this loop, leading to uncontrolled proliferation. Our results provide a mechanistic explanation of why FBXW7 inactivation (by deletion, mutation, and promoter hyper-methylation) was observed frequently in human cancers (Wang et al., 2012; Welcker and Clurman, 2008), as well as a mechanism by which FBXW7 acts as a tumor suppressor by targeting SHOC2 for degradation. On the other hand, the SHOC2-Raptor binding inactivates mTORC1, whereas the Raptor-SHOC2 binding inactivates RAS/MAPK, establishing a negative cross-talk between two dominant growth-survival signals, which is subject to FBXW7 regulation. Thus, the FBXW7-SHOC2-Raptor axis plays a key role in coordination of proliferation and autophagy (Figure 7G).
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yi Sun (sunyi@umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
H1299 cell lines (from lung of a 43 years male human), H358 cell lines (from lung of a male human), A549 cell lines (from lung of a 58 years old male human), HEK293 cell lines (from embryonic kidney of a female human fetus), MiaPaCa-2 cell lines (from pancreas of a 65 years of male human), and HCT116 cell lines (from colon of a male human) were purchased from the American Type Culture Collection (ATCC). Huh7 cell lines (from liver of a male human) were obtained from the Japanese Collection of Research Bioresources (JCRB). H1299, Huh7, HEK293 and MiaPaCa-2 cells were cultured in DMEM medium, whereas H358, HCT116 and A549 cells in RPMI1640 medium. All experiments were carried out in complete medium containing 10% FBS at 37°C.
METHOD DETAILS
RNA isolation and RT-PCR
Total RNA was isolated by Trizol (Life Technologies; 15596018) according to the manufacturer’s instructions. RT-PCR was performed using PrimeScript III First-Stand synthesis system (Invitrogen; 18080-051). To detect the mRNA levels of FBXW7 and SHOC2, the primers used for RT-PCR assays were listed in Table S1. GAPDH was used as internal control.
Lentivirus or siRNA transfection
The siRNA oligoes were purchased from Thermo Fisher Scientific listed in Table S2. The H1 promoter-driven shRNA targeting ERK1: 5′-CATGAAGGCCCGAAACTAC-3′; ERK2: 5′-GCGCTTCAGACATGAGAAC-3′ (Xie et al., 2015). Raptor siRNA (SC-44069) was obtained from Santa Cruz. Briefly, cells were transfected with various siRNAs or shRNAs in MEM medium using DharmaFECT transfection reagent following the manufacturer’s protocol.
Cell proliferation and clonogenic survival
Cells were transfected with various plasmids, siRNA oligoes or shRNA for 48 hr by Lipofectamine 2000 transfection reagent. Cells were seeded and cultured. Cell proliferation was analyzed using the ATPlite Luminescence Assay Kit (PerkinElmer, 6016943). Cell numbers were counted 48 hr later or colonies with 50 or more cells were counted 9-13 days later after being stained with crystal violet (0.5% w/v) and imaged.
Detection of apoptotic cells
Cells were transfected with various plasmids or siRNA oligoes for 48 hr by Lipofectamine 2000 transfection reagent. Cells were stained with 5 μl Annexin V-FITC and 10 μl PI Staining Solution in the 1× binding buffer for 15 min in the dark. Annexin V positive cells represented apoptotic cells and were analyzed by a flow cytometer (BD).
Autophagic vacuoles staining
Autophagic vacuoles were analyzed using a Cyto-ID autophagy detection kit (Enzo Life Sciences) and LC3 antibody staining as described (Xie et al., 2015). Briefly, for Cyto-ID autophagy detection kit staining, cells were stained with Cyto-ID autophagy detection dye and Hoechst 33342 (Life Technologies) for 20 min at 37°C. For LC3 antibody staining, cells were fixed with 100% ice-cold methanol for 5 min, blocked with 1% BSA for 15 min, incubated with an anti-LC3 Ab (1:250; Cell Signaling) at 4°C overnight, washed 3 times, incubated with an Alexa Fluor 488-conjugated secondary antibody (1:200; Invitrogen) for 1 h. Slides were mounted with Vectashield mounting medium and imaged under a microscope (Nikon).
Preparation of phospho-SHOC2-T507 specific antibody
A peptide polyclonal Ab against phospho-SHOC2 at codon Thr507 (NH2-CG503ENLLp[Thr]HL510PEEIGT-COOH) within the FBXW7 binding motif (Figure S1C) was generated, followed by two sequential rounds of affinity purification by Baiqi biotechnology, Inc. (Suzhou, China).
In vivo and in vitro ubiquitylation assays
For in vivo ubiquitylation assay, the HEK293 cells were co-transfected with GST-SHOC2, GST-SHOC2 (T242A), GST-SHOC2 (T507A), HA-FBXW7, HA-FBXW7ΔF, or His-HA-ubiquitin, individually or in indicated combinations. After 48 hr transfection, cells were treated with 10 μM MG132 for 4 h and then harvested for in vivo ubiquitylation as previously described using Ni-beads pull-down (Zhao et al., 2011). For the in vitro ubiquitylation assay, GST-SHOC2 was precipitated from GST-SHOC2 transfected HEK293 cells by GST-beads, serving as the source of substrate. HA-FBXW7 and HA-FBXW7ΔF were precipitated from HA-FBXW7 or HA-FBXW7ΔF transfected HEK293 cells, and eluted with 3xHA peptide (Sigma), serving as the source of E3. The reaction was carried out at 30°C for 1 hr in 30 μL reaction buffer (40 mM Tris-HCl, pH 7.5, 2 mM DTT, 5 mM MgCl2) in the presence of SHOC2 (bound to GST beads), ubiquitin (Boston Biochem), E1 (Boston Biochem), recombinant UbcH5c/E2 (Boston Biochem), ATP, and HA-purified FBXW7 or FBXW7ΔF. After the reaction, the beads were washed three times with cell lysis buffer to remove non-SHOC2 conjugated ubiquitin. The washed beads were then resuspended in 25 μL 2xSDS-PAGE sample buffer for SDS-PAGE and detected by IB with antibodies against GST.
Tissue microarray and immunohistochemistry
Human pancreatic cancer tissue microarrays (TMAs) containing 71 pairs of tumors and matched adjacent tissues and 28 tumors were purchased from Shanghai Outdo Biotech. Co. Ltd (Shanghai, China). Immunohistochemical staining of TMAs using primary antibodies against SHOC2 (Abcam, ab106430), pERK (Cell Signaling, S4370), p4EBP1 (Cell Signaling, S2855) was performed as described previously (Xu et al., 2017). Briefly, after deparaffinization, rehydration, antigen retrieval and blocking, the arrays were incubated overnight at 4°C with indicated antibodies. The slides were developed with DAB and counterstained with hematoxylin. The stained slides were observed under a microscope (Olympus 1X71) and images were acquired using software DP controller (ver. 3.1.1.267, Olympus). Stained tumor tissues and adjacent normal tissues were classified into four groups (0 to 3) according to the staining intensity of each tissue.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using two-tailed Student’s t test for comparison of two groups or one-way analysis of variance for comparison of more than two groups followed by Tukey’s multiple comparison test. The statistical analyses were carried out using the GraphPad Prism software version 5.01 (GraphPad, San Diego, CA). Data were expressed as mean ± standard error of the mean (SEM) of at least 3 independent experiments. A p value < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-SHOC2(T507) | This paper | N/A |
| rabbit anti-S6Kα | Santa Cruz | Cat# sc-230; RRID:AB_632156 |
| mouse anti-β-actin | Santa Cruz | Cat# sc-47778; RRID:AB_626632 |
| rabbit anti-phospho-4EBP1 (Ser65) | Cell Signaling Technology | Cat# 9451; RRID:AB_330947 |
| rabbit anti-phospho-4EBP1 (Thr37 / Thr46) | Cell Signaling Technology | Cat# 2855; RRID:AB_560835 |
| rabbit anti-4EBP1 | Cell Signaling Technology | Cat# 9452;RRID:AB_331692 |
| rabbit anti-phospho-S6K (Thr389) | Cell Signaling Technology | Cat# 9234; RRID:AB_2269803 |
| rabbit anti-ATG5 | Cell Signaling Technology | Cat# 2630; RRID:AB_2062340 |
| rabbit anti-p62 | Cell Signaling Technology | Cat# 5114; RRID:AB_10624872 |
| rabbit anti-ERK1 and ERK2 | Cell Signaling Technology | Cat# 9107; RRID:AB_10695739 |
| rabbit anti-phospho-ERK1 and ERK2 (T202/Y204) | Cell Signaling Technology | Cat# 4376; RRID:AB_331772 |
| rabbit anti-phospho-ERK1 and ERK2 (T202/Y204) | Cell Signaling Technology | Cat# 4370; RRID:AB_2315112 |
| mouse anti-MEK1 | Cell Signaling Technology | Cat# 2352; RRID:AB_10693788 |
| rabbit anti-Raptor | Cell Signaling Technology | Cat# 2280; RRID:AB_561245 |
| rabbit anti-Rictor | Cell Signaling Technology | Cat# 2114; RRID:AB_2179963 |
| rabbit anti-mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 |
| rabbit anti-LC3B | Cell Signaling Technology | Cat# 2775; RRID:AB_915950 |
| rabbit anti-SHOC2 | Abcam | Cat# ab106430;AB_10861003 |
| rabbit anti-FBXW7 | Epitomics | Cat# A301-721A; RRID:AB_1210898 |
| goat anti-GST | GE Lifescience | Cat# 27457701V; RRID:AB_771432 |
| rat anti-HA | Roche | Cat# 11867423001; RRID:AB_390918 |
| mouse anti-Flag | Sigma | Cat# F1804; RRID:AB_262044 |
| Alexa Fluor 488 Polyclonal Antibody | Invitrogen | Cat # A-11094; RRID:AB_221544 |
| Biological Samples | ||
| Human pancreatic cancer tissue microarrays (TMAs) containing 71 pairs of tumors and matched adjacent tissues and 28 tumors | Shanghai Outdo Biotech. Co. Ltd (Shanghai, China) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG132 | Sigma | Cat#: M7449 |
| cycloheximide | Sigma | Cat#: 01810 |
| Hoechst 33342 | ThermoFisher Scientific | Cat#: H1399 |
| Trizol | Life Technologies | Cat#: 15596018 |
| 3xHA peptide | Sigma | Cat#: 92000-76-5 |
| Critical Commercial Assays | ||
| ATPlite Luminescence Assay Kit | PerkinElmer | Cat#: 6016943 |
| Annexin V-FITC apoptosis detection Kit | BD | Cat #: 556547 |
| Cyto-ID autophagy detection kit | Enzo Life Sciences | ENZ-51031-0050 |
| Deposited Data | ||
| SHOC2 mutation | Genome Reference Consortium | http://cbioportal.org |
| SHOC2 structure | This paper; Phyre2 Data | http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index |
| Experimental Models: Cell Lines | ||
| H1299 | American Type Culture Collection (ATCC) | Cat# NCI-H1299 RRID:CVCL_0060 |
| Huh7 | Japanese Collection of Research Bioresources JCRB | Cat# JCRB0403, RRID:CVCL_0336 |
| HEK293 | American Type Culture Collection (ATCC) | Cat# 300192/p777_HEK293, RRID:CVCL_0045 |
| MiaPaCa-2 | American Type Culture Collection (ATCC) | Cat# ACC-733, RRID:CVCL_0428 |
| H358 | American Type Culture Collection (ATCC) | Cat# NCI-H358, RRID:CVCL_1559 |
| HCT116 | American Type Culture Collection (ATCC) | Cat# HCT-116, RRID:CVCL_0291 |
| A549 | American Type Culture Collection (ATCC) | Cat# A549, RRID:CVCL_0023 |
| Oligonucleotides | ||
| Table S1 | This paper | N/A |
| Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pEBG-GST-SHOC2 | This paper | N/A |
| Plasmid: pEBG-GST-SHOC2 (T507A) | This paper | N/A |
| Plasmid: pEBG-GST-SHOC2 (T242A) | This paper | N/A |
| Plasmid: pEBG-GST-SHOC2 (1-84) | This paper | N/A |
| Plasmid: pEBG-GST-SHOC2 (1-346) | This paper | N/A |
| Plasmid: pEBG-GST-SHOC2 (347-582) | This paper | N/A |
| Plasmid: pcDNA3-3xHA | This paper | N/A |
| Plasmid: pcDNA3-3xFlag | This paper | N/A |
| Plasmid: pcDNA3-His-ubiqutin | This paper | N/A |
| Plasmid: pcDNA3-Flag-FBXW7 | This paper | N/A |
| Plasmid: pIRES2-HA-FBXW7 | This paper | N/A |
| Plasmid: pIRES2-HA-FBXW7ΔF | This paper | N/A |
| Plasmid: pRK5-HA-Raptor | Addgene Plasmid #8513 | |
| Plasmid: pRK5-HA-Raptor (1-350) | This paper | N/A |
| Plasmid: pRK5-HA-Raptor (351-900) | This paper | N/A |
| Plasmid: pRK5-HA-Raptor (901-1335) | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism software version 5.01 | GraphPad | https://www.graphpad.com/support/prism-5-updates/ |
| AlphaEaseFC software | AlphaEaseFC | https://alphaeasefc-v.updatestar.com/en |
| Other | ||
| Vectashield Antifade mounting medium | Vector Laboratories | Cat #: H-1200 |
| DMEM, high glucose | ThermoFisher Scientific | Cat #: 11965-084 |
| RPMI1640 medium | ThermoFisher Scientific | Cat #: 11875-085 |
Highlights.
SHOC2 is a FBXW7 substrate for degradation upon phosphorylation by the MAPK signal
SHOC2 binds to Raptor to inactivate the mTORC1 signal and induces autophagy
Raptor binds to SHOC2 to inactivate the RAS-ERK signal and inhibits growth
SHOC2 is altered in lung and pancreatic cancers with correlation of poor survival
ACKNOWLEDGMENTS
This work was supported by National Key R&D Program of China (2016YFA0501800), a National Natural Science Foundation of China grant (81630076), and by NCI grants (CA171277 and CA156744) to Y.S. We would like to thank Ms. Zaiming Tang for her help in the preparation of the phospho-SHOCT507 antibody from Baiqi Biotechnology, Inc.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.052.
REFERENCES
- Badea L, Herlea V, Dima SO, Dumitrascu T, and Popescu I (2008). Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 55, 2016–2027. [PubMed] [Google Scholar]
- Berger AW, Schwerdel D, Ettrich TJ, Hann A, Schmidt SA, Kleger A, Marienfeld R, and Seufferlein T (2017). Targeted deep sequencing of circulating tumor DNA in metastatic pancreatic cancer. Oncotarget 9, 2076–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernis C, Vigneron S, Burgess A, Labbé JC, Fesquet D, Castro A, and Lorca T (2007). Pin1 stabilizes Emi1 during G2 phase by preventing its association with SCF(betatrcp). EMBO Rep. 8, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, and Heitman J (1999). The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 13, 3271–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, and Roux PP (2008). Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 18, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, Fingar DC, and Roux PP (2011). ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 286, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Xiong WC, and Mei L (2006). Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J. Biol. Chem. 281, 927–933. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, and Joazeiro CA (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–34. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Matsumoto A, Inuzuka H, Zhai B, Lau AW, Wan L, Gao D, Shaik S, Yuan M, Gygi SP, et al. (2012). SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep. 1, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Azmi AS, Aboukameel A, Kauffman M, Shacham S, Abou-Samra AB, and Mohammad RM (2014). Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget 5, 3444–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q, Bowden GT, Normolle D, and Sun Y (2007). SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J. Cell Biol. 178, 1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, and Sabatini DM (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, and Sabatini DM (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 11, 859–871. [DOI] [PubMed] [Google Scholar]
- He D, Huang C, Zhou Q, Liu D, Xiong L, Xiang H, Ma G, and Zhang Z (2017). HnRNPK/miR-223/FBXW7 feedback cascade promotes pancreatic cancer cell growth and invasion. Oncotarget 8, 20165–20178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. (2011). SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang ER, Shi P, Bryant J, Chen J, Dukhande V, Gentry MS, Jang H, Jeoung M, and Galperin E (2014). HUWE1 is a molecular link controlling RAF-1 activity supported by the Shoc2 scaffold. Mol. Cell. Biol. 34, 3579–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang ER, Jang H, Shi P, Popa G, Jeoung M, and Galperin E (2015). Spatial control ofShoc2-scaffold-mediated ERK1/2 signaling requires remodeling activity of the ATPase PSMC5. J. Cell Sci. 128, 4428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung M, Abdelmoti L, Jang ER, Vander Kooi CW, and Galperin E (2013). Functional Integration of the Conserved Domains of Shoc2 Scaffold. PLoS One 8, e66067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung M, Jang ER, Liu J, Wang C, Rouchka EC, Li X, and Galperin E (2016). Shoc2-tranduced ERK1/2 motility signals–Novel insightsfrom functional genomics. Cell. Signal. 28, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S, Qin Y, Shi S, Liu X, Hu H, Zhou H, Gao J, Zhang B, Xu W, Liu J, et al. (2015). ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res. 25, 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, and Kim DH (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaduwal S, Jeong WJ, Park JC, Lee KH, Lee YM, Jeon SH, Lim YB, Min do S, and Choi KY (2015). Sur8/Shoc2 promotes cell motility and metastasis through activation of Ras-PI3K signaling. Oncotarget 6, 33091–33105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, and Elledge SJ (2001). Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294, 173–177. [DOI] [PubMed] [Google Scholar]
- Kwon YW, Kim IJ, Wu D, Lu J, Stock WA Jr., Liu Y, Huang Y, Kang HC, DelRosario R, Jen KY, et al. (2012). Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Mol. Cancer Res. 10, 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Klionsky DJ (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477. [DOI] [PubMed] [Google Scholar]
- Li W, Han M, and Guan KL (2000). The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 14, 895–900. [PMC free article] [PubMed] [Google Scholar]
- Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JD, and Jackson PK (2003). Prophase destruction of Emi1 by the SCF(betaTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 4, 813–826. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Levine B (2010). Methods in mammalian autophagy research. Cell 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Young AR, and Narita M (2009). Autophagy facilitates oncogene-induced senescence. Autophagy 5, 1046–1047. [DOI] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. (2007). FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 204, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, and Wang L (2009). FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, and McCormick F (2006). A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol. Cell 22,217–230. [DOI] [PubMed] [Google Scholar]
- Sabatini DM (2006). mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 6, 729–734. [DOI] [PubMed] [Google Scholar]
- Selfors LM, Schutzman JL, Borland CZ, and Stern MJ (1998). soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl. Acad. Sci. USA 95, 6903–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, and Klionsky DJ (2004). Autophagy in health and disease: a double-edged sword. Science 306, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth DS, Sun Q, and Han M (1998). SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94, 119–130. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736. [DOI] [PubMed] [Google Scholar]
- Tan M, Zhao Y, Kim SJ, Liu M, Jia L, Saunders TL, Zhu Y, and Sun Y (2011). SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev. Cell 21, 1062–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, and Wei W (2012). Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 586, 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu P, Inuzuka H, and Wei W (2014). Roles of F-box proteins in cancer. Nat. Rev. Cancer 14, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maitra A, and Wang H (2016). The emerging roles of F-box proteins in pancreatic tumorigenesis. Semin. Cancer Biol. 36, 88–94. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, and Kaelin WG Jr. (2005). The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8, 25–33. [DOI] [PubMed] [Google Scholar]
- Welcker M, and Clurman BE (2008). FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, and Clurman BE (2004). The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 101, 9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. (2011). Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114. [DOI] [PubMed] [Google Scholar]
- Xie CM, Wei D, Zhao L, Marchetto S, Mei L, Borg JP, and Sun Y (2015). Erbin is a novel substrate of the Sag-βTrCP E3 ligase that regulates KrasG12D-induced skin tumorigenesis. J. Cell Biol. 209, 721–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhou W, Yang F, Chen G, Li H, Zhao Y, Liu P, Li H, Tan M, Xiong X, and Sun Y (2017). The β-TrCP-FBXW2-SKP2 axis regulates lung cancer cell growth with FBXW2 acting as a tumour suppressor. Nat. Commun. 8, 14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, and Nakayama KI (2004). Phosphorylation-dependent degradation of c-Myc is mediated bythe F-box protein Fbw7. EMBO J. 23, 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LC, Hartig N, Muñoz-Alegre M, Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Vietri Rudan M, Gewinner C, Henderson S, et al. (2013). An MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol. Cell 52, 679–692. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Karnak D, Tan M, Lawrence TS, Morgan MA, and Sun Y (2016a). FBXW7 Facilitates Nonhomologous End-Joining via K63-Linked Polyubiquitylation of XRCC4. Mol. Cell 61, 419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang Y, Parsels JD, Lohse I, Lawrence TS, Pasca di Magliano M, Sun Y, and Morgan MA (2016b). Fbxw7 Deletion Accelerates KrasG12D-Driven Pancreatic Tumorigenesis via Yap Accumulation. Neoplasia 18, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Xiong X, and Sun Y (2011). DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell 44, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.