

Abstract
Amyloidosis is a spectrum of diseases, in which various proteins which are usual components of plasma are deposited as insoluble beta-pleated sheets extracellularly, disrupting function of various organs. Amyloid light-chain amyloidosis occurs due to the deposition of proteins, derived from immunoglobulin light chains, routinely manifesting with multisystem involvement. Pulmonary involvement is seen in about 50% of cases. Three common patterns of pulmonary amyloidosis on computed tomography (CT) chest are tracheobronchial, nodular parenchymal, and diffuse alveolar septal variety. We hereby report two cases of pulmonary amyloidosis, one being a case of diffuse alveolar septal pulmonary amyloidosis, which is an extremely rare pattern of involvement, with a very poor prognosis, and the other one being tracheobronchial pattern of involvement, which usually results due to the localized deposition of amyloid in the tracheobronchial tree. Knowledge about pulmonary amyloidosis is important due to its poor prognosis and nonspecific findings in CT chest.
KEY WORDS: Amyloid light-chain amyloidosis, apple-green birefringence, chemotherapy, diffuse alveolar septal pattern, diffuse parenchymal pattern, pulmonary amyloidosis, tracheobronchial amyloidosis, video-assisted thoracoscopic surgery-guided lung biopsy
INTRODUCTION
Amyloidosis is a group of disease entities, characterized by abnormal extracellular protein deposition, in various tissues of the body. A patient often presents with a wide range of clinical manifestations, depending on site, amount, and type of protein deposited. We report two cases of pulmonary amyloidosis, diagnosed in our hospital over a period of 10 years. Amyloid light-chain (AL) amyloidosis with alveolar septal pattern of pulmonary involvement is an extremely rare case and the least common variety of pulmonary amyloidosis[1] usually seen in only 3% cases of pulmonary amyloidosis,[2] and its diagnosis antemortem is unusual.[3] The other one is more common variety, tracheobronchial amyloid, which usually results due to the localized deposition of amyloid in the tracheobronchial tree and often misdiagnosed as obstructive airway disease.
CASE REPORTS
Case 1: Alveolar septal pulmonary amyloidosis
A 70-years-old male known diabetic presented with dyspnea and weight loss for 4 months in June 2015 to a pulmonologist elsewhere and was evaluated to have bilateral diffuse centrilobular nodules on computed tomography (CT) chest [Figure 1], treated as panbronchiolitis empirically with erythromycin for 6 months, after ruling out infectious etiology from bronchial wash samples and transbronchial lung biopsy being inconclusive. He reported back in January 2016, with worsening symptoms for further evaluation. Pulmonary function test showed severe restrictive pattern, and CT showed worsening of diffuse bilateral centrilobular nodules [Figure 2]. Video-assisted thoracoscopic surgery (VATS)-guided lung biopsy was done for definitive diagnosis which revealed nodular, perivascular, and interstitial deposits with Apple-green birefringence to polarized light and congo red positivity suggestive of nonamyloid A (AA) amyloidosis [Figure 3]. Urine analysis showed raised urine spot total protein – 543 mg (normal <12 mg). Urine protein-to-creatinine ratio was elevated – 3.22 (normal <0.2, nephrosis >3.5). Serum-free kappa levels were elevated − 719 mg/L, and serum lambda levels were normal − 8.94 mg/L. Serum kappa/lambda ratio was deranged (80.4). Serum immunotyping revealed the absence of monoclonal gammopathy. Bone marrow biopsy done to rule out multiple myeloma showed no specific lesion, and bone marrow aspiration cytology revealed hypercellular marrow and mild plasmacytosis with plasma cells – 5%. Echo showed no cardiac involvement. After extensive workup, he was treated as AL amyloidosis with pulmonary and renal involvement with cyclophosphamide, dexamethasone, and bortezomib along with symptomatic treatment. In 1-year regular follow-up, his symptoms improved and are in good compliance with treatment.
Figure 1.
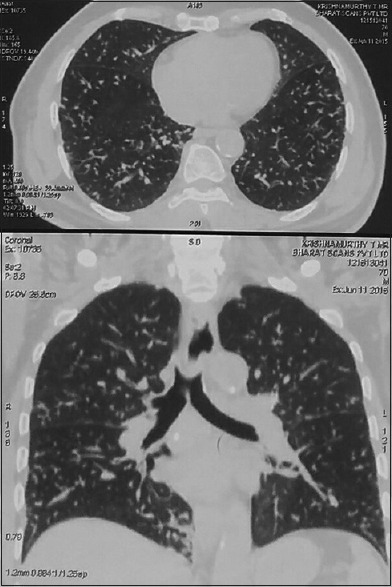
Computed tomography chest showing diffuse centrilobular nodules
Figure 2.
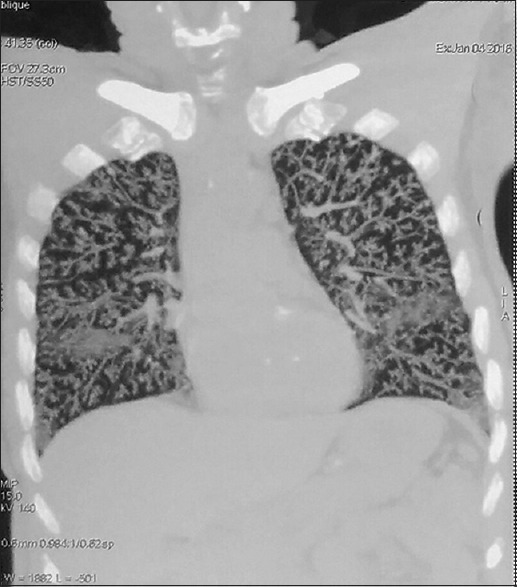
Computed tomography chest repeated after 6 months showing worsening centrilobular nodules bilaterally
Figure 3.
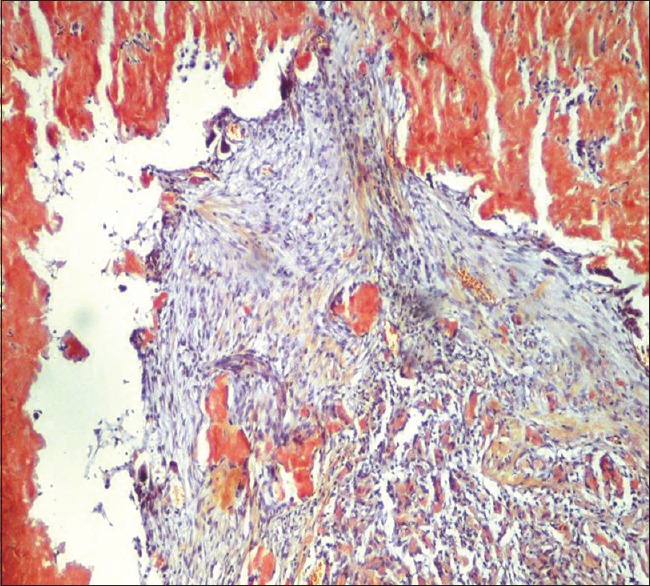
Lung biopsy showing Congo red-positive infiltrates
Case 2: Tracheobronchial amyloidosis
A 42-year-old female with multiple comorbidities presented to our pulmonology outpatient department in 2006. She is a known diabetic and hypertensive on regular treatment and was earlier diagnosed to have right submandibular pleomorphic adenoma and mesangial proliferative nephropathy (renal biopsy proven) and presented with complaints of productive cough and dyspnea on exertion for 3 months. She had a history of similar complaints in 2005 when chest X-ray findings were suggestive of right lower lobe (RLL) collapse and CT chest confirmed collapse consolidation of RLL [Figure 4] which was treated with a course of antibiotics. Chest X-ray in 2006 was normal and CT revealed RLL consolidation consistent with that of 2005 CT scan. Routine blood investigations revealed anemia and elevated erythrocyte sedimentation rate. Bronchoscopy was done in view of recurrent, nonresolving pneumonia, which showed partially occluded left lower lobe segmental bronchi and narrowed RLL segmental bronchi with edematous mucosa [Figure 5]. Histopathology of bronchial biopsy showed apple-green birefringence under polarized light, consistent with non-AA-type amyloidosis. She was treated symptomatically with nebulized bronchodilators and intravenous antibiotics, improved and discharged home, and advised for regular follow-up. During her subsequent visits to the hospital for the past 11 years, she had one episode of lower respiratory tract infection in 2007, which was managed conservatively, and CT chest showed RLL bronchiectasis. Pulmonary function test was done which showed severe obstruction with no reversibility. Bronchoscopy done twice during her follow-up period showed no significant disease progression and occlusion. Bone marrow biopsy was done while evaluating for her anemia showed mildly hypercellular marrow with no evidence of amyloidosis. She was in regular follow-up till recently, her respiratory symptoms were under control with symptomatic treatment, but her renal function worsened requiring dialysis. In this case, there was localized involvement of the tracheobronchial tree, which is the most common type of involvement in the tracheobronchial pattern.
Figure 4.
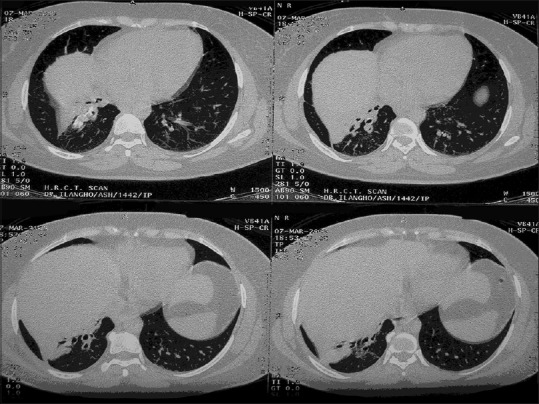
Computed tomography chest confirming right lower lobe collapse
Figure 5.
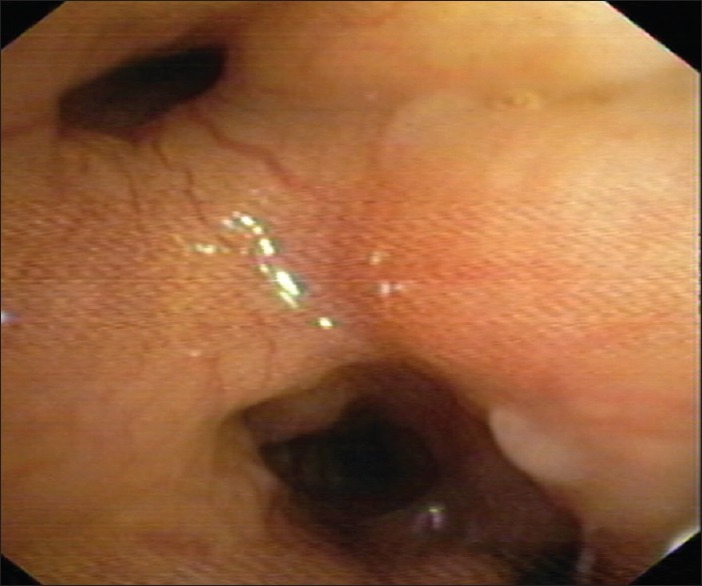
Bronchoscopy showed partially occluded left lower lobe segmental bronchi and narrowed right lower lobe segmental bronchi with edematous mucosa
DISCUSSION
AL/primary amyloidosis usually has male preponderance with median age at diagnosis being 64 years[4,5,6] and usual incidence of 6–10 cases/million/year.[7] It usually occurs in plasma cell dyscrasias. In case 1, our patient is a 70-year-old male without any evidence suggestive of plasma cell dyscrasias. Systemic involvement is commonly seen with kidney being most commonly affected organ. Pulmonary involvement includes three main patterns – tracheobronchial, nodular parenchymal, and diffuse parenchymal/diffuse alveolar septal pattern.[8] In our case, urine analysis showed raised urine spot total protein and elevated urine protein-to-creatinine ratio suggesting renal involvement. Bilateral diffuse centrilobular nodules worsened on CT chest in a span of 6 months, and PFT showed severe restriction suggesting pulmonary involvement proven by VATS-guided lung biopsy which showed nodular, perivascular, and interstitial deposits with Congo red positivity and apple-green birefringence to polarized light-confirming amyloidosis. Deranged serum kappa:lambda ratio and elevated serum kappa levels made AL amyloidosis with alveolar septal pattern on CT chest as an obvious diagnosis. Histological confirmation by Congo red under polarized light showing apple-green birefringence is gold standard for amyloid diagnosis.[9] It is usually followed by immunohistochemistry to find out fibril type, but light-chain deposits often do not stain. The presence of systemic syndrome related to amyloid, amyloid staining by Congo red being positive, and monoclonal plasma cell proliferative disorder being evident due to abnormal serum-free light chain ratio supports our diagnosis.
Tracheobronchial amyloidosis typically occurs in the 5th and 6th decade. Most of the patients are usually asymptomatic. Other presentations which are common are wheeze, stridor, cough, and recurrent pneumonia. The tracheobronchial pattern of involvement is most common variety seen in 53% of cases of pulmonary amyloidosis. Although airway involvement is common in systemic AL amyloidosis, it is usually localized if symptomatic.[10] HRCT chest shows that localized nodules, circumferential thickening of the trachea, and narrowing of the tracheobronchial tree lumen may be present which leads to recurrent infections, atelectasis, and bronchiectasis.[11] In case 2, our patient is a 42-year-old female presented with shortness of breath and cough. CT chest showed collapse consolidation of medial and posterior basal segments of RLL consistent with nonresolving pneumonia, and diagnosis was confirmed by a bronchoscopic biopsy. In our case 2, isolated pulmonary involvement was seen, as there was no evidence of renal, cardiac, and bone involvement.
Amyloidosis is classified into various subtypes as per the WHO classification based on the type of fibrillar protein deposited. As many as 30 varieties are described in human amyloidosis, the most important and common types being (1) AL amyloidosis due to immunoglobulin and light-chain deposition, (2) AA amyloidosis/secondary amyloidosis due to serum AA protein deposition, (3) dialysis-related amyloidosis due to beta 2-microglobulin deposition, (4) heritable amyloidoses due to mutant transthyretin (prealbumin) deposition, and (5) senile amyloidosis due to wild-type transthyretin deposition.
Diffuse alveolar septal pattern of pulmonary amyloidosis is extremely rare.[12,13] Very few case series were published earlier. Thompson et al. reported four cases and Hui et al. reported six cases. Average survival period in patients with systemic amyloidosis with lung involvement is 16 months only.[3] Chemotherapy is the treatment of choice for AL amyloidosis, and regression has been demonstrated earlier systematically.[14] Our patient was initiated with cyclophosphamide, dexamethasone, and bortezomib and is doing well so far with symptomatic improvement in 6-month follow-up.
The treatment of tracheobronchial amyloidosis is still under trial. It is usually managed symptomatically. However, if there is a significant occlusion of the tracheobronchial tree, bronchoscopic recanalization with Nd:YAG laser and CO2 laser is the most successful treatment so far. Stents also have a role if occlusion is complete. Colchicine and systemic steroids were not proved to be effective. Cases were reported with good response to local radiation.[15] Follow-up with close watch on new respiratory symptoms, bronchoscopy, and pulmonary function tests helps in further management once diagnosis is established. In our case, the patient was symptomatically treated since occlusion was not severe, and she was in regular follow-up with bronchoscopy done twice in follow-up period which showed no significant disease progression and occlusion.
CONCLUSION
Amyloidosis should be considered as one of the differentials along with other common diagnoses such as malignancy, infections, and vasculitis when multisystem involvement is seen or when nonresolving pneumonia is encountered. Knowledge about pulmonary amyloidosis is important due to nonspecific findings in CT chest, and poor prognosis and treatment depend on the pattern of involvement. Although the prognosis of tracheobronchial amyloidosis is usually good, follow-up is crucial to act appropriately, if significant endobronchial occlusion occurs.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Urban BA, Fishman EK, Goldman SM, Scott WW, Jr, Jones B, Humphrey RK, et al. CT evaluation of amyloidosis: Spectrum of disease. Radiographics. 1993;13:1295–308. doi: 10.1148/radiographics.13.6.8290725. [DOI] [PubMed] [Google Scholar]
- 2.Berk JL, O'Regan A, Skinner M. Pulmonary and tracheobronchial amyloidosis. Semin Respir Crit Care Med. 2002;23:155–65. doi: 10.1055/s-2002-25304. [DOI] [PubMed] [Google Scholar]
- 3.Poh SC, Tjia TS, Seah HC. Primary diffuse alveolar septal amyloidosis. Thorax. 1975;30:186–91. doi: 10.1136/thx.30.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyle RA, Greipp PR. Amyloidosis (AL).Clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665–83. [PubMed] [Google Scholar]
- 5.Kyle RA, Gertz MA. Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45–59. [PubMed] [Google Scholar]
- 6.Kyle RA, Greipp PR, O'Fallon WM. Primary systemic amyloidosis: Multivariate analysis for prognostic factors in 168 cases. Blood. 1986;68:220–4. [PubMed] [Google Scholar]
- 7.Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O'Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted county, Minnesota, 1950 through 1989. Blood. 1992;79:1817–22. [PubMed] [Google Scholar]
- 8.Kanada DJ, Sharma OP. Long-term survival with diffuse interstitial pulmonary amyloidosis. Am J Med. 1979;67:879–82. doi: 10.1016/0002-9343(79)90748-4. [DOI] [PubMed] [Google Scholar]
- 9.Pinney JH, Lachmann HJ. Amyloidosis and the lung. Eur Respir Mon. 2011;54:152–70. [Google Scholar]
- 10.Gillmore JD, Hawkins PN. Amyloidosis and the respiratory tract. Thorax. 1999;54:444–51. doi: 10.1136/thx.54.5.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung MJ, Lee KS, Franquet T, Müller NL, Han J, Kwon OJ, et al. Metabolic lung disease: Imaging and histopathologic findings. Eur J Radiol. 2005;54:233–45. doi: 10.1016/j.ejrad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Gómez AT, Alonso RM, García MM, Fabbricatore AA, García-Tejedor JL. Thoracic amyloidosis: High-resolution computed tomographic findings in 3 cases. J Comput Assist Tomogr. 2008;32:926–8. doi: 10.1097/RCT.0b013e31815a3420. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Chen H, Wang S. Laryngo-tracheobronchial amyloidosis: A case report and review of literature. Int J Clin Exp Pathol. 2014;7:7088–93. [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy TJ, Myerowitz RL, Bender BL. Diffuse parenchymal amyloidosis of lungs and breast. Its association with diffuse plasmacytosis and kappa-chain gammopathy. Arch Pathol Lab Med. 1979;103:583–5. [PubMed] [Google Scholar]
- 15.Kurrus JA, Hayes JK, Hoidal JR, Menendez MM, Elstad MR. Radiation therapy for tracheobronchial amyloidosis. Chest. 1998;114:1489–92. doi: 10.1378/chest.114.5.1489. [DOI] [PubMed] [Google Scholar]