

Abstract
Abnormalities of the D2R gene (DRD2) play a role in the pathogenesis of human essential hypertension; variants of the DRD2 have been reported to be associated with hypertension. Disruption of Drd2 (D2−/−) in mice increases blood pressure and may cause salt sensitivity. The hypertension of D2−/− mice has been related, in part, to increased sympathetic activity and renal oxidative stress and endothelin B receptor expression. We tested in D2−/− mice the effect of etamicastat, a reversible peripheral inhibitor of dopamine-β-hydroxylase that reduces the biosynthesis of norepinephrine from dopamine and decreases sympathetic nerve activity. Blood pressure was measured in anesthetized D2−/− mice treated with etamicastat by gavage, (10 mg/kg), conscious D2−/− mice and D2+/+ littermates, with the D2R selectively silenced in the kidney, treated with etamiscastat in the drinking water (10 mg/kg/day). Tissue and urinary catecholamines and renal expression of selected G protein-coupled receptors, enzymes related to the production of reactive oxygen species, and sodium transporters were also measured. Etamicastat decreased blood pressure both in anesthetized and conscious D2−/− mice and mice with renal-selective silencing of D2R to levels similar or close to those measured in D2+/+ littermates. Etamicastat decreased cardiac norepinephrine and increased cardiac and urinary dopamine levels in D2−/− mice. It also normalized the increased renal protein expressions of ETB, NADPH oxidase isoenzymes, NHE3, and NCC, and increased the renal expression of D1R but not D5R in D2−/− mice. In conclusion, etamicastat is effective in normalizing the increased blood pressure and some of the abnormal renal biochemical alterations of D2−/− mice.
Keywords: hypertension, kidney, G-protein couple receptors, dopamine
INTRODUCTION
Inhibition of dopamine β-hydroxylase (DBH) may provide significant clinical improvement in patients suffering from cardiovascular disorders, such as hypertension and chronic heart failure. The rationale for the use of DBH inhibitors is based on their ability to inhibit the biosynthesis of norepinehrine (NE), via inhibition of the enzymatic hydroxylation of dopamine (DA) (1).
Direct inhibition of sympathetic nerve function by reducing the biosynthesis of NE, preventing the conversion of DA to NE in sympathetic nerves, and possibly by increasing the release of DA, can induce renal vasodilation, diuresis, and natriuresis. β-adrenergic blockers are no longer recommended as primary therapy for hypertension except for patients with coexisting conditions, such as coronary heart disease or left ventricular dysfunction. a-adrenergic blockers are also not recommended as first-line therapy for hypertension because they may be associated with increased incidence of adverse cerebrovascular and cardiovascular outcomes (2). Therefore, inhibitors of DBH may provide significant clinical advantages over other drug treatments, especially those that affect the sympathetic nervous system.
Etamicastat [(R)-5-(2-aminoethyl)-1-(6,8-difluorochroman-3-yl)-1,3-dihydroimidazole-2-thione hydrochloride] is a potent reversible inhibitor of peripheral DBH with limited access to the brain (1). In spontaneously hypertensive rats (SHR) but not in normotensive control rats, oral administration of etamicastat lowered both systolic and diastolic blood pressures in a dose-dependent manner without affecting the heart rate, (3). Etamicastat, chronically administered in drinking water, also significantly reduced both blood pressure and urinary NE excretion but increased urinary DA excretion in SHR (4). This DBH inhibitor has also been shown to decrease blood pressure in hypertensive patients (5).
The intrarenal dopaminergic system plays an important role in the normal regulation of renal sodium excretion and blood pressure (6). Human essential hypertension and some rodent models of genetic hypertension are associated with decreased renal dopamine production and receptor function (6,7). Both dopamine D2-like (Drd2, Drd3 and Drd4) and D1-like (Drdl and Drd5) receptors have been shown to regulate arterial blood pressure. Abnormalities of the D2R gene (DRD2) play a role in the pathogenesis of human essential hypertension; several variants of the human DRD2 have been reported to be associated with hypertension (8,9) and disruption (D2−/−) or renal selective silencing of Drd2 in mice increases systolic and diastolic blood pressures (10–12) and may cause salt sensitivity (13). The hypertension of D2−/− mice has been related to increased sympathetic and vascular smooth muscle endothelin B receptor (ETBR) activities (10), as well as increased reactive oxygen species (ROS) (11).
In this study we determined the effects of inhibition of DBH and subsequently NE formation with etamicastat on blood pressure in D2−/− mice. We measured blood pressure in conscious and anesthetized D2−/−mice and D2+/+ littermates after acute and short-term administration of etamicastat, catecholamine levels in the heart and urine, and renal expression of selected G protein-coupled receptor and enzymes related to ROS production. In addition, the effect of etamicastat on the elevated blood pressure of mice in which renal cortical Drd2 was silenced by the renal subcapsular infusion of Drd2-specific siRNA via an osmotic minipump (12). We also determined the expression of sodium transporters, exchangers, channels, and pump in the kidney of D2−/−mice and D2+/+ littermates before and after treatment with etamicastat.
MATERIAL AND METHODS
D2 Receptor-Deficient Mice
The original F2 hybrid strain (129/SvXC57BL/6J, Oregon Health Sciences University, Portland) that contained the mutated D2R allele (D2−/−) was backcrossed into wild-type C57BL/6J for more than 20 generations and genotyped (10). All mice were bred in the Animal Care Facility of the University of Maryland School of Medicine and The George Washington University School of Medicine & Health Sciences. Male D2−/− mice and D2+/+ littermates fed 0.6% NaCl were studied at 4 to 6 months of age. All studies were approved by the Animal Care and Use Committees of the University of Maryland School of Medicine and The George Washington University School of Medicine & Health Sciences.
Treatment with etamicastat and blood pressure measurements
Acute treatment
Etamicastat (10 mg/kg), synthesized in the Department of Chemistry, BIAL-Portela & Ca, S.A. Portugal, at a purity of 99.5%, or vehicle (tap water) was administered by gastric gavage (200 μL) to D2−/− and D2+/+ mice. The mice were individually housed in metabolic cages for collection of urine samples before measurement of blood pressure. Blood pressure was measured 9 or 18 h after drug administration. Systolic and diastolic blood pressures were measured (Cardiomax II, Instruments, Columbus, OH) from the aorta, via the femoral artery, under pentobarbital anesthesia (50 mg/kg, administered intraperitoneally). Blood pressures were recorded 1 h after the induction of anesthesia when blood pressures were stable. The mice were euthanatized (pentobarbital 100 mg/kg) at the conclusion of the study. The hearts were harvested, frozen in isopentane at −30°C on dry ice, and stored at −80°C until studied. Tissue and urine catecholamines were quantified, as reported (1,14,15)
Short-term etamicastat treatment in conscious mice
TA-PAC20 transmitters (Data Sciences International, St. Paul, MN) were implanted into the carotid artery of D2−/− and D2+/+ mice under isoflurane anesthesia, and blood pressures were measured on individual platforms one week after the surgery (16,17). Etamicastat (10 mg/kg/day) or vehicle (tap water) was added in the drinking water after baseline blood pressure measurement. The mice were monitored for 5 days after starting drug treatment. Blood pressure and heart rate were recorded every 10 min throughout the study. Data were collected and stored automatically in a dedicated computer that ran and analyzed the data (Dataquest). Thereafter, the mice were euthanized, kidneys were harvested and the renal expression of selected G protein-coupled receptors, ROS-related enzymes and sodium transporters, exchanges, channel, and pump were quantified, as reported (10,11,16–22).
In another study, mice implanted with TA-PAC20 transmitters were treated with etamicastat (10 mg/kg/day in the drinking water) for 5 days and fed a normal salt (0.6% NaCl) diet. At the end of this period the dose of etamicastat was increased to 50 mg/kg/day for another 5 days. Blood pressure was recorded (one reading per h) on the last day on each treatment.
Acute renal-specific downregulation of D2R
Renal cortical Drd2 was silenced by the renal subcapsular infusion of Drd2-specific siRNA via an osmotic minipump (12,18,19). Adult male C57BL/6J mice were uninephrectomized one week before the implantation of the minipump. Osmotic minipumps (ALZET® Osmotic Pump, 100 μl; flow rate 0.5 μl/h. for 7 days) were filled with previously validated Drd2-specific siRNA (delivery rate 3μ,g/day) or non-silencing siRNA as control. The siRNAs were dissolved in an in vivo transfection reagent (TransIT® In Vivo Gene Delivery System, Mirus) under sterile conditions. The minipumps were fitted with a polyethylene delivery tubing (Alzet #0007701) and the tip of the tubing was inserted within the subcapsular space of the remaining kidney. Etamicastat treatment (10 mg/kg/day in the drinking water) was started immediately after pump implantation. Blood pressure was measured under pentobarbital anesthesia, as described, 7 days after pump implantation.
Immunoblotting
Whole kidney lysates were prepared in lysis-buffer supplemented with protease inhibitors, as previously reported (10,11,16–22). Samples with equal amounts of proteins were separated by 10% SDS-polyacrylamide gel (Bio-Rad) electrophoresis and transferred onto nitrocellulose membranes. The membranes were sequentially probed with the primary antibodies (1:5000) at 4°C overnight and corresponding horseradish peroxidase-conjugated secondary antibodies (1:10000, Pierce at room temperature for 1 h). Chemiluminescence was detected using SuperSignal West Dura Substrate (Thermo Fisher Scientific, Waltham, MA), followed by autoradiography. The band densities of the proteins of interest were quantified by the NIH Image J and normalized by corresponding total actin bands. Alternatively, for infrared detection of protein signal, the membranes were probed with IR-dye 680- or 800-labeled secondary antibodies (LI-COR Bioscience, Lincoln, NE). The band densities of the proteins of interest were quantified using the Odyssey Infrared imaging system (Li-COR) and normalized by corresponding total actin bands.
The rabbit polyclonal antibodies against dopamine receptors D1R (DRD1), D3R (DRD3) D4R (DRD4), and D5R (DRD5) were generated in our laboratory while rabbit polyclonal antibodies against D2R (EMD Millipore, Billerica, MA) and actin (Sigma-Aldrich, St. Louis, MO) were purchased. We have reported the specificity of our D1R antibody (17,19), D3R antibody (18), D4R antibody (21), D5R antibody (20), D1R antibody from Origene (Rockville, MD) (21), and D2R from EMD Millipore (12). The NOX4 (NADPH oxidase 4) affinity-purified antibody used in these studies was raised in rabbit against the peptide KVPSRRTRRLLDKSKT, which is 100% homologous to both rat (NCBI accession no. NP-445976) and mouse (NCBI accession no. NP- 056575) Nox4 and 93% homologous to human Nox4 (NCBI accession no. AAH40105) at sequence 88–103 of 578 amino acids. The specificity of this antibody has already been reported (23). The sources of other antibodies used were: NHE3, NKCC2, NCC, αENaC, βENaC, γENac, αNaKATPase, generous gifts of Dr. Mark Knepper (20); ETBR (Alomone Labs, Jerusalem, Israel); NOX1 (NADPH Oxidase 1, Santa Cruz Biotechnologies, Dallas, TX), NOX2 (gp91 phox, Upstate Biotech/Thermo Fisher Scientific), HO-1 (Enzo Life Sciences, Farmingdale, NY) and HO- 2 (Enzo Life Sciences).
Assay of catecholamines
Catecholamines in urine and tissue (DA and NE) were assayed by high performance liquid chromatography with electrochemical detection (HPLC-ED), as previously described. The lower limit of detection of dopamine and norepinephrine is 350 fmol (1,14).
Statistical Analysis
Data are reported as mean ± SEM. Comparisons between 2 groups used the Student’s t-test. Oneway ANOVA, followed by Holm-Sidak test, was used to assess significant differences among three or more groups. P<0.05 was considered statistically significant.
RESULTS
Etamicastat decreases blood pressure in mice with germline deletion of D2R or renal-silenced D2R.
Systolic blood pressure measured under anesthesia was higher in D2−/− mice than in D2+/+ littermates treated with vehicle (122±1 vs. 96±6 mmHg, n=4–5/group). Systolic blood pressure also measured under anesthesia, 9 or 18 h after gavage administration of 10 mg/kg etamicastat, was decreased in D2−/− mice to levels similar to those in D2+/+ littermates (Figure 1A). Diastolic blood pressures, which were increased in D2−/− mice relative to their D2+/+ littermates (91±1 vs 68±2 mmHg; P<0.05), were normalized at 9 h (D2−/−: 72±5; D2+/+: 72±1 mmHg) and 18 h (D2−/−: 75±3; D2+/+: 76±4 mmHg) after the administration of etamicastat.
Figure 1. Effect of etamicastat on blood pressure in anesthetized and conscious D2+/+ and D2−/−mice.
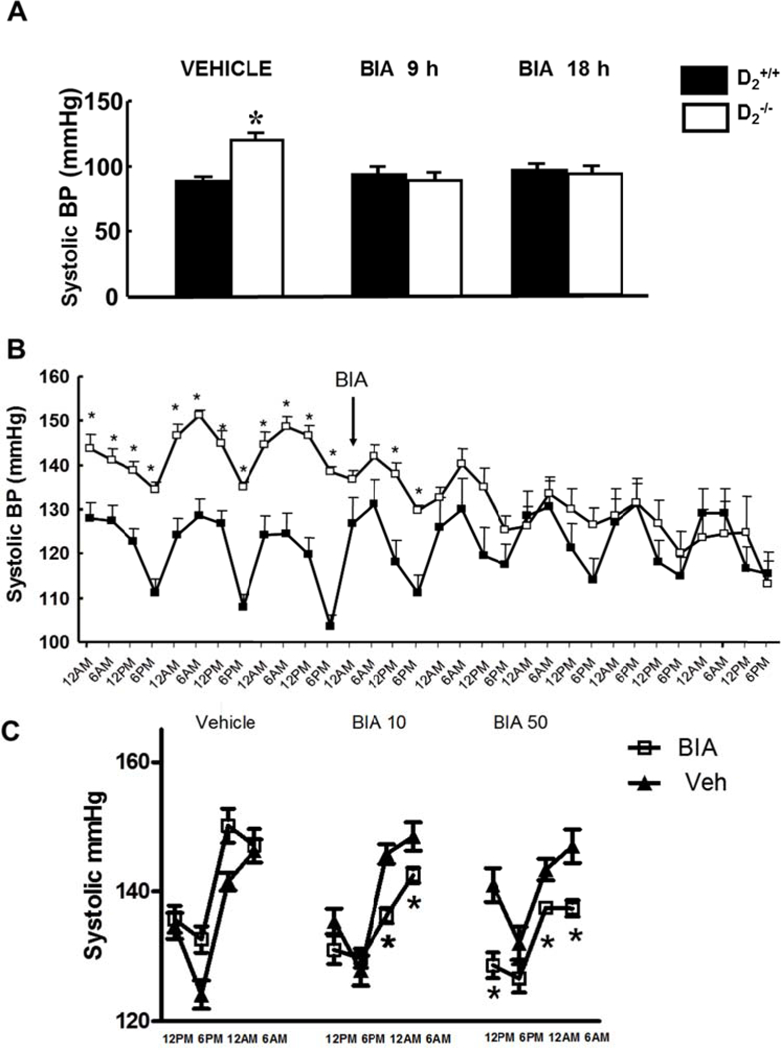
A. Etamicastat (Etam, 1O mg/kg) or vehicle was administered by gavage. Blood pressure was measured under pentobarbital anesthesia 9 and 18 h after etamicastat administration in different groups of D2+/+ and D2−/− mice. n=4–5/group; *P<O.O5 vs all others, one-way ANOVA followed by Holm-Sidak test
B. Etamicastat (Etam, 1O mg/kg/day) or vehicle was added to the drinking water. Blood pressure was measured by telemetry in conscious mice. Values are means of 6 h measurements (one per h). D2+/+ n=5; D2−/− n=4. *P<O.O5 vs D2+/+ one-way ANOVA followed by Holm-Sidak test.
C. Etamicastat (1O or 5O mg/kg/day) was added to the drinking water. Blood pressure was measured by telemetry in conscious mice. Values shown are means of 6 hr measurements during the day and night (one reading per h after 5 days on each treatment). D2+/+ n=4; D2−/− n=3. * P<O.O5 vs D2−/− vehicle or same treatment D2+/+, two-way ANOVA followed by Holm-Sidak test.
Systolic blood pressure measured by telemetry in conscious mice was also higher D2−/− than in D2+/+ mice (Figure 1B). Administration of etamicastat (10 mg/kg/day, n=4–5/group), added to the drinking water, also decreased systolic blood pressure in D2−/− mice but had no significant effect in D2+/+ littermates. The decrease in systolic blood pressure was noted 24 h after starting treatment and persisted throughout the duration of the study (Figure 1B). The decrease in systolic blood pressure was more marked during the night when the mice are awake, feeding, and drinking water (Figure 1C). The conscious systolic blood pressure during the day or night was similar in mice treated for five days with a 10 or 50 mg/kg/day dose of etamicastat.
Renal cortical-selective silencing of the D2R increases blood pressure in mice (12). As shown in Figure 2, mice implanted with an osmotic minipump for the continuous renal subcapsular infusion of D2R siRNA, which decreased renal D2R expression by 70–80% (12), had increased systolic blood pressure under anesthesia. The increase in blood pressure resulting from the D2R siRNA infusion was prevented in mice treated with etamicastat in the drinking water (10 mg/kg/day, n=5/group) during the 7 days of D2R siRNA infusion (Figure 2).
Figure 2. Effect of etamicastat on blood pressure in mice with renal-selective silencing of the D2R.
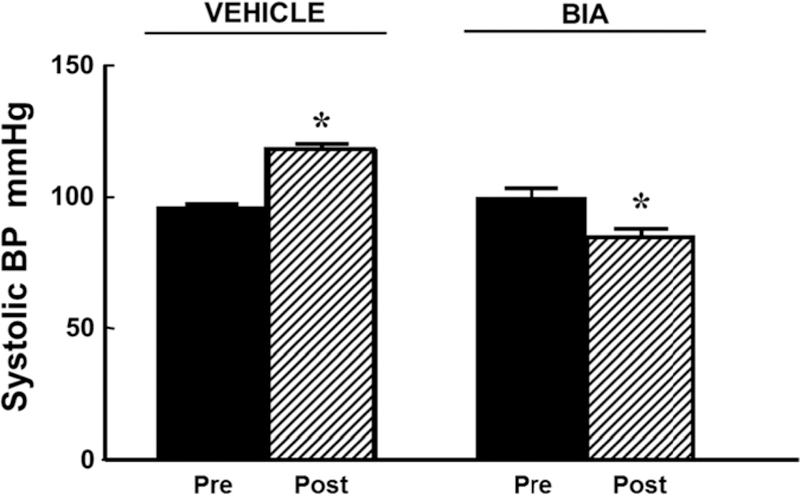
Renal cortical Drd2 was silenced by the renal subcapsular infusion of Drd2 siRNA, via an osmotic minipump for seven days in uninephrectomized adult male C57BL/6J mice (see Methods). Etamicastat (Etam, 1O mg/kg/day in the drinking water) was started immediately after pump implantation. Blood pressure was measured under anesthesia before and 7 days after pump implantation. n=5/group; *P<O.O5 vs all others; one-way ANOVA followed by Holm-Sidak test. Two-way ANOVA positive for effect of D2R siRNA and etamicastat.
Effect of etamicastat on catecholamines in tissue and urine
The cardiac NE content was higher in vehicle-treated D2−/− than D2+/+ mice. Eighteen hours after the stomach-gavage of etamicastat, cardiac NE content decreased in D2−/− mice but was minimally affected in D2+/+ mice. By contrast, urinary excretion of NE was similar in vehicle-treated D2+/+ and D2−/− mice; etamicastat decreased NE excretion in D2+/+ mice only (Figure 3A, n=5–8/group).
Figure 3. Effect of etamicastat on norepinephrine (NE) and dopamine (DA) content in heart, and urine of D2+/+ and D2−/− mice.
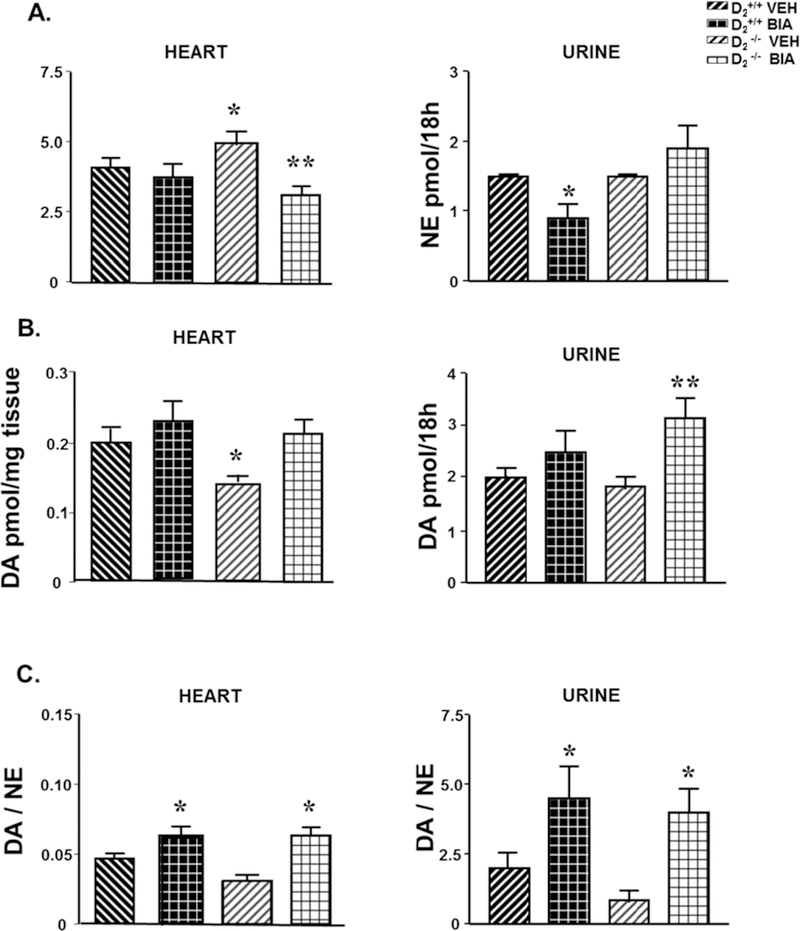
Etamicastat (Etam, 10 mg/kg/dose) or vehicle was administered by gavage. Urine collection was started immediately after gavage and lasted for 18 h. Tissues were collected after ending the urine collection. Urine and tissue NE (A) and DA (B) were measured by HPLC-ED, n=5–8/group. The tissue or urine DA/NE ratio (C) was calculated. *P<0.05 vs all others; **P<0.05 vs same group vehicle-treated mice; one-way ANOVA followed by Holm-Sidak test.
The cardiac DA content, which was lower in vehicle-treated D2−/− than D2+/+, was increased by etamicastat in D2−/− but not in D2+/+ mice. Urinary DA was similar in untreated mice of both strains but was increased by etamicastat in D2−/− but not D2+/+ mice (Figure 3B). The DA/NE ratio was lower, although not significantly, in tissues and urine of untreated D2−/− than in D2+/+ mice and increased by etamicastat in heart and urine of both D2+/+ and D2−/− mice (Figure 3C).
Effect of etamicastat on the renal expression of dopamine and endothelin B receptors
In D2−/− mice, relative to D2+/+ littermates, the renal protein expressions of D3R and ETBR were increased. Etamicastat treatment did not alter the increased renal expression of D3R, but increased D1R expression, normalized ETBR expression, and decreased D5R expression (Figures 4A and B, n=4–5/group).
Figure 4. Effect of etamicastat on the renal expression of dopamine, angiotensin II type 1, and endothelin B receptors.
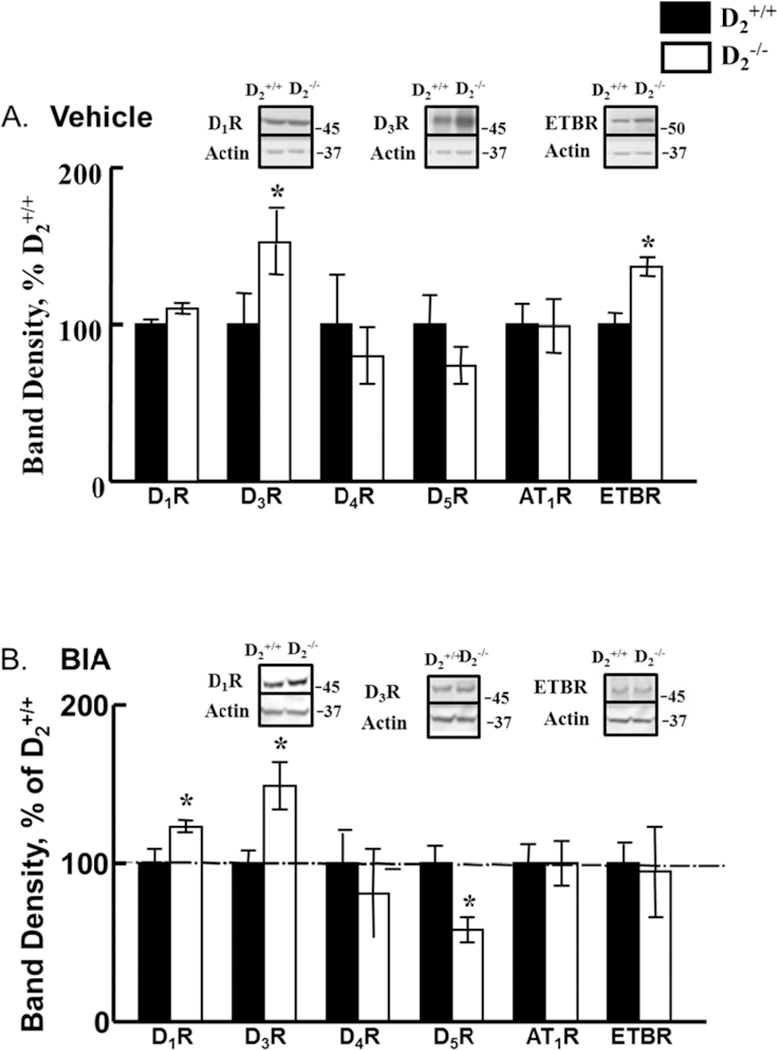
Etamicastat (Etam, 10 mg/kg/day) was added to the drinking water for 5 days. Receptor expression was determined by immunoblotting. A. Receptor expression in vehicle-treated mice; n=8– 10/group; B. Receptor expression in etamicastat-treated mice; n=4–5/group. *P<0.05 vs D2+/+ mice; one-way ANOVA followed by Holm-Sidak test.
Effect of etamicastat on the renal expression of ROS-related enzymes.
In agreement with our previous report (11) the renal expression of heme oxygenase-2 (HO-2) was decreased and the renal expressions of NADPH oxidase isoforms NOX1, NOX2, and NOX4 were increased in D2−/− mice, relative to D2+/+ littermates. Treatment with etamicastat normalized the expression of these NOX isoforms but did not affect the decreased HO-2 expression in D2−/− mice (Figures 5A and B, n=5/group).
Figure 5. Effect of etamicastat on the expression of ROS-related enzymes.
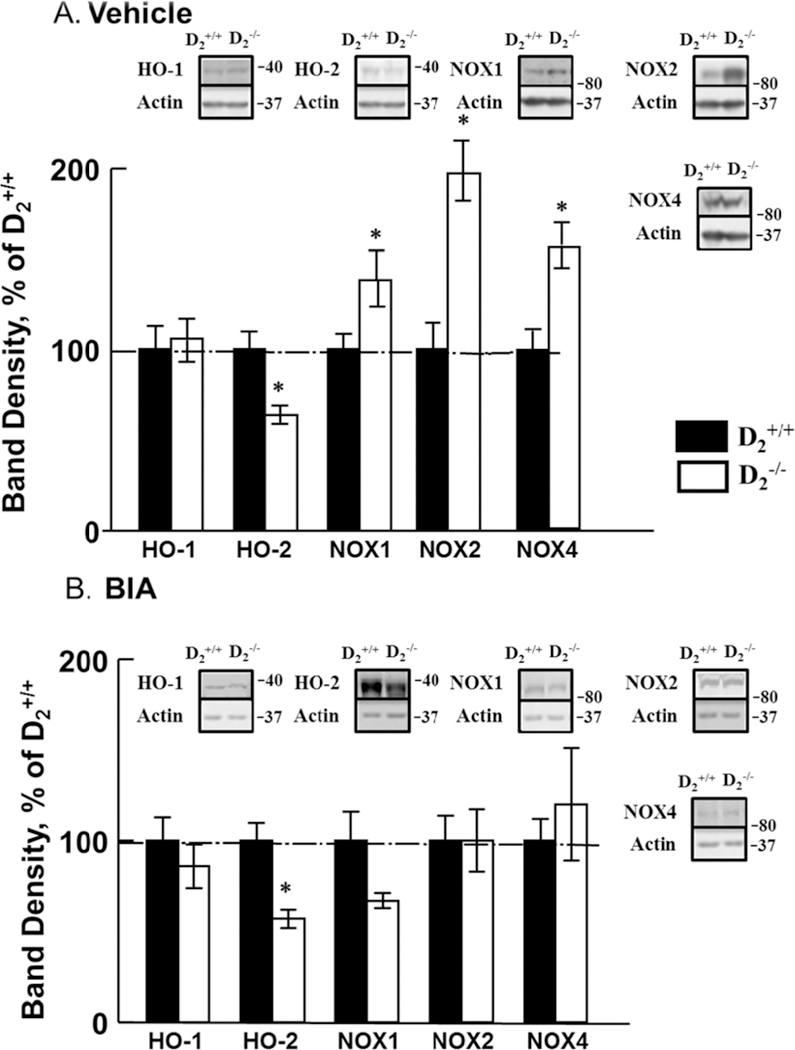
Etamicastat (Etam, 10 mg/kg/day) was added to the drinking water for 5 days. Expression of HO-1, HO-2, NOX1, NOX2, and NOX4 was determined by immunoblotting. A. Expression of HO and NOX isoforms in vehicle-treated mice; n=5/group; B. Expression of HO and NOX isoforms in etamicastat-treated mice; n=5/group. *P<0.05 vs D2+/+ mice; one-way ANOVA followed by Holm-Sidak test.
Effect of etamicastat on the expression of renal sodium cotransporters, exchangers, channels, and pump D2−/− mice.
The renal protein expressions of NHE3 and NCC were increased but the renal protein expression of aNaKATPase was decreased in D2−/− mice, relative to D2+/+ littermates. Etamicastat normalized the renal protein expressions of sodium hydrogen exchanger type 3 (NHE3) and aNaKATPase and decreased that of sodium chloride cotransporter (NCC) (Figures 6A and B, n=4–5/group).
Figure 6. Effect of etamicastat on the expression of renal sodium transporters, exchangers, channels, and pump.
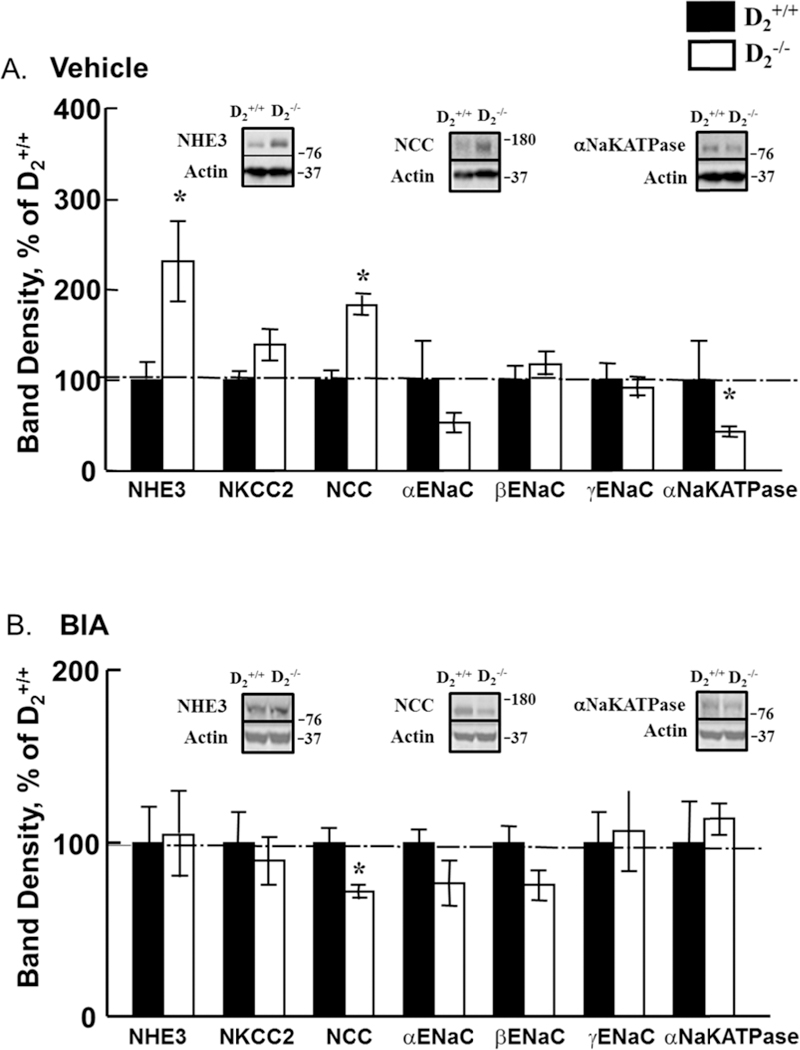
Etamicastat (Etam, 10 mg/kg/day) was added to the drinking water for 5 days. Renal expression of sodium transporters, exchangers, channels, and pump was determined by immunoblotting. A. Expression in vehicle-treated mice; n=4–5/group; B. Expression in etamicastat-treated mice; n=4/group. *P<0.05 vs D2+/+ mice; one-way ANOVA followed by Holm-Sidak test.
DISCUSSION
The results of this study show that either the acute- or short-term administration of the peripheral DBH inhibitor, etamicastat, decreases blood pressure in both conscious and anesthetized mice with germline deletion of Drd2 or renal-selective silencing of Drd2 (renal subcapsular infusion of Drd2 siRNA in D2+/+ mice) but not in wild-type littermates or D2+/+ mice that received renal subcapsular infusion of non-silencing siRNA. The reduction in blood pressure in D2−/− mice caused by etamicastat correlates with an increase in the DA/NE content in heart and urine and D1R expression in the kidney, as well as a normalization of renal Nox isoforms (Nox1, Nox2, and Nox4) and proximal tubule NHE3 and a decrease in distal convoluted tubule NCC.
The reduction in blood pressure in hypertensive D2−/− mice is in agreement with the reported ability of etamicastat to decrease blood pressure in hypertensive rats and humans (3–5). The hypertension in D2−/− mice is, in part, related to increased sympathetic activity proved by the ability of α1-adrenergic receptor blockade and adrenalectomy to decrease the elevated blood pressure of D2−/− mice to the same level as that noted in D2+/+ littermates (10). The NE content of the heart was increased in vehicle-treated D2−/− mice which may reflect the increased sympathetic activity in these mice (10, 24). Etamicastat treatment of these D2−/− mice significantly decreased cardiac NE and increased cardiac DA. In wild-type littermates treated with etamicastat there was a trend for NE to decrease and DA to increase but the effects were not statistically significant. In previous studies using etamicastat, the decrease in DBH activity and sympathetic drive was associated with a decrease in the NE in the heart in wild-type mice (1) and urine in healthy human subjects (25). In the current study cardiac NE content was decreased by 40% in D2−/− mice but minimally in D2+/+, 18 h after administration of a 10 mg/kg dose of etamicastat. This modest decrease may be due to differences in sympathetic activity between D2−/− and D2+/+mice (10,26). However, it has been shown that in normal mice, a 100 mg/kg dose of etamicastat decreased heart NE content by 75% (1). Deletion of the Dbh gene in mice is associated with NE deficiency. However, the extracellular levels of DA are also decreased in the nucleus accumbens and caudate putamen but not in the prefrontal cortex and increased in adrenergic neurons, cerebellum, liver, lung, retina, skeletal muscle, and spleen of Dbh−/− mice on mixed C57BL/6 and 129/SvEv background (27, 28).
Urine NE and DA levels, were similar in D2−/− and their wild-type littermates, in agreement with our previous report (10) and with the failure of others to find differences in dopamine levels in the brain striatum of D2−/− and D2+/+ mice (29). Basal DA efflux in the striatum was reported to be similar in D2−/− and D2+/+ mice (29), although an increase in DA metabolites was found by others (30). In D2+/+ mice etamicastat decreased urinary NE. Because about 35% of urinary NE is derived from the kidney (24) this result could be taken to suggest that the inhibitory effect of etamicastat is more pronounced or of longer duration in the kidney than in the heart.
The increase in blood pressure in D2−/− mice is not due to an increase in the activity of the renin-angiotensin system (10). However, we have reported that the hypertension in D2−/− mice is associated with increased production of ROS, accompanied by increased expression of NOX enzymes, as well as decreased expression of HO-2, results that were corroborated in the present study (11). Treatment with etamicastat normalized the renal expression of Nox enzymes but did not reverse the decrease in HO-2 expression. In D2−/− mice, treatment with spironolactone decreased blood pressure, and normalized the increased expression of NOX1 and NOX4 but had no effect on the increased NOX2 or the decreased HO-2 expressions. Thus it is possible that the increase in blood pressure in D2−/− mice drives the increase in NOX enzyme expression in D2−/− mice. However, the decrease in HO-2 appears to be at least, in part, a cause rather than a consequence of the increase in blood pressure.
In tissues outside the central nervous system, the inhibition of DBH increases DA release (31,32); DA has vasorelaxant effects that cause an increase in renal blood flow and abet the sodium excretion caused by inhibition of renal tubular sodium transport (6). Independent of innervation, the kidney synthesizes DA that is not metabolized to NE and prevents the ability of moderate sodium load to increase blood pressure by inducing diuresis and natriuresis (6,33). Cardiac and urinary DA levels are increased in D2−/− mice after etamicastat administration. In spite of the fact that DA produced by the kidney from L-DOPA is not metabolized to NE because DBH is not expressed in renal tubules (34), inhibition of DBH could still increase renal DA because of NE production from renal nerves (35). Most if not all of the DA in the urine is synthesized in the kidney under normal conditions (6,35). However, an increase in DA release in peripheral tissues, caused by inhibition of DBH by etamicastat, may substantially increase DA in the glomerular filtrate. The increase in the DA/NE ratio in the urine in both groups could be a reflection of an increase in filtered DA rather than an increase in renal DA production. DA increases renal sodium excretion, in part, by inhibiting renal NHE3, sodium phosphate cotransporters (NaPi-IIa, NaPi-IIc), Cl-/HCO3- exchanger, sodium bicarbonate exchanger (NBCe1), NaKATPase, NCC, ENac, and potassium channel (6). Treatment with etamicastat reversed the increased renal protein expressions of NHE3, NCC, and ETBR but increased NaKATPase protein in D2−/− mice suggesting that these changes were secondary to either the increased sympathetic activity or the increase in blood pressure. However, etamicastat also increased renal D1R expression, did not alter the increased D3R expression and decreased D5R expression in D2−/− mice, indicating some specific effect on renal dopamine receptor expression when renal/urinary DA is increased.
In conclusion, etamicastat is effective in normalizing blood pressure in D2−/− mice, in which hypertension is caused, in part, by increased activity of the sympathetic nervous system. In D2−/− mice, etamicastat normalized the increased renal expression on NHE3, decreased the renal expression of NCC, but increased the renal expression of NaKATPAse. Etamicastat also increased the renal expression of D1R and normalized the increased renal expression of ETBR, decreased the renal expression of D5R without affecting the increased renal expression of D3R. Etamicastat also normalized the increased renal expression of NOX isoenzymes. Whether or not these effects are primary or secondary to the etamicast-induced decrease in blood pressure in D2−/− mice remains to be determined. However, we have reported that decreasing the blood pressure of D2−/− mice with spironolactone does not normalize the increased renal NOX2 expression in D2−/− mice suggesting that the negative regulation of NOX expression by etamicastat may be blood pressure independent.
ACKNOWLEDGMENTS
BIAL - Portela & Ca, S.A. supported this study. BIAL had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
PSS is an employee of BIAL-Portela & Ca, S.A. (the sponsor of the study).
Footnotes
CONFLICT OF INTEREST
REFERENCES
- 1.Beliaev A, Learmonth DA, Soares-da-Silva P. Synthesis and biological evaluation of novel, peripherally selective chromanyl imidazolethione-based inhibitors of dopamine beta hydroxylase. JMed Chem. 2006: 49:1191–1197 [DOI] [PubMed] [Google Scholar]
- 2.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS, Ortiz E. 2014. evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014: 311:507–520. [DOI] [PubMed] [Google Scholar]
- 3.Igreja B, Pires NM, Bonifacio MJ, Loureiro AI, Fernandes-Lopes C, Wright LC, Soares-da-Silva P. Blood pressure-decreasing effect of etamicastat alone and in combination with antihypertensive drugs in the spontaneously hypertensive rat. Hypertens Res 2015;38:30–38. [DOI] [PubMed] [Google Scholar]
- 4.Igreja B, Wright L, Soares-da-Silva P. Sustained high blood pressure reduction with etamicastat, a selective peripheral dopamine-β-hydroxylase inhibitor. J Am Soc Hypertens 2016;10:207–216.. [DOI] [PubMed] [Google Scholar]
- 5.Almeida L, Nunes T, Costa R, Rocha JF, Vaz-da-Silva M, Soares-da-Silva P. Etamicastat, a novel dopamine ß-hydroxylase inhibitor: tolerability, pharmacokinetics, and pharmacodynamics in patients with hypertension. Clin Ther. 2013:35:1983–1996. [DOI] [PubMed] [Google Scholar]
- 6.Armando I, Villar VA, Jose PA (2011). Dopamine and renal function and blood pressure regulation. Compr Physiol. 2011:1:1075–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banday AA, Lokhandwala MF Dopamine receptors and hypertension. Curr Hypertens Rep. 2008; 10, 268–275 [DOI] [PubMed] [Google Scholar]
- 8.Fang YJ, Thomas GN, Xu ZL, Fang JQ, Critchley JA, Tomlinson B. An affected pedigree member analysis of linkage between the dopamine D2 receptor gene TaqI polymorphism and obesity and hypertension. Int J Cardiol. 2005;102:111–116. [DOI] [PubMed] [Google Scholar]
- 9.Thomas GN, Tomlinson B, Critchley JA. Modulation of blood pressure and obesity with the dopamine D2 receptor gene TaqI polymorphism. Hypertension 2000;36:177–182. [DOI] [PubMed] [Google Scholar]
- 10.Li XX, Bek M, Asico LD, Yang Z, Grandy DK, Goldstein DS, Rubinstein M, Eisner GM, Jose PA. Adrenergic and endothelin B receptor-dependent hypertension in dopamine receptor type-2 knockout mice. Hypertension 2001;38:303–308. [DOI] [PubMed] [Google Scholar]
- 11.Armando I, Wang X, Villar VA, Jones JE, Asico LD, Escano C, Jose PA. Reactive oxygen species dependent hypertension in dopamine D2 receptor-deficient mice. Hypertension 2007;49:1–7. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Cuevas S, Asico LD, Escano C, Yang Y, Pascua AM, Wang X, Jones JE, Grandy D, Eisner G, Jose PA, Armando I. Deficient dopamine D2 receptor function causes renal inflammation independently of high blood pressure. PLoS One 2012;7:e38745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozono R1, Ueda A Oishi Y, Yano A, Kambe M, Katsuki M, Oshima T (2003). Dopamine D2 receptor modulates sodium handling via local production of dopamine in the kidney. J Cardiovasc Pharmacol. 2003; 42:S75–S79. [DOI] [PubMed] [Google Scholar]
- 14.Soares-da-Silva P, Fernandes MH, Pinto-do-O PC. Cell inward transport of l-DOPA and 3-O-methyl-l-DOPA in rat renal tubules. Br J Pharmacol. 1994; 112:611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soares-da-Silva P, Pestana M, Fernandes MH. Involvement of tubular sodium in the formation of dopamine in the human renal cortex. J Am Soc Nephrol. 1993;3:1591–99. [DOI] [PubMed] [Google Scholar]
- 16.Bek MJ, Wang X, Asico LD, Jones JE, Zheng S, Li X, Eisner GMK, Grandy Dk, Carey RM, Soares-da-Silva P, Jose PA. Angiotensin-II type 1 receptor-mediated hypertension in D4 dopamine receptor-deficient mice. Hypertension 2006;47:288–95. [DOI] [PubMed] [Google Scholar]
- 17.Escano CS, Armando I, Wang X, Asico LD, Pascua A, Yang Y, Wang Z, Lau YS, Jose PA.. Renal dopaminergic defect in C57Bl/6J mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1660–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armando I, Villar VA, Jones JE, Lee H, Wang X, Asico LD, Yu P, Yang J, Escano CS Jr, Pascua-Crusan AM, Felder RA, Jose PA. Dopamine D3 receptor inhibits the ubiquitin-specific peptidase 48 to promote NHE3 degradation. FASEB J 2014:28:1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villar VA, Armando I, Sanada H, Frazer LC, Russo CM, Notario PM, Lee H, Comisky L, Russell HA, Yang Y, Jurgens JA, Jose PA, Jones JE. Novel role of sorting nexin 5 in renal D1 dopamine receptor trafficking and function: implications for hypertension. FASEB J 2013:27:1808–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Luo Y, Escano CS, Yang Z, Asico L, Li H, Jones JE, Armando I, Lu Q, Sibley DR, Eisner GM, Jose PA. Upregulation of renal sodium transporters in D5 dopamine receptor-deficient mice. Hypertension 2010;55:1431–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Escano CS, Asico L, Jones JE, Barte A, Lau YS, Jose PA, Armando I. Upregulation of renal D5 dopamine receptor ameliorates the hypertension in D3 dopamine receptor-deficient mice. Hypertension 2013;62:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng C, Wang Z, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA (2005). Aberrant ETB receptor regulation of AT 1 receptors in renal proximal tubule cells of spontaneously hypertensive rats. Kidney Int 2005;68:623–631. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Armando I, Asico LD, Escano C, Wang X, Lu Q, Felder RA, Schnackenberg CG, Sibley DR, Eisner GM, Jose PA. The elevated blood pressure of human GRK4gamma A142V transgenic mice is not associated with increased ROS production. Am J Physiol Heart Circ Physiol. 2007;29:H2083H2092. [DOI] [PubMed] [Google Scholar]
- 24.Kopin IJ. Catecholamine metabolism: basic aspects and clinical significance. Pharmacol Rev 1985;37:333–364. [PubMed] [Google Scholar]
- 25.Nunes T, Rocha JF, Vaz-da-Silva M, Falcao A, Almeida L, Soares-da-Silva P. Pharmacokinetics and tolerability of etamicastat following single and repeated administration in elderly versus young healthy male subjects: an open-label, single-center, parallel-group study. Clin Ther 2011; 33:776–791. [DOI] [PubMed] [Google Scholar]
- 26.Pires M, Igreja B, Moura E, Wright LC, Serrao MP, Soares-da-Silva P. Blood pressure decfease in spontanueously hypertensive rats following renal denervation or dopamine-β-hydroxylase inhibition with Etamicastat. Hypertens Res 2015; 38:605–612.. [DOI] [PubMed] [Google Scholar]
- 27.Schank JR, Ventura R, Puglisi-Allegra S, Alcaro A, Cole CD, Liles LC, Seeman P, Weinshenker D. Dopamine beta-hydroxylase knockout mice have alterations in dopamine signaling and are hypersensitive to cocaine. Neuropsychopharmacology 2006;31:2221–2230. [DOI] [PubMed] [Google Scholar]
- 28.Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature 1995;374:643–646. [DOI] [PubMed] [Google Scholar]
- 29.Rouge-Pont F, Usiello A, Benoit-Marand M, Gonon F, Piazza PV, Borrelli E. Changes in extracellular dopamine induced by morphine and cocaine: crucial control by D2 receptors. J Neurosci 2002;22:3293–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung MY, Skryabin BV, Arai M, Abbondanzo S, Fu D, Brosius J, Robakis NK, Polites HG, Pintar JE, Schmauss C. Potentiation of the D2 mutant motor phenotype in mice lacking dopamine D2 and D3 receptors. Neuroscience 1999;91:911–24. [DOI] [PubMed] [Google Scholar]
- 31.Soares-da-Silva P Evidence for a non-precursor dopamine pool in noradrenergic neurones of the dog mesenteric artery. Naunyn Schmiedebergs Arch Pharmacol 1986;333:219–223. [DOI] [PubMed] [Google Scholar]
- 32.Soares-da-Silva P A comparison between the pattern of dopamine and noradrenaline release from sympathetic neurones of the dog mesenteric artery. Br J Pharmacol 1987; 90:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussain T, Lokhandwala MF (2003). Renal dopamine receptors and hypertension. Exp Biol Med (Maywood) 2003;228:134–42. [DOI] [PubMed] [Google Scholar]
- 34.Hartman BK. Immunofluorescence of dopamine-β-hydroxylase. Application of improved methodology to the localization of the peripheral and central noradrenergic nervous system. J Histochem Cytochem 1973;21:312–332. [DOI] [PubMed] [Google Scholar]
- 35.Morgunov N, Baines AD (1981). Renal nerves and catecholamine excretion. Am J Physiol 1981;240:F75–F81. [DOI] [PubMed] [Google Scholar]