

ABSTRACT
Rho signaling is a conserved mechanism for generating forces through activation of contractile actomyosin. How this pathway can produce different cell morphologies is poorly understood. In the Drosophila embryonic epithelium, we investigate how Rho signaling controls force asymmetry to drive morphogenesis. We study a distinct morphogenetic process termed ‘alignment’. This process results in striking columns of rectilinear cells connected by aligned cell–cell contacts. We found that this is driven by contractile actomyosin cables that elevate tension along aligning interfaces. Our data show that polarization of Rho effectors, Rok and Dia, directs formation of these cables. Constitutive activation of these effectors causes aligning cells to instead invaginate. This suggests that moderating Rho signaling is essential to producing the aligned geometry. Therefore, we tested for feedback that could fine-tune Rho signaling. We discovered that F-actin exerts negative feedback on multiple nodes in the pathway. Further, we present evidence that suggests that Rok in part mediates feedback from F-actin to Rho in a manner independent of Myo-II. Collectively, our work suggests that multiple feedback mechanisms regulate Rho signaling, which may account for diverse morphological outcomes.
KEY WORDS: Rho signaling, Epithelial morphogenesis, Actomyosin contractility, Developmental mechanics, Non-muscle Myosin-II, Zipper, Drosophila melanogaster
Summary: A distinctive morphological process of cell alignment in the Drosophila embryonic epithelium reveals that multiple feedback mechanisms in the Rho pathway can fine-tune tissue morphology during development.
INTRODUCTION
Tissue function frequently relies on the underlying shapes and organization of its constituent cells. Diverse processes are responsible for coordinating the morphology and arrangement of cells into complex geometries. Failure of these morphogenetic processes can lead to developmental defects and cancer (Wallingford et al., 2013; Wang, 2009).
Common to all morphogenetic processes is the use of mechanical force in order to generate high-energy arrangements of cells. Specifically, contractile force produced by actomyosin assemblies can be used to induce various morphological changes: apical constriction, cell intercalation, cell migration, and many others (Lecuit and Lenne, 2007; Levayer and Lecuit, 2012).
We study a distinctive morphological process in the Drosophila embryonic epithelium that our lab has termed ‘alignment’ (Simone and DiNardo, 2010). This process occurs in the anterior region of each abdominal segment during mid-embryogenesis. Before alignment, the cells exhibit irregular packing where the angles separating cell–cell contacts average close to 120°. This is characteristic of a simple epithelium in a low-energy state (Classen et al., 2005; Gibson et al., 2006). In each segment, alignment occurs at two interfaces of cell–cell contacts along the dorsal-ventral axis (Fig. 1A,B) (Marcinkevicius and Zallen, 2013; Simone and DiNardo, 2010). After two hours, these two interfaces straighten significantly relative to non-aligning regions of the epithelium (Fig. 1A,B). Quantitatively, this means that angles between contacts approach 180°, which is a high-energy configuration.
Fig. 1.

Alignment is driven by local tension asymmetry generated by actomyosin contractility. (A) Schematic of the ventral face of a Drosophila embryo after alignment. Arrows indicate anterior (A)–posterior (P) and dorsal (D)–ventral (V) axes. In each abdominal segment, the two aligning interfaces are indicated (purple). (B) Tension (peak velocity) was elevated along aligning (purple) cell–cell contacts relative to orthogonal (green) and non-aligning (orange) contacts. Cell junctions were visualized with E-cad::GFP. Orthogonal: n=21 junctions from 15 embryos; aligning: n=31 junctions, 21 embryos; non-aligning: n=46 junctions, 26 embryos. (C) Depletion of MLC along one interface caused alignment defects (red arrowheads, dashed line). Cell junctions were visualized with E-cadherin (E-cad::tdTomato), and deGradFP-expressing cells identified by nuclear localization signal (NLS::mCherry) signal. Blue arrowheads mark bi-nucleated cells caused by MLC knockdown. Depletion was restricted to the anterior interface and did not affect the morphology of the posterior interface (white arrowheads, dashed line). Wild-type control (WT CTRL): n=15 interfaces, 6 embryos; deGradFP+: n=17 interfaces, 6 embryos. (D) MLC knockdown (MLC KD, red) caused a significant decrease in alignment compared to that of embryos expressing untagged MLC as a control (WT CTRL, yellow). (E) MLC knockdown resulted in a significant decrease in tension compared to wild-type (WT) control along aligning contacts (purple), but no significant change along orthogonal contacts (green). The net result is a reduction in tension asymmetry (orange arrows). WT CTRL: n=13 orthogonal junctions, 7 embryos, n=17 aligning junctions, 5 embryos; deGradFP+: n=12 orthogonal junctions, 5 embryos, n=18 aligning junctions, 6 embryos. ***P<0.0001, Mann–Whitney U-test; bars show mean±s.d. Scale bars: 4 μm.
Our lab previously reported the requirement for non-muscle Myosin II (Myo-II) in alignment (Simone and DiNardo, 2010). Furthermore, we showed that enrichments of non-muscle Myo-II and F-actin arise along interfaces, forming supracellular actomyosin cables (Röper, 2013; Simone and DiNardo, 2010). This evidence strongly indicates a role for asymmetric force generation. However, the polarity signals that direct the actomyosin cytoskeleton in alignment are unknown. In this report, we show a key function for Rho signaling in spatial regulation of actomyosin and cortical tension during alignment.
The Rho family of small GTPases are master regulators of the actin cytoskeleton. Rho1 is the sole homolog of RhoA in Drosophila (hereafter Rho) (Sasamura et al., 1997). Two effectors of Rho are key players in promoting contractile actomyosin: Rho kinase (Rok) and Diaphanous (Dia). Rok has many phosphorylation targets, the most significant of which is the regulatory myosin light chain (MLC) (Amano et al., 2010). This phosphorylation event is essential to the activation of Myo-II mini filaments (Amano et al., 1996). Dia is the sole Diaphanous family formin in Drosophila (Breitsprecher and Goode, 2013). It promotes actin filament nucleation and elongation, classically within linear structures of actin such as stress fibers and filopodia (Breitsprecher and Goode, 2013).
Rho signaling is required in many morphogenetic programs, but how this pathway allows for production of distinct cell and tissue morphologies is still poorly understood. One contributing factor is spatial regulation of Rho activation. In this paper, we demonstrate that planar polarization of Rho activation and its effectors to aligning junctions is critical for the formation of actomyosin cables, thereby driving alignment.
Planar polarization of Rho can, however, result in cell geometries different from alignment. Two such examples are convergent extension and mechanical tissue boundaries. During convergent extension, Rho signaling becomes upregulated in junctions along the dorsal-ventral axis (Kerridge et al., 2016; Munjal et al., 2015; Simoes et al., 2014), initiating the formation of actomyosin asymmetry (Bertet et al., 2004; Zallen and Wieschaus, 2004). At mechanical boundaries, actomyosin cables also increase force along cell–cell contacts in order to prevent cell mixing between tissue compartments (Aliee et al., 2012; Landsberg et al., 2009; Ly et al., 2017; Major and Irvine, 2006; Monier et al., 2010). Studies suggest that Rho signaling is also polarized to guide the formation of these boundaries in many tissues (K.O. and S.D., unpublished results) (Aliee et al., 2012; Landsberg et al., 2009; Ly et al., 2017; Major and Irvine, 2006).
These two processes strongly resemble alignment in the manner that Rho signaling and actomyosin remodeling is polarized to create local force asymmetry in the tissue (Aliee et al., 2012; Fernandez-Gonzalez et al., 2009; Landsberg et al., 2009; Ly et al., 2017; Rauzi et al., 2008). Yet, the final resulting morphologies of these events are quite different from one another. During convergent extension, junctions activated for Rho and actomyosin will shrink and remodel (Bertet et al., 2004; Zallen and Wieschaus, 2004). At tissue boundaries, cell–cell contacts also shorten in length and partly align with one another (Aliee et al., 2012; Landsberg et al., 2009). In contrast, alignment produces very straight interfaces of cell–cell contacts where individual contacts elongate relative to neighboring, orthogonal contacts (Fig. 1A,B). Indeed, the observation that the contacts under the most tension are competent to lengthen is contrary to most current in silico models (Classen et al., 2005; Gibson et al., 2006), highlighting a key distinguishing feature of alignment.
Given that these systems do not appear to exhibit differences in the spatial deployment of Rho or its effectors, other mechanisms must account for these disparate morphological outcomes. In this report, we present evidence that pathway feedback could allow for modification of Rho signaling outcomes. We explored the interactions among the major components in the Rho pathway, revealing both positive and negative feedback. Some of these mechanisms are consistent with reports in other systems (Bement et al., 2015; Graessl et al., 2017; Segal et al., 2018; Staus et al., 2011), while others represent novel interactions. Additionally, in contrast to observations in other systems, we found no evidence for a role for Myo-II activity in feedback regulation of Rho (Graessl et al., 2017; Munjal et al., 2015; Priya et al., 2015; Priya et al., 2017). These results suggest that numerous feedback interactions can fine-tune Rho signaling to allow for plasticity in morphological outcomes.
RESULTS
Local actomyosin contractility generates force asymmetry to drive alignment
We previously proposed that alignment was caused by shrinkage of select cell–cell contacts along the interfaces (Fig. S1A). However, analyzing the cell dynamics of this process at higher temporal resolution revealed no correlation between the remodeling events and the emergence of alignment (Fig. S1C). Therefore, we considered alternatively whether alignment may be caused by collective cell shape changes driven by actomyosin contractility along the length of the interface (Fig. S1B).
To test this, we used laser ablation to map the forces in the aligning epithelium. We ablated individual junctions within the aligning interface (Fig. 1B, magenta; Fig. S2A) and orthogonal junctions intersecting the aligning interfaces (Fig. 1B, green; Fig. S2B). As a control, we ablated junctions within non-aligning interfaces (Fig. 1B, orange; Fig. S2C). Tension along aligning interfaces was over threefold higher compared with that of orthogonal junctions (Fig. 1B), revealing a large local force asymmetry. Furthermore, relative to non-aligning junctions, tension was about twofold higher along aligning junctions (Fig. 1B) demonstrating that this asymmetry is not a global property of the epithelium.
To determine whether this tension asymmetry is actomyosin dependent, we used ‘deGradFP’ to deplete MLC (Caussinus et al., 2012) and disrupt contractility in aligning cells. In brief, this approach utilizes a construct (deGradFP) that targets GFP-tagged proteins for degradation (see Materials and Methods). The presence of deGradFP in an MLC mutant embryo (sqhAX3) that only expresses a transgenic GFP-tagged MLC would result in knockdown of Myo-II activity. We used a spatially-restricted engrailed-GAL4 line to express UAS-deGradFP (Fig. S2E). This resulted in knockdown of MLC in the cells at only the anterior aligning interface, resulting in alignment defects (Fig. 1C,D, red arrowheads). This was in contrast to sibling controls that expressed wild-type, untagged MLC from a sqh+ allele and were, therefore, not susceptible to deGradFP (Fig. 1D, WT CTRL). Depletion of Myo-II activity also led to binucleated cells, indicating disrupted cytokinesis (Fig. 1C, blue arrowheads). To rule out the possibility that blocking cytokinesis caused the alignment defect, we knocked down Pavarotti (Pav), a kinesin-like protein involved in cytokinetic ring assembly (Adams et al., 1998). Pav knockdown using RNAi led to the appearance of binucleated cells but did not affect the planar polarization of F-actin and Myo-II to aligning interfaces, nor did knockdown disrupt alignment (Fig. S2D,E).
We then used deGradFP knockdown of MLC proteins to test whether force asymmetry is generated by the actomyosin cables. We found that tension was significantly decreased along aligning junctions of the MLC-depleted interface in comparison to those in control embryos (Fig. 1E). This resulted in a 30% reduction in tension asymmetry between aligning and orthogonal junctions (Fig. 1E). These results demonstrate that cells at the aligning interface generate cortical tension by upregulating actomyosin contractility. Furthermore, these local forces are responsible for alignment, rather than extrinsic forces from tissues distal to the aligning epithelium.
We gained further insight into the mechanics of alignment by capitalizing on variability in the engrailed-GAL4 expression pattern. This line primarily expresses in the first column of cells comprising the anterior aligning interface (Fig. S2F,G). However, there was occasional GAL4 expression in cells on the posterior side of the interface (Fig. S2G). This yielded two scenarios: unilateral knockdown in which depletion only occurred on one side of the interface (Fig. S2G, salmon) and bilateral knockdown in which cells on both sides of the interface were depleted for MLC (Fig. S2G, red). Unilateral knockdown caused a decrease in tension along aligning junctions relative to that of sibling controls (Fig. S2G). With bilateral knockdown, tension was reduced even further compared to unilateral knockdown (Fig. S2G). This demonstrates that cortical tension along the aligning interfaces comes from actomyosin in the cells on each side of the interface. Interestingly, alignment was equally defective with either unilateral or bilateral knockdown of MLC (Fig. S2H), suggesting that tensile forces must be applied from both sides of the interface for alignment to occur.
Polarization of Rho signaling coordinates alignment
We next sought to identify the polarity signals that guide alignment. Given its role in regulating contractile actomyosin, we examined the localization of Rho pathway factors. A fluorescent sensor for activated, GTP-bound Rho exhibited a consistent, subtle enrichment along aligning contacts relative to orthogonal contacts (Fig. 2A, purple vs green) (Munjal et al., 2015). Corroborating this, we found that the Rho effectors Rok and Dia were also enriched along aligning contacts (Fig. 2B,C).
Fig. 2.

Alignment is regulated by polarized Rho signaling. (A) Right panel: A GFP sensor for Rho-GTP enriched along aligning junctions (purple in graph) relative to orthogonal junctions (green in graph). Left panel: Rho-GTP signal intensity is displayed using the Fire lookup table in ImageJ (calibration bar shows low to high signal). n=34 interfaces, 7 embryos. (B) GFP::RokK116A enriched along aligning interfaces relative to orthogonal junctions. n=38 interfaces, 7 embryos. RokK116A avoids potential artifacts from overactivation (Simões et al., 2010). (C) Anti-Dia antibody staining was enriched along aligning junctions relative to orthogonal junctions. n=98 interfaces, 8 embryos. (D) rho loss-of-function mutants (rho72O/72F, red) had significant alignment defects compared to wild-type sibling controls (yellow). Anti-pTyr antibody staining was used to visualize cell outlines. Control: n=76 interfaces, 6 embryos; rho72O/72F: n=75 interfaces, 5 embryos. (E) Pharmacological inhibition of Rok with either Y-27632 or H-1152 caused significant decreases in alignment. Y-27632: n=21 interfaces, 5 embryos; H-1152: n=11 interfaces, 3 embryos; H2O: n=26 interfaces, 6 embryos. (F) Pharmacological inhibition of Dia disrupted alignment. White arrowheads indicate cell junctions that became convoluted after drug treatment. SMIFH2: n=27 interfaces from 6 embryos; DMSO: n=16 interfaces from 3 embryos. Par3::mCherry was used to mark AJs in A,B, magenta. Anti-phospho-Tyrosine (pTyr) antibody staining was used as a marker for cell outlines in C,D. Each line in graphs of A–C represents one interface and matches average aligning (purple) and orthogonal (green) junction measurements. Fluorescently tagged E-cadherin was used in E,F to visualize cell outlines before (yellow) and after (red) drug treatments. Each line in the graphs of E,F represents one interface and matches measurements before (yellow) and after drug treatment (red) or vehicle treatment (gray). Control injections with either H2O (for Rok inhibitors) or DMSO (for SMIFH2) did not significantly reduce alignment. ***P<0.0001; N.S., not significant; Mann–Whitney U-test, bars show mean±s.d. Scale bars: 4 μm.
To test whether the Rho pathway is required for alignment, we disrupted these three components. First, analysis of strong loss-of-function rho mutants had significant alignment defects (Fig. 2D). Second, we tested the necessity for Rok. Genetic depletion was problematic due to the maternal and zygotic contribution of Rok, coupled to the relatively late stage during which alignment takes place. Instead, we injected either of two Rok inhibitors, Y-27632 and H-1152, each of which caused a rapid loss of alignment in contrast to embryos injected with solvent alone (Fig. 2E). Finally, to evaluate the function of Dia, we used complementary genetic and pharmacological approaches. The epithelium of embryos homozygous for a null allele of dia was frequently too disrupted to analyze alignment (data not shown). However, heterozygous embryos (dia2/+) had significant alignment defects compared to homozygous wild-type (+/+) embryos (Fig. S3A,B,D, yellow vs pink). To further deplete Dia activity, we analyzed dia2/+ embryos laid by dia2/+ females. These embryos had more severe alignment defects relative to dia2/+ embryos laid by +/+ females, showing that depletion of maternally-contributed Dia compromises alignment further (Fig. S3B,C,D, pink vs red). Detection of alignment phenotypes even with mild perturbations of Dia suggested a critical role for this formin in alignment.
To achieve stage-specific disruption of Dia during alignment, we treated embryos with the formin inhibitor SMIFH2 (Rizvi et al., 2009). Analysis of expression data for all formin family members indicated that Dia is the only formin present and cortically enriched to aligning interfaces (Fig. S3E). Therefore, the effects observed from SMIFH2 treatment can be attributed to the inhibition of Dia.
SMIFH2 treatment caused a significant loss of alignment (Fig. 2F). Additionally, individual cell junctions across the epithelium became convoluted in morphology (Fig. 2F, arrowheads), further suggesting a role for Dia in regulating cortical tension (Hara et al., 2016; Tornavaca et al., 2015).
Taken together, our data indicate that alignment requires planar polarized Rok and Dia activation via Rho.
Dia and Rok are each required for both F-actin and Myo-II planar polarization
Rok and Dia have well-studied roles in regulating Myo-II and F-actin, respectively. We sought to test whether these factors are necessary for formation of actomyosin cables. As such, we monitored the localization of fluorescently-labeled MLC and an F-actin sensor in response to pharmacological inhibition of Rok or Dia. Before and after treatment, we measured the ratio of fluorescence between aligning and orthogonal junctions and used this as a metric of local planar polarity.
For inhibition of Rok, we used the drug Y-27632 (referred to in this and subsequent experiments as ‘Rok inhibitor’). Injection of the Y-27632 inhibitor caused an immediate depletion of MLC from cell junctions and a loss in its planar polarity (Fig. 3A). This is consistent with the known role of Rok in activating Myo-II through phosphorylation.
Fig. 3.

Rho effectors are required for actomyosin planar polarity. (A,B) Rok inhibition with Y-27632 treatment caused loss in planar polarity of MLC::GFP (A) (Y-27632: n=12 interfaces, 3 embryos; H2O: n=11 interfaces, 3 embryos) and F-actin (B) (representative of 7 embryos). (C,D) Dia inhibition by SMIFH2 treatment caused loss in planar polarity of F-actin (C) (SMIFH2: n=12 interfaces, 3 embryos; DMSO: n=21 interfaces, 5 embryos) and MLC::GFP (D) (SMIFH2: n=17 interfaces, 4 embryos; DMSO: n=26 interfaces, 5 embryos). The actin-binding domain of Utrophin, Utr-ABD::GFP, was used to visualize F-actin in B,C. Planar polarity was quantified as a ratio of fluorescence intensity between aligning and orthogonal regions. Each line in the graphs in A,C,D represents one interface, matching measurements before (yellow) and after drug treatment (red) or vehicle treatment (gray). Dashed orange line indicates a planar polarity value of 1 on the y-axis for reference. Control injections with H2O (for Y-27632) or DMSO (for SMIFH2) did not result in any statistically significant changes. ***P<0.0001; **P<0.001; N.S., not significant; Mann–Whitney U-test. Scale bars: 4 μm.
We next sought to test whether phosphorylation by Rok was sufficient for proper localization of Myo-II. Since inhibitor treatment should block MLC phosphorylation, we tested whether artificially restoring phosphorylation could rescue cortical enrichment and polarization. Phosphomimetic MLC does not rescue the enzymatic activity of Myo-II, but it does allow insight into the phosphorylation dependence of Myo-II localization (Heissler and Sellers, 2015; Vasquez et al., 2016). We examined embryos exogenously expressing a GFP-tagged form of the phosphomimetic mutant MLC in addition to endogenous, wild-type untagged MLC. Phosphomimetic MLC enriched along aligning junctions at levels comparable to wild-type MLC expressed with a similar transgenic strategy (Fig. S4A–D). This is likely the result of co-assembly of phosphomimetic MLC and endogenous MLC into mini filaments that can be recruited normally to cell junctions.
Upon inhibition of Rok, wild-type MLC-containing mini filaments would disassemble, whereas phosphomimetic MLC would still form mini filaments with myosin heavy chain. These mini filaments would be recruited to the cortex, but accumulate in a non-polarized manner given the loss of polarized Rok activity. Such symmetric recruitment is precisely what is observed in embryos treated earlier in development with Rok inhibitor (Simões et al., 2010). Surprisingly, during the alignment, treatment with the Rok inhibitor not only led to a loss of planar polarity but to depletion from all cortices, as demonstrated by a dramatically reduced fluorescence intensity measured at both aligning and orthogonal junctions (Fig. S4A,C,D,E,G,H). These changes were comparable to those exhibited on expression of non-phosphomimetic MLC (Fig. S4A,B,E,F). As expected, expression of the phosphomimetic construct did not rescue alignment defects in embryos treated with Rok inhibitor (Fig. S4I). These observations suggest that Rok regulates Myo-II targeting to cell junctions in a manner independent of its ability to phosphorylate MLC. We also observed that F-actin became severely disorganized upon Rok inhibition (Fig. 3B). In turn, this suggests that Rok has a separate function in remodeling F-actin during alignment, in addition to Myo-II activation.
Dia disruption also had profound effects on F-actin and Myo-II distribution. Upon SMIFH2 treatment, F-actin planar polarity at aligning interfaces was lost (Fig. 3C). This indicates that Dia is the primary driver of actin polymerization in the contractile assemblies that promote alignment. Additionally, planar polarity of MLC was compromised with SMIFH2 treatment (Fig. 3D). This demonstrates that Dia-mediated actin remodeling is required to properly incorporate Myo-II into the actomyosin cable.
Our findings show that each of these Rho effectors is necessary to assemble both F-actin and Myo-II into the supracellular cables.
Constitutive activation of Rho effectors changes the morphogenetic activity of actomyosin cables
To further probe the function of the Rho pathway during alignment, we tested the effect of overactivating Rho effectors. We expressed constitutively activated Rok (Rok-CA) (Amano et al., 2010; Simões et al., 2010) or Dia (Dia-CA) (Homem and Peifer, 2008) along a broad patch of the epithelium (Fig. 4A,D, opa-GAL4). Activating either effector caused ectopic furrows that coincided with aligning regions, while adjacent non-aligning regions remained flat (Fig. 4D). To confirm that the invaginations correspond with aligning regions, we examined the localization of the marker Cubitus interruptus, which accumulates in a segmental pattern beginning in the cells posterior to the first aligning interface (Fig. S5A). We always observed the border of this expression domain in the ectopic furrow (Fig. S5B,C, arrowheads). Additionally, upon expression of the constitutively active effectors in alternating segments (Fig. 4B, eve-GAL4), the aligning interfaces were no longer apparent in the expressing segments because they were hidden in the furrows (Fig. 4E, red vs yellow bracketed regions). These ectopic furrows were previously described by other groups and attributed to aberrant persistence of grooves that form normally early in development (Homem and Peifer, 2008; Mulinari et al., 2008). However, our live imaging showed that the early appearing grooves regressed normally despite constitutively active effector expression (Fig. 4F, first two panels; Fig. 4J). Thus, the furrows arise de novo at aligning interfaces.
Fig. 4.

Overactivation of Rho signaling changes the morphological outcome from alignment to apical constriction. (A,B) Schematics show expression pattern (green) of opa-GAL4 (A) and eve-GAL4 (B) over the ventral face of the embryonic epithelium. Magenta lines indicate aligning interfaces in each abdominal segment. Fluorescent images show fixed embryos in a similar orientation expressing UAS-NLS::mCherry with opa-GAL4 (A) or eve-GAL4 (B). (C,D) In wild-type embryos (C), the epithelium is flat within the plane of adherens junctions (AJs). opa-GAL4>Dia-CA (D) drives invagination only in aligning regions. Arrowheads indicate the location of the anterior aligning interface within each segment. The epithelium is shown in cross-section along the apical–basal axis with apical side on top. Wild-type control: representative of 5 embryos; eve-GAL4>Dia-CA: representative of 4 embryos. (E) Aligning interfaces invaginate with constitutive activation of Rho effectors. Anterior aligning interfaces in non-expressing, control segments (yellow brackets) are pseudo-colored yellow. In a surface view, the aligning interfaces are no longer detected in eve-GAL4>Dia-CA-expressing segments (red brackets), as they are located within furrows. Representative of 5 embryos. (F–I) Anisotropic apical constriction in eve-GAL4>Dia-CA-expressing cells was observed by live imaging. (F) E-cad::tdTomato marked cell outlines. Dashed red line marks the anterior aligning interface affected by Dia-CA expression. As the ectopic furrow formed, furrow depth (G), apical area (H), and anisotropy (I) were measured for tracked cells (pseudo-colored cyan in F). Area (H) and anisotropy (I) measurements for all the tracked cells were averaged at each time point in the graphs for the same interface: either Dia-CA-expressing (red) or a control interface from a non-expressing segment in the same embryo (yellow). We observed identical trends for three other embryos that were analyzed in this manner (not shown). (J–L) F-actin planar polarity was retained, but levels increased by constitutive expression of Dia. (J) Dia-CA was expressed with eve-GAL4. F-actin (green) distribution was detected by phalloidin staining. pTyr marked cell outlines (magenta). (K) F-actin planar polarity was not significantly different in Dia-CA-expressing segments (red arrowheads and data points) compared to control, non-expressing segments (yellow arrowheads and data points). Dashed orange line indicates a planar polarity value of 1 on the y-axis for reference. (L) Dia-CA expression caused elevation of F-actin levels along aligning junctions compared to control interfaces. Control and Dia-CA: n=20 interfaces, 5 embryos. Anti-pTyr antibody staining was used to mark AJs in A–E,J. *P<0.01; N.S., not significant; Mann–Whitney U-test, bars show mean±=s.d. Scale bars: 12 μm in C,E; all others are 4 μm.
We found that Rok-CA expression caused additional, irregular changes across the epithelium (Fig. S5D). These cell shape changes do not appear to be relevant to furrow formation, as we did not observe this in Dia-CA expressing embryos (Fig. 4F,J). Therefore, we focused on Dia-CA expression to investigate how furrows form.
Initially, we hypothesized that furrows result from the absence of the spatial cue that promotes actomyosin assembly. Expression of constitutively active effectors that accumulate along all cell junctions would lead to apolar upregulation of actomyosin. Apolar cytoskeletal forces would cause isotropic apical constriction, leading to furrow formation (Martin, 2010).
However, we were surprised to observe apical constriction was anisotropic in cells expressing Dia-CA (Fig. 4F). Using eve-GAL4 to express Dia-CA in alternating segments (Fig. 4B,F, eve-GAL4), we indeed observed decreases in cell surface area in aligning cells as the furrow deepened (Fig. 4F–H; Fig. S5E, red). Surface area increased somewhat among cells in non-expressing segments within the same embryo (Fig. 4G,H; Fig. S5E, yellow) as well as in wild-type embryos (Fig. S5E, gray). As apical surface area decreased, cells became more anisotropic along the axis of alignment, the result of contraction along the orthogonal axis (Fig. 4H,I; Fig. S5F, red vs yellow and gray). This asymmetric behavior is not consistent with the hypothesis that depolarized Rho signaling drives furrow formation.
In fact, when we examined the distribution of F-actin just prior to when aligning interfaces fully ingressed into furrows, we observed that planar polarity was maintained at a level comparable to that of control interfaces (Fig. 4J,K). This was observed when expressing Dia-CA with either eve-GAL4 (Fig. 4J) or opa-GAL4 (data not shown). As expected from constitutive activation of a formin, F-actin levels were significantly elevated at interfaces with Dia-CA expression relative to control interfaces (Fig. 4L). Surprisingly, constitutive activation of Dia did not significantly increase MLC recruitment to aligning interfaces, and its planar polarity was also retained (Fig. S5G–I).
Given that there is no apparent change to the polarized distribution of actomyosin, the data suggest that furrow formation is caused instead by a change in the activity of those polarized assemblies. That change might be linked to the increase in F-actin level driven by Dia-CA, or possibly in the organization of that actin (see Discussion).
F-actin mediates negative feedback to multiple levels of Rho signaling
The generation of these ectopic furrows suggests strongly that there are mechanisms in place to temper Rho pathway activity during alignment. Feedback is one such mechanism that could moderate Rho signaling and regulate morphological outcomes. Given the correlation between furrow formation and increased F-actin polymerization, we next asked whether F-actin itself could feedback to Rho. Indeed, recent studies have shown that F-actin can negatively feedback to Rho in other systems (Bement et al., 2015; Segal et al., 2018). Therefore, we assessed the response of the Rho pathway after treatment with latrunculin B (LatB). Injection of LatB caused a rapid loss of F-actin from cell junctions, indicating increased depolymerization (Fig. 5A; Movie 1). The remaining signal was found in foci, indicating severe filament disorganization as well (Fig. 5A; Movie 1).
Fig. 5.

F-actin negatively regulates Rho signaling at multiple points in the pathway. (A) LatB-induced depolymerization of F-actin. Representative of 4 embryos. (B–D) LatB treatment reveals negative feedback regulation of Rho-GTP. (B) Rho-GTP was visualized with a GFP-labeled sensor. Fluorescence intensity is displayed using the Fire lookup table (calibration bar shows low to high signal). Yellow arrowheads mark position of aligning interfaces prior to drug treatment. Rho-GTP levels increased significantly along aligning junctions after LatB treatment (C), whereas planar polarity was unaffected (D). LatB: n=16 interfaces, 4 embryos; DMSO: n=16 interfaces, 4 embryos. (E) CytoD treatment had profound effects on F-actin organization, resulting in loss of planar polarity. Yellow arrowheads mark interfaces prior to drug treatment. Planar polarity of F-actin is no longer evident at interfaces (red arrowheads) after CytoD exposure. However, this drug did not induce net depolymerization of cortical F-actin. CytoD: n=26 interfaces, 5 embryos; DMSO: n=22 interfaces, 7 embryos. (F–H) CytoD treatment reveals negative feedback to Dia. (F) Dia::GFP was used to visualize its distribution at interfaces before (yellow arrowheads) and after (red arrowheads) drug treatment. CytoD treatment caused a dramatic increase in total Dia levels at aligning junctions (G) and a slight enhancement of planar polarity (H). Dashed orange line indicates a planar polarity value of 1 on the y-axis for reference. CytoD: n=22 interfaces, 4 embryos; DMSO: n=22 interfaces, 7 embryos. (I) Model of F-actin negative feedback within the Rho pathway. Utr-ABD::GFP was used to visualize F-actin in A,E. Each line in graphs represents one interface, matching measurements taken before (yellow) and after drug treatment (red) or vehicle treatment (gray). Control injections with DMSO did not result in statistically significant changes. ***P<0.0001; **P<0.001; *P<0.01; N.S., not significant; Mann–Whitney U-test. Scale bars: 4 μm.
Consistent with previous reports, LatB treatment led to upregulation of Rho-GTP, as high-intensity puncta of Rho-GTP appeared rapidly after treatment (Fig. 5B,C; Movie 2). These puncta were located in the plane of adherens junctions (AJs), indicating that these represent cortical overactivation of Rho rather than artifactual aggregation of the sensor (Fig. 5B; Movie 2). Quantification showed that total Rho-GTP at aligning junctions increased significantly (Fig. 5C). Assessment of planar polarity was complicated by the punctate, heterogeneous signal, but our measurements showed no significant differences in relative enrichment at aligning junctions (Fig. 5D). This suggested to us that the unknown, upstream signal that orients Rho activation is unperturbed by this manipulation.
As expected from aberrant overactivation of Rho, both Rok and Dia distribution were disrupted with LatB treatment. While there were no consistent changes in Rok planar polarity or levels at aligning junctions, its distribution became more punctate and heterogeneous at cell junctions (Fig. S6A–C, Movie 3). Dia signal also became more punctate (Fig. S6D, Movie 4). In contrast to Rok, the amount of Dia at aligning junctions increased consistently whereas planar polarity was not disturbed (Fig. S6E,F, Movie 4). The dissimilar responses by Dia and Rok to LatB treatment may reflect differences in the manner by which Rho activates each effector.
To understand mechanistically what mediates negative feedback between F-actin and Rho, we used cytochalasin D (CytoD), which had a different effect on F-actin in our system. Upon CytoD treatment, F-actin levels did not decrease, but qualitatively appeared to increase, indicating no net depolymerization (Fig. 5E; Movie 5). However, F-actin distribution was broadly disrupted. There was an obvious loss of planar polarization to aligning interfaces along with formation of aberrant foci at cell edges (Fig. 5E; Movie 5). This likely reflects ectopic F-actin assemblies and dramatic changes in filament organization. Overall, this led to a loss of F-actin enrichment at aligning interfaces (Fig. 5E).
Surprisingly, CytoD treatment caused no significant changes in recruitment or planar polarity for Rho-GTP and Rok (Fig. S6G–L). However, this inhibitor dramatically increased Dia levels at aligning junctions (Fig. 5F,G). Dia planar polarity was retained with a small increase in enrichment along aligning interfaces (Fig. 5H). We considered the possibility that the effect on Dia was simply a result of non-specific binding to the ectopic F-actin aggregates generated by CytoD. However, Capping protein beta (CapB, also known as Cpb), another barbed-end binding protein, exhibited no increased or decreased recruitment to aligning cell junctions (Fig. S6M). Therefore, the upregulation of Dia recruitment to cell junctions suggests that there is selective negative feedback from F-actin to the Dia branch of Rho signaling (Fig. 5I). The organization of actin filaments may be the cue for this feedback, as net depolymerization of F-actin did not correlate with Dia upregulation.
Cumulatively, our observations suggest negative feedback from F-actin to multiple points in the Rho pathway, engaging distinct mechanisms for each of these interactions.
Positive crosstalk between F-actin and Myo-II in actomyosin cables
We next tested whether the two cytoskeletal components regulate each other. MLC depletion by deGradFP caused a decrease in F-actin levels along aligning interfaces resulting in attenuation of planar polarity (Fig. 6A–C). Reciprocally, exposure to either CytoD or LatB caused an immediate decrease in MLC planar polarity (Fig. 6D,E). Thus, mutual positive feedback between Myo-II and F-actin is critical to formation of actomyosin cables.
Fig. 6.
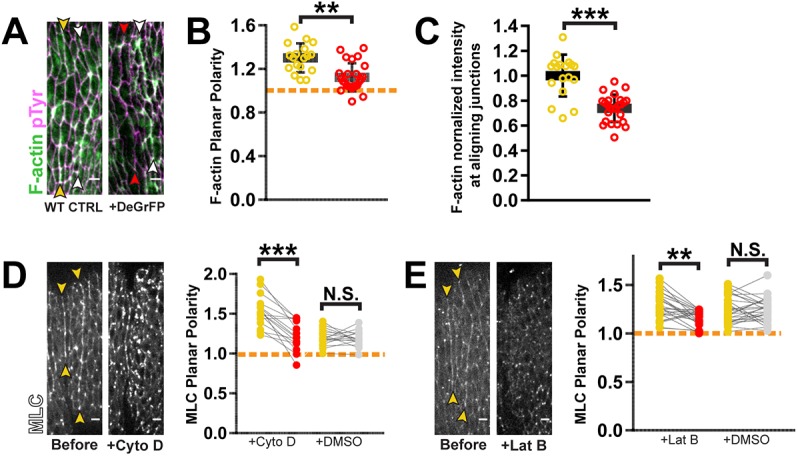
F-actin and myosin positively regulate one another within contractile cables. (A–C) F-actin was depleted from actomyosin cables upon MLC knockdown. (A) Red arrowheads indicate the interface where MLC was depleted. Knockdown condition was compared to sibling wild-type control (WT CTRL, yellow arrowheads). Phalloidin staining was used for F-actin detection. Anti-pTyr antibody staining marked cell outlines. (B) F-actin planar polarity was reduced with MLC depletion. (C) F-actin levels at aligning junctions were also decreased with MLC knockdown. To account for variability between samples, the fluorescence of the affected interface was normalized to that of the unaffected, posterior interface (white arrowheads in A). WT CTRL: n=18 interfaces, 4 embryos; deGradFP+: n=25 interfaces, 5 embryos. (D) MLC::GFP enrichments along aligning interfaces were lost with CytoD treatment. CytoD: n=16 interfaces, 3 embryos; DMSO: n=16 interfaces, 5 embryos. (E) MLC::GFP enrichments along aligning interfaces were lost with LatB treatment. LatB: n=25 interfaces, 6 embryos; DMSO: n=27 interfaces, 5 embryos. Yellow arrowheads in D,E mark aligning interfaces prior to drug treatment. Each line in graphs D,E represents one interface, matching measurements taken before (yellow) and after drug treatment (red) or vehicle treatment (gray). Dashed orange line indicates a planar polarity value of 1 on the y-axis for reference. No significant changes were observed with DMSO control injections. ***P<0.0001; **P<0.001; N.S., not significant; Mann–Whitney U-test, bars show mean±s.d. Scale bars: 4 μm.
Contractility and Myo-II do not regulate Rho signaling
Myo-II itself has also been shown to feedback to Rho. Additionally, we demonstrated that F-actin levels decrease with MLC knockdown. This suggests that Myo-II could also regulate the Rho pathway through F-actin.
We could not assess the effect of Myo-II disruption on Rho activation directly, nor analyze Rok, because these assays require a GFP-tagged sensor or reporter, respectively. Since deGradFP-mediated knockdown targets the GFP moiety, this would also interfere with analysis of any other GFP-tagged factors in the cells being investigated. However, we were able to analyze Dia levels using antibody staining. Surprisingly, MLC knockdown affected neither the level of Dia along aligning junctions, nor its polarized enrichment to those junctions (Fig. 7A–C). This suggests that there is no feedback from Myo-II to Rho signaling in the context of alignment, with the caveat that we could not directly assay Rho activity. If feedback is indeed absent, this contrasts to other cases in which Myo-II has been shown to feedback to Rho either positively or negatively (Graessl et al., 2017; Munjal et al., 2015; Priya et al., 2015; West et al., 2017).
Fig. 7.

Independent of actomyosin contractility, Rok regulates the Rho pathway. (A–C) MLC and contractility do not feedback to the Rho pathway. (A) Dia was detected by antibody staining. MLC knockdown did not change Dia planar polarity (B) or levels at aligning interfaces (C). Fluorescence measurements at interfaces with MLC depletion (red arrowheads and data points) were compared to those of wild-type sibling controls (WT CTRL, yellow arrowheads and data points). White arrowheads mark the posterior aligning interfaces where DeGradFP is not expressed. WT CTRL: n=57 interfaces, 11 embryos; deGradFP+: n=34 interfaces, 6 embryos. (D–F) Rok negatively regulates Rho activation. (D) Rho-GTP is visualized with a GFP-labeled Rho sensor. Yellow arrowheads mark the aligning interfaces before drug treatment and red indicates their position after treatment with Rok inhibitor Y-27632. Fluorescence intensity is displayed using the Fire lookup table. Planar polarity of Rho-GTP was not affected by this treatment (E), but the total amount at aligning junctions increased significantly (F). Y-27632: n=34 interfaces, 7 embryos; H2O: n=15 interfaces, 4 embryos. (G–I) Rok positively regulates Dia planar polarity. (G) Dia::GFP distribution is altered with Rok inhibition. (H) Planar polarity of Dia is decreased significantly with Rok inhibition. Yellow arrowheads mark aligning interfaces prior to drug treatment. (I) Drug treatment did not significantly decrease Dia levels at aligning interfaces. Y-27632: n=18 interfaces, 4 embryos; H2O: n=17 interfaces, 4 embryos. (J) Schematic for model of Rok-mediated negative feedback through F-actin. For graphs E,F,H,I, each line represents one interface, matching measurements taken before drug exposure (yellow) and after Y-27632 injection (red) or vehicle treatment (gray). Rho-GTP levels were found to significantly decrease with control water injection. This likely reflects photobleaching during the course of the experiment. However, given that Y-27632 treatment resulted in the opposite change (increase in Rho-GTP levels), the change in signal from control injection does not impact the conclusions made from the experiment. In all other instances, control water injection did not cause statistically significant changes. Dashed orange line indicates a planar polarity value of 1 on the y-axis for reference. ***P<0.0001; **P<0.01; N.S., not significant; Mann–Whitney U-test, bars show mean±s.d. Scale bars: 4 μm.
Collectively, our data indicates not only that F-actin-mediated negative feedback is independent of contractility, but that it also does not rely on total actin filament levels. This latter insight follows from the observation that a reduction of F-actin caused by MLC knockdown did not correlate with a change in Rho pathway activity. Instead, we hypothesize that actin filament organizational changes are the cue for feedback (see Discussion).
Rok positively regulates Dia in a Myo-II independent manner
While Myo-II did not appear to feedback to Rho signaling, we wanted to test whether Rok has any feedback interactions with the pathway independent of its ability to activate Myo-II. Indeed, after Rok inhibitor treatment, we observed a dramatic elevation in Rho-GTP levels (Fig. 7D,F). Planar polarity of Rho-GTP was maintained, indicating that the underlying polarity signal orienting Rho activation does not rely on Rok (Fig. 7E). Given that F-actin is significantly disorganized with Rok inhibition (Fig. 3B) and that LatB-mediated disruption of F-actin also resulted in an upregulation in Rho-GTP (Fig. 5B), we hypothesize that an F-actin remodeling activity of Rok mediates negative feedback to Rho.
While Rok inhibition increased Rho-GTP levels, it did not increase Dia levels at aligning junctions (Fig. 7G,I). However, Dia planar polarity was lost (Fig. 7H), despite the maintenance of Rho polarity (Fig. 7D,E). This indicates that Rok has a separate role in maintaining Dia enrichment at aligning interfaces (Fig. 7J).
DISCUSSION
We have shown that alignment constitutes a tissue-scale, coordinated cell shape change driven by local increases in contractile force. These forces derive from actomyosin supracellular cables, the assembly of which is guided by polarized Rho signaling. Furthermore, we have revealed feedback acting at different levels of the pathway that may allow precise control over morphological outcomes downstream from Rho.
Actomyosin-generated, bilateral cortical tension is required for alignment
Depletion of actomyosin-based forces lowered the tension along aligning junctions, consequentially reducing the force asymmetry relative to orthogonal junctions (Fig. 1E). It is worth noting that there was still a significant force asymmetry with myosin depletion (Fig. 1E). One explanation for this decreased tension is that polarized Rho activity along aligning interfaces sequesters Myo-II away from orthogonal junctions. However, it is also possible that other mechanisms actively decrease tension along orthogonal junctions.
Two cells contribute actomyosin to each of the junctions in the aligning interface. Therefore, it was not surprising to observe an additional decrease in tension from bilateral versus unilateral knockdown of MLC (Fig. S2G). Intriguingly, unilateral depletion of MLC was sufficient to disrupt the aligned morphology (Fig. S2H). This implies that the minimum force required for alignment necessitates the contribution of both cells (bilateral contractility). Alternatively, there may be a need for symmetric contractility – unilateral application of force could be as counter-productive to alignment as full depletion of Myo-II activity.
The plasticity of Rho signaling outcomes
We have demonstrated an essential role for polarization of Rho signaling as a spatial cue. Both branches of the Rho pathway – Rok and Dia – are critical for recruitment of Myo-II and F-actin into the supracellular cables that drive alignment.
At both cytoskeletal and signaling levels, alignment strongly resembles two other morphogenetic processes – convergent extension and mechanical boundary formation. These two processes produce a cell geometry distinct from alignment. Convergent extension results in junctions that shrink completely (Bertet et al., 2004; Zallen and Wieschaus, 2004). Junctions also shorten along mechanical boundaries (Aliee et al., 2012; Landsberg et al., 2009; Umetsu et al., 2014). In contrast, junctions elongate during alignment despite being under cortical tension at a level comparable to both convergent extension and mechanical boundary formation (Aliee et al., 2012; Fernandez-Gonzalez et al., 2009; Landsberg et al., 2009; Ly et al., 2017; Rauzi et al., 2008; Umetsu et al., 2014). In all three cases, the force asymmetry is driven by supracellular actomyosin cables (Aliee et al., 2012; Fernandez-Gonzalez et al., 2009; Landsberg et al., 2009; Major and Irvine, 2006; Rauzi et al., 2008) that are assembled downstream of Rho planar polarization (Munjal et al., 2015; Kerridge et al., 2016; Simoes et al., 2014). How Rho is able to orchestrate these very different cell shape changes while initiating the same downstream events is perplexing and was the primary motivation for our studies on feedback.
Overactivation of the Rho pathway alters the morphogenetic output of actomyosin assemblies
Overactivation of Rho effectors in the aligning epithelium provided the first hint that feedback regulation allows for plasticity in pathway outcomes. Constitutive activation of either Dia or Rok altered the activity of actomyosin cables to yield a distinct tissue morphology – tissue invagination.
It is unclear how the structure or activity of actomyosin cables is modified to produce furrows. Increased Myo-II activity can induce ectopic furrowing in the early embryo (Urbano et al., 2018). However, we did not observe increased MLC recruitment in our system. It remains possible that changes in the distribution of mono- and di-phosphorylated MLC, and therefore some change in Myo-II activation that we could not assess, might account for furrowing. However, in the aforementioned study in early embryos, the increased recruitment of MLC did not correlate with additional contractile force along the plane of AJs (Urbano et al., 2018). It is notable that the furrows we observed correlated with an increase in F-actin polymerization. Perhaps this changes the organization and/or activity of contractile assemblies leading to furrowing. For example, a reorganization of assemblies might shift the direction of contractile force generated rather than its magnitude. Another reasonable hypothesis is that the additional filaments allow actomyosin cables to mechanically engage with other contractile assemblies, such as medio-apical actomyosin. Connectivity of the medio-apical actomyosin network to junctions is critical for ventral furrow formation in the early embryo, and the emergence of this furrow is also coupled to anisotropic apical constriction as we observe in our system (Martin et al., 2010). Further exploration of this question will provide insight into why moderation of Rho effectors is important for differentiating the function of actomyosin in morphogenesis.
Self-regulation of Rho signaling through feedback
Our study suggests a number of feedback interactions in the pathway that could temper Rho activity during alignment (Fig. S7). Consistent with other works (Bement et al., 2015; Graessl et al., 2017; Segal et al., 2018), LatB treatment revealed a potential role for negative feedback from F-actin to Rho activation. While some Rho GTPase activating proteins (RhoGAPs) have been implicated in this regulatory loop, the underlying mechanisms are mostly unknown (Graessl et al., 2017; Segal et al., 2018). The broad conservation of this feedback suggests that many relevant players are still to be identified (Graessl et al., 2017; Segal et al., 2018). While corroboration of our findings with genetic approaches to manipulating F-actin would extend the power of our study, our data yield three significant insights into this potential mechanism.
First, it is not mediated by contractile force, as disruption of Myo-II did not appear to increase Rho pathway activity. This contrasts with several morphogenetic events where Myo-II activity positively regulates Rho (Graessl et al., 2017; Munjal et al., 2015; Priya et al., 2015). The lack of feedback between Myo-II and Rho during alignment is supported by the fact that we observed no increase in Myo-II levels upon Rho pathway overactivation. Indeed, there have been recent reports of oscillating Rho activity in other systems that are Myo-II independent (Michaux et al., 2018; Segal et al., 2018), corroborating the idea that contractile feedback is not a universal property of the Rho pathway. The activation state of Rho is primarily determined by RhoGAPs that turn off Rho guanine exchange factors (RhoGEFs) that promote its activation. It is possible that only a subset of RhoGAPs and/or RhoGEFs are responsive to feedback from Myo-II and that this subset does not participate in alignment. If this is the case, context-dependent control over specific RhoGAPs and/or RhoGEFs may be a strategy for differentiating signaling outcomes. For example, one mediator of negative feedback between Myo-II and Rho has recently been identified in the Arf-GEF, Steppke (West et al., 2017). We found that alignment occurred normally in steppke mutant embryos (data not shown), supporting the idea that regulatory links between Myo-II and Rho utilized for one morphogenetic process might not be engaged in others.
Second, it is likely that the negative feedback circuit is not simply monitoring the total level of F-actin. Recall that LatB-induced depolymerization set off Rho activation whereas redistribution of F-actin by CytoD did not. This initially indicated to us that the total amount of actin filaments may be the cue for negative feedback. However, MLC depletion caused a 30% reduction of F-actin at aligning interfaces, but this did not affect the polarized recruitment of Dia. While it is possible that more significant depletion is needed to attenuate negative feedback, it is more probable that some aspect of actin organization is the cue for inhibitory regulation and that this organization is unaffected by Myo-II depletion. Indeed, the organization of filaments can determine binding specificity of different actin regulators (Winkelman et al., 2016). Such regulators could interact with RhoGAPs and/or RhoGEFs in a manner that antagonizes Rho.
Finally, we have presented evidence that Rok facilitates this negative feedback. This must be a Myo-II independent function of Rok, as MLC depletion did not appear to effect Rho activity whereas Rok inhibition upregulated the Rho-GTP sensor. Rok can regulate the actin cytoskeleton through phosphorylation of other targets such as Moesin, which itself can suppress Rho via a RhoGAP (Amano et al., 2010; Hipfner et al., 2004; Neisch et al., 2013; Speck et al., 2003).
Other factors besides Rok may also participate in negative feedback, and in turn allow for different actin remodeling activities that could affect morphological outcomes. For example, Rho can be antagonized by Rho family members Rac proteins and Cdc42, although the mechanisms underlying this antagonism are not fully elucidated (Guilluy et al., 2011). Dynamic interchange between Rho and Rac and/or Cdc42 activation could strike a balance between promoting linear actin and branched actin. Linear actin would allow the generation of contractile forces within supracellular cables while branched actin would strengthen cell adhesions (Del Signore et al., 2018; Efimova and Svitkina, 2018). This could explain how junctions in aligning interfaces are able to elongate while under contractile force.
Novel to this study, our data also suggest that a second tier of negative feedback control over Dia exists and that this feedback might be directly engaged by F-actin. It is logical that F-actin feedback would target the branch of Rho signaling responsible for filament polymerization to maintain appropriate levels of actin remodeling, particularly since we demonstrate that the formation of ectopic furrows is correlated to an elevation in F-actin levels. However, because the mechanism underlying this reaction remains unclear, establishing further genetic support for this feedback is warranted. Still, we can glean some information from that fact that after CytoD treatment, Dia accumulated at both membrane and in larger foci (Fig. 5F). The latter likely corresponds to the aggregates of F-actin observed with drug treatment (Fig. 5E). Therefore, we hypothesize that this feedback mechanism suppresses recruitment of Dia to both the cell membrane and to actin filaments. Analysis of transgenic lines expressing forms of Dia deficient in either actin- or membrane-binding would give additional insight into this mechanism.
Additionally, we have demonstrated that Rok has a separate function in positively regulating Dia planar polarity. In mammalian cells, it has been shown that Rok can phosphorylate Dia to activate it directly (Staus et al., 2011). It is unknown whether this phosphorylation site is conserved in Drosophila, so genetics-based approaches to manipulating putative phosphorylation residues as well as Rok activity would allow us to test this hypothesis further.
Regulation of feedback as a strategy for diversifying morphogenesis
Combinatorial control over feedback could modify signaling outcomes of the Rho pathway. In many contexts where positive feedback on Rho has been demonstrated, the GTPase cycles between activation and inactivation (i.e. activator–inhibitor systems) (Bement et al., 2015; Graessl et al., 2017; Munjal et al., 2015; Segal et al., 2018). The consequences of this are spatio-temporally dynamic actomyosin assemblies. While the temporal resolution at which we analyzed Rho-GTP and actomyosin at aligning junctions is limited in this report, these enrichments appear to be more stable than that observed in excitable systems. This may be a product of the apparent lack of a positive feedback control over Rho activity coupled to multiple tiers of negative feedback. This idea would require a more robust sensor for Rho-GTP that would allow analysis of its dynamics at higher temporal resolution.
Negative feedback also appears important for suppression of epithelial invaginations. Additionally, we demonstrated positive feedback between Myo-II and F-actin as well as from Rok to Dia. This positive feedback amongst downstream components in the pathway may allow for amplification of the relatively subtle enhancements of Rho-GTP we have observed in alignment. Additionally, positive feedback may buffer against small fluctuations in Rho activity that may arise from negative feedback (Priya et al., 2015, 2017).
Since Rho is central to numerous morphogenetic programs, further investigation into the factors that dictate feedback regulation will provide insight into the extraordinary versatility of this signaling pathway.
MATERIALS AND METHODS
Fly stocks and husbandry
For a full list of fly stocks and reagents, please see Table S1. Flies were raised on standard cornmeal medium at 25°C. For knockdown of Pav with RNAi, the cross was kept at 28°C to boost GAL4-mediated expression of the short hairpin RNA. Embryos were collected on apple juice agar plates supplemented with a dab of fresh yeast paste. After allowing adult flies to lay fertilized eggs for approximately 12 h, the embryos were transferred to a nylon mesh strainer by rinsing with de-ionized water and using a paintbrush. Embryos were de-chorionated with 50% bleach for no longer than 5 min, and then processed as described in subsections below.
Immunofluorescence
For Dia staining, embryos were processed with a heat-fixing protocol (Müller and Wieschaus, 1996). This begins with immersion of dechorionated embryos in 3 ml of boiling E-Wash buffer (0.3% Triton X-100, 0.4% NaCl) in a glass vial. The vial is then quickly filled with ice-cold E-wash buffer (approximately 17 ml), capped and submerged in ice. The embryos are washed once with phosphate buffered saline (PBS; 7 mM NaHPO4, 3 mM KH2PO4, 150 mM NaCl) before being devitellinized with a 2:1 methanol:heptane mixture with vigorous shaking for 30 s. Devitellinized embryos were washed three times with 100% methanol and stored at 4°C for at least two days prior to beginning the immunofluorescence staining.
For phalloidin staining, embryos were fixed in a glass vial at 23°C in a 1:1 mixture of 40% paraformaldehyde (PFA):heptane for 12 min while on a rocker (Theurkauf, 1992). The PFA was removed and replaced with PBS solution. Embryos were transferred onto double-sided tape adhered to a glass dish and covered with a small volume of PBS. Embryos were manually removed from the vitelline membrane using a tungsten needle. Embryos were transferred to a 0.1% Triton X-100 in PBS (PBS-TX) solution before proceeding immediately with immunofluorescence staining.
For all other stains, embryos were fixed in a glass vial at 23°C in a 1:1 mixture of 4% PFA in PBS:heptane for 20 min while on a rocker (Mitchison and Sedat, 1983). The PFA was removed and replaced with an equal volume of 90% methanol, 50 mM EGTA. The vial was vigorously shaken for 30 s to devitellinize the embryos. The heptane was removed and devitellinized embryos were washed with 100% methanol three times. Embryos were transferred to 100% ethanol for storage or to PBS-TX for immediate immunofluorescence staining.
For the remainder of the immunofluorescence staining protocol, all incubations occurred on a rocker. Embryos were incubated in PBS-TX for 10 min at 23°C. Embryos were then incubated in block solution (1% Normal Donkey Serum, 1% Normal Goat Serum, 0.1% Triton X-100 in PBS) for 1 h at 23°C. Embryos were incubated overnight at 4°C in primary antibody diluted in block. Antibodies used were as follows: anti-pTyr (1:1000), anti-Dia (1:500), chicken anti-GFP (1:2000) and/or rabbit anti-GFP (1:1000), anti-RFP (1:500). For full details on antibodies please see Table S1. Embryos were washed three times with PBS-TX with the last wash lasting at least 1 h at 23°C. They were then incubated in an appropriate mix of secondary antibodies conjugated to Alexa Fluor 488 or 647, or Cy3. All secondary antibodies were diluted in block solution at a concentration of approximately 2 μg/ml. After a 1 h incubation at 23°C, embryos were washed as described above with PBS-TX.
For F-actin imaging, embryos were incubated in approximately 0.05 μM Rhodamine–phalloidin or 0.1 μM Alexa Fluor 647–phalloidin diluted in PBS-TX for 20 min at 23°C. Embryos were then washed with PBS-TX as previously described.
For DNA staining, embryos were incubated in ∼1 μg/ml Hoechst 33258 in PBS-TX for five minutes at 23°C. This was followed by four washes of PBS-TX without rocking.
Embryos were transferred to a 2% n-propyl gallate:80% glycerol solution prior to being mounted on glass slides, covered with 23×30 mm glass coverslips and sealed with clear nail polish.
Fixed and stained embryos were imaged on one of two systems. First, an Axioplan Zeiss widefield microscope equipped with either a 40×0.75 NA water immersion objective or a 20×0.75 NA dry objective using AxioVision software. For imaging with the 40× objective, a structured illumination system (Zeiss Apotome) was used in order to remove out-of-focus information. The second system used was an IX7 Olympus spinning disk confocal microscope equipped with a 100× oil immersion 1.4 NA objective. Images were acquired with a Hamamatsu Photonics electron multiplying charge-coupled device camera (EMCCD, model C9100-13) controlled using MetaMorph software (Molecular Devices). Z-stacks were acquired with a 0.5 μm step size.
Live imaging
Embryos were sorted and oriented on an agar plate using a tungsten needle. A weak adhesive was made from dissolving double-sided tape in heptane (heptane glue). Embryos were transferred to a strip of dried heptane glue on a glass slide such that the dorsal side made contact with the glue. Embryos were covered with approximately 3 μl of Halocarbon 27 oil. Two 18×18 mm coverslips were glued on either side of the embryos to create spacers. A 23×30 mm coverslip was glued to the spacers to cover the embryos (Mason et al., 2013).
Time-lapse imaging was performed on either a Leica DM16000 B spinning disk confocal with a 63×1.2 NA water immersion objective or an IX7 Olympus spinning disk confocal microscope with a 60×1.2 NA water immersion objective. Images were acquired with an EMCCD camera (Andor iXon 3 897E or Hamamatsu photonics, model C9100-13) controlled by MetaMorph software. Z-stacks were acquired with a 0.5 μm step size and a time interval of either 2.5, 5 or 10 min.
Laser ablation
Embryos were mounted as described above. Fluorescently labeled E-cadherin was used to visualize cell junctions. Either MLC–mCherry or engrailed-GAL4>NLS::mCherry was used as a marker to identify the position of aligning interfaces. Embryos were imaged using the IX7 Olympus spinning disk with a 100×1.4 NA objective (see above). A Micropoint nitrogen-pumped laser with a 405 nm dye cell was used for laser ablation. The ablation laser was controlled using Andor iQ3 software that interfaced with MetaMorph. A single time-point, two-color z-stack image was taken initially to allow for unambiguous, post-acquisition identification of the ablated junction. Then, a single z-plane, single-color time lapse was initiated to image E-cadherin with an interval of 0.25 s for a total of 75 s. Within the first 10 s of the time lapse, a single point ROI was placed on the target junction and a single pulse of the laser was fired. The remaining approximately 60 s of the time lapse captured the subsequent retraction of the adjacent membranes around the ablation site.
Drug injections
For drug injection experiments, embryos were mounted as described for live imaging but with their ventral side adhered to a heptane glue strip on a 23×30 mm coverslip. Embryos were covered with Halocarbon 700 oil. The coverslip was glued to a transparent plastic frame that fit into our microscope specimen holder. Imaging pre- and post-injection was performed on the Leica DM16000 B spinning disk system with a 63×1.2 NA objective (see above). A micromanipulator was mounted onto the sample holder stage using an optical post apparatus (Thorlabs). A single time-point, z-stack image with a step size of 0.5 μm was taken of each embryo prior to injection. Femtotip needles (Eppendorf, 930000043) were backfilled with drug or control vehicle solution using capillary loading tips before being installed onto the micromanipulator. A time-lapse, z-stack series was immediately acquired after injections were complete (between 5 and 15 min from the start of injecting the first embryo).
Drugs were diluted as follows: Cyto D, 5 mM in 1:1 DMSO:MilliQ-filtered water; Lat B, 5 μg/μl in 100% DMSO; SMIFH2, 20 μg/μl in 100% DMSO; Y-27632, 10–20 mM in MilliQ-filtered water; H-1152, 10 mM in MilliQ-filtered water. Alignment and fluorescent protein localization was analyzed prior to injection and at the following time points after the beginning of injection: Cyto D, 10 min; Lat B, 10–20 min, SMIFH2, 90 min; Y-27632 and H-1152, 10 min. As a negative control, embryos of the same genotype were injected with solvent to rule out any defects caused by injection or exposure to the vehicle.
Cyto D binds to the barbed ends of actin filaments to block the addition of monomers (Brenner and Korn, 1979; Goddette and Frieden, 1986). It can also induce actin dimers, which causes ATP hydrolysis, depleting the pool of actin monomers competent for incorporation into filaments. Effectively, this has been reported to induce actin depolymerization in some physiological contexts (Casella et al., 1981) while only blocking polymerization in others (Morris and Tannenbaum, 1980).
Lat B binds to actin monomers, sequestering them from filaments to block polymerization and promote depolymerization (Yarmola et al., 2000).
DeGradFP-mediated knockdown
We used the UAS-driven deGradFP construct (Caussinus et al., 2012). The construct contains a GFP-recognizing nanobody fused to an F-box domain. This allows for recruitment of GFP-labeled substrates to the E2 ligase complex for eventual ubiquitin-mediated degradation. Males expressing both engrailed-GAL4 and UAS-DeGradFP were crossed to females that had a null MLC allele, sqhAX3, on their X chromosome and a transgene expressing GFP-labeled MLC. Embryos that inherited a Y chromosome from the male only expressed MLC::GFP and were therefore functionally MLC-depleted in the GAL4 expressing regions. Embryos that inherited the X chromosome from the male were used as controls as they received a wild-type, untagged allele of MLC that rescued cells from deGradFP knockdown. In both deGradFP knockdown and control embryos, the anterior aligning interface was analyzed in each abdominal segment.
Constitutive activation of Rok and Dia
The UAS-Rok-CA line expresses Rok that is missing the C-terminus responsible for auto-inhibition (Simões et al., 2010). The UAS-Dia-CA line expresses Dia that has truncations at both the N- and C-termini, leading to deletion of the GTPase interacting and autoinhibitory domains, respectively (Homem and Peifer, 2008).
Quantification and statistical analysis
The degree of alignment was determined by measuring the angles between adjacent cell contacts using the angle tool in ImageJ. The angles of a single interface were averaged and normalized to a range of 120° to 180° to give a percent alignment. Angles of 180° are by definition 100% aligned and 120° is defined as 0% aligned.
Fluorescence measurements along cell junctions were manually measured using the segmented line tool in Fiji with the width set to 3 pixels. To test for differences in fluorescence intensity between aligning and orthogonal junctions (Fig. 2), measurements were taken of each aligning and orthogonal junction in a given interface. A ROI covering a small area of the embryo sample was measured for background fluorescence. This average background fluorescence was subtracted from all junctional measurements to correct for background. The corrected mean gray values (MGVs) were averaged for both types of junction. The two averages were treated as a paired sample for each interface and were compared using a Wilcoxon Rank test to determine statistical significance (Fig. 2). For comparing the fluorescence intensity between aligning and orthogonal junctions (as in Fig. 2), the average MGV was compared between that of aligning junctions and orthogonal junctions within the same interface. A Wilcoxon Rank test was used to determine whether there was a significance difference in fluorescence levels between aligning and orthogonal junctions, with the measurements for each interface being treated as a paired sample.
For comparing the average fluorescence between embryos processed with immunofluorescence staining, we accounted for variability in staining by normalizing the MGV at the experimentally perturbed interface to that of the unaffected interface (in other words, the signal at control or MLC-depleted interfaces was normalized to the regions indicated with white arrowheads in Fig. 6C and Fig. 7C). For all other experiments, the MGV was used to compare fluorescence intensity at aligning interfaces between different embryos.
To measure planar polarity, the MGVs of aligning junctions and orthogonal junctions were averaged separately. The ratio between these averages was then used to represent planar polarity. A value >1 indicates relative enrichment along aligning junctions with respect to orthogonal junctions.
Measurements of fluorescence intensity, planar polarity and alignment were compared between sample groups of different genotypes or before and after drug treatment using non-parametric statistical tests. In most cases, an unpaired Mann–Whitney test was performed. In cases of drug treatment where the same cells could be precisely tracked and analyzed before and after drug exposure, a paired, Wilcoxon Rank test was performed (as in Fig. S6E,F).
For analysis of cell morphology during furrow ingression, each cell flanking the interface was manually traced using the polygon tool in Fiji to create an ROI. Apical surface area (μm2), major axis length and minor axis length were extracted from the ROI. Cell anisotropy was calculated as a ratio between the major and minor axes. Furrow depth was determined by counting the number of z-slices between the top and bottom of the furrow at each time point. The average apical surface area, average cell anisotropy, and furrow depth were plotted for each time point. These parameters were compared between a Dia-CA-expressing segment and an adjacent, non-expressing control segment for the same embryo over the same time points. The anterior-most interface was analyzed in each case, as eve-GAL4 is expressed more strongly in the cells along the anterior interface compared to the posterior interface.
Maximum velocity as inferred from laser ablation experiments was used as a metric of cortical tension. The length of the ablated junction was manually measured by drawing a line with the line tool in Fiji. This measurement was taken prior to ablation and every 5 s after ablation up to a total of 60 s. Maximum velocity was calculated from these measurements.
For comparing cell junction remodeling events to the degree of alignment, embryos expressing E-cad::GFP and MLC::mCherry were live-imaged as described above at a time interval of 2.5 min. The duration of the live imaging included before, during and after completion of alignment. Interfaces were manually tracked using Fiji software. Alignment and the number of three-way and four-way vertices were measured for each time point. A four-way vertex is formed when a junction between two three-way vertices is remodeled away (Fig. S1A). Therefore, the number of cell junction shrinking events was represented by the fraction of four-way vertices present in the interface. For each time point and each of three interfaces analyzed, the percent alignment was plotted against the percent four-way vertices. A Pearson correlation test was used to assess the relationship between these two metrics, and a linear regression was fitted to the data (Fig. S1C).
Error bars for all graphs represent standard deviation (s.d.). All statistical tests and curve fitting were executed with GraphPad Prism software.
Supplementary Material
Acknowledgements
We thank Jennifer Zallen, Thomas Lecuit, Adam Martin, Yang Hong, Yohannes Bellaiche, Steven Wasserman, Amin Ghabrial, Roger Karess and the Bloomington Drosophila Stock Center for kindly providing reagents. We thank members of Stephen DiNardo's, Shawn Little's and Maya Capelson's lab for helpful discussions. We are grateful to Erfei Bi, Tatyana Svitkina, Erika Holzbaur and Michael Granato for feedback on the manuscript. We appreciate Andrea Stout and the Cell and Developmental Biology Department's Microscopy core for assistance with live imaging and laser ablation.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.O., S.D.; Methodology: K.O., S.D.; Formal analysis: K.O., C.C.; Investigation: K.O., C.C.; Writing - original draft: K.O.; Writing - review & editing: K.O., C.C., S.D.; Supervision: S.D.; Funding acquisition: K.O., S.D.
Funding
Funding sources include National Institutes of Health grants GM117708 (K.O.), HD083185 (K.O.), GM007229 (K.O.), GM45747 (S.D.), and GM60804 (S.D.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.224378.supplemental
References
- Adams R. R., Tavares A. A. M., Salzberg A., Bellen H. J. and Glover D. M. (1998). pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev. 12, 1483-1494. 10.1101/gad.12.10.1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliee M., Röper J.-C., Landsberg K. P., Pentzold C., Widmann T. J., Jülicher F. and Dahmann C. (2012). Physical mechanisms shaping the Drosophila dorsoventral compartment boundary. Curr. Biol. 22, 967-976. 10.1016/j.cub.2012.03.070 [DOI] [PubMed] [Google Scholar]
- Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y. and Kaibuchi K. (1996). Phosphorylation and activation of myosin by Rho-associated Kinase (Rho-kinase). J. Biol. Chem. 271, 20246-20249. 10.1074/jbc.271.34.20246 [DOI] [PubMed] [Google Scholar]
- Amano M., Nakayama M. and Kaibuchi K. (2010). Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton 67, 545-554. 10.1002/cm.20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement W. M., Leda M., Moe A. M., Kita A. M., Larson M. E., Golding A. E., Pfeuti C., Su K.-C., Miller A. L., Goryachev A. B. et al. (2015). Activator-inhibitor coupling between Rho signalling and actin assembly makes the cell cortex an excitable medium. Nat. Cell Biol. 17, 1471-1483. 10.1038/ncb3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertet C., Sulak L. and Lecuit T. (2004). Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature 429, 667-671. 10.1038/nature02590 [DOI] [PubMed] [Google Scholar]
- Breitsprecher D. and Goode B. L. (2013). Formins at a glance. J. Cell Sci. 126, 1-7. 10.1242/jcs.107250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. L. and Korn E. D. (1979). Substoichiometric concentrations of cytochalasin D inhibit actin polymerization. Additional evidence for an F-actin treadmill. J. Biol. Chem. 254, 9982-9985. [PubMed] [Google Scholar]
- Casella J. F., Flanagan M. D. and Lin S. (1981). Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 293, 302-305. 10.1038/293302a0 [DOI] [PubMed] [Google Scholar]
- Caussinus E., Kanca O. and Affolter M. (2012). Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19, 117-121. 10.1038/nsmb.2180 [DOI] [PubMed] [Google Scholar]
- Classen A.-K., Anderson K. I., Marois E. and Eaton S. (2005). Hexagonal packing of Drosophila wing epithelial cells by the planar cell polarity pathway. Dev. Cell 9, 805-817. 10.1016/j.devcel.2005.10.016 [DOI] [PubMed] [Google Scholar]
- Del Signore S. J., Cilla R. and Hatini V. (2018). The WAVE regulatory complex and branched F-actin counterbalance contractile force to control cell shape and packing in the Drosophila eye. Dev. Cell 44, 471- 483.e4 10.1016/j.devcel.2017.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimova N. and Svitkina T. M. (2018). Branched actin networks push against each other at adherens junctions to maintain cell-cell adhesion. J. Cell Biol. 217, 1827-1845. 10.1083/jcb.201708103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R., Simoes S. d. M., Röper J.-C., Eaton S. and Zallen J. A. (2009). Myosin II dynamics are regulated by tension in intercalating cells. Dev. Cell 17, 736-743. 10.1016/j.devcel.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson M. C., Patel A. B., Nagpal R. and Perrimon N. (2006). The emergence of geometric order in proliferating metazoan epithelia. Nature 442, 1038-1041. 10.1038/nature05014 [DOI] [PubMed] [Google Scholar]
- Goddette D. W. and Frieden C. (1986). Actin polymerization. The mechanism of action of cytochalasin D. J. Biol. Chem. 261, 15974-15980. [PubMed] [Google Scholar]
- Graessl M., Koch J., Calderon A., Kamps D., Banerjee S., Mazel T., Schulze N., Jungkurth J. K., Patwardhan R., Solouk D. et al. (2017). An excitable Rho GTPase signaling network generates dynamic subcellular contraction patterns. J. Cell Biol. 216, 4271-4285. 10.1083/jcb.201706052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C., Garcia-Mata R. and Burridge K. (2011). Rho protein crosstalk: another social network? Trends Cell Biol. 21, 718-726. 10.1016/j.tcb.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y., Shagirov M. and Toyama Y. (2016). Cell boundary elongation by non-autonomous contractility in cell oscillation. Curr. Biol. 26, 2388-2396. 10.1016/j.cub.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Heissler S. M. and Sellers J. R. (2015). Four things to know about myosin light chains as reporters for nonmuscle myosin-2 dynamics in live cells. Cytoskeleton 72, 65-70. 10.1002/cm.21212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipfner D. R., Keller N. and Cohen S. M. (2004). Slik Sterile-20 kinase regulates Moesin activity to promote epithelial integrity during tissue growth. Genes Dev. 18, 2243-2248. 10.1101/gad.303304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem C. C. F. and Peifer M. (2008). Diaphanous regulates myosin and adherens junctions to control cell contractility and protrusive behavior during morphogenesis. Development 135, 1005-1018. 10.1242/dev.016337 [DOI] [PubMed] [Google Scholar]
- Kerridge S., Munjal A., Philippe J.-M., Jha A., de las Bayonas A. G., Saurin A. J. and Lecuit T. (2016). Modular activation of Rho1 by GPCR signalling imparts polarized myosin II activation during morphogenesis. Nat. Cell Biol. 18, 261-270. 10.1038/ncb3302 [DOI] [PubMed] [Google Scholar]
- Landsberg K. P., Farhadifar R., Ranft J., Umetsu D., Widmann T. J., Bittig T., Said A., Jülicher F. and Dahmann C. (2009). Increased cell bond tension governs cell sorting at the Drosophila anteroposterior compartment boundary. Curr. Biol. 19, 1950-1955. 10.1016/j.cub.2009.10.021 [DOI] [PubMed] [Google Scholar]
- Lecuit T. and Lenne P.-F. (2007). Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat. Rev. Mol. Cell Biol. 8, 633-644. 10.1038/nrm2222 [DOI] [PubMed] [Google Scholar]
- Levayer R. and Lecuit T. (2012). Biomechanical regulation of contractility: spatial control and dynamics. Trends Cell Biol. 22, 61-81. 10.1016/j.tcb.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Ly D., Resch E., Ordiway G. and DiNardo S. (2017). Asymmetrically deployed actomyosin-based contractility generates a boundary between developing leg segments in Drosophila. Dev. Biol. 429, 165-176. 10.1016/j.ydbio.2017.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major R. J. and Irvine K. D. (2006). Localization and requirement for Myosin II at the dorsal-ventral compartment boundary of theDrosophila wing. Dev. Dyn. 235, 3051-3058. 10.1002/dvdy.20966 [DOI] [PubMed] [Google Scholar]
- Marcinkevicius E. and Zallen J. A. (2013). Regulation of cytoskeletal organization and junctional remodeling by the atypical cadherin Fat. Development 140, 433-443. 10.1242/dev.083949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A. C. (2010). Pulsation and stabilization: contractile forces that underlie morphogenesis. Dev. Biol. 341, 114-125. 10.1016/j.ydbio.2009.10.031 [DOI] [PubMed] [Google Scholar]
- Martin A. C., Gelbart M., Fernandez-Gonzalez R., Kaschube M. and Wieschaus E. F. (2010). Integration of contractile forces during tissue invagination. J. Cell Biol. 188, 735-749. 10.1083/jcb.200910099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason F. M., Tworoger M. and Martin A. C. (2013). Apical domain polarization localizes actin–myosin activity to drive ratchet-like apical constriction. Nat. Cell Biol. 15, 926-936. 10.1038/ncb2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T. J. and Sedat J. (1983). Localization of antigenic determinants in whole Drosophila embryos. Dev. Biol. 99, 261-264. 10.1016/0012-1606(83)90275-0 [DOI] [PubMed] [Google Scholar]
- Monier B., Pélissier-Monier A., Brand A. H. and Sanson B. (2010). An actomyosin-based barrier inhibits cell mixing at compartmental boundaries in Drosophila embryos. Nat. Cell Biol. 12, 60-65. 10.1038/ncb2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A. and Tannenbaum J. (1980). Cytochalasin D does not produce net depolymerization of actin filaments in HEp-2 cells. Nature 287, 637-639. 10.1038/287637a0 [DOI] [PubMed] [Google Scholar]
- Mulinari S., Barmchi M. P. and Häcker U. (2008). DRhoGEF2 and diaphanous regulate contractile force during segmental groove morphogenesis in the Drosophila embryo. Mol. Biol. Cell 19, 1883-1892. 10.1091/mbc.e07-12-1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H. A. and Wieschaus E. (1996). armadillo, bazooka, and stardust are critical for early stages in formation of the zonula adherens and maintenance of the polarized blastoderm epithelium in Drosophila. J. Cell Biol. 134, 149-163. 10.1083/jcb.134.1.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munjal A., Philippe J.-M., Munro E. and Lecuit T. (2015). A self-organized biomechanical network drives shape changes during tissue morphogenesis. Nature 524, 351-355. 10.1038/nature14603 [DOI] [PubMed] [Google Scholar]
- Neisch A. L., Formstecher E. and Fehon R. G. (2013). Conundrum, an ARHGAP18 orthologue, regulates RhoA and proliferation through interactions with Moesin. Mol. Biol. Cell 24, 1420-1433. 10.1091/mbc.e12-11-0800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R., Gomez G. A., Budnar S., Verma S., Cox H. L., Hamilton N. A. and Yap A. S. (2015). Feedback regulation through myosin II confers robustness on RhoA signalling at E-cadherin junctions. Nat. Cell Biol. 17, 1282-1293. 10.1038/ncb3239 [DOI] [PubMed] [Google Scholar]
- Priya R., Gomez G. A., Budnar S., Acharya B. R., Czirok A., Yap A. S. and Neufeld Z. (2017). Bistable front dynamics in a contractile medium: travelling wave fronts and cortical advection define stable zones of RhoA signaling at epithelial adherens junctions. PLoS Comput. Biol. 13, e1005411 10.1371/journal.pcbi.1005411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauzi M., Verant P., Lecuit T. and Lenne P.-F. (2008). Nature and anisotropy of cortical forces orienting Drosophila tissue morphogenesis. Nat. Cell Biol. 10, 1401-1410. 10.1038/ncb1798 [DOI] [PubMed] [Google Scholar]
- Rizvi S. A., Neidt E. M., Cui J., Feiger Z., Skau C. T., Gardel M. L., Kozmin S. A. and Kovar D. R. (2009). Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol. 16, 1158-1168. 10.1016/j.chembiol.2009.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaux J. B., Robin F. B., McFadden W. M. and Munro E. M. (2018). Excitable RhoA dynamics drive pulsed contractions in the early C. elegans embryo. J. Cell Biol. 217, 4230-4252. 10.1083/jcb.201806161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röper K. (2013). Supracellular actomyosin assemblies during development. Bioarchitecture 3, 45-49. 10.4161/bioa.25339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasamura T., Kobayashi T., Kojima S., Qadota H., Ohya Y., Masai I. and Hotta Y. (1997). Molecular cloning and characterization of Drosophila genes encoding small GTPases of the rab and rho families. Mol. Gen. Genet. MGG 254, 486-494. 10.1007/s004380050443 [DOI] [PubMed] [Google Scholar]
- Segal D., Zaritsky A., Schejter E. D. and Shilo B.-Z. (2018). Feedback inhibition of actin on Rho mediates content release from large secretory vesicles. J. Cell Biol. 217, 1815-1826. 10.1083/jcb.201711006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões S. D. M., Blankenship J. T., Weitz O., Farrell D. L., Tamada M., Fernandez-Gonzalez R. and Zallen J. A. (2010). Rho-kinase directs Bazooka/Par-3 planar polarity during Drosophila axis elongation. Dev. Cell 19, 377-388. 10.1016/j.devcel.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes S. D. M., Mainieri A. and Zallen J. A. (2014). Rho GTPase and Shroom direct planar polarized actomyosin contractility during convergent extension. J. Cell Biol. 204, 575-589. 10.1083/jcb.201307070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone R. P. and DiNardo S. (2010). Actomyosin contractility and Discs large contribute to junctional conversion in guiding cell alignment within the Drosophila embryonic epithelium. Development 137, 1385-1394. 10.1242/dev.048520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck O., Hughes S. C., Noren N. K., Kulikauskas R. M. and Fehon R. G. (2003). Moesin functions antagonistically to the Rho pathway to maintain epithelial integrity. Nature 421, 83-87. 10.1038/nature01295 [DOI] [PubMed] [Google Scholar]
- Staus D. P., Taylor J. M. and Mack C. P. (2011). Enhancement of mDia2 activity by Rho-kinase-dependent phosphorylation of the diaphanous autoregulatory domain. Biochem. J. 439, 57-65. 10.1042/BJ20101700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurkauf W. E. (1992). Behavior of structurally divergent α-tubulin isotypes during Drosophila embryogenesis: evidence for post-translational regulation of isotype abundance. Dev. Biol. 154, 205-217. 10.1016/0012-1606(92)90060-T [DOI] [PubMed] [Google Scholar]
- Tornavaca O., Chia M., Dufton N., Almagro L. O., Conway D. E., Randi A. M., Schwartz M. A., Matter K. and Balda M. S. (2015). ZO-1 controls endothelial adherens junctions, cell–cell tension, angiogenesis, and barrier formation. J. Cell Biol. 208, 821-838. 10.1083/jcb.201404140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umetsu D., Aigouy B., Aliee M., Sui L., Eaton S., Jülicher F. and Dahmann C. (2014). Local increases in mechanical tension shape compartment boundaries by biasing cell intercalations. Curr. Biol. 24, 1798-1805. 10.1016/j.cub.2014.06.052 [DOI] [PubMed] [Google Scholar]
- Urbano J. M., Naylor H. W., Scarpa E., Muresan L. and Sanson B. (2018). Suppression of epithelial folding at actomyosin-enriched compartment boundaries downstream of Wingless signalling in Drosophila. Development 145, dev155325 10.1242/dev.155325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez C. G., Heissler S. M., Billington N., Sellers J. R. and Martin A. C. (2016). Drosophila non-muscle myosin II motor activity determines the rate of tissue folding. eLife 5, e20828 10.7554/eLife.20828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford J. B., Niswander L. A., Shaw G. M. and Finnell R. H. (2013). The continuing challenge of understanding, preventing, and treating neural tube defects. Science 339, 1222002 10.1126/science.1222002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. (2009). Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol. Cancer Ther. 8, 2103-2109. 10.1158/1535-7163.MCT-09-0282 [DOI] [PubMed] [Google Scholar]
- West J. J., Zulueta-Coarasa T., Maier J. A., Lee D. M., Bruce A. E. E., Fernandez-Gonzalez R. and Harris T. J. C. (2017). An actomyosin-Arf-GEF negative feedback loop for tissue elongation under stress. Curr. Biol. 27, 2260-2270.e5. 10.1016/j.cub.2017.06.038 [DOI] [PubMed] [Google Scholar]
- Winkelman J. D., Suarez C., Hocky G. M., Harker A. J., Morganthaler A. N., Christensen J. R., Voth G. A., Bartles J. R. and Kovar D. R. (2016). Fascin- and α-actinin-bundled networks contain intrinsic structural features that drive protein sorting. Curr. Biol. 26, 2697-2706. 10.1016/j.cub.2016.07.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmola E. G., Somasundaram T., Boring T. A., Spector I. and Bubb M. R. (2000). Actin-Latrunculin A structure and function differential modulation of actin-binding protein function by Latrunculin A. J. Biol. Chem. 275, 28120-28127. 10.1074/jbc.M004253200 [DOI] [PubMed] [Google Scholar]
- Zallen J. A. and Wieschaus E. (2004). Patterned gene expression directs bipolar planar polarity in Drosophila. Dev. Cell 6, 343-355. 10.1016/S1534-5807(04)00060-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.