

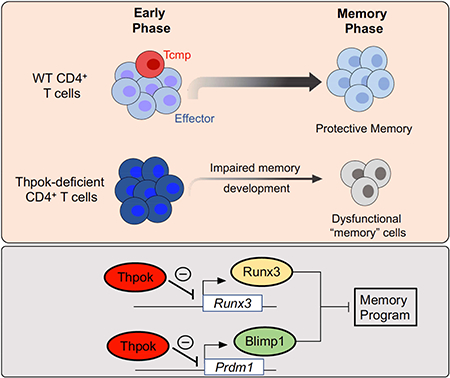
Summary
Memory CD4+ T cells mediate long term immunity, and their generation is a key objective of vaccination strategies. However, the transcriptional circuitry controlling the emergence of memory cells from early CD4+ antigen-responders remains poorly understood. Here, using single-cell RNAseq to study the transcriptome of virus-specific CD4+ T cells, we identified a gene signature distinguishing potential memory precursors from effector cells. We found that both that signature and the emergence of memory CD4+ T cells required the transcription factor Thpok. We further demonstrated that Thpok cell-intrinsically protected memory cells from a dysfunctional, effector-like transcriptional program, similar to but distinct from the exhaustion pattern of cells responding to chronic infection. Mechanistically, Thpok bound genes encoding the transcription factors Blimp1 and Runx3, and acted by antagonizing their expression. Thus, a Thpok-dependent circuitry both promotes the differentiation and functional fitness of memory CD4+ T cells, two previously unconnected critical attributes of adaptive immunity.
eTOC Blurb
Memory CD4+ T cells contribute to long-term immunity against pathogens. Ciucci et al. show that the transcription factor Thpok is essential to safeguard the memory potential of early pathogen-responding CD4+ T cells, and serves by antagonizing a Blimp1- and Runx3-driven dysfunction transcriptional circuitry.
Graphical Abstract
Introduction
Immunological memory, the ability to mount a fast secondary effector response to a new challenge by a previously encountered pathogen, is a defining property of the adaptive immune system, and relies on long-lived antigen-specific B and T cells. T cells responding to an initial infection undergo extensive proliferation, giving rise to effector populations that promote pathogen clearance. Following a contraction phase when most effector T cells die by apoptosis, memory T cells survive and provide long-lasting protection to reinfection.
In addition to promoting CD8+ T cell and B cell memory responses, CD4+ T cells give rise to memory populations essential for durable immunity (Laidlaw et al., 2016; MacLeod et al., 2009), including against human influenza malaria, and Leishmania infection (Mordmuller et al., 2017; Wilkinson et al., 2012; Zaph et al., 2004). Although most antigen-responding CD4+ T cells have the potential to give rise to short-lived effector and long-lived memory cells (Cho et al., 2017; Tubo et al., 2016), what controls the emergence and functional fitness of memory CD4+ cells remains poorly understood. Unlike for memory CD8+ T cells, whose precursors can be identified early during the immune response (e.g. through their expression of interleukin-7 receptor (IL-7Rα), identifying putative precursors of memory CD4+ T cells has been challenging (Ahmed et al., 2009; Marshall et al., 2011; Pepper and Jenkins, 2011). One perspective holds that ‘memory precursors’ diverge from short-term effectors early during the immune response, notably by restraining expression of differentiation programs specific of effector fates (including interferon-γ (IFNγ)-expressing T helper-1 (Th1) cells) and maintaining expression of genes characteristic of undifferentiated cells; supporting this idea, studies have identified specific CD4+ T cell subsets exhibiting higher potential for memory differentiation (Cho et al., 2017; Luthje et al., 2012; Marshall et al., 2011; Nish et al., 2017; Pepper et al., 2011). However, there is support for the possibility that differentiated effectors can give rise to long-lived memory cells (Fazilleau et al., 2009; Harrington et al., 2008; Marshall et al., 2011; McKinstry et al., 2014).
Here, we have taken advantage of single-cell RNA sequencing (scRNAseq) to characterize the gene expression programs underpinning the heterogeneity of T cell responses and the development of memory T cells. Analyzing CD4+ T cells at the peak of the response to an acute viral infection, we identified a transcriptional signature characteristic of potential memory cell precursors. We found that both the emergence of this signature and the generation of a durable and effective memory CD4+ response required the transcription factor Thpok, which notably prevented the establishment of a dysfunctional gene expression pattern characteristic of short-lived effector cells. Mechanistically, Thpok protected memory development by restraining the expression of the transcription factors Runx3 and Blimp1 and bound to chromatin regions within or near these genes. Thus, a Thpok-dependent transcriptional circuitry controls the emergence of a memory program in early CD4+ T cell responders and is critical for the establishment of durable and effective CD4+ T cell memory.
Results
Single cell RNAseq identifies a CD4+ T cell memory precursor signature
We performed scRNAseq to dissect early T cell responses against the Armstrong strain of Lymphocytic ChorioMeningitis Virus (LCMV Arm), which in wild-type (WT) mice is cleared by innate and CD8+ T cells (Matloubian et al., 1994). We used the droplet-based GemCode Chromium technology (Fig. 1A) (Zheng et al., 2017) and, at 7 days (d) post infection (p.i.), captured non-naive (CD44hi) spleen T cells, which included LCMV-reactive cells and pre-existing memory cells of unrelated specificity. We first analyzed 7006 cells, each expressing an average of 1446 genes (Table S1). Visualization with t-distributed stochastic neighbor embedding (t-SNE), which positions cells based on similarity in gene expression, revealed a clear demarcation between CD4+ and CD8+ T cells, and negligible contamination by non-T cells (Fig. S1A). Subsequent analyses were carried on CD4+ T cells only and aimed at identifying cell subsets defined by specific gene expression signatures. Unsupervised clustering of CD44hi CD4+ T cells (2782 cells) identified 6 clusters (clusters A1-F1) matching the known heterogeneity of this population (Hale et al., 2013; Marshall et al., 2011) (Fig. 1B and Fig. S1B). Clusters A1, B1 and E1 included cells expressing Tbx21 (encoding T-bet, characteristic of Th1 cells), Bcl6 (T follicular helper (Tfh) cells), and Foxp3 (regulatory T (Treg) cells) (Fig. 1C). Cluster D1 cells expressed genes typical of memory cells, including Il7r, Bcl2 and Ccr7 (Kaech et al., 2002; Marshall et al., 2011). Its high expression of Il7r, which was minimally expressed in d7 p.i. virus-specific CD4+ T cells (Purton et al., 2007), suggested that it mostly included pre-existing memory cells (Tmem) unrelated to LCMV infection. For each cluster, we computed signatures based on genes differentially expressed between that cluster and all others.
Figure 1: Identification of CD4+ T cell effector signatures by scRNAseq.

(A) Schematic of experimental procedures.
(B-D) scRNAseq of WT CD4+ CD44hi T cells at d7 p.i. LCMV. (B) t-SNE plot (experiment 1); each dot is a CD4+ T cell and colors highlight cell clusters. (C) Differential expression of select genes in clusters shown in (B). (D) Expression of signature components among CD4+ T cell clusters defined in both replicates (A-E, replicate 1 on left of each pair), or unique to each replicate (F-H).
(E-G) scRNAseq of d7 p.i. WT GP66:I-Ab+ CD4+ T cells. (E) Purification strategy. (F) t- SNE plot; colors highlight the 8 clusters. (G) Expression of selected genes and signature scores among clusters shown in (F).
(H) CCR7 vs. CXCR5 expression on spleen T cells (left) defines Th1-, TFH- and Tcmp-cell subsets, which are analyzed for CXCR6 expression (right). Data are from one representative experiment out of three. Please see also Figure S1.
Using the same approach on an independent replicate of 956 d7 p.i. CD44hl CD4+ spleen T cells (Table S1), we identified 8 clusters (Fig. S1C); five of these (A2-E2) had gene expression signatures closely matching those of clusters A1-E1 identified in the first replicate, highlighting the robustness of this analysis. There was less similarity between clusters F1 and F2-H2, which could not be assigned a specific signature (Fig. S1D). Thus, we defined Th1, Tfh, Treg, and Tmem cell signatures as the intersection of corresponding gene signatures defined in each replicate (Fig. 1D and Table S2). In addition, the signature defined by clusters C1 and C2 comprised genes typical of memory cells, including Tcf7 (encoding TCF-1), Bcl2 or Ccr7 but not Il7r, and excluded typical Tfh and Th1 cell effector genes. Because CCR7-expressing CD4+ T cell responders have an increased potential to become memory cells (Cho et al., 2017; He et al., 2013; Marshall et al., 2011; Pepper and Jenkins, 2011; Sallusto et al., 1999) we speculated that this signature, which we designated T central memory precursor (Tcmp), identified a gene expression program related to memory potential.
To unambiguously assess virus-specific cells, we performed scRNAseq on d7 p.i. CD4+ spleen T cells specific for the LCMV glycoprotein-derived GP66 peptide complexed to I-Ab MHC-II molecules (Fig. 1E and Table S1). We clustered this LCMV-specific population and scored CD4+ T cells in every cluster for each gene signature we previously defined (Fig. 1F, G). GP66-specific cells separated into clusters with high scores for Th1 (Cluster I-III), Tfh (Cluster V) and Tcmp (Cluster VIII) cell signatures (Fig. 1G and S1E). In contrast, GP66-specific cells showed little or no enrichment for the Treg cell signature, consistent with the lack of a Foxp3+ Treg cell component in the GP66-specific response (Crawford et al., 2014) (Fig. S1F, G). GP-66-specific cells also scored low for the Tmem cell signature (Fig. S1H), in agreement with the idea that this signature identified memory or memory-like cells that were not reactive against and pre-existing infection with LCMV. Additional clusters partially matched these well-defined signatures but were not robustly identified across experiments (Fig. 1D,G), suggesting functional plasticity among early CD4+ T cell responders. However, high scores for Tfh and Tcmp cell signatures were mutually exclusive (Figs. 1D, G and S1E, I), even though expression of specific genes (e.g. Cxcr5 and Tcf7) was observed in clusters corresponding to each signature. Taken together, these scRNAseq analyses identified a specific Tcmp cell transcriptomic signature, distinct from that of differentiated effector cells, and highly enriched in a CCR7-expressing subset known to have elevated CD4+ T cell memory potential. The demarcation between Tfh and Tcmp cell signatures matched the separation between CCR7– CXCR5+ (Tfh) and CCR7+ CXCR5+ cells in flow cytometry analyses (Fig. 1H); thus, subsequent experiments used co-expression of both receptors to identify cells with elevated memory potential. Of note, some Th1 cells had positive scores for genes of the Tcmp cell signature (Fig. S1D, I), highlighting that expression of Th1 cell-associated genes does not exclude conservation of memory potential, as previously noted (Harrington et al., 2008; Marshall et al., 2011; Nish et al., 2017; Pepper et al., 2011; Tubo et al., 2016).
The Tcmp cell signature is Thpok-dependent
We sought genetic evidence to support the functional significance of the Tcmp cell signature. We noted that there are substantial differences between CD4+ and CD8+ memory T cells (Crawford et al., 2014; Homann et al., 2001; Schiemann et al., 2003); accordingly, no d7 p.i. LCMV CD8+ T cell cluster scored high for the Tcmp cell signature (data not shown). Because the transcription factor Thpok promotes CD4+ T cell differentiation in the thymus and its expression remains largely specific of CD4+ T cells after thymic egress (Carpenter and Bosselut, 2010; He et al., 2005; Sun et al., 2005), we inquired whether Thpok was necessary for the expression of the Tcmp cell signature. We analyzed the single-cell transcriptome of LCMV-specific CD4+ T cells deficient for Zbtb7b (encoding Thpok), using Zbtb7bfl/fl mice carrying a cre cDNA genetically targeted into Tnfrsf4 (encoding Ox40) and designated Zbtb7bAD (for ‘activation-induced deletion’). In these mice, Cre was preferentially expressed in antigen-activated post-thymic CD4+ T cells (Fig. S2A), therefore leaving naive conventional CD4+ T cells untouched. These mice carried a Rosa26YFP allele to identify Cre-expressing cells (Srinivas et al., 2001), and cells subject to scRNAseq were sorted for YFP expression.
Thpok deletion in Zbtb7bAD mice affected neither viral clearance nor expansion of antigen-specific cells (Fig. S2B-E). However, t-SNE analyses of scRNAseq data obtained from 1330 Zbtb7bAD and 2154 control d7 p.i. GP66-specific T cells showed minimal overlap between the two genotypes (Fig. 2A). Unsupervised clustering identified clusters with high scores for Th1-,Tfh-and Tcmp cell signatures among control cells (control clusters d7 Ctrl1–2, Ctrl3–4, and Ctrl8, respectively). In contrast, the Thpok-deficient response was dominated by clusters with a Th1 cell signature (clusters d7 Zbtb7bAD1–4, Fig. 2B), in line with the high frequency of IFNγ- producing cells in response to GP66 peptide stimulation (Fig. S2F). All Thpok-deficient clusters showed low scores for the Tcmp cell signature: although cluster d7 Zbtb7bAD5 showed limited expression of Bcl2 and Ccr7, it differed from the control Tcmp cell cluster d7 Ctrl8 by its low expression of Tcf7 and Id3 and high expression of Th1 cell-associated genes Id2 or Tbx21. In agreement with these results, the number of CCR7-expressing cells was greatly reduced in Zbtb7bAD compared to control mice at d7 p.i. (Fig. 2C). Finally, the Thpok-deficient response had no Tfh component, suggesting that Thpok was needed for Tfh cell differentiation, which we confirmed in separate experiments (Vacchio et al., submitted). Thus, Thpok is necessary for the emergence of the Tcmp cell gene signature.
Figure 2: Thpok controls the emergence of the Tcmp signature.

(A, B) scRNAseq of Zbtb7bAD or control (Tnfrsf4-cre+ Zbtb7b+/+) GP66:I-Ab+ Rosa26YFP+ spleen T cells at d7 p.i. LCMV Arm. (A) Top t-SNE plot shows cells colored by genotype. Bottom t-SNE plots show cells from each genotype, independently clustered and color-coded by cluster. (B) Signature scores (top three rows) and expression of selected genes (middle panel) among clusters of Zbtb7bAD and control cells displayed in (A). Percentages (bottom row) indicate cluster size relative to the total population.
(C) Contour plots (left) and quantification (right) of CCR7 expression on spleen GP66:I- Ab+ T cells. Data are from 11 (Zbtb7bAD) and 5 (Ctrl) mice, obtained in two experiments representative of three. Please see also Figure S2.
Cell-intrinsic Thpok is required for the generation of CD4+ memory T cells
The absence of cells with a high Tcmp signature score in Thpok-deficient mice prompted us to analyze the role of Thpok in memory CD4+ T cell development and maintenance. Wild-type mice clear LCMV Arm by d7-d8 p.i., and generate populations of virus-specific CD4+ and CD8+ memory T cells (Matloubian et al., 1994). Analyses at d90 after LCMV Arm infection showed lower numbers of CD4+ but not CD8+ T cells in Zbtb7bAD than in control animals (we evaluated the CD8+ T cell response by staining for the virus-derived GP33 peptide complexed to H-2Db MHC-I molecules, Fig. 3A, B). This suggested that Thpok was needed for CD4+ T cell memory. Normally, LCMV Arm infection results in durable LCMV immunity, including against the Clone 13 strain that establishes chronic infection in non-immune mice. To evaluate the role of Thpok in immune memory, we compared the responses of Zbtb7bAD and control mice to re-challenge by LCMV Clone 13 more than 90 days after infection with LCMV Arm. Clone 13 re-challenged Thpok-deficient mice showed impaired viral clearance (Fig. 3C), similar to MHC II-deficient mice that lack CD4+ T cells (Fig. S2G), and they had reduced numbers of both CD4+ and CD8+ LCMV-responsive T cells (Fig. 3D). Although germline Zbtb7b disruption had been reported to directly affect CD8+ T cell memory responses (Setoguchi et al., 2009), it was unlikely that the effect on CD8+ T cells from Zbtb7bAD mice was cell-intrinsic as only 25–30% of them expressed Cre (Fig. S2C). To address this, we generated mixed bone marrow chimeras combining allelically marked ‘tester’, either Zbtb7bAD or control (Zbtb7b+/+ Tnfrsf4-cre+), and WT competitor components (Fig. 3E). Zbtb7bAD CD4+ T cells were out-competed by their WT counterparts at d90 p.i. (Fig. 3F) and remained so when chimeric mice were re-challenged with LCMV Clone 13 (Fig. 3G), indicating a cell-intrinsic requirement for the generation of memory CD4+ T cells. In contrast, Zbtb7bAD CD8+ T cells normally competed with WT cells; this indicated that, in addition to its cell-intrinsic effect on CD4+ T cell memory, Thpok is necessary for CD4+ T cell help to CD8+ T cell responses (Laidlaw et al., 2016).
Figure 3: Thpok promotes the development of memory CD4+ T cells.

(A) Schematic of the experiment shown in (B-D).
(B) GP66:I-Ab vs. CD44 expression on spleen T cells at d90 p.i. LCMV Armstrong (left). Numbers of GP66:I-Ab+ and GP33:H-2Db+ spleen T cells from 7 Zbtb7bAD and 10 control mice (right). Data are from one experiment representative of three independent experiments.
(C) Serum virus titers 5 days after LCMV Clone 13 rechallenge of LCMV Arm-immunized mice (except black squares: naive WT mice d5 p.i. with Clone 13 as controls). Data summarize 4 independent experiments, each symbol represents a separate mouse (Zbtb7bAD, n=15; Ctrl, n=23; Naive WT, n=9). Bars denote geometric mean; LOD: limit of detection.
(D) GP66:I-Ab+ and GP33:H-2Db+ T cells numbers in immune Zbtb7bAD (n=9) or control (n=10) mice and in naive WT mice (n=7) at d5 p.i. with LCMV Clone 13. Data summarize 2 independent experiments.
(E) Schematic of the experiment shown in (F, G). Mixed bone marrow chimera made from WT ‘competitor’ and either Zbtb7bAD or Ctrl ‘tester’ components were infected with LCMV Arm and analyzed at d90 p.i. (F) or at d5 post LCMV Clone 13 infection performed after d90 p.i. with LCMV Armstrong (G). Each panel shows one out of two independent experiments.
(F) Tester:Competitor ratios among GP66:I-Ab+ and GP33:H-2Db+ spleen T cells for Zbtb7bAD:WT (red) and Ctrl:WT (empty circles) chimera (each symbol represents a distinct chimera, n=8 per group).
(G) Tester:Competitor ratios shown as in (F) at d5 after LCMV Clone 13 rechallenge (n=3 per group). P-values from two-way ANOVA.
(H, I) Spleen T cells from indicated mice, analyzed at d90 p.i. with LCMV Arm. (H) CD8α vs. CD4 expression (right) gated on GP66:I-Ab+ cells (left plots). (I) Numbers of GP66:I- Ab+ cells (left) and percentage of CD4lo cells (right) in Zbtb7bAD (n=5), Zbtb7bAD Cd4silAD (n=5) and control (n=9) animals. Data are from one out of two independent experiments. Please see also Figure S2 and Figure S3.
We next analyzed the Thpok-deficient antigen-specific ‘memory’ cells that persisted several weeks after resolution of LCMV Arm infection. These cells had reduced CD4 surface expression (Fig. S3A), consistent with Thpok promoting Cd4 expression by antagonizing the Cd4 transcriptional silencer (Muroi et al., 2008; Wildt et al., 2007). Indeed, disrupting the Cd4 silencer in Zbtb7bAD cells efficiently sustained Cd4 expression; however, it failed to reconstitute normal memory populations (Fig. 3H, I). Thus, although CD4 expression is important for CD4+ T cell homeostasis in vivo (Wang et al., 2001), the impact of Thpok on memory CD4+ T cell generation is independent of its effect on CD4 expression. We also noted that the GP66-specific Thpok-deficient population included an IL-7Rαlo KLRG1hi component typical of short-lived terminally differentiated effectors (Kaech et al., 2003) (Fig. 4A). This skewing toward effector differentiation was cell-intrinsic, as it was observed both in mixed bone marrow chimera, and in Zbtb7bAD CD4+ T cells expressing the I-Ab-GP66 specific SMARTA TCR transgene adoptively transferred into LCMV-infected WT hosts (Fig. S3B). It was not restored by enforced CD4 expression in Zbtb7bAD Cd4silAD cells (Fig. S3C). Additionally, skewing towards an IL-7Rαlo KLRG1hi phenotype was amplified during the recall response to clone 13 rechallenge (Fig. S3D); this indicated that Thpok disruption not only reduced the number of memory T cells, but impaired their function on a single cell basis.
Figure 4: Early impact of Thpok on CD4+ T cell memory differentiation.

(A) Contour plots (left) and quantification (right, n=5 per group) of IL-7Rα and KLRG1 expression on GP66:I-Ab+ spleen T cells from Zbtb7bAD:WT or Ctrl:WT mixed bone marrow chimera, at d90 p.i. with LCMV Arm. Data are from one representative experiment out of two.
(B-E) RNAseq analysis of gene expression on GP66:I-Ab+ Zbtb7bAD (CD4 Zbtb7bAD) or control (CD4 Ctrl), and GP33:H-2Db+ (CD8 Ctrl) spleen T cells at d30 p.i. with LCMV Arm. (B) Principal Component Analyses (PCA). (C) Heat map of differential gene expression (as defined in Fig. S3F). (D, E) Expression (fragments per million, FPM) of selected genes in indicated populations. P-values are from one-way ANOVA. (B, D, E) each symbol represents a biological replicate.
(F) Contour plots (left) and quantification (right, n=5 per group) of CCR7 and CXCR5 expression on GP66:I-Ab+ spleen T cells from Zbtb7bAD:WT mixed bone marrow chimera analyzed at d30 p.i. with LCMV Arm. Data are from one representative experiment out of two.
(G) (left) Mixed bone marrow chimera (Cd4-creERT2+ Zbtb7bfl/fl: Cd4-creERT2+ Zbtb7b+/+) were infected with LCMV Arm after (ΔEarly) or 3 weeks before (ΔLate) tamoxifen treatment and analyzed at d100 p.i. (right) Percentage of KLRG1hi IL-7Rαlo cells among Thpok-deleted and Zbtb7b+/+ controls (from the same chimera) GP66:I-Ab+ spleen T cells (ΔEarly, n=4; ΔLate, n=3; Ctrl, n=7). Data are from one experiment and representative of 2 independent mixed bone marrow experiments and 3 similar experiments in non-chimeric mice; P-values from unpaired one-sided Welch’s t-test. Please see also Figure S3 and Figure S4.
To further analyze the impact of Thpok on CD4+ T cell memory, we performed RNAseq on LCMV-specific T cell populations sorted on d30 p.i. with LCMV Arm, at which timepoint the number of antigen-specific T cells was already lower in Zbtb7bAD than control mice (Fig. S3E). Principal component analyses showed that d30 Thpok-deficient CD4+ T cells differed from both CD4+ and CD8+ control memory T cells (Fig. 4B). In addition to expressing CD8-lineage genes, consistent with previous reports (Mucida et al., 2013; Vacchio et al., 2014; Wang et al., 2008a) (Fig. S3F, G), Thpok-deficient cells showed impaired expression of prototypical memory genes, including Ccr7, Cxcr5, Tcf7, Bcl2 or Il7r (Hale et al., 2013; Marshall et al., 2011) (Fig. 4C-E), and of CCR7 and CXCR5 proteins (Fig. 4F). Conversely, they inappropriately expressed genes typical of Th1 cells, including Ifng, Tbx21, Id2 or Prdm1 (encoding the transcription factor Blimp1) (Fig. S3H). Consistent with this Th1 cell effector diversion, Thpok-deficient cells had increased expression of Granzyme B (Fig. S3H, I). Thus, Thpok is necessary for the appropriate expression of a memory program in the CD4+ T cells persisting after resolution of infection.
Thpok is required early during activation for the emergence of the memory program
Because scRNAseq analyses indicated that Thpok was needed for the acquisition of the Tcmp cell signature at d7 p.i. (Fig. 2B, C), we examined whether Thpok supported the maintenance of CD4+ T memory cells, or preserved memory potential at the initial phase of the response. To this end, we deleted Thpok either before or three weeks after LCMV Arm infection, using a tamoxifen-inducible cre transgene driven by Cd4 cis-regulatory elements (Cd4-creERT2). To eliminate the possibility of host-specific or indirect effects, we used chimeric mice containing Zbtb7bfl/fl and control Zbtb7b+/+ components, both carrying the Cd4-creERT2 transgene. While loss of Thpok before infection impaired memory cell development, loss of Thpok after viral clearance had no detectable impact (Fig. 4G and S4). Thus, Thpok regulates an early checkpoint in CD4+ T cell responders protecting their memory potential, in line with its impact on the Tcmp cell signature.
Thpok restrains a dysfunctional, exhaustion-like circuitry
RNAseq analyses showed that, in addition to expressing CD8+ T cell-lineage genes and failing to establish a memory program, d30 p.i. Thpok-deficient CD4+ T cells expressed genes characteristic of dysfunctional T cells responding to chronic infection (Crawford et al., 2014; Wherry and Kurachi, 2015), including Lag3, Havcr2 and Cd244 (respectively encoding Lag3, Tim3 and 2B4; Fig. 5A). Because of this mixed gene expression pattern, we explored the heterogeneity of the d30 p.i. population by scRNAseq. t-SNE visualization of Zbtb7bAD and control spleen GP66-specific T cells showed limited overlap between the two genotypes (Fig. 5B). Unsupervised clustering identified four groups of control cells, of which three (clusters d30 Ctrl1–3) expressed markers typical of memory cells, including Id3, Tcf7, Il7r and Bcl2. The fourth cluster (d30 Ctrl4) showed attributes of memory Tfh cells, including expression of Bcl6 but not Ccr7 (Fig. 5C). None of the five Thpok-deficient clusters (d30 Zbtb7bAD 1–5) expressed these memory markers or had a Tfh cell-memory profile similar to d30 Ctrl4. These findings indicated that Thpok is important for the development of memory CD4+ T cells with Tfh-and Thl-cell skewing. Three Thpok-deficient clusters (d30 Zbtb7bAD 1–3) expressed effector genes, including Prdm1 and Tbx21. Cluster d30 Zbtb7bAD5, which included 18% of Thpok-deficient cells, additionally showed expression of Havcr2, Lag3, and Pdcd1 (encoding PD-1) dysfunction genes. The remaining Thpok-deficient cluster, d30 Zbtb7bAD4, associated Tfh cell (Bcl6), memory (Sell) and dysfunction (Eomes and Lag3) gene expression. Flow cytometry of d30 p.i. Thpok-deficient cells did not reproducibly detect expression of PD-1, in line with its low expression by population RNAseq (data not shown); however, it confirmed higher expression of Tim3 and 2B4 relative to control cells (Fig. 5D). Importantly, d30 p.i. Thpok-deficient cells showed impaired IL-2 production (Fig. 5E), a hallmark of T cell dysfunction (Crawford et al., 2014; Wherry and Kurachi, 2015). This dysfunctional profile was maintained during recall responses as Thpok-deficient LCMV-specific CD4+ T cells showed reduced production of IL-2 and increased expression of Tim3 and 2B4 after Clone13 re-challenge (Fig. S5A,B). We conclude from these analyses that Thpok restrains a dysfunctional program resembling, but distinct from, that of exhausted T cells in chronic infections.
Figure 5: Thpok is needed for the functional fitness of CD4+ memory T cells.

(A) RNAseq expression of selected genes in Zbtb7bAD or Ctrl GP66:I-Ab+ spleen T cells as in Fig. 4D; P-values from one-way ANOVA.
(B, C) scRNAseq of Zbtb7bAD or control (Tnfrsf4-cre+ Zbtb7b+/+) spleen GP66:I-Ab+ Rosa26YFP+ T cells at d30 p.i. with LCMV Arm. (B) t-SNE plots shows all cells (top, colored by genotype) and each cell population after independent clustering [bottom, color code for clusters in (C, bottom)]. (C) Signature scores (top three rows) and expression of selected genes (bottom rows) among clusters of Zbtb7bAD and control cells displayed in (B).
(D) Contour plots (left) and MFI (right) of Tim3 and 2B4 expression on GP66:I-Ab+ spleen T cells from Zbtb7bAD (n=7) and control (n=5) mice at d30 p.i. LCMV Arm. Data are from two experiments and representative of three independent experiments.
(E-G) Naive Zbtb7bAD and control SMARTA TCR transgenic cells were adoptively transferred into WT recipient one day before LCMV Arm infection. (E) Cytokine production by spleen T cells at d30 p.i. after peptide stimulation (left). Percentage of IL-2-producers among rFNγ+ cells (right, n=5 recipient mice per genotype). Please see also Figure S5.
(F) Overlaid histograms (left) and MFI (right) of Tim3 and 2B4 expression on Zbtb7bAD (red line) or control (black line) SMARTA spleen T cells at d7 p.i. LCMV Arm; gray-filled histograms show staining on CD44lo CD4+ T cells from the same mouse (n=5 per group).
(G) Percentage of IL-2 producers among IFNγ+ Zbtb7bAD and control SMARTA spleen T cells after GP66 peptide stimulation at d7 p.i. LCMV Arm (n=5 per genotype). (E-G) data are gated on donor-derived cells and are from one experiment representative of two independent experiments per timepoint.
To examine if Thpok targets the dysfunction program during the initial phase immune response, we defined a dysfunction signature on d30 Thpok-deficient (Zbtb7bAD) cells analyzed by scRNAseq (Table S2) and used this signature to query d7 p.i. scRNAseq results. We found higher scores for this signature in Thpok-deficient than in control cells (Fig. S5C). Accordingly, adoptive transfer experiments showed cell-intrinsic increases in the expression of Tim3 and 2B4 and reduction in the production of IL-2 by d7 p.i. Thpok-deficient CD4+ T cells (Fig. 5F, G). Thus, during the initial response of CD4+ T cells to infection, Thpok both promotes the functional ‘fitness’ of CD4+ T cells, by restraining a dysfunction transcriptional program, and preserves their memory potential.
Thpok repression of Prdm1 and Runx3 promotes memory and functional fitness in CD4+ T cells
To obtain mechanistic insight into Thpok control of the memory and dysfunction circuitries, we performed Thpok chromatin immunoprecipitation (ChIP) from effector CD4+ T cells. We generated, by CRISPR-Cas9 editing, a gene-targeted mouse carrying a Zbtb7bBio allele encoding a Thpok protein with a biotin acceptor site (Fig. S6A). The Zbtb7bBio allele was functional, as it supported CD4+ T cell development in homozygous Zbtb7bBio/Bio mice (Fig. S6B); furthermore, the encoded protein was efficiently biotinylated in CD4+ T cells carrying the Zbtb7bBio allele and expressing the BirA biotin ligase from the Rosa26 locus (Fig. S6C). Analyses of triplicate ChIP-seq samples obtained from Zbtb7bBio Rosa26BirA+ CD4+ T cells identified approximately 11,000 Thpok-bound genes in effector CD4+ T cells, most of which recruited Thpok at promoter regions (Fig. S6D). Consistent with a previous report (Muroi et al., 2008), Thpok molecules bound the Zbtb7b and Cd4 loci (Fig. S6E and data not shown).
ChIP-seq detected Thpok binding to most Thpok-regulated genes identified in d7 p.i. LCMV-specific CD4+ T cells by scRNAseq (Fig. S6F). These notably included Prdm1 and Id2, encoding two transcription factors promoting T cell effector against memory differentiation (Crotty et al., 2010; Rutishauser et al., 2009; Yang et al., 2011) (Fig. 6A). We focused on Prdm1 as a potential functional target of Thpok because its product, Blimpl, acts upstream of Id2 in T cells (Miyazaki et al., 2014; Rutishauser et al., 2009; Shaw et al., 2016). Analyses of Zbtb7bAD mice carrying a PrdmYFP reporter allele confirmed that Thpok restrains Blimp1 expression in LCMV-specific CD4+ T cells (Fig. 6B, C). To examine if Thpok protects memory potential or prevents dysfunction through Prdm1 repression, we assessed the differentiation of Zbtb7bfl/fl Prdm1fl/fl Tnfrsf4-cre (Zbtb7bAD Prdm1AD) CD4+ T cells. To identify cell-intrinsic effects, most analyses were conducted in mixed bone marrow chimeras. Inactivating Prdm1 in Thpok-deficient cells restored a d90 p.i. IL-7Rαhi KLRG1lo profile typical of WT memory CD4+ T cells, and enhanced IL-2 production (Fig. 6D, E). Mirroring these late effects on functional fitness, Prdm1 disruption fully reverted the Tim3 up-regulation observed at d7 p.i. in Thpok-deficient cells (Fig. S7A), with a similar but incomplete effect on 2B4. In contrast, Prdm1 disruption only modestly enhanced CCR7 expression at d7 p.i. and failed to restore long-term memory populations (Fig. S7B, C). These findings suggested that Thpok repression of Blimp1 expression contributes to maintain the functional fitness of memory cells, but that it does not mediate the impact of Thpok on the generation of memory precursors.
Figure 6: Thpok maintains memory CD4+ T cell fitness by restraining Blimp1 expression.

(A) Thpok ChIP-seq in activated CD4+ T cells from Zbtb7bBio/+ Rosa26BirA+ (Zbtb7bBio) and Zbtb7b+/+ Rosa26BirA+ (Ctrl) mice. Grey shading highlights peaks detected in 3 independent replicates.
(B) Histogram plots of YFP expression on GP66:I-Ab+ spleen T cells from Zbtb7bAD and control animals carrying the Prdm1YFP reporter, at d7 p.i. with LCMV Arm. Quantification (C) shows YFP expression on all (total) or YFP+ GP66:I-Ab+ T cells. Data are from one experiment (n=3 mice per genotype) representative of two independent experiments.
(D, E) Zbtb7bAD:WT and Zbtb7bAD Prdm1AD: WT mixed bone marrow chimeras were analyzed d90 p.i with LCMV Arm. (D) Contour plots (left) and quantification (right) of IL-7Rα vs. KLRG1 surface expression on GP66:I-Ab+ spleen T cells. (E) Percentage of IL-2 producing cells among IFNγ+ spleen T cells of Zbtb7bAD (n=4), Zbtb7bAD Prdm1AD (n=4) and WT (n=8) genotype after GP66 peptide stimulation. Data are from one bone marrow chimera experiment and representative of 3 independent non-chimeric experiments. Please see also Figure S6 and Figure S7.
In addition to Blimp1, d7 p.i. Thpok-deficient CD4+ T cells had increased expression of Runx3 (Fig. S7D), a factor expressed in CD8+ and Th1 CD4+ T cells (Djuretic et al., 2009) and previously shown to be a functional target of Thpok (Carpenter and Bosselut, 2010; Egawa and Littman, 2008; Muroi et al., 2008; Wang et al., 2008a). Consistent with direct transcriptional control, we found that Thpok bound the Runx3 locus (Fig. 7A). Moreover, we noted from published ChIP-seq datasets (Mackay et al., 2016; Shan et al., 2017) that several genes characteristic of memory cells, including Tcf7 and Ccr7, could recruit Blimpl and Runx molecules in addition to Thpok (Fig. S7E). Consequently, we speculated that maintaining memory potential required the repression of both Prdm1 and Runx3 by Thpok. To assess this possibility, we generated Zbtb7bfl/fl Prdm1fl/fl Cbfbfl/fl Tnfrsf4-cre (Zbtb7bAD Prdm1AD CbfbAD) animals, in which deletion of the obligatory Runx cofactor Cbfβ overcomes potential overlap of functions between Runx1–3 proteins. We could not evaluate long-term memory responses in these mice as Runx activity is needed for long-term survival of CD4+ T cells (Egawa et al., 2007), including of memory CD4+ T cells (Fig. S7F). Thus, we assessed d7 p.i. LCMV-specific CD4+ T cells for markers of memory potential (CXCR5 and CCR7) and of dysfunction (Tim3 and 2B4). We performed these analyses in mixed bone marrow chimeras to avoid indirect effects of gene disruption. Triple-deficient cells differentiated normally into CCR7hi CD4+ T cells, unlike their Zbtb7bAD or Zbtb7bAD Prdm1AD counterparts (Fig. 7B, C), whereas a partial rescue was observed in Zbtb7bAD CbfbAD cells. Moreover, inactivation of both Blimpl and Cbfβ fully reverted the up-regulation of inhibitory receptors Tim3 and 2B4 seen in Thpok-deficient cells (Fig. 7D). We conclude from these experiments that Thpok promotes the emergence and functional fitness of memory CD4+ T cell precursors by restraining the expression or activity of Blimpl and Runx molecules.
Figure 7: Memory CD4+ T cell differentiation involves Thpok antagonism of Blimp1 and Runx3.

(A) Thpok ChIP-seq on the Runx3 locus, displayed as in Fig. 6A.
(B-D) Bone marrow chimeras combining WT competitor and either Zbtb7bAD, Zbtb7bAD Prdm1AD, Zbtb7bAD CbfbAD, Zbtb7bAD Prdm1AD CbfbAD or Ctrl ‘tester’ cells were analyzed at d7 p.i. with LCMV Arm. (B) CCR7 and CXCR5 expression on GP66:I-Ab+ tester spleen T cells.(C) Tester:Competitor ratios among spleen GP66:I-Ab+ CCR7+ T cells (n=5 per group). (D) Tim3 and 2B4 expression on GP66:I-Ab+ cells (n=3 per group). (C, D) Data are representative of one (D) or 2 separate (C, D) experiments on one set of mixed chimeras and 3 similar experiments on non-chimeric animals of the same genotype. Please see also Figure S6 and Figure S7.
Altogether, our study demonstrates that the transcription factor Thpok antagonizes Blimpl and Runx3 expression to support CD4+ T cell memory potential and restrain a gene expression pattern characteristic of dysfunctional T cells. These functions of Thpok identify a shared mechanistic basis for two previously unconnected attributes of CD4+ helper T cells, ensuring their functional fitness to support long-lived immunity.
Discussion
Memory CD4+ T cells are essential for durable immunity and are targeted by current vaccination strategies, including against influenza and malaria (Mordmuller et al., 2017; Valkenburg et al., 2018). Thus, it is important to elucidate the mechanisms that control their development. Here, starting from single-cell transcriptome analyses of virus-specific CD4+ T cells, we identified a regulatory switch controlling both the emergence of memory cells and their functional fitness. Generating transcriptome-wide gene expression signatures from large numbers of cells captured during the initial and memory phases of the immune response, we have provided a high-resolution analysis of effector and memory CD4+ T cell differentiation that had not been accessible to previous single-cell (Lonnberg et al., 2017) and population-level studies (Hale et al., 2013; Marshall et al., 2011).
At the initial phase of the response, we identified characteristic Th1 and Tfh cell transcriptomic signatures, which included genes encoding prototypical fate markers (e.g. T-bet and Id2 for Th1 cells or CXCR5 and Icos for Tfh cells); these signatures were largely mutually exclusive, consistent with the idea that Th1 and Tfh effector cell fates show little functional overlap (Crotty, 2018). The Tcmp cell signature associated markers of memory cells (including Ccr7, Tcf7, or Bcl2) and excluded genes characteristic of Th1 and Tfh effector cell genes (Tbx21, Prdm1, Bcl6), supporting its assignment to cells with increased memory potential. Accordingly, that signature mapped to a subset of CD4+ T cells expressing CCR7 (and also CXCR5, although in intermediate amounts) and previously reported to contain memory precursor activity (Pepper et al., 2011). Partial activation of the Tcmp cell signature was detected in Th1 but not Tfh cell subsets, suggesting that it identifies cells skewed towards a Th1- rather than Tfh-memory cell potential. Consistent with this idea, two components of Tcmp signature, Klf2 and Ccr7, inhibit Tfh cell differentiation (Haynes et al., 2007; Lee et al., 2015). The possibility of an early divergence of central and Tfh memory cell precursors does not exclude that their differentiation programs share transcriptional regulators. Bcl6, which is needed for the generation of memory Tfh cells (He et al., 2013), promotes the survival of all memory CD4+ T cell subsets (Ichii et al., 2007). Additionally, Bcl6 is needed for the generation of the CXCR5+ subset during the initial CD4+ T cell response (Pepper et al., 2011); while this may simply reflect the importance of Bcl6 for CXCR5 expression, it could also indicate its involvement in the emergence of CD4+ T cell memory, in addition to long-term memory cell survival.
In addition to its cell-intrinsic effect on CD4+ T cell memory, Thpok is needed for CD4+ T cell help to CD8+ T cell memory, a function critical for long-term immunity (Shedlock and Shen, 2003; Sun and Bevan, 2003). Several mechanisms, non-mutually exclusive, have been proposed to account for CD4+ T cell help to CD8+ T cells; these include IL-2 production and the ‘licensing’ of dendritic cells, through CD40 signaling, for the provision of memory-inducing signals to CD8+ T cells (Laidlaw et al., 2016; MacLeod et al., 2009). Both IL-2 secretion and expression of CD154, the ligand for CD40, depend on Thpok, accounting at least in part for its contribution to CD4+ T cell help for the generation of memory CD8+ T cells.
Thpok protects the functional fitness of CD4+ memory cells by restraining a ‘dysfunction’ gene expression program that shares features with the ‘exhausted’ state of T cells subject to chronic antigen stimulation, including impaired IL-2 production and dependence on Blimp1 (Crawford et al., 2014; Shin et al., 2009; Wherry and Kurachi, 2015). However, this dysfunction pattern differs from conventional exhaustion as it emerges despite antigen clearance, e.g. in mixed bone marrow chimera or adoptively transferred cells; accordingly, it is not accompanied by PD1 expression, a hallmark of recent antigen engagement (Agata et al., 1996). Additionally,the Thpok-repressed dysfunction pattern is distinct from the cytotoxic diversion we and others previously identified in Thpok-deficient CD4+ T cells (Mucida et al., 2013; Reis et al., 2013; Vacchio et al., 2014; Wang et al., 2008a), most notably because the former depends on Blimpl, whereas the latter depends on Runx activity.
Repression of Blimpl- and Runx3-encoding genes mediated the impact of Thpok on the emergence and functional fitness of memory CD4+ T cells. Our demonstration that Thpok bound within or near both loci supports the possibility that their repression by Thpok is at least in part direct. Thpok repression of Blimpl feeds into a negative regulatory loop previously characterized in differentiating CD4+ effectors, whereby Bcl6 and Blimpl inhibit each other’s expression (Crotty et al., 2010). In addition, we found that Thpok binds the Bcl6 gene (Vacchio et al., submitted), whereas Runx3 was reported to shift the Bcl6-Blimp1 equilibrium towards Blimp1 expression (Shan et al., 2017), so that its repression could reinforce directs effects of Thpok on the Bcl6-Blimp1 loop. Together, these observations support the idea that Thpok serves at multiple entry points to shift the Bcl6-Blimp1 loop to restrain Blimp1.
In summary, the present study identified a Thpok-operated checkpoint controlling both the emergence of CD4+ memory cells and their functional fitness, two previously unconnected aspects of CD4+ T cells responses. While Thpok is expressed in all conventional CD4+ T cells, it is sensitive to both antigenic stimulation (He et al., 2008; Wang et al., 2008a) and environmental factors, as recently shown of gut microbial metabolites (Cervantes-Barragan et al., 2017; Reis et al., 2013). Thus, the importance of Thpok in CD4+ T cell memory and functions identify this factor as a potential target to enhance responses to immunization strategies against infections or cancer.
STAR Methods
Lead author
Rémy Bosselut, remy.bosselut@nih.gov
Mice
Mice carrying floxed alleles for Zbtb7b (Wang et al., 2008a), Cbfb (Naoe et al., 2007), Prdm1 (Shapiro-Shelef et al., 2003), Cd4sil (Zou et al., 2001), Rosa26YFP (Srinivas et al., 2001), or Rosa26BirA (Driegen et al., 2005) were previously described as were Zbtb7b–/– (Wang et al., 2008b), H2-Ab1b–/– (Grusby et al., 1991), SMARTA (Oxenius et al., 1998), Cd4-creERT2 (Aghajani et al., 2012) and Tnfrsf4-cre (Zhu et al., 2004) animals. Prdm1YFP mice were from the Jackson laboratory and CD45.1 and CD45.2 C57BL/6 mice from Charles River laboratories. All transgenic mice were heterozygous for the transgene they express. The Tnfrsf4-cre allele was maintained heterozygous and only female Tnfrsf4-cre+ mice were used for breeding. Except where specified otherwise, control mice included in experimental designs were Tnfrsf4-cre+ Zbtb7b+/+ or Tnfrsf4-cre– animals from the same line as experimental mice. Mice were housed in specific pathogen-free facilities. Animal procedures were approved by the NCI Animal Care and Use Committee.
Infectious Models, mixed bone marrow chimera and induced gene deletion
Non-chimeric 6–16 weeks old animals were infected by intra-peritoneal injection of 2 × 105 pfu of LCMV Armstrong. For re-challenge experiments mice were infected intra-venously with 2 × 106 pfu of LCMV Clone 13.
For adoptive transfer experiments, 1000 spleen CD4+ T cells (unseparated or sorted CD44lo Rosa26YFP–) from SMARTA TCR transgenic mice (Zbtb7bAD or controls) were intra-venously injected into CD45 congenic recipients, which were infected with LCMV Arm 24 hours later.
For mixed bone marrow chimera experiments, T cell-depleted (Pan T Dynal kit, Invitrogen) bone marrow cells were prepared from CD45 disparate mice, mixed together at various ratios (from 1:1 to 1:3), and injected into lethally irradiated (900 rads) CD45.1 recipients. Chimeric animals were infected eight weeks after reconstitution. In experiments using Cd4ERT2Cre animals, gene disruption was induced by tamoxifen gavage (5mg/mouse/day for 4 consecutive days) before or 3 weeks after LCMV Arm infection. Chimerism was calculated as the ratio of tester over competitor cells within the population of interest.
Determination of virus titers
Serum from infected animals was isolated from peripheral blood by centrifugation and stored at −80°C. Viral RNA was extracted with PureLink Viral RNA column (Invitrogen) according to the manufacturer instructions, reverse-transcribed (Thermoscript, Invitrogen), and quantified by real-time PCR with SYBR green dye (Invitrogen) on a QuantStudio 6 Flex system (Applied Biosystems), using primers specific for the LCMV GP gene (McCausland and Crotty, 2008) (Fwd-GCAACTGCTGTGTTCCCGAAAC, Rev-CATTCACCTGGACTTTGTCAGACTC). For each experiment, the limit of detection (LOD) of the assay was determined using the serum of non-infected animals.
Antibodies
Antibodies for the following specificities were purchased either from Becton-Dickinson Pharmingen, Biolegend or ThermoFisher Ebiosciences: CD4 (RM4.4 or GK1.5), CD8α (53–6-7), CD45.2 (104), CD45.1 (A20), TCRβ (H57–597), CD5 (53–7.3), B220 (RA3–6B2), IFNγ (XMG1.2), IL-2 (JES6–5H4), CD44 (IM7), IL-7Rα (A7R34), KLRG1 (2F1), CCR7 (4B12), CXCR5 (SPRCL5), Tim3 (RMT3–23), 2B4 (R244F4), CXCR6 (SA051D1), streptavidin. MHC tetramers loaded with the LCMV GP33 or GP66 peptides were obtained from the NIH Tetramer Core Facility.
Cell preparation and staining
Spleen cells were prepared and stained as previously described (Ciucci et al., 2017). Surface staining with GP66:I-Ab tetramer or for CCR7 or CXCR5 was performed at 37°C, and staining with GP33:H-2Db tetramer at 4°C, for 1 hour prior to staining with other antibodies. Cytokine staining was performed after 5 hours on splenocytes incubated in the presence of GP66 (GLKGPDIYKGVYQFKSVEFD, 2μg/ml) or GP33 (KAVYNFATM, 0.20μg/ml) peptides (Anaspec) and Golgi Stop (Crawford et al., 2014).
Flow cytometry data was acquired on LSR II or LSR Fortessa cytometers (BD Biosciences) and analyzed with FlowJo software (TreeStar). Dead cells and doublets were excluded by LiveDead staining (Invitrogen) and forward scatter height by width gating. Except if otherwise mentioned, numbers in the cytometry plot are percentages of cells in the gates. Cell sorting was performed on a FACSAria or FACS Fusion (BD Biosciences). Because of the effect of Thpok-deletion on the expression of CD4 and CD8α, analyses and purification of GP66- specific cells were performed without gating for CD4 and CD8α expression.
Population RNAseq
RNA from GP66:I-Ab+ and GP33:H-2Db+ T cells, sorted from the spleen of LCMV-infected mice at d30 p.i., was isolated using the RNeasy Plus Micro kit (Qiagen). Quality control was performed by bioanalyzer (Agilent), and RNA samples with a RNA integrity number (RIN) > 8 were processed for library preparation using SMARTer Ultra Low Input reagent (Takara) and Nextera XT DNA (Illumina) library preparation kits. Libraries were sequenced with paired-end reads of 126bp on a HiSeq2500 sequencer (Illumina) to reach 50 million read pairs per sample. For each cell subset and genotype, data are derived from three distinct mice, with separate processing from cell sorting to sequencing. Raw RNAseq fastq reads were trimmed with Trimmomatic and aligned to mouse genome (mm10) using STAR (v. 2.4.0h). Gene-assignment and count of RNA reads were performed with HTseq. Further analyses were performed with R software and differentially expressed genes were identified using DESeq2 using the Wald test (FDR <0.01).
Generation of a Zbtb7bBio genetically targeted allele
Specific sgRNAs targeting the 3’ end of the Zbtb7b open reading frame were designed using the online tool MIT CRISPR Design (crispr.mit.edu) and produced in vitro using MEGAshortscript T7 transcription Kit (Thermo Fisher Scientific). A 153bp double stranded template containing the composite linker-biotin acceptor sequence agcatgcgctcgggtggaGGCCTGAACGACATCTTCGAGGCTCAGAAAATCGAATGGCACGA A (one-letter code amino acid sequence smrsggGLNDIFEAQKIEWHE, biotin acceptor peptide sequence in uppercase letters) flanked by 45bp left and right Thpok homology regions was synthesized as Ultramer DNA oligo (Integrated DNA Technologies). sgRNAs, dsDNA donor template and Cas9 mRNA (TriLink Biotechnologies) were microinjected into zygotes from C57BL/6Ncr mice at the one-cell stage to generate the Zbtb7bBio allele.
ChIP-Sequencing
Splenic CD4+ T cells from Zbtb7bBio/+ Rosa26BirA or Rosa26BirA animals were enriched using Dynabeads Untouched Mouse CD4 cells kit (Invitrogen) and stimulated with anti-CD3 (1μg/ml), anti-CD28 (3μg/ml) and IL-12 (10ng/ml) for 3 days and then with IL-2 (100ng/ml) for another day. Live cells were sorted, washed in PBS and fixed in 1% formaldehyde-containing PBS for 5 min at 37 degrees. Following quenching with 0.125M glycine, cells pellets were snap-frozen on dry ice and stored at −80°C. Frozen fixed cells were then lysed in 1% SDS RIPA buffer (20 mM Tris-HCl pH 7.6, 2 mM EDTA, 150 mM NaCl, 0.1% sodium deoxycholate, 1% TritonX100) for 30 minutes at 4°C, spun down and sonicated in 0.1% SDS RIPA buffer using a Qsonica Q800R sonicator (30s on, 59s off, 85% amplitude) to obtain a sheared chromatin with an average size of 200bp. Sheared chromatin was pre-cleared with protein-A magnetic beads (Invitrogen 10001D) followed by immunoprecipitation with M280 Streptavidin beads (Invitrogen 11205D). After washing, immunoprecipitated chromatin was reverse-crosslinked by overnight incubation at 70°C in 50nM 50mM Tris-HCl pH 8., 10mM EDTA, 1% SDS buffer. DNA was subsequently treated with proteinase K and RNase (both at 0.2mg/ml) before purification using the QIAquick PCR purification kit (Qiagen).
DNA was processed for library preparation using the Accel-NGS 2S DNA reagent (Swift). Libraries were sequenced (75bp single-ended reads) on a NextSeq sequencer (Illumina). Raw fastq reads were trimmed based on quality score (Phred >32) and aligned to mouse genome (mm10) using Bowtie on Partek Flow on the National Institutes of Health high-performance computing Biowulf cluster. Peak calling was performed with MACS2 comparing the ChIP samples from the Zbtb7bBio Rosa26BirA chromatin to the Rosa26BirA chromatin (broad peak, qvalue<0.1). Further analyses were performed with R software with the ChIPseeker package.
The Cbfβ (Shan et al., 2017) and Blimp1 (Mackay et al., 2016) datasets were obtained from the gene expression omnibus (respectively accession GSE81888 and GSE79339) and aligned to the mouse genome (mm10 release) using the Bowtie package on the Partek Flow server.
Single cell RNAseq
5000–10000 splenic T cells (either a mix of CD4+ and CD8+ T cells or GP66:I-Ab+ T cells, as indicated in Figures) were sorted from LCMV infected mice, loaded onto a 10X Chromium platform to generate cDNAs carrying cell- and transcript-specific barcodes that were used to construct sequencing libraries using the Chromium Single Cell 3′ Library & Gel Bead Kit v2 according to the manufacturer instructions. Libraries were sequenced on multiple runs of Illumina NextSeq using paired-end 26×98bp or 26×57bp in order to reach a sequencing saturation greater than 70%, resulting in at least 50,000 reads/cell. Single-cell sequencing files were processed, and count matrixes extracted using the Cell Ranger Single Cell Software Suite (v1.3.1). Further analyses were performed in R using the Seurat package (Butler et al., 2018) (2.3.0).
Data was pre-processed by removing genes expressed in fewer than 2 cells and excluding cells expressing fewer than 500 genes, or more than 10% mitochondrial genes. Reduction of data dimensionality was performed on the first 20 principal components (PC) calculated on the highly variable genes for each pre-processed dataset.
To define gene expression signatures, we first clustered d7 p.i CD44hi datasets (including both CD8+ and CD4+ T cells) and defined as CD4+ clusters those clusters scoring high for Cd4 and negative or low for Cd8a. Cells from these clusters were extracted from the whole set. Highly variable genes were re-defined on the selected cells and Cd4-expressing populations were clustered. For each cluster, a cluster-specific gene signature was defined using the FindAllMarkers function selecting only positively enriched genes. The final Th1, Tfh, Tcmp, Treg and memory cell signatures were defined as the intersection of matching cluster-specific gene signatures identified in the two independent replicates. Gene signature scores were calculated on regressed datasets (regression of the number of UMI and percentages of mitochondrial genes with ScaleData function) using the AddModuleScore function.
For analyses of Thpok-deficient and control cells, clustering was performed separately for each sample, and further analyses, including t-SNE visualization, were performed after merging datasets using the MergeSeuratObjects function. Clusters representing less than 1% of each population (which corresponds to the expected duplicate capture rate) where excluded from downstream analyses.
Statistical analyses
Except for deep-sequencing data, statistical significance was calculated with Prism software. Except where otherwise indicated in figure legends, error bars in graphs indicate standard deviation and statistical comparisons were done by unpaired two-sided Welch’s t-test.
Data availability
Sequence data are deposited in the NCBI Gene Expression Omnibus under accession number GSE116506, GSE116519 and GSE121002. All other data and code are available upon request.
Supplementary Material
Highlights.
Single-cell RNA sequencing characterizes CD4+ central memory precursor T cells.
CD4+ central memory precursor T cells depend on the transcription factor Thpok.
Protective memory responses are impaired by disruption of Thpok in CD4+ T cells.
Thpok represses a dysfunction, exhaustion-like, program driven by Blimp1 and Runx3
Acknowledgments
We thank E. Castro and H. Kwak for animal care and genotyping and Q. Xiao for technical assistance; D. Swing and R. Koogle for help in generating the Zbtb7bBio allele; M. Jenkins for helpful discussions; Y. Belkaid, J. Zhu, J. O’Shea, Ming Li, F. Gounari and the NIH tetramer facility for mice and reagents; Anne Gégonne, the CCR Flow Cytometry Core and the NIH High performance computing cluster for assistance; G. Abou Ezzi, J. Ashwell, D. Barber, A. Bhandoola, L. Chopp, J. Cowan, C. Harly and A. Magen for critical reading of the manuscript; and D. Goldstein, M. Malik and the NCI Office of Science and Technology Resources for their support. This work was supported by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, and Honjo T (1996). Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 8, 765–772. [DOI] [PubMed] [Google Scholar]
- Aghajani K, Keerthivasan S, Yu Y, and Gounari F (2012). Generation of CD4CreER(T(2)) transgenic mice to study development of peripheral CD4-T-cells. Genesis 50, 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Bevan MJ, Reiner SL, and Fearon DT (2009). The precursors of memory: models and controversies. Nat Rev Immunol 9, 662–668. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AC, and Bosselut R (2010). Decision checkpoints in the thymus. Nat Immunol 11, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J, Cortez VS, Caparon MG, Donia MS, Gilfillan S, et al. (2017). Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 357, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YL, Flossdorf M, Kretschmer L, Hofer T, Busch DH, and Buchholz VR (2017). TCR Signal Quality Modulates Fate Decisions of Single CD4+ T Cells in a Probabilistic Manner. Cell Rep 20, 806–818. [DOI] [PubMed] [Google Scholar]
- Ciucci T, Vacchio MS, and Bosselut R (2017). A STAT3-dependent transcriptional circuitry inhibits cytotoxic gene expression in T cells. Proc Natl Acad Sci U S A 114, 13236–13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, and Wherry EJ (2014). Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 40, 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S (2018). Do Memory CD4 T Cells Keep Their Cell-Type Programming: Plasticity versus Fate Commitment? Complexities of Interpretation due to the Heterogeneity of Memory CD4 T Cells, Including T Follicular Helper Cells. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Johnston RJ, and Schoenberger SP (2010). Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol 11, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuretic IM, Cruz-Guilloty F, and Rao A (2009). Regulation of gene expression in peripheral T cells by Runx transcription factors. Adv Immunol 104, 1–23. [DOI] [PubMed] [Google Scholar]
- Driegen S, Ferreira R, van Zon A, Strouboulis J, Jaegle M, Grosveld F, Philipsen S, and Meijer D (2005). A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res 14, 477–482. [DOI] [PubMed] [Google Scholar]
- Egawa T, and Littman DR (2008). ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nat Immunol 9, 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa T, Tillman RE, Naoe Y, Taniuchi I, and Littman DR (2007). The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med 204, 1945–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazilleau N, McHeyzer-Williams LJ, Rosen H, and McHeyzer-Williams MG (2009). The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol 10, 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grusby MJ, Johnson RS, Papaioannou VE, and Glimcher LH (1991). Depletion of CD4+ T cells in major histocompatibility complex class Il-deficient mice. Science 253, 1417–1420. [DOI] [PubMed] [Google Scholar]
- Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, Wu T, Iyer SS, and Ahmed R (2013). Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38, 805817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Janowski KM, Oliver JR, Zajac AJ, and Weaver CT (2008). Memory CD4 T cells emerge from effector T-cell progenitors. Nature 452, 356–360. [DOI] [PubMed] [Google Scholar]
- Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, and Cyster JG (2007). Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol 179, 5099–5108. [DOI] [PubMed] [Google Scholar]
- He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, Sun X, Vandenberg K, Rockman S, Ding Y, et al. (2013). Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 39, 770–781. [DOI] [PubMed] [Google Scholar]
- He X, He X, Dave VP, Zhang Y, Hua X, Nicolas E, Xu W, Roe BA, and Kappes DJ (2005). The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature 433, 826–833. [DOI] [PubMed] [Google Scholar]
- He X, Park K, Wang H, He X, Zhang Y, Hua X, Li Y, and Kappes DJ (2008). CD4- CD8 lineage commitment is regulated by a silencer element at the ThPOK transcription-factor locus. Immunity 28, 346–358. [DOI] [PubMed] [Google Scholar]
- Homann D, Teyton L, and Oldstone MB (2001). Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med 7, 913–919. [DOI] [PubMed] [Google Scholar]
- Ichii H, Sakamoto A, Arima M, Hatano M, Kuroda Y, and Tokuhisa T (2007). Bcl6 is essential for the generation of long-term memory CD4+ T cells. Int Immunol 19, 427–433. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Hemby S, Kersh E, and Ahmed R (2002). Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, and Ahmed R (2003). Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4, 1191–1198. [DOI] [PubMed] [Google Scholar]
- Laidlaw BJ, Craft JE, and Kaech SM (2016). The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 16, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Skon CN, Lee YJ, Oh S, Taylor JJ, Malhotra D, Jenkins MK, Rosenfeld MG, Hogquist KA, and Jameson SC (2015). The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity 42, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonnberg T, Svensson V, James KR, Fernandez-Ruiz D, Sebina I, Montandon R, Soon MS, Fogg LG, Nair AS, Liligeto U, et al. (2017). Single-cell RNA-seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, and Nutt SL (2012). The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol 13, 491–498. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. [DOI] [PubMed] [Google Scholar]
- MacLeod MK, Clambey ET, Kappler JW, and Marrack P (2009). CD4 memory T cells: what are they and what can they do? Semin Immunol 21, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, and Kaech SM (2011). Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Concepcion RJ, and Ahmed R (1994). CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68, 8056–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCausland MM, and Crotty S (2008). Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J Virol Methods 147, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, and Swain SL (2014). Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Commun 5, 5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu LF, Varki N, Chang AN, Broide DH, and Murre C (2014). Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol 15, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordmuller B, Surat G, Lagler H, Chakravarty S, Ishizuka AS, Lalremruata A, Gmeiner M, Campo JJ, Esen M, Ruben AJ, et al. (2017). Sterile protection against human malaria by chemoattenuated PfSPZ vaccine. Nature 542, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, Reis BS, Huang Y, Lambolez F, Docherty M, et al. (2013). Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class Il-restricted cytotoxic T lymphocytes. Nat Immunol 74, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroi S, Naoe Y, Miyamoto C, Akiyama K, Ikawa T, Masuda K, Kawamoto H, and Taniuchi I (2008). Cascading suppression of transcriptional silencers by ThPOK seals helper T cell fate. Nat Immunol 9, 1113–1121. [DOI] [PubMed] [Google Scholar]
- Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F, Littman DR, and Taniuchi I (2007). Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med 204, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH, Yen B, Rothman NJ, Bhandoola A, Xue HH, et al. (2017). CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J Exp Med 274, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenius A, Bachmann MF, Zinkernagel RM, and Hengartner H (1998). Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol 28, 390–400. [DOI] [PubMed] [Google Scholar]
- Pepper M, and Jenkins MK (2011). Origins of CD4(+) effector and central memory T cells. Nat Immunol 72, 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, and Jenkins MK (2011). Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35, 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton JF, Tan JT, Rubinstein MP, Kim DM, Sprent J, and Surh CD (2007). Antiviral CD4+ memory T cells are IL-15 dependent. J Exp Med 204, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, and Mucida D (2013). Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat Immunol 74, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, and Kaech SM (2009). Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 37, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, and Lanzavecchia A (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 407, 708712. [DOI] [PubMed] [Google Scholar]
- Schiemann M, Busch V, Linkemann K, Huster KM, and Busch DH (2003). Differences in maintenance of CD8+ and CD4+ bacteria-specific effector-memory T cell populations. Eur J Immunol 33, 2875–2885. [DOI] [PubMed] [Google Scholar]
- Setoguchi R, Taniuchi I, and Bevan MJ (2009). ThPOK derepression is required for robust CD8 T cell responses to viral infection. J Immunol 183, 4467–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, Kurup SP, Van Braeckel- Budimir N, Su Y, Martin MD, et al. (2017). The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol 18, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, and Calame K (2003). Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 19, 607–620. [DOI] [PubMed] [Google Scholar]
- Shaw LA, Belanger S, Omilusik KD, Cho S, Scott-Browne JP, Nance JP, Goulding J, Lasorella A, Lu LF, Crotty S, and Goldrath AW (2016). Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat Immunol 17, 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock DJ, and Shen H (2003). Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300, 337–339. [DOI] [PubMed] [Google Scholar]
- Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, and Wherry EJ (2009). A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Liu X, Mercado P, Jenkinson SR, Kypriotou M, Feigenbaum L, Galera P, and Bosselut R (2005). The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nat Immunol 6, 373–381. [DOI] [PubMed] [Google Scholar]
- Sun JC, and Bevan MJ (2003). Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300, 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo NJ, Fife BT, Pagan AJ, Kotov DI, Goldberg MF, and Jenkins MK (2016). Most microbe-specific naive CD4(+) T cells produce memory cells during infection. Science 351, 511514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacchio MS, Wang L, Bouladoux N, Carpenter AC, Xiong Y, Williams LC, Wohlfert E, Song KD, Belkaid Y, Love PE, and Bosselut R (2014). A ThPOK-LRF transcriptional node maintains the integrity and effector potential of post-thymic CD4+ T cells. Nat Immunol 15, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkenburg SA, Li OTW, Li A, Bull M, Waldmann TA, Perera LP, Peiris M, and Poon LLM (2018). Protection by universal influenza vaccine is mediated by memory CD4 T cells. Vaccine 36, 4198–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wildt KF, Castro E, Xiong Y, Feigenbaum L, Tessarollo L, and Bosselut R (2008a). The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity 29, 876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wildt KF, Zhu J, Zhang X, Feigenbaum L, Tessarollo L, Paul WE, Fowlkes BJ, and Bosselut R (2008b). Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4(+) T cells. Nat Immunol 9, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Strong J, and Killeen N (2001). Homeostatic competition among T cells revealed by conditional inactivation of the mouse Cd4 gene. J Exp Med 194, 1721–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildt KF, Sun G, Grueter B, Fischer M, Zamisch M, Ehlers M, and Bosselut R (2007). The transcription factor Zbtb7b promotes CD4 expression by antagonizing Runx-mediated activation of the CD4 silencer. J Immunol 179, 4405–4414. [DOI] [PubMed] [Google Scholar]
- Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, Lambkin- Williams R, Gilbert A, Oxford J, Nicholas B, et al. (2012). Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 18, 274–280. [DOI] [PubMed] [Google Scholar]
- Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, et al. (2011). The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol 12, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaph C, Uzonna J, Beverley SM, and Scott P (2004). Central memory T cells mediate longterm immunity to Leishmania major in the absence of persistent parasites. Nat Med 10, 11041110. [DOI] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF Jr., Guo L, and Paul WE (2004). Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol 5, 1157–1165. [DOI] [PubMed] [Google Scholar]
- Zou YR, Sunshine MJ, Taniuchi I, Hatam F, Killeen N, and Littman DR (2001). Epigenetic silencing of CD4 in T cells committed to the cytotoxic lineage. Nat Genet 29, 332336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data are deposited in the NCBI Gene Expression Omnibus under accession number GSE116506, GSE116519 and GSE121002. All other data and code are available upon request.