

Abstract
A total of 217 Mycoplasma bovis isolates cultured from clinical cases in Ontario, Canada, over the past 30 y were selected to be characterized by a multi-locus sequence typing (MLST) method. Eleven housekeeping genes were evaluated for suitability for MLST; 2 loci that had been used in prior MLST schemes, dnaN and metS, along with hsp70 were chosen for further sequence analysis. The remaining loci—adk, efp, gmk, gyrB, polC, rpoB, tpiA, and uvrC genes—were not used because they had little to no sequence variation. The sequence data from the chosen loci (dnaN, hsp70, metS) generated 28 sequence types (STs), with the 3 loci having 15, 5, and 7 alleles, respectively. These molecular typing results revealed that the STs had a temporal distribution; over the course of 3 decades, some STs disappeared and new STs appeared. Recent isolates had a greater variety of STs, which may indicate that new strains are emerging more rapidly now than in the past.
Keywords: Multi-locus sequence typing, Mycoplasma bovis
Mycoplasma bovis, formerly Mycoplasma agalactiae subsp. bovis, is the most important bovine mycoplasmal pathogen in North America, causing mastitis, pneumonia, arthritis, decubital abscesses, otitis media, and other diseases in both dairy and beef cattle.6 Despite this, the role of the organism in disease development is not completely clear.6 Multi-locus sequence typing (MLST) schemes, which combine sequence data from several housekeeping genes to produce a sequence type (ST) for each strain, have proven useful for understanding population structures with bacterial pathogens.2,3,13 One study4 developed a MLST scheme for M. bovis using 4 genes—translation elongation factor G (fusA), DNA gyrase subunit B (gyrB), translation elongation factor G (lepA), and RNA polymerase B-subunit (rpoB)—but reported little variability among the limited number of sequences (n = 8) examined. However, several other applications have demonstrated that MLST is a useful typing method for the analysis of M. bovis isolates. As examples, host-specific genotypes have been found in cattle versus bison using the housekeeping genes alcohol dehydrogenase-1 (adh), glutamate tRNA ligase (gltX), glycerol-3-phosphate dehydrogenase (gpsA), gyrB, phosphate acetyltransferase-2 (pta), thymidine kinase (tdk), and transketolase (tkt),9 and the presence of genetically distant and divergent clusters was found to be predominantly associated with geographical origins using DNA polymerase III B chain (dnaN), methionyl-tRNA synthetase (metS), DNA recombination/repair protein RecA (recA), elongation factor Tu (tufA), ATP synthase F1 subunit alpha (atpA), RNA polymerase sigma factor (rpoD), and tkt.10 As well, M. bovis isolates from France clustered into 2 subtypes, with recent M. bovis strains being more homogeneous than older ones in the collection of the last 35 y using dnaN, DNA polymerase III subunit alpha (polC), adenosine kinase (adk), rpoB, and gyrB loci.1
We developed a MLST scheme to characterize M. bovis isolates from clinical cases submitted to the Animal Health Laboratory (AHL) Ontario, Canada from 1978 to 2010. Based on availability, ~7 isolates per year were chosen from archived cultures, resulting in a total of 211 unique isolates. A single isolate was chosen per submission to the AHL (a submission is defined as one or more specimens submitted from the same farm or herd, at the same time). Each isolate used in our study represented a single animal and, in addition, isolates were selected to ensure that there was a variety between breed of cattle (simplified as beef, n = 81; dairy, n = 115; or other or unknown, n = 15); affected body system (arthritis or joint, n = 38; mastitis or udder, n = 33; reproductive or abortion, n = 12; respiratory, n = 105; or other or unknown, n = 23); and geographic distribution within the province as determined by the first letter of the farm postal code (K, n = 35; L, n = 24; N, n = 141; P, n = 5; or other or unknown, n = 6). An additional 5 unusual M. bovis isolates, originating from goats (adult lung tissue, n = 2; neonatal lung, n = 1; lung, age unknown, n = 1; fetal stomach content, n = 1), and a single equine isolate (synovium), were also analyzed, bringing the total number of isolates to 217.
Isolates were stored on agar blocks at −70°C then thawed and propagated on Hayflick agar plates containing 15% horse serum11 for 48 h in a 35–39°C incubator with 5.0–6.0% CO2 and 80–100% relative humidity. A single colony was chosen from the original culture and re-plated. Each isolate was passaged 3 times in this manner in order to produce a pure clone, which was then cultured aerobically in Hayflick broth11 for 48 h at 37°C prior to DNA extraction.
The following candidate genes from the then-unpublished M. bovis PG45 genome (Rosenbusch RF, pers. comm., 2009) were chosen for evaluation for MLST: adk, dnaN, translation elongation factor P (efp, elongation factor P), guanylate kinase (gmk), gyrB, metS, rpoB, and triose-phosphate isomerase (tpiA). These candidate genes were chosen because they were homologs of the housekeeping genes evaluated for a M. hyopneumoniae MLST scheme.7 Additional housekeeping genes evaluated included heat shock protein 70 (hsp70, AJ276681), excinuclease ABC subunit C (uvrC, AF003959), along with polC, using previously described primers.5 Primers were designed (Primer3 v.0.4.0, Whitehead Institute for Biomedical Research, Cambridge, MA) to amplify a 300–600-bp segment of the target loci (Table 1). The PCR amplification primers were used to sequence (ABI Prism 3730 or 3100 DNA Sequencer, Thermo Fisher Scientific, Waltham, MA) all products, and the entirety of the amplicon (excluding the primer regions) was used to assign alleles to each locus. These 11 candidate genes were evaluated for use in the MLST scheme by sequencing a portion of each gene: adk (regions 48–650, of 650 bp), efp (regions 45–553, of 563 bp), gmk (regions 24–556, of 587 bp), rpoB (regions 2–502, 482–1057, 1038–1372, and 1377–1895, of 3,635 bp), uvrC (regions 0–225 and 1120–1588, of 1715 bp), gyrB (positions 2–502, 482–1057, 1038–1372, and 1377–1895, of 1,967 bp), polC (positions 3705–3967 of 4,376 bp), and tpiA (positions 97–761 of 782 bp). A selection of strains isolated in multiple years spanning 1978–2010, including laboratory type strain M. bovis 227 (isolated in 1971 from an early case of mycoplasma arthritis in calves)12 and/or 3 different amplified fragment length polymorphism types isolated from beef cattle bronchoalveolar lavage samples were used to screen the candidate genes (see Supplementary Table 1).
Table 1.
Primer sequences and information for loci used in the multi-locus sequence typing scheme for Mycoplasma bovis.
| Locus | Locus tag/GenBank accession | Genome position | Primer name | Primer sequence | Amplified region | Analyzed sequence size (bp) |
|---|---|---|---|---|---|---|
| dnaN | MBOVPG45_RS00010 | 1,543–2,652 | dnaN-F | 5’-GCAGCAAGTAATGGTGCAAA | 439–965 | 486 |
| dnaN-R | 5’-TCAAAAACAAAAGGCGAACC | |||||
| metS | MMB_RS01065 | 246,830–243,680 | metS-F | 5’-AAATGGCGATGAAATTGTGC | 242–758 | 517 |
| metS-R | 5’-CGCTGGTTAATGTCCAAGGT | |||||
| hsp70 | MBOVPG45_RS00785 | 179,470–181,248 | Hsp70-Mb227-F47 | 5’-GGAACATTTGACGTTTCAATTC | 520–966 | 405 |
| Hsp70-Mb227-R493 | 5’-AACAGCGGGCATTCTAGTTG |
Several of the housekeeping genes evaluated for use in a MLST scheme were not suitable given lack of genetic variability, despite the fact that the majority of each gene sequence was evaluated (with the exception of polC, for which only 262 of 4,376 bp were examined). Based on the regions we sequenced, the adk (502 of 650 bp of the gene were examined), efp (508 of 563 bp), gmk (532 of 587 bp), rpoB (1,950 of 3,635 bp), and uvrC (693 of 1,715) amplicons had no sequence variation; gyrB, polC, and tpiA generated only 2 alleles. Similar to these findings, no single nucleotide polymorphisms (SNPs) were detected in the adk and rpoB loci in M. bovis strains from France.1 We examined a region of the 650-bp adk gene similar to the French study (we used region 168–650 for allele assignment vs. an unspecified 476-bp region of 49–623), but completely different regions of rpoB (1649–2053, 2356–2771, and 2887–3320 vs. 1075–1880). The latter result suggests that the region of the rpoB gene examined in the French isolates and in the Ontario M. bovis collection is highly invariant. A contrasting result was found in a prior M. bovis MLST scheme that found 4 alleles in the rpoB gene in a limited number (n = 8) of European and African isolates.4 This may suggest that either the region of the gene that was examined (which includes ~200 bp of sequence that was not used in our study) may contain more sequence variation, or that these isolates were more genetically diverse. This phenomenon of invariant housekeeping genes found in our study was also seen in the recA, tpiA, and uvrC genes of the closely related mollicute M. agalactiae.8 The lack of genetic variability in the 8 housekeeping genes in M. bovis that were evaluated but not chosen for use in our study may indicate that these highly conserved genes are under strong selective pressure that prevents sequence polymorphisms. Alternatively, there may be little variability in these genes because the isolates are from a limited geographic area (Ontario, Canada). There have been previous reports describing other loci used in M. bovis MLST schemes (e.g., there is a scheme based on dnaN, metS, recA, atpA, rpoD, and tkt,10 another based on adh-1, gltX, gpsA, gyrB, pta-2, tdk, and tkt,9 and a third based on adk, dnaN, polC, and gyrB).1 The region of dnaN used to determine allele assignment in our study overlaps the dnaN amplicon used to examine isolates from France1 (position 459–945 vs. an unspecified 376-bp region of 611–1,101). The dnaN, hsp70, and metS loci generated more than 3 alleles and were chosen for our MLST scheme.
The DNA extract of each isolate was PCR amplified, and then sequenced (ABI Prism 3730 or 3100 DNA Sequencer, Thermo Fisher Scientific) in both directions using the PCR primers. The forward and reverse sequences of the dnaN, hsp70, and metS loci from 217 isolates were assembled into contigs and the primer sequences removed (SeqMan Pro v.8.1.5(3), DNASTAR, Madison. WI). The resulting sequences were analyzed (BioNumerics v.6.1 and MLST plug-in, Applied Maths, Austin, TX), which automatically assigned an allele number to each unique allele for each of dnaN, hsp70, and metS. Finally, a ST was assigned to each isolate based on the alleles from each of the 3 loci included in the analysis. Statistical analysis was then performed (SPSS Statistics v.23, IBM, Armonk, NY).
A total of 217 isolates (211 bovine, 5 caprine, and 1 equine) were evaluated. A total of 1,409 bp was examined (487 bp, 517 bp, and 405 bp, respectively, for dnaN, hsp70, and metS) to determine the ST of the M. bovis strains. AHL type strain M. bovis 227 isolated in 197112 was designated as the reference strain, and assigned ST-1 which consisted of allele-1 for each of the dnaN, hsp70, and metS loci. A total of 14 dnaN alleles (GenBank accessions KY224348– KY224361), 5 hsp70 alleles (KY224362–66), and 7 metS alleles (KY224367–73) were identified. A total of 28 ST resulted, of which the bovine isolates were chosen for detailed examination. One of the most common dnaN alleles, dnaN-01, was found in 37% of all bovine isolates (79 of 211) but if this number is broken down by decade, an interesting pattern emerges: dnaN-01 makes up most (91%, 63 of 69) of isolates from the 1970s and 1980s, but only 23% (16 of 70) of isolates from the 1990s, and none of isolates from the 2000s. The most common hsp70 and metS alleles, hsp70-1 and metS-1, have a similar pattern (Fig. 1A). The frequency of allele-1 from all 3 genes sharply declined around 1990 and was replaced by another common allele: dnaN-8 (50% of all isolates), hsp70-3 (39%), and metS-3 (24%; Fig. 1B), and replaced again by dnaN-12, hsp70-5, and metS-7 after year 2000 (Fig. 1C).
Figure 1.

Frequency of the most common alleles for the dnaN, hsp70, and metS loci, by year of isolation.
Although most of the STs contained only a small number of SNPs when compared to the reference strain, a handful contained multiple SNPs. For the dnaN locus, allele-1 was the second most common and is only found in ST-1 (78 of 79) and ST-7 (1 of 79). Allele-1 is very similar to most other dnaN alleles, differing by only one (allele-2 through allele-9) or 2 bases (allele-10, 11, and 15). However, there are multiple (18–19) nucleotide substitutions between allele-1 and alleles-12, -13, and -14 (data not shown). These latter alleles are only found in more recent isolates (2001–2009), and show the most sequence variability, but comprise only 6% (13 of 211) of bovine isolates. All of the sequence changes in the 3 loci chosen for our MLST analysis were base pair substitutions; no insertions and no deletions were found. This is as expected, as indels would cause frameshift mutations that would disrupt the function of the essential protein encoded by the housekeeping gene.
Many of the 28 ST found in our study were represented by only a single isolate (n = 14 bovine strains, plus caprine isolate ST-3) or by 2 isolates (6 STs; Table 2). The 3 most common STs (ST-1, ST-10, and ST-21) made up the bulk of the bovine isolates (73%, 154 of 211) as well as 3 of 5 caprine isolates and the single equine isolate. Fifteen of the STs (7% of isolates) were represented by a single isolate; 6 were represented by only 2 isolates each (Table 2).
Table 2.
Sequence type (ST) allelic profiles of Mycoplasma bovis isolates.
| Allelic profile |
n | Year(s) isolated | |||
|---|---|---|---|---|---|
| dnaN | hsp70 | metS | |||
| ST-1* | 1 | 1 | 1 | 82 | 1978–1995, 1997, 1998 |
| ST-2 | 2 | 1 | 1 | 1 | 1982 |
| ST-3† | 3 | 1 | 1 | 1 | 1982 |
| ST-4 | 5 | 1 | 1 | 1 | 1986 |
| ST-5 | 4 | 1 | 1 | 2 | 1983, 1986 |
| ST-6 | 6 | 1 | 1 | 1 | 1986 |
| ST-7 | 1 | 1 | 2 | 1 | 1988 |
| ST-8 | 7 | 1 | 1 | 1 | 1988 |
| ST-9 | 9 | 1 | 1 | 1 | 1991 |
| ST-10 | 8 | 3 | 3 | 47 | 1990–1998, 2000–2001, 2003–2005 |
| ST-11 | 8 | 3 | 4 | 1 | 1995 |
| ST-12 | 8 | 1 | 1 | 1 | 1995 |
| ST-13 | 15 | 5 | 7 | 1 | 2009 |
| ST-14 | 10 | 3 | 5 | 2 | 1996, 1999 |
| ST-15 | 11 | 3 | 3 | 1 | 1999 |
| ST-16 | 8 | 3 | 5 | 10 | 1997, 1999–2002 |
| ST-17 | 8 | 3 | 1 | 1 | 2000 |
| ST-18 | 13 | 3 | 8 | 1 | 2006 |
| ST-19 | 10 | 3 | 7 | 2 | 2000 |
| ST-20 | 8 | 3 | 8 | 2 | 2008 |
| ST-21 | 8 | 5 | 7 | 29 | 2000–2009 |
| ST-22‡ | 8 | 3 | 7 | 7 | 2001, 2004, 2006–2007, 2009–2010 |
| ST-23 | 12 | 3 | 8 | 10 | 2001–2002, 2004–2009 |
| ST-24 | 8 | 5 | 8 | 5 | 2002–2003, 2007–2009 |
| ST-25 | 8 | 6 | 7 | 2 | 2003 |
| ST-26 | 12 | 5 | 8 | 1 | 2008 |
| ST-27 | 14 | 3 | 8 | 1 | 2009 |
| ST-28 | 8 | 7 | 3 | 2 | 2007 |
ST-1 includes 3 caprine and 1 equine sample.
ST-3 is a caprine sample.
ST-22 includes 1 caprine sample.
The most common ST, ST-1, represented 37% (78 of 211) of all bovine isolates, and was found primarily in the late 1970s and 1980s. Beginning in 1990, ST-1 became less common and was not found in isolates from 1999 or later (Tables 2, 3; Fig. 2). ST-10 was the second most common type representing 22% (47 of 211) of bovine isolates. This type appeared in 1990 and was common from 1990–1998 but declined in frequency beginning in 2000 and was not seen after 2005 (Table 2; Fig. 2). ST-21 was the third most common type, representing 14% (29 of 211) of isolates. This type was not seen until 2000, when it appeared at a frequency of 14% (1 of 7) up to 71% (5 of 7) yearly up until the present day (Table 2; Fig. 2).
Table 3.
Frequency of the 7 most common sequence types (STs) by year of isolation expressed as a percentage of all isolates examined from the given time frame.
| Years isolated | ST-1 |
ST-10 |
ST-16 |
ST-21 |
ST-22 |
ST-23 |
ST-24 |
Total* (n) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % isolates | n | % isolates | n | % isolates | n | % isolates | n | % isolates | n | % isolates | n | % isolates | ||
| 1971–1982† | 18 | 94.7 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 19 |
| 1983–1987 | 31 | 88.6 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 35 |
| 1988–1992 | 24 | 66.7 | 9 | 25.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 36 |
| 1993–1997 | 4 | 11.1 | 28 | 77.8 | 1 | 2.8 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 36 |
| 1998–2002 | 1 | 2.9 | 7 | 20.6 | 9 | 26.5 | 7 | 20.6 | 2 | 5.9 | 2 | 5.9 | 1 | 2.9 | 34 |
| 2003–2007 | 0 | 0.0 | 3 | 8.3 | 0 | 0.0 | 17 | 47.2 | 3 | 8.3 | 6 | 16.7 | 2 | 5.6 | 36 |
| 2008–2010 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 5 | 33.3 | 1 | 6.7 | 2 | 13.3 | 2 | 13.3 | 15 |
Only bovine isolates are included.
Includes type strain M. bovis 227 isolated in 1971. All other strains are from 1978 to 1982.
Figure 2.
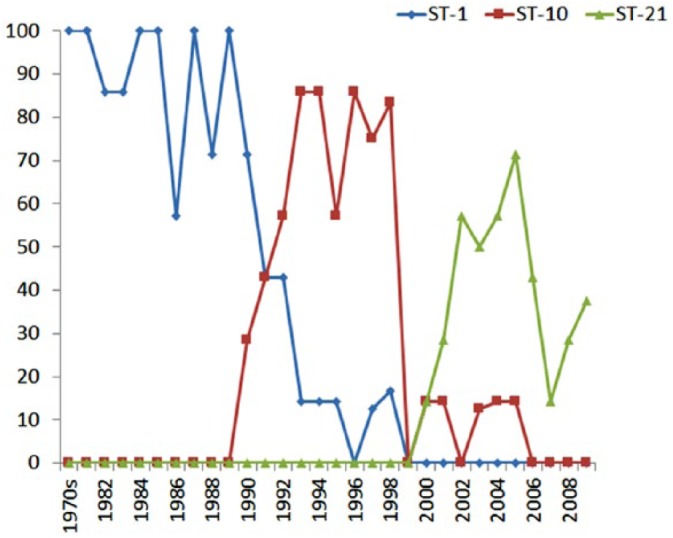
Frequency of the 3 most common sequence types, ST-1, -10, and -21, by year of isolation.
There was a greater number of STs in the more recent isolates than in older isolates. If the data are examined chronologically, there were 8 STs in the 1978–1989 isolates, 8 in the 1990–1999 isolates, and 15 in the 2000–2009 isolates. Of these 15, 5 were represented by single isolates and 4 were represented by only 2 isolates.
The unique ST-3 was found in a single caprine isolate, and was not found in bovine isolates. Three of the M. bovis strains isolated from the 5 caprine cases, and the single equine isolate, were ST-1, the most common type in cattle. The remaining caprine isolate was ST-22, found in 3% of the bovine isolates.
The 5 most common types, STs, -1, -10, -16, -21, and -23, account for 81% (159 of 196) of isolates and were examined in more detail. When these data were analyzed with the Fisher exact chi-square test (SPSS Statistics), there was no significant difference between the number of beef versus dairy isolates for any of the STs (data not shown), nor was the distribution among geographic areas of the province significant (data not shown). Although our study found 15 dnaN alleles out of 217 isolates examined, only 4 alleles were found out of 60 French isolates.1 If we had used the same forward primer as the French study (position 611 instead of 439), only a single allele would have been missed. A study10 that encompassed a very geographically diverse set of 137 isolates found 12 metS alleles, whereas our study found only 7. Again, this may be the result of the limited geographic range of our isolates, or to the different regions chosen for allele assignment (position 242–758 in our study, versus 439–1009). The differences in ST distribution among affected body systems (generalized as arthritis or joint, mastitis or udder, reproductive or abortion, or respiratory) seemed significant; however, we cannot rule out that the data were biased because of variation in the time distribution of the isolates.
Our MLST data indicate that M. bovis has evolved chronologically. Over the course of 30 plus years, some STs have disappeared whereas new types have appeared. These chronologic changes may be related to the pathogenicity of M. bovis, but more investigation is required to determine this. More recent isolates had a greater variety of STs, which may indicate that new strains are emerging more rapidly now than in the past. This finding is somewhat different from the M. bovis isolated in France1 where recent strains were more homogeneous than older ones in the collection of the last 35 y. Whether the difference is the result of different selection pressure in Ontario and Europe, or because of the different MLST schemes, needs to be further investigated.
In our study, there was no obvious geographic grouping of M. bovis, and there was no significant difference found among isolates from different postal codes (data not shown). However, our M. bovis study was limited to strains isolated from the province of Ontario, Canada and so may not have included a wide enough geographic area to see any separation between isolates.
A 2015 study found that the sequence types were host specific in cattle versus bison.9 In our study, we only found ST-3 in caprine isolates. Analysis of a larger collection of ST-3 is required to show if this MLS type is unique to caprine isolates.
Supplementary Material
Acknowledgments
Dr. Ricardo F. Rosenbusch (Veterinary Microbiology and Preventive Medicine, College of Veterinary Medicine, Iowa State University) kindly provided sequences of 11 essential genes of the then-unpublished M. bovis PG45 genome for primer selection.
Footnotes
Declaration of conflicting interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: This research was financially supported by the Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA)–Animal Health Laboratory, Animal Health Strategic Investment (AHSI) program.
References
- 1. Becker CA, et al. Loss of diversity within Mycoplasma bovis isolates collected in France from bovines with respiratory diseases over the last 35 years. Infect Genet Evol 2015;33:118–126. [DOI] [PubMed] [Google Scholar]
- 2. Enright MC, Spratt BG. Multilocus sequence typing. Trends Microbiol 1999;7:482–487. [DOI] [PubMed] [Google Scholar]
- 3. Maiden MC. Multilocus sequence typing of bacteria. Ann Rev Microbiol 2006;60:561–588. [DOI] [PubMed] [Google Scholar]
- 4. Manso-Silván L, et al. Phylogeny and molecular typing of Mycoplasma agalactiae and Mycoplasma bovis by multilocus sequencing. Vet Microbiol 2012;161:104–112. [DOI] [PubMed] [Google Scholar]
- 5. Marenda MS, et al. Suppression subtractive hybridization as a basis to assess Mycoplasma agalactiae and Mycoplasma bovis genomic diversity and species-specific sequences. Microbiol 2005;151:475–489. [DOI] [PubMed] [Google Scholar]
- 6. Maunsell FP, et al. Mycoplasma bovis infections in cattle. J Vet Intern Med 2011;25:772–783. [DOI] [PubMed] [Google Scholar]
- 7. Mayor D, et al. Multilocus sequence typing (MLST) of Mycoplasma hyopneumoniae: a diverse pathogen with limited clonality. Vet Microbiol 2008;127:63–72. [DOI] [PubMed] [Google Scholar]
- 8. McAuliffe L, et al. Multilocus sequence typing of Mycoplasma agalactiae. J Med Microbiol 2011;60:803–811. [DOI] [PubMed] [Google Scholar]
- 9. Register KB, et al. Multilocus sequence typing of Mycoplasma bovis reveals host-specific genotypes in cattle versus bison. Vet Microbiol 2015;175:92–98. [DOI] [PubMed] [Google Scholar]
- 10. Rosales RS, et al. Global multilocus sequence typing analysis of Mycoplasma bovis isolates reveals two main population clusters. J Clin Microbiol 2015;53:789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruhnke H, Rosendal S. Useful protocols for diagnosis of animal mycoplasmas. In: Whitford HW, et al., eds. Mycoplasmosis in Animals: Laboratory Diagnosis. Ames, IA: Iowa State University Press, 1994:141–144. [Google Scholar]
- 12. Singh UM, et al. Mycoplasma arthritis in calves. Can Vet J 1971;12:183–185. [PMC free article] [PubMed] [Google Scholar]
- 13. Turner KM, Feil EJ. The secret life of the multilocus sequence type. Int J Antimicrob Agents 2007;29:129–135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.