

Abstract
The expression of MHC class II molecules (MHCII) on tumor cells correlates with survival and responsiveness to immunotherapy. However, the mechanisms underlying these observations are poorly defined. Using a murine breast tumor line, we showed that MHCII-expressing tumors grew more slowly than controls and recruited more functional CD4+ and CD8+ T cells. In addition, MHCII-expressing tumors contained more TCR clonotypes expanded to a larger degree than control tumors. Functional CD8+ T cells in tumors depended on CD4+ T cells. However, both CD4+ and CD8+ T cells eventually became exhausted, even in MHCII-expressing tumors. Treatment with anti-CTLA4, but not anti-PD-1 or anti-TIM-3, promoted complete eradication of MHCII-expressing tumors. These results suggest tumor cell expression of MHCII facilitates the local activation of CD4+ T cells, indirectly helps the activation and expansion of CD8+ T cells, and, in combination with the appropriate checkpoint inhibitor, promotes tumor regression.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2262-5) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, MHC class II, T-cell exhaustion, TCR repertoire
Introduction
The capacity of the immune system to recognize and respond to tumors is now generally accepted [1, 2], although these responses are often counteracted by a variety of local immune-inhibitory mechanisms, including signaling through T-cell-inhibitory receptors [3]. In fact, the therapeutic inhibition of these receptors with antibodies, known as checkpoint inhibitors, can elicit remarkable clinical responses [4, 5]. However, for many tumor types, only a minority of patients experiences a therapeutic benefit, a phenomenon ascribed to variations in the tumor microenvironment as well as the magnitude of the local immune response prior to treatment [6, 7]. Thus, it is important to understand how variations in gene expression by tumor cells and the tumor microenvironment contribute to clinical outcomes.
The expression of MHC class II (MHCII) molecules by tumor cells is one variable that strongly predicts clinical outcomes. Although the expression of MHCII is normally restricted to professional antigen-presenting cells, such as dendritic cells, macrophages, and B cells, our group and others have shown that the expression of MHCII on tumor cells is associated with improved patient outcomes [8–15], and more vigorous immune responses. In fact, the expression of the class II transcriptional activator (CIITA), the master regulator of MHCII expression [16], is independently associated with PFS in breast cancer [14]. These data suggest that the presentation of antigen to CD4+ T cells by tumor cells is a critical component of functional anti-tumor immune responses.
CD4+ T cells are typically not cytolytic, but instead make cytokines that promote inflammation and cellular activation. Moreover, CD4+ T cells provide “help” to CD8+ T cells by promoting dendritic cell activation and cross-presentation [17, 18]. Thus, the expression of MHCII on tumor cells likely promotes the local activation of CD4+ T cells, which indirectly help CD8+ T cells to kill tumor cells [19, 20]. T cells responding to persistent antigens, such as those expressed by tumor cells, often become exhausted [21], compromised in their ability to express cytokines or kill target cells [22], permitting tumor outgrowth despite the initial immune activation. Thus, MHCII expression on tumor cells may promote successful anti-tumor immunity by enhancing CD4+ functionality, creating a more inflamed tumor microenvironment, and preventing the exhaustion of CD8+ T cells.
Here, we tested how ectopic expression of MHCII on murine TS/A breast tumor cells altered tumor-specific CD4+ and CD8+ T-cell responses. We found that local MHCII expression increased the magnitude and duration of both CD4+ and CD8+ T-cell responses in tumors. We also observed alterations in the T-cell repertoire, with more clonotypes expanded to a larger extent in MHCII-expressing tumors. Interestingly, although effector functions of T cells were significantly augmented and prolonged in MHCII-expressing tumors, both CD4+ and CD8+ T cells eventually became exhausted. The administration of anti-CTLA4, but not anti-PD-1 or anti-TIM3, further augmented immune responses and promoted the eradication of MHCII-expressing tumors. Thus, MHCII expression on tumor cells expands the T-cell repertoire and delays T-cell exhaustion, thereby allowing checkpoint inhibitors like anti-CTLA4 to facilitate tumor regression.
Materials and methods
Cell culture and transfection
TS/A cells were cultured in DMEM (Corning) supplemented with 10% FBS (HyClone Laboratories), dissociated with 0.05% trypsin, 0.53 mM EDTA (Corning), and recultured in DMEM-FBS. Cells were transfected with pcDNA3.1 with or without the hCIITA cDNA (complete coding sequence, accession number AY699071) using Xfect Transfection Reagent (Clontech) and selected in 500 µg/mL geneticin (Life Technologies). MHCII-expressing clones were selected by cell sorting. Western blot for hCIITA, MHCII, and CD74 was performed with antibodies against hCIITA (E-12, Santa Cruz Biotechnology), MHCII (M5/114, Millipore), and CD74 (R&D Systems).
RNA isolation and nanostring analysis
Total RNA was purified from excised tumors using the RNeasy Mini kit (Qiagen). RNA from whole blood was isolated using the Paxgene blood RNA kit (Qiagen). RNA quality was assessed using Bioanalyzer 2100 (Agilent) and final concentration determined by Qubit (Life Technologies). For nanostring analysis, 100 ng of purified RNA was added to 3 µL of Reporter CodeSet and 2 µL Capture ProbeSet using an nCounter master kit as recommended (NanoString Technologies). Samples were processed the following day using the nCounter DxPrep Station and nCounter Dx Digital Analyzer (NanoString Technologies). Data were analyzed using the nSolver 2.6 software. Heat maps were created using Cluster 3.0 and Java TreeView-1.1.6r4.
Mice, tumor administration, and antibody treatments
Mice were injected with 1 × 105 TS/A or TS/A-hCIITA tumor cells into the mammary fat pad. The length and width of tumors were measured by calipers and tumor volume was calculated using the formula, 0.4 × length × width2. For depletion experiments, 200 µg of CD4-depleting antibody (GK1.5) or isotype control (LTF-2) were administered prior to tumor cell injection (day − 1) and again on day 3. For antibody blockade, 200 µg of anti-PD-1 (RMP1-14), anti-CTLA4 (9H10), or anti-TIM3 (RMT3-23) were administered on days 7, 10, 13, 16, and 19. All antibodies were from BioXcell.
Tumor disassociation and T-cell restimulation
Tumors were excised using a scalpel and weighed using an AL54 analytical balance (Mettler Toledo). Tumors were diced and incubated in RPMI1640 media (Lonza) supplemented with 5% FBS, 1.25 mg collagenase (c7657, Sigma) and 150 U DNase (d5025, Sigma), followed by shaking at 200 rpm for 35 min at 37 °C. Cell suspensions were filtered through 70 µm nylon cell strainers (Corning). T cells were restimulated in RPMI1640, 5% FBS, 5 ng/mL phorbol 12-myristate 13-acetate (Sigma), 65 ng/mL ionomycin (Thermo Fisher Scientific), and 10 µg/mL brefeldin A (Sigma) at 37 °C for 5 h.
Flow cytometry and antibodies
Cell cycle analysis was performed by fixing single-cell suspensions of TS/A and TS/A-hCIITA cells overnight with 70% ethanol at 4 °C overnight and incubating them with 50 µg/mL propidium iodide (Thermo Fisher Scientific) in PBS, 0.1% Triton-X100 (Sigma), and 50 µg/mL RNase at 37 °C for 20 min in the dark. DNA content was determined using a BD FACSCalibur (BD Biosystems) flow cytometer and analyzed by ModFit LT version 3.3 (Verity Software House).
Immunophenotyping was performed by blocking cell suspensions in PBS with 2% donor calf serum and 10 µg/mL FcBlock (2.4G2-BioXCell) for 10 min on ice, followed by staining with fluorochrome-conjugated antibodies for 30 min. Stained cells were fixed in 10% neutral buffered formalin (Sigma) and permeabilized with 0.1% IGEPAL CO-630 (Sigma) in staining solution for 45 min. All staining was performed in the dark at 4 °C. Antibodies against I-A/I-E (M5/114.15.2) and CD4 (GK1.5) were obtained from BioLegend. Antibodies against IFNγ (XMG1.2) and CD8 (53-6.7) were obtained from BD Biosciences. Antibodies against CD3 (17A2), KLRG1 (2F1), granzyme B (NGZB), CD45.2 (104), and PD-1 (J43), were obtained from eBioscience. The MuLV class I tetramer-H2Ld (SPSYVYHQF) was obtained from the NIH Tetramer Core Facility at Emory University. The LIVE/DEAD red fixable dye was obtained from Life Technologies.
Samples were run on a BD FACSCanto II system (BD Biosciences) and data were analyzed using FlowJo version 9.9.
T-cell repertoire sequencing and analysis
TCR sequencing was performed using the Mouse TCR beta, Illumina, V-C genes kit from iRepertoire, Huntsville, AL. In brief, 500 ng of RNA was reverse transcribed using a one-step reverse transcription and amplification kit (Qiagen). The PCR product was purified using Ampure XP magnetic beads (Agencourt), and amplified again (TopTaq PCR Kit, Qiagen), to add adaptor sequences. Libraries were purified with Ampure XP magnetic beads and sequenced using Illumina MiSeq 150 nt paired-end read-length. The TCR CDR3 sequences were extracted from the raw sequencing data by iRepertoire. Briefly, raw paired-end fastq files were first demultiplexed based on barcode and merged reads were mapped using a Smith-Waterman algorithm to germline V, D, J, and C reference sequences from the IMGT website (http://www.imgt.org). To define the CDR3 region, the position of CDR3 boundaries of reference sequences from the IMGT database was migrated onto reads through mapping results, and the resulting CDR3 regions were extracted and translated into amino acids. Reading frames not containing a stop codon were filtered and error-corrected using iRepertoire proprietary SMART algorithm.
Statistical analysis
Hypothesis of difference testing between pooled replicate group means at each time point was done using multiple, unpaired independent t tests with the Holm–Sidak method and assuming α = 0.05. Statistical analyses were calculated using GraphPad Prism version 7.0a and all reported p values are two-tailed. The TCR high-throughput sequencing data were analyzed in R environment using tcR package and common R routines. Diversity was measured using the Inverse Simpson Index. CDR3 sequence similarity between repertoires was assessed using overlap index and weighted Horn’s index available in the tcR package.
Results
Transfection with hCIITA promotes MHCII expression on TS/A breast cancer cells
To test the role of MHCII on anti-tumor immunity, we transfected the murine breast cancer cell line, TS/A, with the human class II transcriptional activator (hCIITA) or with empty vector. By Western blot, we found that TS/A cells transfected with hCIITA expressed hCIITA as well as murine CD74 (invariant chain) and MHCII (Supplemental Fig. 1a), whereas cells transfected with empty vector did not. We also found that hCIITA-transfected cells expressed high levels of murine MHCII on the cell surface (Supplemental Fig. 1b). Importantly, we found that the cell cycling times of hCIITA-expressing and control TS/A cells were nearly equivalent (Supplemental Fig. 1c).
Surface expression of MHCII impairs tumor growth in vivo
We next injected TS/A and TS/A-hCIITA cells into the mammary fat pads of BALB/c mice and used flow cytometry to assess the expression of MHCII on CD45neg cells from disassociated tumors 14 days later. Similar to our findings in vitro, we observed that the majority of CD45neg cells from TS/A-hCIITA tumors expressed high levels of MHCII (Fig. 1a), whereas CD45neg cells from control tumors did not. However, in contrast to their similar growth kinetics in vitro, we found that TS/A-hCIITA tumors grew more slowly than control TS/A tumors, as measured by tumor volume (Fig. 1b) and excised tumor mass (Fig. 1c). To test whether this difference in tumor growth was dependent on adaptive immunity, we injected tumor cells into BALB/c.scid mice and found that the growth of TS/A-CIITA tumors was indistinguishable from the growth of control TS/A tumors over the same period (Fig. 1d). These results suggested that the observed difference in growth of MHCII-positive and MHCII-negative tumors in wild-type mice is mediated by adaptive immunity.
Fig. 1.

MHCII expression on tumor cells impairs tumor growth via adaptive immunity. a MHCII surface expression on live, CD45neg cells from disassociated tumors was measured by flow cytometry on day 14. b Tumor volume was monitored over time in at least 20 mice BALB/c per group per timepoint. c Tumor mass was determined in ten samples per group per timepoint. d Tumor volume was monitored over time in ten BALB/c.scid mice per group. e RNA was extracted from TS/A and TS/A-CIITA tumors (harvest day listed across bottom) and analyzed by Nanostring assay. Heat map shows fold increases in red and fold decreases in green. f Schematic of sequential tumor injections into contralateral mammary pads of the same mouse. g Growth of primary tumors. h Growth of secondary tumors. Error bars represent standard error of the mean. Statistical differences are expressed as *p < 0.05, **p < 0.005, #p < 0.0005
To test changes in gene expression, we excised whole tumors, prepared mRNA, and analyzed the expression of 48 genes using a NanoString assay. We found that counts of mRNAs encoding MHCII α and β chains were more numerous in TS/A-hCIITA tumors relative to control tumors (Fig. 1e), whereas counts of mRNAs encoding MHC class I molecules were largely similar. We also observed that IFNγ, granzyme B, and perforin were upregulated in TS/A-hCIITA tumors relative to control tumors (Fig. 1e). Interestingly, we found that the master transcription factor for Tregs, FoxP3, was expressed similarly in TS/A-hCIITA and control tumors. Together, these data demonstrated that MHCII expression on TS/A tumors promoted adaptive immune responses, altered the tumor microenvironment, and impaired tumor growth.
We next tested whether the local expression of MHCII promoted a systemic immune response by first injecting mice with TS/A or TS/A-hCIITA tumors in the left mammary pad, and 7 days later injecting a second tumor into the right mammary pad (Fig. 1f). We found that primary tumors grew at rates similar to that seen above (Fig. 1g). However, secondary tumors grew more slowly in all the groups (Fig. 1h), with the expression of MHCII on either the primary or secondary tumors conferring a reduction of tumor growth and, when both primary and secondary tumors express MHCII, 5/5 mice completely rejected the secondary tumor (Fig. 1h). Thus, tumor expression of MHCII benefits anti-tumor immunity both locally and systemically.
MHCII-expressing tumors have increased CD+ T-cell activation
To test whether MHCII expression bolstered the local CD4+ T-cell response, we harvested TS/A-hCIITA and control tumors at the indicated times, restimulated the TILs for 5 h, and measured the expression of IFNγ, IL-17, and IL-4. We found very little IL-17 or IL-4 made by T cells from either tumor type (Supplemental Fig. 2). In contrast, CD4+ T cells in control tumors made some IFNγ, whereas CD4+ T cells from MHCII-expressing tumors produced strikingly greater amounts of IFNγ (Fig. 2a), even after normalizing the numbers of T cells to tumor mass (Fig. 2b). Interestingly, CD4+ T cells in TS/A-hCIITA tumors also produced substantially more granzyme B (Supplemental Fig. 3), raising the possibility that CD4+ T cells may be directly cytolytic to tumor cells. Nevertheless, CD4+ T cells in both groups experienced a progressive loss in the ability to produce IFNγ and granzyme B (Fig. 2a and Supplemental Fig. 3), suggesting that they eventually became exhausted.
Fig. 2.

MHCII-expressing tumors promote CD4+ TIL activation. a IFNγ production by CD4+ T cells was assayed by flow cytometry. Plots are gated on live, CD3+CD4+ cells. b The number of IFNγ-producing CD4+ T cells was normalized to tumor mass. c The total number of CD4+ T cells was normalized to tumor mass. This experiment contained five mice per group per timepoint and was performed two times with similar results. d Tumor-infiltrating Tregs were determined by flow cytometry. Plots gated on live, CD3+CD4+ cells. e Total number of Tregs normalized to mass. f MFI of PD-1 on Tregs. g Intratumoral CD8-to-Treg ratio. Error bars represent standard error of the mean. Statistical differences are expressed as *p < 0.05, **p < 0.005, #p < 0.0005
Since CD4+ T-cell stimulation without appropriate costimulation can lead to the differentiation of Tregs, we next enumerated Tregs. We found that the frequency of Tregs was similar in both tumor types (Fig. 2d), but that there was a significant reduction of total Tregs per gram in MHCII-expressing tumors (Fig. 2e). Moreover, Tregs infiltrating MHCII-expressing tumors expressed lower levels of PD1, suggesting a decrement in suppressive capacity (Fig. 2f). Importantly, the CD8:Treg ratio was increased in MHCII-expressing tumors (Fig. 2g). These data indicate that tumor expression of MHCII promotes CD4+ T-cell effector function without promoting Treg accumulation.
Tumor expression of MHCII enhances the CD8+ T-cell response
Help from CD4+ T cells enhances CD8 T-cell responses [23, 24]. Therefore, we next enumerated CD8+ TILs in control and MHCII-expressing tumors. Given that TS/A tumor cells harbor an immunogenic murine leukemia virus (MuLV) [25], we initially enumerated MuLV-specific CD8+ T cells. We found similar frequencies of total and tumor-specific CD8+ T cells in both TS/A-CIITA and control tumors at the early times (Fig. 3a), but much higher frequencies of total and MuLV-specific CD8+ T cells in TS/A-hCIITA tumors on days 17 and 21. The higher frequencies of T cells were reflected in the numbers of total CD8+ T cells (Fig. 3b) and the numbers of tumor-specific CD8+ T cells (Fig. 3c).
Fig. 3.

MHCII-expressing tumors maintain functional, tumor-specific CD8+ T cells. a The frequency of MuLV env-specific CD8+ T cells was determined by tetramer binding and flow cytometry. Plots are gated on live, CD3+CD8+ cells. b The number of CD8+ T cells was normalized to tumor mass. c The number of MuLV env-specific CD8+ T cells was normalized to tumor mass. d The ability of CD8+ T cells to make IFNγ and granzyme B (GZB) following restimulation ex vivo was determined by intracellular staining and flow cytometry. Plots are gated on live, CD3+CD8+ cells. e The number of CD8+IFNγ+GZB+ cells was normalized to tumor mass. (f) The expression of PD-1 and TIM3 on CD8+ TILs was analyzed by flow cytometry. Histograms are gated on live, CD3+CD8+ cells. g The volume of TS/A and TS/A-hCIITA tumors was measured over an extended time frame and the arrow indicates a change in tumor growth rate corresponding to T-cell exhaustion. These experiments contained five mice per group per timepoint and were performed at least two times. Error bars represent standard error of the mean. Statistical differences are expressed as *p < 0.05, **p < 0.005, #p < 0.0005
We next assayed the ability of CD8+ T cells to produce IFNγ and granzyme B following restimulation ex vivo. We observed dramatically increased production of both IFNγ and granzyme B by CD8+ TILs from TS/A-hCIITA tumors relative to those in control tumors at all timepoints (Fig. 3d). However, as we observed with CD4+ T cells, the CD8+ T cells in both groups progressively lost their ability to produce IFNγ and granzyme B (Fig. 3d–e). Consistent with the idea that these cells are becoming exhausted, we observed that the expression of the inhibitory receptors, PD-1 and TIM3, increased over time on CD8+ T cells (Fig. 3f). To demonstrate the functional significance of CD8+ TIL exhaustion, we injected TS/A and TS/A-hCIITA cells into BALB/c mice and allowed tumors to grow for an extended period (Fig. 3g). We found that although TS/A-hCIITA tumors initially grew more slowly than control tumors, they grew at similar rates following day 21 (Fig. 3g), a time that coincides with the loss in T-cell effector functions.
Given the expression of PD-1 on CD8+ T cells, particularly those in TS/A-hCIITA tumors, we hypothesized that PD-1 blockade would preserve CD8+ T-cell function and further impair tumor growth. However, we found minimal difference in tumor growth following treatment with anti-PD-1 (Fig. 4a). To investigate why PD-1 blockade had no impact on tumor growth, we assayed the expression of the PD-1 ligands, PD-L1 and PD-L2, on TS/A and TS/A-hCIITA tumor cells. We found that PD-L1 and PD-L2 were similarly expressed by control and CIITA-expressing tumors (Fig. 4b). As a control for PD-L1 staining, we showed that PD-L1 was highly expressed by a portion of CD45+ cells in both groups (Fig. 4c). This result led us to test anti-CTLA4 and anti-TIM3—alone and in combination with anti-PD-1. We found that anti-CTLA4 reduced the growth of control tumors and eradicated MHCII-expressing tumors (Fig. 4d), either alone or in combination with anti-PD1. In contrast, anti-TIM3, either alone or in combination with anti-PD1, failed to alter tumor growth (Fig. 4e). These data demonstrate the expression of MHCII on tumor cells potentiates the effects of at least some checkpoint inhibitors.
Fig. 4.

Checkpoint blockade variably affects tumor control. a Tumor growth in anti-PD-1 treated mice. b, c Expression of PD-L1 and PD-L2 on cell suspensions from disassociated tumors was analyzed by flow cytometry. Plots are gated on live, CD45neg cells (b) or live, CD45+ cells (c). d Tumor growth in mice treated with anti-CTLA4 alone or in combination with anti-PD-1 (combo). e Tumor growth in mice treated with anti-TIM3 alone or in combination with anti-PD-1 (combo). These experiments each contained five mice per cohort per timepoint. Error bars represent standard error of the mean. Statistical difference is expressed as *p < 0.05, **p < 0.005, and #p < 0.0005
CD4+ T-cell depletion diminishes the benefit of MHCII expression
To directly test whether CD4+ T cells supported CD8+ T-cell responses, we depleted CD4+ cells prior to tumor cell implantation and evaluated tumor growth as well as the number and function of CD8+ T cells. As expected, we found that CD4 depletion restored the growth of MHCII-positive tumors to nearly that of MHCII-negative tumors in non-depleted mice (Fig. 5a). However, we also found that CD4 depletion modestly increased the growth of MHCII-negative tumors, suggesting that CD4+ T cells have some role in promoting anti-tumor immunity via interactions with other, non-tumor, MHCII-expressing cells. Importantly, CD4 depletion notably reduced the frequency of CD8+ T cells that produced IFNγ and granzyme B (Fig. 5b), albeit not to the levels found in CD8+ T cells from MHCII-negative tumors. Surprisingly, the numbers of CD8+ T cells were increased in both MHCII-positive and MHCII-negative tumors following CD4 depletion (Fig. 5c). Consequently, the numbers of CD8+ T cells producing IFNγ and granzyme B were also greatest in CD4-depleted TS/A-hCIITA tumors (Fig. 5d). We confirmed the elimination of CD4+ T cells following CD4 depletion (Fig. 5e). These data suggest that, despite the ability of CD4+ T cells to provide help to CD8+ T cells and facilitate the functional control of tumor growth, their absence does not entirely negate CD8+ T-cell function and may even allow additional CD8+ T-cell expansion via homeostatic mechanisms [26, 27].
Fig. 5.
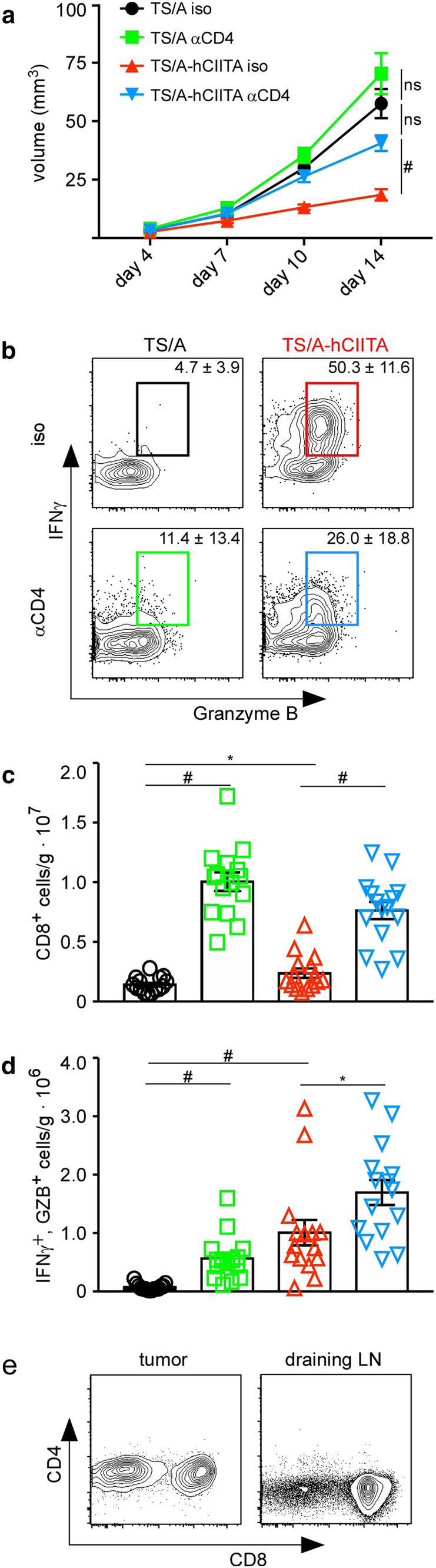
CD4 depletion impairs tumor growth and CD8+ T-cell activity. a Tumor volume was measured over 14 days. b T cells from tumors were restimulated ex vivo and the expression of IFNγ and granzyme B was assayed by flow cytometry on day 14. Plots shown are gated on live, CD3+, and CD8+ lymphocytes. c The number of CD8+ T cells was normalized to tumor mass. d The number of CD8+IFNγ+GZB+ cells was normalized to tumor mass. This experiment had five mice per group and was performed three times with similar results. Error bars represent standard error of the mean. Statistical difference is expressed as *p < 0.05, **p < 0.005, and #p < 0.0005
Expression of MHCII expands the responding T-cell clones in tumors
To test whether local MHCII expression promoted T-cell clonal expansion or accumulation, we harvested whole tumor and matched blood samples and sequenced the CDR3 region of the TCRβ chain to identify unique T-cell clones. We found fewer unique sequences in tumors relative to blood, but there were no differences between groups (Fig. 6a). Although the proportions of individual clones were uniformly distributed in blood, we found many expanded clones in tumors (Fig. 6b). Moreover, we observed more clones expanded to a greater magnitude in TS/A-hCIITA tumors than in control tumors, whereas no differences were observed between blood samples from these same mice (Fig. 6b).
Fig. 6.

MHCII expression increases the number and magnitude of expanded TCR clonotypes in tumors, but not blood. a The numbers of unique CDR3 reads were compared and horizontal lines denote group mean. b Tree-map plots show relative sizes of individual clonotypes in matched blood and tumor samples on day 14. Each colored shape corresponds to a unique clone and its relative area indicates its representation in the repertoire. c Inverse Simpson Index quantifies the expansion of detected clones in blood and tumor. Statistical significance assessed by the non-parametric Mann–Whitney test. d Analysis of TIL CDR3 amino acid sequence similarity in all the samples using pairwise overlap indexes, where legend values represent indexes. e Quantification of pairwise comparison is demonstrated by plotting the overlap indexes. f Overlap indexes were weighted by abundance of shared clones using Horn’s index. In e and f, error bars and horizontal lines indicate standard deviation and mean, respectively. g Proportion of CDR3 amino acid sequences in each repertoire that are shared by a number (mice) of other repertoires in the same group (data represents 12 mice). Horizontal bars represent the mean and error bars are standard deviation. Statistical difference is expressed as *p < 0.05, **p < 0.005, and #p < 0.0005
Using the Inverse Simpson Index as a normalized measure of diversity, we found that blood samples had high index scores that were not significantly different between TS/A-hCIITA and control tumors, whereas tumor samples had low diversity measures and MHCII-expressing tumors were significantly lower than control tumors (Fig. 6c). This result can be attributed to the local accumulation of the most abundant clonotypes, especially of the top ten clones (data not shown). These data suggested that expanded T-cell clones accumulated in tumors, but did not recirculate.
To determine which T-cell compartment contained expanded clones, we sorted CD4+ and CD8+ T cells from tumors on day 17 and performed repertoire analysis. Although we found some clonal expansion of CD4 T cells in TS/A and TS/A-hCIITA tumors (Supplemental Fig. 4a), it was more evident in CD8+ T cells from TS/A-hCIITA tumors. These differences were not due to the total CDR3 reads (Supplemental Fig. 4b), but were more evident in the number of unique reads (Supplemental Fig. 4c). However, we found that, after normalization with the Inverse Simpson Index, reduced diversity was evident in only the CD8+ T-cell compartment (Supplemental Fig. 4d).
We next tested whether clonotypes of identical amino acid sequence were expanded in multiple mice. By performing pairwise comparisons of normalized numbers of sequences, we found increased sharing of CDR3 sequences between TILs within MHCII-expressing tumors (Fig. 6d). In addition, overlap indexes showed a significantly greater number of common sequences in TS/A-hCIITA tumors relative to controls (Fig. 6e). Moreover, weighting sequence overlap by shared clones’ abundance using Horn’s pairwise overlap index also yielded a significant difference (Fig. 6f). Finally, comparing sequences common to multiple mice (rather than two in pairwise comparison) revealed a significant increase in the number of shared sequences amongst TILs from MHCII-expressing tumors (Fig. 6g). Collectively, these data suggested that MHCII expression on TS/A-hCIITA tumors increased intratumoral T-cell clonal expansion, including clonotypes shared between mice, suggesting that they are responding to particular tumor antigens.
Discussion
Consistent with the previous studies [20], our data show that the expression of MHCII on breast cancer cells expands local CD4+ and CD8+ T cells and impairs tumor growth. Our data extend those studies to show that T cells infiltrating MHCII-expressing tumors produce more effector molecules for longer periods, suggesting that locally stimulated CD4+ T cells promote CD8+ T-cell accumulation and function. Surprisingly, clonal expansion is primarily observed in CD8+ T cells, rather than CD4+ T cells. Despite the local activation of both CD4+ and CD8+ T cells in MHCII-expressing tumors, both cell types eventually become exhausted—a process that can be circumvented using antibodies against CTLA4, but not PD-1 or TIM3. These findings suggest that MHCII expression on tumor cells is an important factor in the success of anti-tumor immune responses.
MHCII molecules are typically expressed by professional antigen-presenting cells, like dendritic cells, which activate naïve CD4+ T cells in lymphoid organs. As a result, many tumors contain relatively few MHCII-expressing cells other than immunosuppressive cells of the myeloid lineage [28, 29]. However, if tumor cells themselves express MHCII, then they should be able to directly stimulate CD4+ T cells. In fact, the previous studies show that MHCII expression enables tumor cells to directly present intracellular antigens to CD4+ T cells [20, 30] and promotes dendritic cell accumulation, which may also enhance T-cell stimulation. Together, MHCII-expressing tumor cells and dendritic cells should allow CD4+ T cells to recognize cryptic antigens and provide help to CD8+ T cells [31, 32]. Interestingly, CD4+ T cells in MHCII-expressing tumors strongly expressed GZB, suggesting that they may also be directly cytotoxic to tumor cells. However, the previous studies from Motara et al. [20] show that both CD4+ and CD8+ T cells are required to eliminate CIITA-expressing tumors, suggesting that CD4+ and CD8+ T cells have non-redundant activities. Therefore, the most likely explanation is that stimulation of CD4+ T cells by tumor-specific MHCII expression provides local help to CD8+ T cells, which expand and more effectively kill tumor cells.
The enhanced clonal expansion and effector functions of CD8+ T cells in MHCII-expressing tumors, despite unchanged expression of MHC class I molecules, suggest that locally activated CD4+ T cells produce factors that enhance CD8+ T-cell responses [32]. The expanded T-cell repertoire in MHCII-expressing tumors may be due to epitope spreading [33], or to the local presentation of neoantigens or cryptic antigens [34]. Although T cells may be responding to hCIITA, which is only about 75% identical to mouse CIITA, this possibility is unlikely to explain the dramatic increase in T-cell function. Moreover, immunity against hCIITA-expressing tumors enhances the clearance of tumors that lack hCIITA, suggesting that T cells are primarily responding to endogenous tumor antigens, such as MuLV gp70 [25]. Importantly, CD4 depletion largely negated the benefits of MHCII expression by tumor cells. Paradoxically, CD4 depletion also increased the number of CD8+ T cells in tumors, possibly due to homeostatic expansion [35]. However, CD8+ T-cell activation without help from CD4+ T cells can compromise their effector and memory functions [18, 36] and lead to exhaustion [21]—phenotypes that are consistent with the poor control of tumor growth that we observed following CD4+ T-cell depletion.
Despite increased activation of both CD4+ and CD8+ T cells in MHCII-expressing tumors, both cell types eventually lose effector functions and become exhausted. This exhausted phenotype is likely due to prolonged TCR signaling in the absence of inflammation or costimulation [37]. In addition, tumors often acquire immune-inhibitory characteristics. For example, although IFNγ is critical for anti-tumor immunity [20], prolonged IFNγ signaling often leads to “adaptive resistance”, in which tumor cells upregulate T cell-inhibitory proteins like indoleamine oxidase (IDO) or PD-L1 [38]. As a result, the augmented IFNγ production by T cells responding to MHCII-expressing tumors may initially promote anti-tumor immunity, but later lead to immune suppression [39].
Although T-cell exhaustion can sometimes be overcome with blocking antibodies to inhibitory receptors, like PD-1, in most cases, only a subset of patients respond to therapy. For example, patients whose melanoma tumors express MHCII are more likely to clinically benefit from PD-1/PD-L1 blockade [8]. However, despite the increased expression of PD-1 on CD8+ T cells in our experiments, PD-1 blockade failed to preserve T-cell effector functions or promote tumor clearance, regardless of MHCII expression. This result is not entirely unexpected, as we failed to find substantial PD-L1 expression on tumor cells at any time point, a predictor of response to anti-PD-1 blockade [40]. Similarly, anti-TIM3 was also unable to promote tumor control, despite the expression of TIM3 on CD8+ TILs. In contrast, anti-CTLA4 antibody led to significantly impaired growth of control tumors and complete rejection of MHCII-expressing tumors. Hence, the efficacy of checkpoint blockade is mediated by numerous factors, including tumor phenotype, pre-treatment immune response, and timing of administration.
In summary, our data show that MHCII expression on tumor cells enhances and prolongs both CD4+ and CD8+ T-cell responses and slows tumor growth. Although T cells eventually become exhausted, even in MHCII-expressing tumors, their functions can be maintained by checkpoint blockade with anti-CTLA4. Given the enhanced immune responses and better clinical outcomes in both mice and humans with MHCII-expressing tumors [8, 10, 13, 15], we believe that that therapies triggering MHCII expression on tumor cells in vivo should be developed and used in combination with other immunity-enhancing strategies to prevent T-cell exhaustion and promote tumor clearance.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Uma Mudunuru and Scott Simpler for animal husbandry, Eddy Yang and Debbie Della Manna of the NanoString Laboratory and Enid Keyser of the Comprehensive Flow Cytometry Core for lending respective expertise. The TS/A murine mammary adenocarcinoma cell line was provided by Roberto S. Accolla, Department of Clinical and Biological Sciences, University of Insubria, Italy.
Abbreviations
- gp70
Glycoprotein 70
- GZB
Granzyme B
- hCIITA
Human class II transcriptional activator
- MHCII
MHC class II
- MuLV
Murine leukemia virus
Author contributions
TRM designed, performed, and interpreted experiments, and wrote the manuscript. ML designed, performed, and interpreted experiments. DS performed TCR repertoire sequencing and analysis. SJC performed TCR repertoire sequencing and analysis. ML designed, performed, and interpreted experiments. SM-P designed, performed, and interpreted experiments. RCA designed and interpreted experiments and edited the manuscript. DJB designed and interpreted experiments and edited the manuscript. AF designed and interpreted experiments and edited the manuscript. TDR designed and interpreted experiments and edited the manuscript.
Funding
This work was supported by the University of Alabama at Birmingham Comprehensive Cancer Center (P30 CA013148), National Institutes of Health Grant CA216234, and by the Breast Cancer Research Foundation of Alabama.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures involving animals were performed in accordance with the guidelines of the National Research Council (United States) Committee for the Update of the Guide for the Care and Use of Laboratory Animals and were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC) in protocol 09854.
Research involving human participants and animals
BALB/c mice were purchased from Charles River Laboratories International, Inc. BALB/c.scid mice (CBySmn.CB17-PrkdcscidIJ) were purchased from The Jackson Laboratory.
Cell line authentication
TS/A cells were obtained at passage 22 and passaged 2 times prior to freezing archival samples. TS/A cells were authenticated by assessing MHC haplotype via flow cytometry and by detection of antigens from murine leukemia virus. Transfected and control TS/A cells were also confirmed by gene expression of MHCII pathway gene products using nanostring assay and Western blot. Both cell lines tested negative for mycoplasma (and 13 other mouse pathogens) via PCR performed by Charles River Research Animal Diagnostic Services on June 2015.
References
- 1.Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29(8):1093–1102. doi: 10.1038/onc.2009.416. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting-integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 6.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in NSCLC. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsushita K, Takenouchi T, Shimada H, Tomonaga T, Hayashi H, Shioya A, et al. Strong HLA-DR antigen expression on cancer cells relates to better prognosis of colorectal cancer patients: possible involvement of c-myc suppression by interferon-gamma in situ. Cancer Sci. 2006;97(1):57–63. doi: 10.1111/j.1349-7006.2006.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schonocchia G, Eppenberger-Castori S, Zlobec I, Karamitopoulou E, Arriga R, Coppola A, et al. HLA class II antigen expression in colorectal carcinoma tumors as a favorable prognostic marker. Neoplasia. 2014;16(1):31–42. doi: 10.1593/neo.131568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diepstra A, van Imhoff GW, Karim-Kos HE, van den Berg A, te Meerman GJ, Niens M, et al. HLA class II expression by Hodgkin Reed–Sternberg cells is an independent prognostic factor in classical Hodgkin’s lymphoma. J Clin Oncol. 2007;25(21):3101–3108. doi: 10.1200/JCO.2006.10.0917. [DOI] [PubMed] [Google Scholar]
- 12.Tada K, Maeshima AM, Hiraoka N, Yamauchi N, Maruyama D, Kim SW, et al. Prognostic significance of HLA class I and II expression in patients with diffuse large B cell lymphoma treated with standard chemoimmunotherapy. Cancer Immunol Immunother. 2016;65(10):1213–1222. doi: 10.1007/s00262-016-1883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callahan MJ, Nagymanyoki Z, Bonome T, Johnson ME, Litkouhi B, Sullivan EH, et al. Increased HLA-DMB expression in the tumor epithelium is associated with increased CTL infiltration and improved prognosis in advanced-stage serous ovarian cancer. Clin Cancer Res. 2008;14(23):7667–7673. doi: 10.1158/1078-0432.CCR-08-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forero A, Li Y, Chen D, Grizzle WE, Updike KL, Merz ND, et al. Expression of the MHC class II pathway in triple-negative breast cancer tumor cells is associated with a good prognosis and infiltrating lymphocytes. Cancer Immunol Res. 2016;4(5):390–399. doi: 10.1158/2326-6066.CIR-15-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oldford SA, Robb JD, Codner D, Gadag V, Watson PH, Drover S. Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int Immunol. 2006;18(11):1591–1602. doi: 10.1093/intimm/dxl092. [DOI] [PubMed] [Google Scholar]
- 16.Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol. 2005;5(10):793–806. doi: 10.1038/nri1708. [DOI] [PubMed] [Google Scholar]
- 17.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of IL-12 and enhances T cell stimulatory capacity- T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 19.Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol. 2003;33(5):1183–1192. doi: 10.1002/eji.200323712. [DOI] [PubMed] [Google Scholar]
- 20.Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, et al. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res. 2006;12(11 Pt 1):3435–3443. doi: 10.1158/1078-0432.CCR-06-0165. [DOI] [PubMed] [Google Scholar]
- 21.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188(12):2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77(8):4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bevan MJ. Helping the CD8+ T-cell response. Nat Rev Immunol. 2004;4(8):595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 24.Bourgeois C, Veiga-Fernandes H, Joret A, Rocha B, Tanchot C. CD8 lethargy in the absence of CD4 help. Eur J Immunol. 2002;32:2199–2207. doi: 10.1002/1521-4141(200208)32:8<2199::AID-IMMU2199>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 25.Rosato A, Santa SD, Zoso A, Giancomelli S, Milan G, Macino B, et al. The cytotoxic T-lymphocyte response against a poorly immunogenic mammary adenocarcinoma is focused on a single immunodominant class I epitope derived from the gp70 env product of an endogenous retrovirus. Cancer Res. 2003;63:2158–2163. [PubMed] [Google Scholar]
- 26.Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–192. doi: 10.1172/JCI0215175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennaceur K, Chapman J, Brikci-Nigassa L, Sanhadji K, Touraine JL, Portoukalian J. Dendritic cells dysfunction in tumour environment. Cancer Lett. 2008;272(2):186–196. doi: 10.1016/j.canlet.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qi L, Rojas J, Ostrand-Rosenberg S. Tumor cells present MHC class II-restricted nuclear and mitochondrial antigens and are the predominant antigen presenting cells in vivo. J Immunol. 2000;165:5451–5461. doi: 10.4049/jimmunol.165.10.5451. [DOI] [PubMed] [Google Scholar]
- 31.Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor-self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70(21):8368–8377. doi: 10.1158/0008-5472.CAN-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology. 2012;1(5):670–686. doi: 10.4161/onci.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol Res. 2016;4(8):669–678. doi: 10.1158/2326-6066.CIR-15-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prlic M, Blazar BR, Khoruts A, Zell T, Jameson SC. Homeostatic expansion occurs independently of costimulatory signals. J Immunol. 2001;167:5664–5668. doi: 10.4049/jimmunol.167.10.5664. [DOI] [PubMed] [Google Scholar]
- 36.Voehringer D, Liang H, Locksley RM. Homeostasis and effector function of lymphopenia-induced memory like T cells in constitutively T cell depleted mice. J Immunol. 2008;180:4742–4753. doi: 10.4049/jimmunol.180.7.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013;5(200):1–10. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167(6):1540–1554 e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.