

Abstract
Single-electron reduction of a carbonyl to a ketyl enables access to a polarity-reversed platform of reactivity for this cornerstone functional group. However, the synthetic utility of the ketyl radical is hindered by the strong reductants necessary for its generation, which also limit its reactivity to net reductive mechanisms. Herein, we report a strategy for net redox-neutral generation and reaction of ketyl radicals. The in situ conversion of aldehydes to α-acetoxy iodides lowers their reduction potential (Δ 1200 mV), allowing for milder access to the corresponding ketyl radicals and an oxidative termination event. Upon subjecting these iodides to a Mn2(CO)10 pre-catalyst and visible light irradiation, an atom transfer radical addition (ATRA) mechanism affords a broad scope of vinyl iodide products with high Z-selectivity.
One Sentence Summary:
An atom transfer radical addition strategy enables mild, catalytic access to a redox-neutral, ketyl radical coupling mechanism.
Main Text:
The ketyl coupling of carbonyls offers a mechanistically inverted approach to construct C-C bonds versus classic, polar mechanisms (1). However, a major limitation of this valuable method is its reliance on strong, stoichiometric reductants (e.g. Na, K, Ti) (2–4) to overcome the large reduction potential of carbonyls (Ep,c > −2 V vs SCE) (5, 6). As shown in Figure 1a, a powerful tool used to overcome the thermodynamic barrier for ketyl generation is Kagan’s reagent (SmI2) (7, 8). This single-electron reductant can even be used catalytically when coupled with strong, stoichiometric reductants (e.g. Zn/Hg) (9). Recently, photochemical approaches have been applied to address this fundamental thermodynamic challenge in a complementary manner. For example, Knowles and coworkers developed a proton-coupled electron-transfer (PCET) strategy to reduce ketones to ketyl radicals, employing Ir or Ru photocatalysts (< −1.5 V), Brønsted acids, and a milder stoichiometric reductant (e.g. Hantzsch ester) (10, 11). The groups of Yoon, Ngai, and Ye (12–15) have also shown that concerted use of Lewis acids enables photocatalytic reduction of carbonyls to access ketyl radicals and their vinylogous analogs. Additional metal-catalyzed strategies for carbonyl-alkyne coupling promote complementary reactivity (by Ni, Ru, or Ir) with alternate reductants (e.g. Et3B, Mg, H2) (16–18). However, in each of the ketyl-based strategies, a redox-neutral approach remains mechanistically unfeasible because catalysts capable of overcoming the high Ered of a carbonyl are necessarily also prone to reducing the resulting ketyl-coupling adduct.
Figure 1. Discovery of a redox-neutral ketyl radical coupling.

(A) Current synthetic approaches to ketyl radicals rely on strong reductants that reduce both the carbonyl and subsequent radical intermediates. (B) An alternate strategy based on atom transfer of an α-acetoxy iodide enables milder reductive initiation, coupled with oxidative termination, to provide a redox-neutral, ketyl coupling mechanism. (C) Several catalysts enable this atom transfer strategy, including [CpFe(CO)2]2, Ru(bpy)3Cl2, Ir(ppy)2(dtbbpy)PF6, and Mn2(CO)10, which provide a range of chemo- and stereoselectivities. (D) Proposed mechanism: Photoinitiated homolysis of Mn2(CO)10 occurs with visible light (blue LED). The Mn(CO)5 catalyst then formally reduces the in situ formed α-acetoxy iodide via inner-sphere atom transfer. The resulting ketyl radical combines with an alkyne to form a vinyl radical, which is oxidized by the Mn catalyst to form the vinyl iodide product and regenerate Mn(CO)5. This catalyst turnover step precludes an alternate reductive pathway, wherein a second reduction of the vinyl radical affords the classic, non-iodo product.
Our complementary strategy, outlined in Figure 1, addresses this challenge by replacing the carbonyl reduction step with a halogen abstraction event via an atom transfer radical addition (ATRA) mechanism. In this case, a Mn catalyst promotes mild, ketyl radical formation from an in situ generated intermediate containing a weak C-I bond. The resulting radical is capable of coupling with a range of alkynes. Catalyst turnover occurs (via formal oxidation of the ensuing vinyl radical intermediate) by atom transfer (19, 20) with Mn-I, in a net, redox-neutral mechanism – enabling access to synthetically versatile (21) Z-vinyl halides.
In our synthetic design, we focused on addressing the challenge of ketyl radical generation from aliphatic aldehydes. We envisioned that their conversion to α-oxy iodides would benefit from hyperconjugative donation of the non-bonding oxygen electrons into the C-I anti-bonding orbital (n→σ*) to further weaken this bond. Among several α-iodo radical precursors we investigated, the α-acetoxy derivative proved simplest to generate under mild conditions. This in situ activation is performed by combining AcI (AcCl, NaI) with carbonyls (5% Zn(OTf)2, 0°C, 15 min) in a modified version of Adams’ nearly century-old procedure (22). The resulting aldehyde derivatives are conveniently handled in an aerobic atmosphere at room temperature, and are stable to basic, aqueous washes, without elimination. This practical synthetic accessibility allowed us to electrochemically validate our hypothesis that α-acetoxy iodides (Ep,c −1.1 V; Figure S2) are more easily reduced than their aldehyde precursors (> −2 V) (5) as well as other alkyl iodides (23–26). Next, we explored the generation of ketyl radicals – and their redox-neutral coupling with alkynes – by employing a variety of atom transfer catalysts.
In accordance with our design plan, we observed that several photocatalysts promote this redox-neutral alkyne coupling with ketyl radicals derived from aliphatic aldehydes (1), as illustrated in Figure 1c. Expecting the nucleophilicity of α-OAc ketyl radicals to be attenuated, we explored their combination with several alkynes of varying electronic character. In particular, coupling of silyl acetylene 2 affords silyl-vinyl-iodide 3 (containing orthogonal handles for further synthetic manipulation) in a streamlined approach that obviates an alkynylation, Al-reduction, then iodination sequence. Among the atom transfer catalysts investigated, several photocatalysts afforded new, redox-neutral coupling adduct 3 preferentially over the classic, reduced coupling product 4. These catalysts included complexes of earth abundant, first row metals (e.g. Mn, Fe) as well as more reducing photocatalysts (e.g. Ru, Ir; see Table S1 for further details). Ultimately, a Mn2(CO)10 catalyst was found to provide excellent selectivity for redox-neutral coupling (3:4, >20:1) along with high Z:E diastereoselectivity (>20:1). The latter feature is notable as there are few methods to access vinyl iodides with high Z-selectivity (27). Although Mn-mediated reactions are typically associated with oxidative mechanisms, atom transfer pathways are accessible via Mn2(CO)10 (28–30). Unlike typical photocatalysts, a photon is not necessary for turnover of the Mn catalyst. However, we observed that continual irradiation is necessary to access high efficiency and selectivity, likely due to an equilibrium between the pre-catalyst dimer and the active catalyst. Given the high chemo- and stereoselectivity afforded by the Mn catalyst, we decided to further explore its synthetic potential in this redox-neutral mechanism.
In our mechanistic proposal, shown in Figure 1d, Mn2(CO)10 precatalyst is homolyzed to Mn(CO)5 (5, Mn•) by irradiation with a blue LED. This 17-electron species is a competent ATRA catalyst, which can abstract I• from the weak C-I bond (BDE: 58 kcal/mol) (31) of in situ generated α-acetoxy iodide 6. Combination of the resultant ketyl radical 7 with alkyne 8 affords vinyl radical 9. This open-shell intermediate is formally oxidized by Mn(CO)5I (10, [Mn]-I ) via atom transfer to regenerate the Mn• catalyst (5), while also forming vinyl iodide 3’ in a redox-neutral mechanism (32). The net conversion of 6 to 3’ is exothermic due to formation of a strong vinyl C-I bond (BDE: 61–68 kcal/mol). An observed, post-reaction, isomerization of the vinyl iodide products – from 1:1 to >20:1 Z:E selectivity – is also thermodynamically favored by up to 3 kcal/mol (33). This Mn-catalyzed isomerization likely occurs via an intermediate vinyl radical, which is consistent with reports of photoinduced, single-electron reduction of aryl iodides for sp2 radical generation (34, 35). This Mn-catalyzed ATRA mechanism precludes an alternate pathway, in which vinyl radical 9 is further reduced to vinyl anion 11 (Ep,c −0.1 V (36), which is at least 2 V more favorable than carbonyl to ketyl reduction) to afford allyl ester 4. Instead, by coupling catalyst-turnover with product formation, the classic reductive mechanism can be overridden by this redox-neutral pathway.
In probing the synthetic utility of this strategy, we were pleased to find that aliphatic aldehydes, which are challenging to reduce to ketyls (Ep,c > −2 V) (5), efficiently combine with a range of alkynes, as shown in Fig 2. Alkynes with broad electronic character are coupled to the stabilized ketyl radicals, providing Z-vinyl iodides in up to 20:1 dr. For example, alkynes with highly electron-releasing substituents, such as SiEt3 or BPin, are competent partners (12, 13), affording versatile vinyl silanes, or boronates (37, 38). Aryl alkynes and 1,3-enynes afford valuable styrene and diene adducts (14, 15), and electron-deficient propiolates also undergo ketyl coupling to merge these electrophiles (16, 17).
Figure 2. Synthetic scope of redox-neutral ketyl radical coupling.

(*) 20% Mn2(CO)10, 1 equiv KOAc; (†) 5% Mn2(CO)10, 0.5 equiv iPr2NEt; (‡) 10% Mn2(CO)10. See Supplementary Materials for experimental details. Isolated yield and Z:E diastereoselectivity indicated below each entry. Abbreviations: Ac, acyl; d.r., diastereoselectivity; Pin, pinacol; Piv, pivaloyl; TBDPS, tert-butyldiphenylsilyl; Phth, phthalyl; Ts, toluenesulfonyl.
In addition to conjugated alkynes, we found propargyl esters to be suitable ketyl radical acceptors that afford Z-vinyl iodide 18, which contains orthogonal allyl esters. An internal competition between alkynes (alkyl- vs ester- substituted) selectively converts the propiolate alkyne to vinyl iodide 19, leaving the unsubstituted alkyne intact. The mild, radical addition conditions are also tolerant of alcohols (20) and ketones (21, 22), illustrating the orthogonality and synthetic utility of this ATRA-based strategy.
To further demonstrate the functional group compatibility of this ketyl-alkyne coupling, a wide range of aliphatic aldehydes was also explored. Aldehyde-derived ketyl radical precursors with both small (23) and large (24) steric footprints provide Z-vinyl iodides in >20:1 selectivity. The tolerated functionality on the aldehyde component includes arenes of varying electronic character (25–27), ethers (28–29), and amides (30–32). Alkyl halides (e.g. Cl, 33; I, 34) are also tolerated in the reaction, illustrating selective reaction of the Mn catalyst with the hyperconjugatively activated α-acetoxy iodides.
Finally, although some ketone-derived ketyl radicals are too hindered for efficient cross-coupling, we found trifluoroacetone provides efficient access to Z-vinyl iodide 35 – bearing an adjacent tertiary ester. Electron-rich alkenes are also capable of ketyl radical coupling, including those substituted with Si (36) or B (37). An anti-oxidant, vitamin E, was also incorporated in the alkyne acceptor (38) without inhibiting this mild, ketyl radical coupling.
Our mechanistic hypothesis for the redox-neutral, catalytic reaction described above is supported by reaction intermediate isolation and resubjection, competition experiments, and kinetic measurements (Figure 3). To probe the origin of the high Z:E diastereoselectivity, we monitored product formation of vinyl iodides 12 and 16 over the reaction course (Fig 3a). In each case, we observed rapid product formation (>70% yield) within 15 min, albeit with modest Z:E selectivity (7:1, 1:1). For the first 2 h, product yields remain within 5–10% of their 15 min values, but Z:E selectivity increases dramatically (>20:1, 14:1), suggesting a product isomerization mechanism. Additional evidence of post-reaction epimerization was found when a 1:1 mixture of vinyl iodide 16 was resubjected to the Mn photocatalyst for 2 h, affording the Z-isomer selectively: 4:1 (10% catalyst) or >20:1 (20% catalyst) (Fig 3b).
Figure 3. Mechanistic experiments.

(A) Z:E selectivity increases over the reaction course (for both alkyne classes) suggesting a product isomerization pathway. (B) A 1:1 mixture of vinyl iodide isomerizes to increased ratios of the Z-isomer correlating with Mn catalyst loading, illustrating the catalyst’s role in isomerization. (C) Selective combination with an electron-deficient alkyne indicates the ketyl radical is nucleophilic. (D) The catalyst turnover step involving sp2 C-I formation is recapitulated by trapping an aryl radical with [Mn]-I to form Ar-I. (E) Selective recombination of the vinyl radical with I• (vs intramolecular traps) suggests this step is rapid. (F) Intramolecular traps with sp2 or sp3 C• intermediates afford either two- or three-component cascade couplings.
Further insights were obtained for each elementary step of the atom transfer radical addition via the mechanistic experiments shown in Figure 3. First, a competition experiment between an electron-poor and electron-rich alkyne resulted in exclusive ketyl radical coupling to the electron-deficient propiolate alkyne (16 vs 12), indicating the α-OAc radical has nucleophilic character (Fig 3c). Next, we validated viability of the key oxidation step – required for catalyst turnover – by stoichiometric trapping of an aryl radical with [Mn]-I to form Ar-I (Fig 3d). The sp2 C• radical precursor, diazonium 39, affords aryl iodide 40 in 70% yield when combined with Mn(CO)5 I and Eosin Y photocatalyst (3% without light, 68% with only thermal initiation). Next, the rate of atom transfer was compared to known radical clocks (39) appended to propiolate (Fig 3e). For each of the three intramolecular traps investigated, only atom transfer (i.e. vinyl C-I termination, 41-43) was observed – in lieu of radical cyclization, which occurs rapidly with these acceptors (> 1×108 s−1). Finally, cascade reactions were facilitated by incorporation of intramolecular traps within the ketyl radical precursors (Fig 3f). Interestingly, unlike the radical clock experiments that did not manifest cyclization, these ring closures occurred selectively, albeit with divergent outcomes dependent on the hybridization of the coupled radical intermediate. For example, alkyne 44, which forms an sp2 C•, selectively traps iodine affording two-component coupling product 45, even in the presence of alkyne traps (e.g. -CO2Et, -SiEt3). Alternatively, an sp3 C• formed by ketyl radical cyclization of alkene 46 selectively combines with Et3Si-alkyne to afford three-component coupling product 47. In this case, intramolecular sp3 to sp3 translocation of the carbon radicals (α-OAc to α-CO2Et) precedes intermolecular trapping of the alkyne. The resultant sp3 C• is long-lived enough (or exists as a living radical) (40) to then combine with an alkyne forming an sp2 C• that is rapidly trapped as the vinyl iodide.
Finally, to demonstrate the synthetic utility of these functionally rich, ketyl-alkyne adducts (bearing a geminal, vinyl iodide/silane), atom transfer product 3 was manipulated, as shown in Figure 4. First, Pd-catalyzed cross-coupling of the vinyl iodide with a range of boronic acids afforded alkylation (48), vinylation (49), and arylation (50–52) products with complete stereoretention. Next, the resulting E-vinyl silane 48 was converted to a Z-vinyl iodide with N-iodosuccinimide (NIS), and subsequent arylation with PhB(OH)2 affords 53 via a modular, five-component coupling.
Figure 4. Synthetic utility of the atom transfer product.
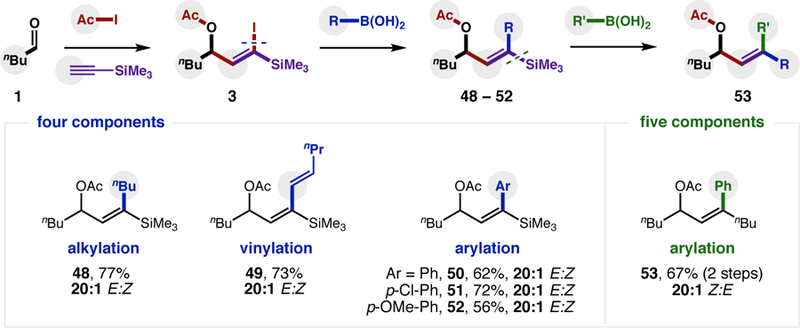
Silyl vinyl iodide 3 was coupled to alkyl, vinyl, and aryl boronic acids with 10% Pd(PPh3)2Cl2 in a net, four component coupling. The resulting E-vinyl silane was converted to a Z-vinyl iodide with NIS, then also coupled with phenyl boronic acid in a net, five component coupling.
We expect this atom transfer method for accessing ketyl radicals – and selectively combining them with an electronically diverse range of π-acceptors – through the use of earth-abundant metal catalysts will have broad utility in various synthetic arenas.
Supplementary Material
Acknowledgments:
We are grateful to Mingfu He and Andrew Chen for assistance with CV experiments and DFT studies. Funding: Financial support was provided by the National Science Foundation (NSF CAREER 1654656), as well as National Institutes of Health (NIH R35 GM119812) and American Chemical Society Petroleum Research Fund. S.C.F. is grateful for an HHMI Gilliam Fellowship.
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability: All data are available in the main text or the supplementary materials.
References and Notes:
- 1.Hart D, Science 223, 883–887 (1984). [DOI] [PubMed] [Google Scholar]
- 2.Kahn BE, Rieke RD, Chem. Rev 88, 733–745 (1988). [Google Scholar]
- 3.McMurry JE, Chem. Rev 89, 1513–1524 (1989). [Google Scholar]
- 4.Szostak M, Fazakerley NJ, Parmar D, Procter DJ, Chem. Rev 114, 5959–6039 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Roth HG, Romero NA, Nicewicz DA, Synlett 27, 714–723 (2016). [Google Scholar]
- 6. All potentials reported vs SCE (Saturated Calomel Electrode).
- 7.Girard P, Namy JL, Kagan HB, J. Am. Chem. Soc 102, 2693–2698 (1980). [Google Scholar]
- 8.Molander GA, Harris CR, Chem. Rev 96, 307–338 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Corey EJ, Zheng GZ, Tetrahedron Lett 38, 2045–2048 (1997). [Google Scholar]
- 10.Tarantino KT, Liu P, Knowles RR, J. Am. Chem. Soc 135, 10022–10025 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR, J. Am. Chem. Soc 135, 17735–17738 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Ischay MA, Anzovino ME, Du J, Yoon TP, J. Am. Chem. Soc 130, 12886–12887 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Du J, Skubi KL, Schultz DM, Yoon TP, Science 344, 392–396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KN, Lei Z, Ngai M-Y, J. Am. Chem. Soc 139, 5003–5006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye C-X et al. , Nat. Commun 9, 410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 354, 300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montgomery J, Angew. Chem. Int. Ed 43, 3890–3908 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Tasker SZ, Standley EA, Jamison TF, Nature 509, 299–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams TM, Stephenson CRJ, in Visible Light Photocatalysis in Organic Chemistry (Wiley-Blackwell, 2018), pp. 73–92. [Google Scholar]
- 20.Theriot JC et al. , Science 352, 1082–1086 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Renata H, Zhou Q, Baran PS, Science 339, 59–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.French H, Adams R, J. Am. Chem. Soc 43, 651–659 (1921). [Google Scholar]
- 23.Nguyen JD, D’Amato EM, Narayanam JMR, Stephenson CRJ, Nat. Chem 4, 854–859 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Wallentin CJ, Nguyen JD, Finkbeiner P, Stephenson CRJ, J. Am. Chem. Soc 134, 8875–8884 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Monks BM, Cook SP, Angew. Chem. Int. Ed 52, 14214–14218 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Shen Y, Cornella J, Juliá-Hernández F, Martin R, ACS Catal 7, 409–412 (2017). [Google Scholar]
- 27.Koh MJ, Nguyen TT, Zhang H, Schrock RR, Hoveyda AH, Nature 531, 459–465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snider BB, Chem. Rev 96, 339–364 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Meyer TJ, Caspar JV, Chem. Rev 85, 187–218 (1985). [Google Scholar]
- 30.Baird MC, Chem. Rev 88, 1217–1227 (1988). [Google Scholar]
- 31. See Table S4 for calculated BDE (bond dissociation energies).
- 32. Radical chain propagation could also provide 3’; however, this pathway is unlikely, as initiation by 50% Et3B/O2 only affords 29% product. Moreover, competition experiments between α-OAc bromides and α-OAc iodides do not provide crossover products. See Supplementary Materials for full experimental details.
- 33. Despite the larger size of iodine (and higher priority in E/Z notation), its longer C-I bond (and smaller A-value) likely contributes to the thermodynamic favorability of the Z-vinyl iodide.
- 34.Creutz SE, Lotito KJ, Fu GC, Peters JC, Science 338, 647–651 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Ghosh I, Ghosh T, Bardagi JI, König B, Science 346, 725–728 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Andrieux CP, Pinson J, J. Am. Chem. Soc 125, 14801–14806 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Zhang L et al. , Science 351, 70–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kischkewitz M, Okamoto K, Mück-Lichtenfeld C, Studer A, Science 355, 936–938 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Beckwith ALJ, O’Shea DM, Tetrahedron Lett 27, 4525–4528 (1986). [Google Scholar]
- 40.Koumura K, Satoh K, Kamigaito M, Macromolecules 41, 7359–7367 (2008). [Google Scholar]; Supplemental References. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.