

Supplemental Digital Content is available in the text
Keywords: biological function enrichment analysis, Kashin-Beck disease, network analysis, the Molecular Complex Detection Algorithm (MCODE)
Abstract
To perform a comprehensive analysis focusing on the biological functions and interactions of Kashin-Beck disease (KBD)-related genes to provide information towards understanding the pathogenesis of KBD.
A retrospective, integrated bioinformatics analysis was designed and conducted. First, by reviewing the literature deposited in PubMed, we identified 922 genes genetically associated with KBD. Then, biological function and network analyses were conducted with Cytoscape software. Moreover, KBD specific molecular network analysis was conducted by Cytocluster using the Molecular Complex Detection Algorithm (MCODE).
The biological function enrichment analysis suggested that collagen catabolic process, protein activation cascade, cellular response to growth factor stimulus, skeletal system development, and extrinsic apoptosis played important roles in KBD development. The apoptosis pathway, NF-kappa B signaling pathway, and the glutathione metabolism pathway were significantly enriched in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway network, suggesting that these pathways may play key roles in KBD occurrence and development. MCODE clusters showed that in top 3 clusters, 54 of KBD-related genes were included in the network and 110 candidate genes were discovered might be potentially related to KBD.
The 110 candidate genes discovered in the current study may be related to the development of KBD. The expression changes of apoptosis and oxidative stress-related genes might serve as biomarkers for early diagnosis and treatment of KBD.
1. Introduction
Kashin-Beck Disease (KBD) is a type of endemic, chronic, and disabling osteoarthropathy with high disability. KBD is also named as endemic osteochondropathy, as it is mainly prevalent at defined areas of North Korea, Russia, and China (mainly in northeastern and southwestern China).[1] By 2015, there were 0.57 million KBD patients in China. Of these patients, nearly 13 thousand were children under 13 years old.[2,3] KBD patients usually exhibit the clinical symptoms, such as enlargement of phalanges or other joints, joint pain, limited movement, deformed limb joints.[4–6] Besides the detrimental effect on all aspects of quality of life, KBD places a heavy economic burden on society as a whole.[7]
Previous studies reported that environmental factors played important roles in KBD development, such as selenium deficiency in the environment of endemic KBD areas and cereal contamination by fungal mycotoxins.[8,9] However, the etiology and pathogenesis have not been fully unraveled. In recent years, growing evidence has implicated that KBD is a complex disease due to a complicated interplay of environmental and genetic factors. Some studies suggested that some genes might contribute to the development of KBD, such as GPX1,[10]GPX4,[11]SELS,[9]SEPP,[12]ADAM12,[13]Col2A1,[14]Bcl-2, Bax, and Fas.[15] But these studies only provided a limited explanation for the KBD occurrence and insufficiently revealed their roles and functions in the development of KBD. In the whole genome-wide, these genes do not play independent roles; instead, these genes form biological function and pathway networks through their intricate interactions, which can give us a more complete picture of their genetic function contributing to the development of KBD.[16] Furthermore, genetic studies have indicated that the pathological molecular mechanisms may be attributed to hundreds of genes and their variants for KBD.[17,18] With the rapid development of bioinformatics, such as biological function enrichment analysis, protein–protein interaction (PPI) and the Molecular Complex Detection Algorithm (MCODE), an increasing number of differentially expressed genes (DEGs) were identified between KBD cases and healthy controls.[19,20] Therefore, a comprehensive analysis of potential genes, which uncovers more detailed information involved in pathways and networks as compared to conventional single-gene analysis, is necessary.
In the present study, we first formed a comprehensive collection of genes that were reported to be associated with KBD. Biological function enrichment analysis, based on the KBD-related genes, was then conducted to identify their significant biological functions. We also constructed a pathway network to explore possible crosstalk among the significant pathways. Moreover, we made a PPI network of these KBD-related genes using MCODE.[21] This study may provide further information for understanding the molecular mechanisms of KBD based on a systematic bioinformatics analysis.
2. Materials and methods
2.1. Collection of KBD-related genes
KBD-related genes identified in human genetic association studies in PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) were collected using the term (Kashin-Beck Disease or KBD) AND (polymorphism [MeSH] or genotype [MeSH] or alleles [MeSH]).[22] KBD-related genes were retrieved and screened under the following criteria:
-
1.
they must be reported in publications which matched the above retrieve strategy;
-
2.
the KBD-related genes should be provided in official symbols;
-
3.
genes that exhibited significant associations (P < .05) with KBD were included and that showed insignificant associations (P > .05) with KBD were excluded.
By July 19, 2017, a total of 58 publications that were matched the criteria were retrieved. We further included genes with a significant association with KBD (P < .05) after reading the full text or abstracts of the original articles. A KBD-related gene set with 922 members that was reported significantly associated with KBD was obtained from 24 studies. For microarray analyses or genome-wide association studies (GWAS) at a genome-wide significance level, the comprehensive and positive genes were obtained from the supplementary data of these studies.
2.2. Enrichment analysis of KBD-related genes
Gene Ontology Term (GO; www.geneontology.org) and Kyoto Encyclopedia of Genes and Genomes (KEGG; www.genome.ad.jp/kegg) pathway enrichment analyses were performed using Cytoscape 3.5.1. GO is usually used to explore the molecular function (MF), biological process (BP) and cellular component (CC) of candidate genes. In our present study, we mainly focused on BP of the candidate genes. KEGG is widely used for extracting the pathway information based on the molecular interactions. Fisher's exact test (two-side) or x2 test were performed to classify the pathway category. The false discovery rate (FDR) was used for the P-value correction based on the Benjamini and Hochberg method.[23,24] Significantly enriched pathways and GO terms (P ≤ .05, number of enrichment genes ≥2) were identified through Cytoscape 3.5.1.
2.3. Establishment of pathway networks among KBD-related genes
The pathway network among KBD-related genes was established to understand their possible interactions and crosstalk. Due to differences in the sensitivity and specificity of detection methods or sources, we constructed the pathway networks based on the gene lists from GWAS, microarray analysis, and exome sequencing using ClueGO, which is a plugin for Cytoscape 3.5.1. The ClueGO network is analyzed with kappa statistics based on the similarity of their associated genes, which reveals the interactions among the pathways. The significance of the terms and groups is automatically calculated from Gene Ontology, KEGG, Reactome, and CluePedia by ClueGO based on Cytoscape 3.5.1.[25–29]
2.4. Construction of KBD-related PPI network via MCODE
In the present study, we first identified the PPI network relationships of the 922 KBD-related genes in the whole network based on the biological network from GeneMANIA, which used many large, publicly available biological datasets to find related genes. MCODE cluster analysis was then performed using Cytocluster software (degree cutoff = 2, node score cutoff = 0.2, K-core = 2, MAX depth = 100) to identify the most important MCODE clusters according to clustering scores. Finally, the screened cluster networks were visualized with Cytoscape 3.5.1 software. The importance of clustering networks was assessed by clustering scores.
3. Results
3.1. Identification of KBD-related genes from the literature
By carefully and comprehensively retrieving articles matching our criteria from the PubMed database, we screened genetic association studies related to KBD and selected only the publications that found gene(s) significantly associated with KBD. Original manuscripts reporting a negative or insignificant association were excluded. Finally, a KBD-related gene set consisting of 922 members reported to be significantly associated with KBD was retrieved from 24 studies (Table 1). The list of 922 genes is provided in Supplemental File 1.
Table 1.
Sources of KBD-related genes.

3.2. Biological function and pathway enrichment analysis towards KBD-related genes
Results showed that the KBD-related genes were involved in 672 biological function terms in total. The top five biological function categories enriched in KBD-related genes detected by exome sequencing, GWAS and microarray analysis are listed in Table 2. The results showed that biological functions (collagen catabolic process, protein activation cascade, cellular response to growth factor stimulus, skeletal system development, extrinsic apoptosis, et al) were enriched among KBD-related genes, which indicates that collagen formation, skeletal development, and cellular programs (growth, differentiation, migration, apoptosis, et al) may affect KBD.
Table 2.
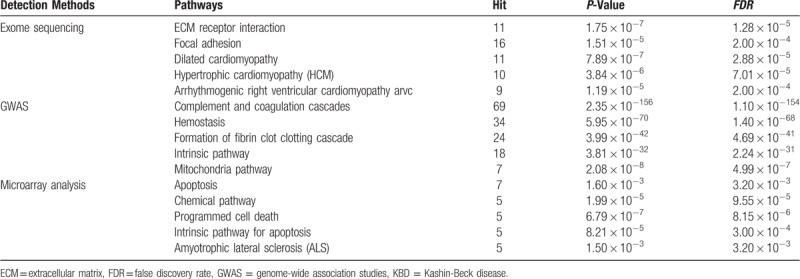
Top five biological functions enriched in KBD-related genes detected by exome sequencing, GWAS and microarray analysis.

Among the significantly enriched pathways for KBD, several pathways related to cell death and apoptosis, for example, Sa_programmed cell death, Reactome_intrinsic pathway for apoptosis, Biocarta_WNT pathway, extracellular matrix (ECM) receptor interaction, were enriched in the KBD-related genes. Also, pathways related to immune or inflammation-related signaling were identified, such as, Adherens junction, Regulation of actin cytoskeleton, and Reactome signaling in immune system, suggesting that the immune system is also involved in the development of KBD. In addition, oxidative stress-related signaling pathways, such as Glutathione metabolism, were also significantly enriched, which indicates that KBD development is related to the body's oxidative stress response. Top five pathways enriched in KBD-related genes detected by exome sequencing, GWAS and microarray analysis were showed in Table 3.
Table 3.
Top five pathways enriched in KBD-related genes detected by exome sequencing, GWAS and microarray analysis.
3.3. KEGG pathway network of KBD-related genes
The pathway network showed that significantly enriched KEGG pathways related to KBD were divided into 15 dependent clusters. Pathways in the same cluster exhibit the same or similar functions; however, the different clusters were relatively independent, suggesting they may exhibit different functions. Top three clusters with most related genes were exhibited in Fig. 1. Apoptosis pathway, NF-kappa B signaling pathway, and glutathione metabolism pathway were enriched in the predominant three clusters.
Figure 1.

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway network for Kashin-Beck disease (KBD)-related genes. A: From genes detected by genome-wide association studies (GWAS); B: From genes detected by microarray analysis; C: From genes detected by exome sequencing; pathways in the same cluster exhibit the same or similar functions; apoptosis pathway, NF-kappa B signaling pathway, and glutathione metabolism pathway were enriched in the predominant three clusters.
3.4. Construction of KBD-specific protein network via MCODE
The topological properties of PPI via pathway-based MCODE cluster analysis were performed to help understand the potential biological mechanisms associated with the network. The higher the clustering scores, the more important the biological function of this clustering in the development of KBD. Therefore, we analyzed the three clusters with the highest clustering scores in detail. The results are shown in Fig. 2. In Fig. 2A, the PPI cluster (MCODE cluster score = 6.114) had 36 genes in total, including 24 (66.67%) gene members in the KBD-related gene list; while 12 (33.33%) genes were not included in the list. In Fig. 2B, the PPI cluster (MCODE cluster score = 5.077) had 40 genes in total, including 25 (62.50%) gene members in the KBD-related gene list, but 15 (37.50%) genes were not in the list. In Fig. 2C, the PPI cluster (MCODE cluster score = 4.184) had 88 genes in total, in which 14 (15.91%) gene members were KBD-related but 74 (84.09%) genes have not been reported before. In these 3 PPI clusters, 54 of the KBD-related genes were included in the human interactome network, among which 110 candidates genes that are likely to be highly associated with KBD based on MCODE clusters. These 110 genes were listed in Table 4 , which provided a list of new potential candidates for KBD.
Figure 2.

Kashin-Beck disease (KBD) top three specific PPI networks. A: From genes detected by genome-wide association studies (GWAS); B: From genes detected by microarray analysis; C: From genes detected by exome sequencing; the red nodes mean genes in the KBD-related gene list, the green nodes are genes not in the KBD-related gene list.
Table 4.
Genes included in KBD top three specific PPI networks but not in the KBD-related gene list.

Table 4 (Continued).
Genes included in KBD top three specific PPI networks but not in the KBD-related gene list.

4. Discussion
By July 19, 2017, there were 24 articles on gene-related studies about KBD, and we assembled a list of 922 genes that were reported statistically associated with KBD. The detection methods used in these studies were mainly exome sequencing, microarray analysis, and GWAS. However, most of the biological effects of the 922 genes potentially involved in KBD development have not yet been evaluated. Therefore, a thorough understanding of the BPs related to the molecular pathogenesis of KBD is still far from complete.[8,30,31] Fortunately, the developments of high-throughput microarray analysis and sequencing techniques in recent years provide an opportunity to understand the pathogenesis of KBD at a systems biology level based on the biomarkers that have been discovered.
4.1. Pathways related to apoptosis play an important role in KBD occurrence and development
Our biological function enrichment analysis identified specific BPs and pathways in which KBD-related genes are involved. For instance, collagen catabolic process, protein activation cascade, cellular response to growth factor stimulus, skeletal system development, and extrinsic apoptosis were significantly enriched among the KBD-related genes, indicating that biological events such as the maintenance of apoptosis, the degradation of ECM, and biology process related to oxidative stress play key roles in the development of KBD. Studies had reported that KBD is characterized as specific pathological changes in cartilage, causing apoptosis and necrosis of chondrocytes in the epiphyseal plate,[3,32–37] which further demonstrated by our results. The current view about what factors lead to articular chondrocyte apoptosis in KBD focuses on varying degrees of oxidative stress occur in the articular chondrocytes, which is primarily caused by articular chondrocyte apoptosis.[10,12,38–40] Our results also provided evidence for this view. Our enrichment results showed that oxidative stress-related signaling pathways such as glutathione metabolism, were also significantly enriched in KBD-related genes, which means that KBD development is also associated with the body's oxidative stress response. Due to the above results, we have more evidence to infer that a reduction in oxidative stress may reduce chondrocyte apoptosis, and thus prevent the occurrence of KBD.[7,41–45] In addition, the agreements between our results and previous work indicated that our results are reliable.
4.2. Inflammation and immune-related pathways may be also associated with KBD occurrence and development
The KEGG network showed that the key pathways enriched in genes identified by GWAS were complement and coagulation cascades pathways, which are central to host defense. The complement pathway is part of innate immune system and the coagulation pathway is essential for clot formation and the prevention of excessive bleeding.[46–48] The complement and coagulation cascades generate crucial enzymatic activities that, in turn, generate the complement effector molecules, with the opsonization of pathogens, the recruitment of inflammatory and immunocompetent cells, and the direct killing of pathogens as the main consequences.[49–51] The results further consolidate ties between the pathology of KBD and immune-specific activities.[46,47] In addition, the KEGG network also showed that dilated cardiomyopathy pathway was enriched in genes from exome sequencing. Dilated cardiomyopathy pathway represents the final common morphofunctional pathway of various pathological conditions, in which a combination of myocyte injury and necrosis associated with tissue fibrosis results in impaired mechanical function. To our knowledge, this is the first time that KBD has been suggested to have a close biological association with heart disease, which still needs further evaluation to confirm.
4.3. Apoptosis, cell structure and oxidative stress-related events might affect KBD occurrence at protein level
Top three important PPI networks of the KBD-related genes were extracted from reference interactome network (GeneMANIA network) by MCODE clustering, which is a density-based non-overlapping clustering algorithm and based on the seed nodes as the center to expand to its neighbor nodes. Then the nodes that may interact with the seed nodes were screened out to construct complexes in a PPI network.[26,27] MCODE can also identify clusters (highly interconnected regions) within a network.[28,29] It is worth noting that 110 extended genes were not included in the KBD-related genes, but appeared in the top three PPI networks, which were plausible genes that previously not been reported to be associated with KBD. For example, BAD (BCL-2 associated agonist of cell death) is a member of the BCL-2 family,[52,53] which is known as a regulator of programmed cell death. Previous studies have reported that BAD can positively regulate cell apoptosis by forming heterodimers with BCL-xL and BCL-2.[42,43]COL1A2, COL4A2, COL4A3, COL4A5, and COL4A6 were identified as potential target genes in KBD, and these are related to the synthesis of type I collagen and strengthening as well as supporting many tissues, including cartilage and bone.[14]MAPK14 is a member of the MAP kinase family, which acts as integration points for multiple biochemical signals, and are involved in cell proliferation, differentiation, transcription regulation, and development. MAPK14 is activated by oxidative stress and proinflammatory cytokines, but this activation requires phosphorylation of MAPK14 by MAP kinase kinases (MKKs).[8,54,55] Genes play their crucial biological roles in the form of proteins. Therefore, the above events or functions, which were reflected by the PPI network, may be closer to the practical biology processes of the KBD-related genes. Our results extend the findings of the previous studies, and provide further information about the pathogenesis of KBD. Meanwhile, our MCODE cluster analysis was based on a reference interactome network, which not only provided a meaningful inferred network of KBD-related genes but also can be used to identify potential candidate biomarkers.
According to our knowledge, our present study is the first report to integrate all the candidate genes from the previous studies together and perform a comprehensively analysis in order to answer the above key questions (which candidate gene is more important in KBD development, what are the possible signal pathways involved when these candidate genes play their roles, et al) as well as to provide more valuable information. Exploring the environmental susceptibility genes of KBD has become a hot topic in this field. The conclusions of this study may help to update the new understanding of the pathogenesis of KBD and to expand the biomarkers of KBD, which helps early identification and treatment of KBD.
In conclusion, 110 candidate genes discovered in the present study may be related to the development of KBD. The expression changes of apoptosis and oxidative stress related genes might serve as biomarkers for early diagnosis and treatment of KBD. We will further verify our findings through rigorous and independent experiments in big KBD populations.
Acknowledgments
We would like to thank Dr Zhenzhong Li from Beijing Compass Biotechnology Co., Ltd (Beijing, China; http://www.kangpusen.com/) for his kind help with the data analysis. Our study was supported by grants from the National Natural Science Foundation of China (grant nos. 81773372 and 81573104).
Author contributions
RQZ and YMX designed the study. RQZ, HG, and XLY conducted the analyses. RQZ, BRL, DDZ, ZFL, and HLJ performed the analyses and prepared the manuscript. AMY and JZ revised the manuscript. RQZ and YMX submitted the study.
Conceptualization: Yongmin Xiong.
Data curation: Rongqiang Zhang, Hao Guo.
Formal analysis: Rongqiang Zhang, Hao Guo, Xiaoli Yang, Dandan Zhang, Baorong Li, Zhaofang Li.
Funding acquisition: Yongmin Xiong.
Methodology: Rongqiang Zhang.
Software: Rongqiang Zhang.
Supervision: Yongmin Xiong.
Writing – original draft: Rongqiang Zhang, Yongmin Xiong.
Writing – review & editing: Rongqiang Zhang, Hao Guo, Xiaoli Yang, Dandan Zhang, Baorong Li, Zhaofang Li, Yongmin Xiong.
Supplementary Material
Footnotes
Abbreviations: ADAM12 = ADAM metallopeptidase domain 12, APAF1 = apoptotic peptidase activating factor 1, ATR = ATR serine/threonine kinase, Bax = BCL2 associated X, apoptosis regulator, Bcl-2 = BCL2, apoptosis regulator, BP = biological process, CC = cellular component, CCNB1 = cyclin B1, CCND1 = cyclin D1, CD82 = CD82 molecule, COL1A1 = collagen type I alpha 1 chain, Col2A1 = collagen type II alpha 1 chain, COL2A1 = collagen type II alpha 1 chain, COL4A1 = collagen type IV alpha 1 chain, COL4A4 = collagen type IV alpha 4 chain, COL6A6 = collagen type VI alpha 6 chain, COL9A1 = collagen type IX alpha 1 chain, DEGs = differentially expressed genes, Fas = Fas cell surface death receptor, FDR = the false discovery rate, FN1 = fibronectin 1, GO = gene ontology term, GPX1 = glutathione peroxidase 1, GPX4 = glutathione peroxidase 4, GWAS = genome-wide association studies, ITGA2B = integrin subunit alpha 2b, ITGA6 = integrin subunit alpha 6, ITGB1 = integrin subunit beta 1, KBD = Kashin-Beck disease, KEGG = the Kyoto Encyclopedia of Genes and Genomes, LAMA2 = laminin, alpha 2, MAPK14 = mitogen-activated protein kinase 14, MCODE = the Molecular Complex Detection Algorithm, MF = the molecular function, MKKs = MAP kinase kinases, PERP = PERP, TP53 apoptosis effector, PPI = protein–protein interaction, SELS = selenoprotein S, SEPP = selenoprotein P, SERPINE1 = serpin family E member 1, THBS1 = thrombospondin 1, TNR = tenascin R, TNXB = tenascin XB.
HG is co-first author.
Ethical approval: Ethical approval was not necessary, because our study is based on the genes from publications about KBD.
Supplemental Digital Content is available for this article.
The authors report no conflicts of interest.
References
- [1].Luo R, Liu G, Liu W, et al. Efficacy of celecoxib, meloxicam and paracetamol in elderly Kashin-Beck disease (KBD) patients. Int Orthop 2011;35:1409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shi Z, Pan P, Feng Y, et al. Environmental water chemistry and possible correlation with Kaschin-Beck disease (KBD) in northwestern Sichuan, China. Environ Int 2017;99:282–92. [DOI] [PubMed] [Google Scholar]
- [3].Cao CX, Zhang YG, Wu SX, et al. Association of clinical features of bone and joint lesions between children and parents with Kashin-Beck disease in Northwest China. Clin Rheumatol 2013;32:1309–16. [DOI] [PubMed] [Google Scholar]
- [4].Guo Y, Li H, Yang L, et al. Trace element levels in scalp hair of school children in Shigatse, Tibet, an Endemic Area for Kaschin-Beck disease (KBD). Biol Trace Elem Res 2017;180:15–22. [DOI] [PubMed] [Google Scholar]
- [5].Li SJ, Yang LS, Wang WY, et al. Determination of trace elements in drinking water of Kashin-Beck disease (KBD) affected and non-affected areas in Tibet by ICP-AES. Guang Pu Xue Yu Guang Pu Fen Xi 2007;27:585–8. [PubMed] [Google Scholar]
- [6].Zhang F, Guo X, Zhang Y, et al. Genome-wide copy number variation study and gene expression analysis identify ABI3BP as a susceptibility gene for Kashin-Beck disease. Hum Genet 2014;133:793–9. [DOI] [PubMed] [Google Scholar]
- [7].Dai X, Li Y, Zhang R, et al. Effects of sodium selenite on c-Jun N-terminal kinase signalling pathway induced by oxidative stress in human chondrocytes and c-Jun N-terminal kinase expression in patients with Kashin-Beck disease, an endemic osteoarthritis. Br J Nutr 2016;115:1547–55. [DOI] [PubMed] [Google Scholar]
- [8].Dai X, Song R, Xiong Y. The expression of ERK and JNK in patients with an endemic osteochondropathy, Kashin-Beck disease. Exp Cell Res 2017;359:337–41. [DOI] [PubMed] [Google Scholar]
- [9].Du XA, Wang HM, Dai XX, et al. Role of selenoprotein S (SEPS1) -105G>A polymorphisms and PI3K/Akt signaling pathway in Kashin-Beck disease. Osteoarthritis Cartilage 2015;23:210–6. [DOI] [PubMed] [Google Scholar]
- [10].Xiong YM, Zou XZ, Chen Q, et al. Relationship between Gpx1 Pro198leu polymorphism and susceptibility of Kashin-Beck disease. Value Health 2015;18:A638. [Google Scholar]
- [11].Du XH, Dai XX, Xia Song R, et al. SNP and mRNA expression for glutathione peroxidase 4 in Kashin-Beck disease. Br J Nutr 2012;107:164–9. [DOI] [PubMed] [Google Scholar]
- [12].Sun W, Wang X, Zou X, et al. Selenoprotein P gene r25191g/a polymorphism and quantification of selenoprotein P mRNA level in patients with Kashin-Beck disease. Br J Nutr 2010;104:1283–7. [DOI] [PubMed] [Google Scholar]
- [13].Hao J, Wang W, Wen Y, et al. A bivariate genome-wide association study identifies ADAM12 as a novel susceptibility gene for Kashin-Beck disease. Sci Rep 2016;6:31792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hao J, Wang W, Wen Y, et al. Genome-wide association study identifies COL2A1 locus involved in the hand development failure of Kashin-Beck disease. Sci Rep 2017;7:40020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang W, Yu Y, Hao J, et al. Genome-wide DNA methylation profiling of articular cartilage reveals significant epigenetic alterations in Kashin-Beck disease and osteoarthritis. Osteoarthritis Cartilage 2017;25:2127–33. [DOI] [PubMed] [Google Scholar]
- [16].Luo M, Chen J, Li S, et al. Changes in the metabolism of chondroitin sulfate glycosaminoglycans in articular cartilage from patients with Kashin-Beck disease. Osteoarthritis Cartilage 2014;22:986–95. [DOI] [PubMed] [Google Scholar]
- [17].Sun LY, Meng FG, Li Q, et al. Effects of the consumption of rice from non-KBD areas and selenium supplementation on the prevention and treatment of paediatric Kaschin-Beck disease: an epidemiological intervention trial in the Qinghai Province. Osteoarthritis Cartilage 2014;22:2033–40. [DOI] [PubMed] [Google Scholar]
- [18].Zhang F, Dai L, Lin W, et al. Exome sequencing identified FGF12 as a novel candidate gene for Kashin-Beck disease. Funct Integr Genomics 2016;16:13–7. [DOI] [PubMed] [Google Scholar]
- [19].Zhang F, Wen Y, Guo X, et al. Genome-wide pathway-based association study implicates complement system in the development of Kashin-Beck disease in Han Chinese. Bone 2015;71:36–41. [DOI] [PubMed] [Google Scholar]
- [20].Zhang F, Wen Y, Guo X, et al. Genome-wide association study identifies ITPR2 as a susceptibility gene for Kashin-Beck disease in Han Chinese. Arthritis Rheumatol 2015;67:176–81. [DOI] [PubMed] [Google Scholar]
- [21].Alli OA, Ogbolu OD, Alaka OO. Direct molecular detection of Mycobacterium tuberculosis complex from clinical samples—an adjunct to cultural method of laboratory diagnosis of tuberculosis. N Am J Med Sci 2011;3:281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hu Y, Pan Z, Hu Y, et al. Network and pathway-based analyses of genes associated with Parkinson's disease. Mol Neurobiol 2017;54:4452–65. [DOI] [PubMed] [Google Scholar]
- [23].Green GH, Diggle PJ. On the operational characteristics of the Benjamini and Hochberg False Discovery Rate procedure. Stat Appl Genet Mol Biol 2007;6:Article27. [DOI] [PubMed] [Google Scholar]
- [24].Xiao S, Jia J, Mo D, et al. Understanding PRRSV infection in porcine lung based on genome-wide transcriptome response identified by deep sequencing. PLoS One 2010;5:e11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25:1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].O’Driscoll P, Merenyi E, Karmonik C, et al. SOM and MCODE methods of defining functional clusters in MRI of the brain. Conf Proc IEEE Eng Med Biol Soc 2014;2014:734–7. [DOI] [PubMed] [Google Scholar]
- [27].Lin N, Jiang J, Guo S, et al. Functional principal component analysis and randomized sparse clustering algorithm for medical image analysis. PLoS One 2015;10:e0132945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pasluosta CF, Dua P, Lukiw WJ. Nearest hyperplane distance neighbor clustering algorithm applied to gene co-expression analysis in Alzheimer's disease. Conf Proc IEEE Eng Med Biol Soc 2011;2011:5559–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Feldt S, Waddell J, Hetrick VL, et al. Functional clustering algorithm for the analysis of dynamic network data. Phys Rev E Stat Nonlin Soft Matter Phys 2009;79(5 Pt 2):056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen Q, Wang ZL, Xiong YM, et al. The Research on The Incidence of Kashin-Beck Disease and The Selenium Level In Children of Xunyi County. Value Health 2015;18:A638. [Google Scholar]
- [31].Chen Z, Li H, Yang L, et al. Hair selenium levels of school children in Kashin-Beck disease endemic areas in Tibet, China. Biol Trace Elem Res 2015;168:25–32. [DOI] [PubMed] [Google Scholar]
- [32].Chen J, Luo M, Wang W, et al. Altered proteolytic activity and expression of MMPs and aggrecanases and their inhibitors in Kashin-Beck disease. J Orthop Res 2015;33:47–55. [DOI] [PubMed] [Google Scholar]
- [33].Gao ZQ, Guo X, Duan C, et al. Altered aggrecan synthesis and collagen expression profiles in chondrocytes from patients with Kashin-Beck disease and osteoarthritis. J Int Med Res 2012;40:1325–34. [DOI] [PubMed] [Google Scholar]
- [34].Guo X, Ma WJ, Zhang F, et al. Recent advances in the research of an endemic osteochondropathy in China: Kashin-Beck disease. Osteoarthritis Cartilage 2014;22:1774–83. [DOI] [PubMed] [Google Scholar]
- [35].Wang S, Duan C, Zhang F, et al. The roles of the interaction of BCL2-Antagonist/Killer 1, apoptotic peptidase activating factor 1 and selenium in the pathogenesis of Kashin-Beck disease. Biol Trace Elem Res 2016;170:17–24. [DOI] [PubMed] [Google Scholar]
- [36].Zhao QM, Guo X, Lai JH, et al. Association of TNF-alpha and Fas gene promoter polymorphism with the risk of Kashin-Beck disease in Northwest Chinese population. Clin Rheumatol 2012;31:1051–7. [DOI] [PubMed] [Google Scholar]
- [37].Zhou X, Yang H, Guan F, et al. T-2 toxin alters the levels of collagen II and its regulatory enzymes MMPs/TIMP-1 in a low-selenium rat model of Kashin-Beck disease. Biol Trace Elem Res 2016;169:237–46. [DOI] [PubMed] [Google Scholar]
- [38].Huang L, Shi Y, Lu F, et al. Association study of polymorphisms in selenoprotein genes and Kashin-Beck disease and serum selenium/iodine concentration in a Tibetan population. PLoS One 2013;8:e71411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ning Y, Wang X, Wang S, et al. Is it the appropriate time to stop applying selenium enriched salt in Kashin-Beck disease areas in China? Nutrients 2015;7:6195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yao YF, Pei FX, Li XB, et al. Preventive effects of supplemental selenium and selenium plus iodine on bone and cartilage development in rats fed with diet from Kashin-Beck disease endemic area. Biol Trace Elem Res 2012;146:199–206. [DOI] [PubMed] [Google Scholar]
- [41].Ma WJ, Guo X, Yu YX, et al. Cytoskeleton remodeling and oxidative stress description in morphologic changes of chondrocyte in Kashin-Beck disease. Ultrastruct Pathol 2014;38:406–12. [DOI] [PubMed] [Google Scholar]
- [42].Wang SJ, Guo X, Zuo H, et al. Chondrocyte apoptosis and expression of Bcl-2, Bax, Fas, and iNOS in articular cartilage in patients with Kashin-Beck disease. J Rheumatol 2006;33:615–9. [PubMed] [Google Scholar]
- [43].Wang SJ, Guo X, Zuo H, et al. Chondrocyte apoptosis and the expression of Bcl-2, Bax, Fas and iNos in articular cartilage in Kashin-Beck disease. Di Yi Jun Yi Da Xue Xue Bao 2005;25:643–6. [PubMed] [Google Scholar]
- [44].Yang HJ, Zhang Y, Wang ZL, et al. Increased chondrocyte apoptosis in Kashin-Beck disease and rats induced by T-2 toxin and selenium deficiency. Biomed Environ Sci 2017;30:351–62. [DOI] [PubMed] [Google Scholar]
- [45].Wang W, Wei S, Luo M, et al. Oxidative stress and status of antioxidant enzymes in children with Kashin-Beck disease. Osteoarthritis Cartilage 2013;21:1781–9. [DOI] [PubMed] [Google Scholar]
- [46].Han J, Wang W, Qu C, et al. Role of inflammation in the process of clinical Kashin-Beck disease: latest findings and interpretations. Inflamm Res 2015;64:853–60. [DOI] [PubMed] [Google Scholar]
- [47].Tian L, Wang W, Hou W, et al. Autoimmune and inflammatory responses in Kashin-Beck disease compared with rheumatoid arthritis and osteoarthritis. Hum Immunol 2011;72:812–6. [DOI] [PubMed] [Google Scholar]
- [48].Kenawy HI, Boral I, Bevington A. Complement-coagulation cross-talk: a potential mediator of the physiological activation of complement by low pH. Front Immunol 2015;6:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kozarcanin H, Lood C, Munthe-Fog L, et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J Thromb Haemost 2016;14:531–45. [DOI] [PubMed] [Google Scholar]
- [50].Kurosawa S, Stearns-Kurosawa DJ. Complement, thrombotic microangiopathy and disseminated intravascular coagulation. J Intensive Care 2014;2:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wiegner R, Chakraborty S, Huber-Lang M. Complement-coagulation crosstalk on cellular and artificial surfaces. Immunobiology 2016;221:1073–9. [DOI] [PubMed] [Google Scholar]
- [52].Cookman CJ, Belcher SM. Estrogen receptor-beta up-regulates IGF1R expression and activity to inhibit apoptosis and increase growth of medulloblastoma. Endocrinology 2015;156:2395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang J, Singh N, Robinson-Taylor KS, et al. Hepatocyte autophagy is linked to C/EBP-homologous protein, Bcl2-interacting mediator of cell death, and BH3-interacting domain death agonist gene expression. J Surg Res 2015;195:588–95. [DOI] [PubMed] [Google Scholar]
- [54].Onwuameze OE, Nam KW, Epping EA, et al. MAPK14 and CNR1 gene variant interactions: effects on brain volume deficits in schizophrenia patients with marijuana misuse. Psychol Med 2013;43:619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Paillas S, Causse A, Marzi L, et al. MAPK14/p38alpha confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy 2012;8:1098–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.