

Abstract
The inflammatory response to infection or injury dramatically increases the hematopoietic demand on the bone marrow in order to replace effector leukocytes consumed in the inflammatory response. In the setting of infection, pathogen-associated molecular patterns induce emergency hematopoiesis, activating hematopoietic stem and progenitor cells to proliferate and produce progeny for accelerated myelopoiesis. Sterile tissue injury due to trauma also increases leukocyte demand, however the effect of sterile tissue injury on hematopoiesis is not well described. We find that tissue injury alone induces emergency hematopoiesis in mice subjected to polytrauma. This process is driven by IL-1/MyD88-dependent production of G-CSF. G-CSF induces expansion of hematopoietic progenitors including hematopoietic stem cells and multi-potent progenitors, and increases the frequency of myeloid-skewed progenitors. These data provide the first comprehensive description of injury-induced emergency hematopoiesis and identify an IL-1/MyD88/G-CSF dependent pathway as the key regulator of emergency hematopoiesis after injury.
Introduction:
All leukocytes are generated through the process of hematopoiesis, a hierarchical progression of differentiation whereby pluripotent hematopoietic stem cells (HSC) progress through a series of more specialized progeny, ultimately culminating in the generation of terminally differentiated effector cells (1). Specialization is associated with a decreased capacity for self-renewal and the acquisition of effector cell functions (2, 3). At rest, there is a basal rate of leukocyte turnover, which is regulated by clearance of senescent cells and replacement of cells consumed in response to infection. At baseline, leukocytes are replaced through proliferation of committed hematopoietic progenitors such as the granulocyte-monocyte progenitor (GMP) cells proliferating to produce additional neutrophils or monocytes (2). In response to local infections, the proliferative activity of committed progenitors is accelerated to provide the effector leukocytes that are required to replace those consumed by senescence or in response to low-level infectious challenges (4, 5).
In contrast, the immune response to severe infections dramatically increases the demand for leukocytes, rapidly outpacing the proliferative capacity of the committed progenitor pool (4). This drives recruitment of pluripotent populations such as multi-potent progenitors (MPP) and HSC into active hematopoiesis. This process is termed Emergency Hematopoiesis (EH) and is characterized by broad-based activation and expansion of hematopoietic stem and progenitor cell (HPSC) populations to generate the downstream leukocyte progeny needed for an effective immune response (4, 5). Prior studies have demonstrated that severe infections such as sepsis induce EH (6, 7), and that this phenotype can be recapitulated by exogenous administration of pathogen-associated molecular patterns (PAMPS) such as LPS (8) or the TLR2 agonist PAM3CSK4 (9).
Traumatic injury alone (in the absence of infection) also creates a hematopoietic demand due to the consumption of leukocytes in the local and systemic inflammatory response to tissue injury (10–12). Sterile injury has been shown to activate committed progenitors to increase granulopoiesis and monocytopoiesis (13, 14), although plasma isolated after injury has been shown to suppress ex-vivo bone marrow proliferation (15, 16). Hemorrhagic shock has been shown to increase the frequency of immature progenitors (17) but other models of sterile injury found no effect of injury on pluripotent short-term hematopoietic stem cells (ST-HSC) in young animals (18). Taken together, these data leave unresolved the effect of sterile traumatic injury on hematopoiesis. To establish the effect of injury on hematopoiesis, we measured hematopoietic stem and progenitor populations in a clinically relevant model of polytrauma. We find that trauma alone induces emergency hematopoiesis characterized by expansion of immature hematopoietic progenitors through IL-1/MyD88-dependent production of G-CSF, resulting in a progenitor population that is skewed toward myeloid cell production.
Methods.
Mice
C57BL/6J and MyD88−/− mice on the C57BL/6 background were obtained from Jackson Laboratory. All studies were conducted in accordance with the institutional guidelines for humane treatment of animals and were approved by the Washington University Animal Studies Committee.
Polytrauma model
Male C57BL/6 WT and MyD88−/− mice at 10–12 weeks of age were subjected to a multisystem injury consisting of bilateral lower extremity pseudofracture, limited hemorrhagic shock, and partial liver crush injury, as detailed below. Mice were maintained under general anesthesia (2% isoflurane) during the entire procedure. Pseudofracture consisted of lower extremity soft tissue crush injury, induced with a hemostat clamp, followed by the injection of a morselized bone suspension from the femurs and tibiae of a donor mouse. Limited hemorrhagic shock was induced by withdrawing 15% of the calculated total blood volume via cardiac puncture. For the liver crush injury, a hemostat clamp was used to apply six consecutive contusions over the entire area of the left liver lobe. All animals received buprenorphine (0.1 mg/kg) and fluids (1 ml saline) subcutaneously immediately after the procedure.
Cytokine blockade experiments
For G-CSF blockade experiments, mice were injected i/p with 25 μg anti-mouse G-CSF antibody (MAB414, R&D Systems) or rat IgG1 isotype control antibody (MAB005, R&D Systems) at 15 hours before the polytrauma procedure. A second dose of 25 μg antibody was given subcutaneously immediately before polytrauma. Bone marrow progenitor populations were analyzed at 24h after polytrauma. IL-1α and IL-1β blockade experiments were performed by injection (at 15 hours prior to polytrauma and at time of injury) of 150 μg anti-IL-1α antibody, clone ALF-161, and/or 150 μg anti-IL-1β antibody clone B122 (both from BioXCell). Control mice received polyclonal hamster IgG) (BioXCell). For IL-1R blockade experiments, mice were treated with 3 mg recombinant IL-1RA (Anikinra, SOBI pharmaceuticals) injected subcutaneously 1 hour before polytrauma; control animals received an equivalent volume of normal saline. In some experiments, 3 doses of IL-1RA were administered, at times −16 h, 0 h, and 8 h of the polytrauma procedure.
Tissue collection and processing
For plasma isolation, blood was taken via cardiac puncture into heparinized syringes and centrifuged at 10,000 rpm (9,400 xg) for 10 minutes at 4°C. The plasma was then stored at −80°C until analysis. Bone marrow was isolated from the femurs of mice by centrifugation of the bones at 6000 rpm (3380 xg) for 3 minutes, followed by red blood cell (RBC) lysis (HybriMax, Sigma Aldrich). Splenocyte suspensions were prepared by dissociating spleens over 70 μm meshes in HBSS buffer containing 10% bovine calf serum, followed by centrifugation (300xg for 10 minutes) and RBC lysis. Peripheral blood leukocytes were prepared from 500 μl whole blood (taken by cardiac puncture) by adding 5 ml RBC lysis buffer and incubating for 7 minutes at room temperature, followed by two washes with PBS. Peritoneal lavage was performed in euthanized mice by injecting 1 ml complete media (RPMI with 10% fetal calf serum) into the peritoneal cavity. The abdomen was briefly massaged before collection of the resulting cell suspension within the peritoneal cavity. Nucleated cell counts and cell viability were determined by diluting cell suspensions with a mixture of acridine orange and propidium iodide and analyzing on a K2 Cellometer (Nexcelom). For measuring IL-1 levels in liver homogenates, sections of injured and uninjured liver lobes were homogenized in PBS containing protease inhibitor (Pierce/Thermo Fisher) with a Tissue Rupture homogenizer (Qiagen). Cell lysates for IL-1 measurement were prepared by resuspending cell pellets in PBS containing protease inhibitor and 0.5% Triton X-100. Homogenates and cell lysates were spun at 10,000 rpm for 10 minutes, and supernatants were then frozen at −80°C.
Cytokine measurements
Plasma G-CSF and tissue IL-1α and IL-1β levels were measured with FlexSet cytometric bead array (CBA) kits (BD Biosciences). Lower detection limit for G-CSF in plasma samples was 25 pg/ml, and 10 pg/ml for IL-1α and IL-1β in cell lysates and tissue homogenates. Samples were acquired on a Canto II flow cytometer and analyzed using the FCAP software (all from BD Biosciences). IL-1 tissue levels were normalized to total protein concentration by Bradford assay (IBI Scientific) according to the manufacturer’s recommendations. For some experiments, cytokines in plasma at 3, 6, 24, and 48h after polytrauma were measured with the Milliplex Cytokine/Chemokine kit (MCYTMAG-70K-PX32; EMD Millipore) using a Bioplex 200 analyzer.
Flow cytometry
For progenitor cell analysis, cells isolated from bone marrow, spleen, and blood were stained with antibodies diluted in PBS with 2% bovine calf serum and 0.05% sodium azide (= FACS buffer). Cells were stained on ice for 20 minutes, followed by incubation with a dead cell dye (Zombie Aqua or Zombie NIR, both from Biolegend). Antibodies used for progenitor analyses were: lineage cocktail consisting of PerCP-Cy5.5-labeled antibodies to CD3, CD8, CD19, CD11b, Gr-1 (all from Biolegend), CD4, and Ter-119 (BD Biosciences); antibodies to progenitor markers Sca-1-FITC, CD117-BV421, CD135-PE, CD16/32-biotin, CD127-biotin, CD150-PE-Dazzle 594 (Biolegend), CD34-A647, CD48-BV510 (BD Biosciences). Biotin antibodies were detected with BV605-conjugated streptavidin (BD Biosciences). Early progenitor cells were gated as live, lineage-negative, CD117+ Sca-1+ (KSL) cells with the following phenotypes: CD150+ CD48− (LT-HSC), CD34+ CD135− (ST-HSC), CD34+ CD135+ (MPP). MPP subsets were gated as KSL cells with the following surface phenotypes: MPP2: CD135− CD150+ CD48+; MPP3: CD135− CD150− CD48+; MPP4: CD135+ CD150− CD48+. CLP were gated as live, lineage-negative, CD117low Sca-1low CD127+ CD135+ cells. Myeloid progenitors were gated as live, lineage-negative, CD117+ Sca-1− (KL) cells with the following phenotypes: CD34+ CD16/32lo (CMP), CD34+ CD16/32hi (GMP), CD34− CD16/32lo (MEP). Representative FACS plots and gating strategy shown in Supplemental Figure 2.
For leukocyte analysis, cells were stained with fluorochrome-conjugated antibodies to CD45, CD3, CD4, CD8, CD11b, CD11c, CD19, CD115, CCR2, NK1.1, Ly6C, Ly6G, TCRγδ, and (all from Biolegend), Alexa Fluor 700-conjugated CCR2 (R&D Systems), PE-CF594-conjugated Siglec F (BD Biosciences), followed by Zombie Aqua dye staining. Gating on individual immune cell subsets is shown in Supplemental Figure 3.
Following staining, cells were fixed with Cytofix buffer (BD Biosciences). Samples were acquired on an LSR Fortessa equipped with four lasers (488, 405, 640, and 552 nm) using the Diva software (BD Biosciences), and data was analyzed with the FlowJo software (Treestar Inc.).
Methylcellulose assays
To evaluate the hematopoietic potential of HSC from naive and injured animals, 10,000 bone marrow cells were mixed with methylcellulose medium containing growth factors (Methocult GF M3434 medium, StemCell Technologies) and plated in 35-mm tissue culture plates. The number of colonies per plate were counted on day 7 of culture.
Results.
Sterile Polytrauma Injury Induces Emergency Hematopoiesis.
To define the role of sterile injury on the hematopoietic system, we first established a clinically relevant model of sterile injury by modifying our published model of polytrauma (19). Young mice were subjected to a bilateral lower extremity pseudofracture injury and a laparotomy followed by 15% blood volume hemorrhage and a blunt liver contusion. This injury resulted in <5% mortality but did cause an approximately 15% loss of body weight by post-injury day 2, slowly returning to baseline weight over the following 7 days (Supplemental Fig. 1A). To characterize the magnitude of systemic inflammation induced by this injury, we measured plasma cytokines at 24 hours after injury as compared to sham-manipulated animals. This polytrauma injury induced low-magnitude systemic inflammatory response that was almost completely resolved by 24 hours (Supplemental Fig. 1B). This model recapitulates the clinical situation of a complex non-lethal multi-system trauma associated with a transient acute inflammatory phase (20).
To establish the effect of injury on hematopoiesis, we measured HSPC populations in the bone marrow after polytrauma. Mice were subjected to trauma as described above, and 24 hours after injury bone marrow was isolated and assayed by flow cytometry. Hematopoietic stem and progenitor cell populations were analyzed using previously described markers (See materials and Supplemental Figure 2) (2, 3, 21). At 24 hours after injury, injured mice had significantly higher numbers of immature progenitors in the bone marrow (Figure 1A). This was manifest in a significant increase in the heterogenous c-kit+/Sca-1+/lineage negative (KSL) progenitor population, and also in the more rigorously defined populations, including long-term hematopoietic stem cells (LT-HSC), short-term hematopoietic stem cells (ST-HSC) and multipotent progenitor cells (MPP). We observed a significant increase in both the absolute number of cells (Figure 1B) and also the frequency of progenitors among bone marrow cells (Figure 1C). We also measured the number and frequency of committed progenitors including common myeloid progenitors (CMP), common lymphoid progenitors (CLP), granulocyte-macrophage progenitors (GMP) and megakaryocyte-erythroid progenitors (MEP). Consistent with past studies (13), we found an increase in the frequency of GMP and MEP, although there was no difference in the absolute numbers of committed progenitor measured.
Figure 1.

Injury Induces Expansion of Immature HSPC Populations. C57BL/6 mice (n=11–12) were subjected to polytrauma. 24 hours after injury, bone marrow was isolated from injured mice and naïve controls and analyzed by flow cytometry. A) Representative FACS plots for identification of hematopoietic progenitor cells. Early progenitor cells were gated as live, lineage-negative, CD117+ (cKit) Sca-1+ (KSL) cells with the following phenotypes: CD150+ CD48− (LT-HSC), CD34+ CD135− (ST-HSC), CD34+ CD135+ (MPP). The total number of cells per femur (B, mean+/− SD) and the frequency of each population among live bone marrow cells are shown (C, individual values shown, horizontal bar/whisker represent mean+/−SD). Data are combined from 3 independent series of experiments. *= p<0.05 by Mann-Whitney U test.
To determine if these injury-induced changes in HSPC populations were durable over time, we repeated these studies at 10 and 21 days after trauma. At 10 days after trauma, we detected persistent increases in KSL, LT-HSC, ST-HSC, MPP and CLP populations. By 21 days, all populations had returned to baseline except a small but statistically significant elevation in ST-HSC frequency (Figure 2).
Figure 2.

Time Course of Injury-Induced Changes of Hematopoietic Progenitor Frequencies. WT C57BL/6 mice were subjected to polytrauma. At 1, 10 and 21 days after injury, bone marrow was isolated (n=11–18 per time point for injured; n=19 for naive). and progenitor frequency assay by FACS as in Figure 1A. The frequency of each population among live bone marrow cells was calculated. *= p<0.05 by Mann-Whitney U test vs. naïve, mean+/−SD shown; data pooled from 2–4 independent experiments at each time point.
Trauma Skews Progenitors to Myeloid-Predominant Progenitors
The LT-HSC pool is a heterogenous population comprised of both myeloid-predominant and lymphoid-predominant HSC clones (22, 23). Myeloid predominant HSC can be identified by expression of the cell surface molecule CD41 (24). Twenty-four hours after injury we measured the frequency of LT-HSC expressing CD41 by flow cytometry. We found that injury resulted in a shift toward myeloid- predominant HSC manifest as a greater than 3-fold increase in the frequency of CD41+ LT-HSC (Figure 3A). These data demonstrate that injury is acutely altering the LT-HSC cohort available for hematopoiesis. It is unclear if the increased CD41 expression reflects expansion of the existing baseline CD41+ LT-HSC or induction of CD41 within the CD41- LT-HSC cohort. However, the increased population of CD41+ LT-HSC suggests a skewing of hematopoiesis toward production of myeloid cells.
Figure 3.

Injury Skews Immature Progenitors Toward Myeloid Cell Production. C57BL/6 mice were subjected to polytrauma. Bone marrow was isolated from naïve animals or injured animals 1 or 10 days after injury and analyzed by flow cytometry. A: left panel, representative histogram of CD41 expression on LT-HSC. Right panel, the frequency of CD41+ cells within LT-HSC. n=5/group; data pooled from 2 independent experiments; individual values shown, horizontal bar/whisker represent mean+/−SD; B. Representative FACS plots for identification of MPP subset populations within KSL; MPP2: CD135− CD150+ CD48+; MPP3: CD135− CD150− CD48+; MPP4: CD135+ CD150− CD48+. C. The frequency of MPP subsets within BM at 0, 1 and 10 days after injury * = p<0.05 by Mann-Whitney U test. D. Absolute number of each MPP subset per femur from naïve (Day 0) mice and at 1, 10, and 21 days after injury. *, #, @ = p<0.05 by Mann-Whitney U test vs. day 0 (naïve). C,D data represented as mean+/−SD. E. The distribution of MPP populations expressed as a frequency of total MPP at 0, 1, 10 and 21 days after injury. C-E n=11–19 per time point, data pooled from 3–4 independent experiments per time point.
The multipotent progenitor population also contains a heterogenous population of cells distributed across a continuum of myeloid vs. lymphoid potential. Several subsets of MPP can be identified on expression of CD150, CD48 and Flk2 (CD135) (21). The MPP2 and MPP3 population predominately contribute to myelopoiesis; MPP4 are primarily lymphopoietic progenitors (21). We assayed MPP populations in the bone marrow at 1, 10 and 21 days after injury (Figure 3B, C). We found increases in the frequency of all three MPP populations at 1 day after trauma (Figure 3C). At 10 days after injury, MPP2 and MPP4 had regressed toward baseline, but remained statistically significantly elevated. In contrast, the frequency of the myeloid-predominate MPP3 population remained consistently elevated 10 days after injury (Figure 3C). At 21 days after injury, both MPP2 and MPP3 frequencies remained slightly elevated over baseline; there was no statistically significant increase in MPP4. We then measured the absolute number of each MPP subset. Consistent with prior studies, in naïve animals, MPP4 comprised greater than 75% of the total MPP pool (Figure 3D, E). One day after injury there was a small expansion in all MPP populations. At 10 days, we found a significant expansion in the myeloid MPP population such that MPP2 and MPP3 combined to make up >40% of the total MPP populations (Figure 3E). By day 21, the MPP4 population distribution had returned to baseline and small but statistically significant increases in the number of myeloid-predominant MPP2 and MPP3 remained detectable (Figure 3D).
To determine if the injury-induced changes in HSPC populations were associated with functional changes in hematopoiesis, we isolated bone marrow, spleen and blood at 10 or 21 days after injury and measured myeloid and lymphoid leukocyte numbers and frequencies by flow cytometry (Supplemental Figure 3). In the bone marrow 10 days after injury, there was an increase in the number of Ly6Ghi (mature) neutrophils and monocytes (Figure 4A). Eosinophil numbers, as well as T- and B-cell numbers were decreased (Figure 4A, Supplemental Figure 4). By 21 days, we found that injury was associated with increased numbers of Ly6Glo (immature) neutrophils and eosinophils in the injured mice (Figure 4A). There was trend toward increased numbers of total monocytes and a statistically significant increase in classical (CCR2hi) monocytes (Figure 4A and Supplementary Figure 4). These changes were associated with a reciprocal decrease in B-cell frequency but no change in absolute B-cell numbers (Supplementary Figure 4).
Figure 4.
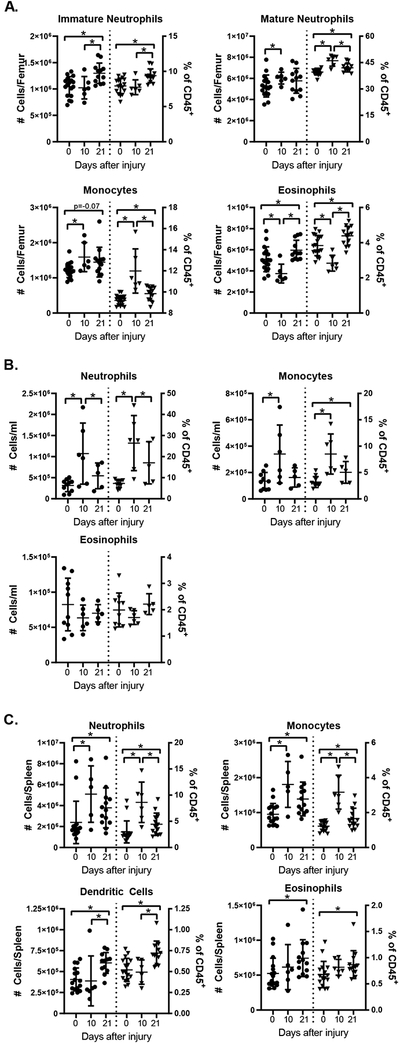
Injury is Associated with Increased Myeloid Cell Numbers in the Bone Marrow and Spleen. WT mice were subjected to polytrauma; bone marrow (A), blood (B) and spleens (C) were harvested 10 or 21 days after injury (or from naïve animals) and analyzed by flow cytometry. Absolute numbers of cells (left y axis) and frequency among CD45+ cells (right y axis) are shown; horizontal bar/whisker represent mean+/−SD). Data are pooled from 2–3 independent experiments per time point. * = p<0.05 by Mann-Whitney U test. Cell subset gating was done as shown in Supplemental Figure 4; splenic dendritic cells were identified as CD3−CD19−NK1.1−CD11b+CD11c+ cells.
We then evaluated the circulating and splenic leukocyte populations. In the circulation 10 days after injury, we found increased numbers of neutrophils and monocytes (Figure 4B); concurrently there was a decrease in B- and T-cell populations (See Supplementary Figure 4). By 21 days after injury, there were no statistically significant differences in myeloid cell populations, however absolute numbers of B- and T-cells were decreased (See Supplementary Figure 4). Splenic leukocytes were also evaluated. 10 days after injury neutrophil and monocyte numbers were increased and B cell numbers were decreased; at 21 days, we detected significantly higher numbers of all analyzed myeloid populations: numbers of neutrophils, monocytes, eosinophils and dendritic cells were increased in injured mice vs. naïve controls (Figure 4C). This was associated with a decrease in CD4+ and CD8+ T-cell frequencies, but there was no difference in absolute T-cell numbers. (Supplementary Figure 4).
IL-1/MyD88 Drives G-CSF Production and Progenitor Expansion after Trauma.
Given that we measured a very limited and rapidly resolving systemic inflammatory response to trauma (Supplementary Figure 1B), we sought to measure a broad panel of circulating mediators in order to determine the mechanisms by which injury induces EH. We isolated plasma from sham- and injured mice 24 hours after manipulation and measured soluble mediators using a multiplexed cytokine assay. We found that there were very limited changes in circulating mediator populations, with the exception of the growth factor granulocyte colony-stimulating factor (G-CSF). We detected a 2-fold increase in G-CSF in injured as compared to sham manipulated mice, with injured animals having >1600 pg/ml G-CSF in the plasma, as compared to <800 pg/ml after sham manipulation (Table I). These data are consistent with previous data demonstrating that trauma is associated with elevated G-CSF levels both in animal model systems (25) and in injured patients (26). To better characterize the G-CSF production induced by injury, we measured G-CSF at 0, 3, 6, 24 and 48 hours after trauma and found that by 3 hours after injury elevated levels of G-CSF were detectable in the plasma, which peaked by 6 hours; G-CSF levels decreased by 24 hours, but remained elevated above baseline at both 24 and 48 hours after injury (Figure 5A).
Table I:
Plasma Cytokines 24 hours after Polytrauma
| Sham (mean+/− SD, n=13) |
Polytrauma (mean+/− SD; n=13) |
|
|---|---|---|
| G-CSF | 769+/−707 | 1610+/−1064* |
| GM-CSF | 65+/−18 | 72+/−27 |
| IL-1α | 77+/−39 | 109+/−72 |
| IL-1β | ND | ND |
| IL-2 | 9+/−2 | 10+/−1 |
| IL-5 | 39+/−41 | 24+/−23 |
| IL-6 | 364+/−790 | 581+/−1324 |
| IL-7 | 12+/−6 | 18+/−11 |
| IL-9 | ND | 80+/−66 |
| IL-10 | 12+/−8 | 31+/−57 |
| IL-12p40 | 9+/−2 | 12+/−8 |
| IL-12p70 | 18+/−19 | 12+/−8 |
| IL-13 | 203+/−104 | 226+/−134 |
| LIX | 355+/−202 | 297+/−122 |
| IL-15 | 123+/−41 | 146+/−84 |
| IP-10 | 185+/−71 | 264+/−164* |
| KC | 1149+/−1160 | 791+/−690 |
| MCP-1 | 157+/−348 | 117+/−118* |
| MIP-1α | 71+/−19 | 82+/−27 |
| MIP-1β | ND | ND |
| M-CSF | 16+/−9 | 27+/−18 |
| MIP-2 | 77+/−37 | 101+/−62 |
| MIG | 52+/−9 | 104+/−95 |
| RANTES | 43+/−18 | 52+/−20 |
| TNF-α | 9+/−3 | 14+/−12 |
p<0.05 vs. Sham by Mann-Whitney U Test
Figure 5.

G-CSF Drives Expansion of Immature Hematopoietic Progenitors after Injury. A. WT mice were subjected to polytrauma and plasma harvested at 3, 6, 24 and 48 hours after injury or from naïve mice (n=4–13/ time-point). Plasma G-CSF was measured by cytometric bead array. Data represented as mean+/−SD; *= p<0.05 vs. naive by ANOVA/Dunn’s Multiple Comparisons Test. Data are pooled from 2–4 independent experiments per time point. B-E: C57BL/6 mice were subjected to polytrauma. 15 hours before injury and at the time of injury, animals were treated with G-CSF blocking antibody or isotype control. 24 hours after injury, bone marrow was harvested and analyzed by flow cytometry for the frequencies of progenitor populations. B. Representative FACS plots showing progenitor subsets in mice with and without G-CSF blockade. C. Progenitor frequency with or without G-CSF blockade. D) MPP subset frequencies with and without G-CSF blockade. C/D: n=7–9, data pooled from 3 independent experiments. E. Frequency of CD41+ LT-HSC (n=3–4, data pooled from 2 independent experiments). Individual values shown, horizontal bar/whisker represent mean+/−SD.
Prior work has demonstrated that exogenous G-CSF alone is sufficient to induce emergency hematopoiesis (12, 13). To establish the role of G-CSF in inducing EH after trauma, we assayed the effect of G-CSF antibody blockade on injury-induced changes in HSPC. Mice were treated with two doses of G-CSF blocking antibody (or isotype-matched control antibody) before injury and progenitor populations analyzed by FACS as before (Figure 5B). We found that G-CSF blockade significantly attenuated the injury-induced expansion of HSPC, blocking expansion of KSL, LT-HSC and ST-HSC populations (Figure 5C). In contrast, we found no difference in committed progenitor frequency in the setting of G-CSF blockade, with an equivalent expansion of GMP and MEP after polytrauma in mice treated with G-CSF blocking antibody. These data suggest that expansion of immature progenitors (i.e. HSC) is driven by G-CSF production, whereas early changes in committed progenitors are G-CSF independent.
To determine the role of G-CSF in the myeloid skewing of early hematopoietic progenitors, we measured the effect of G-CSF blockade on the MPP subset distribution 24 hours after trauma. MPP populations were identified as before (Figure 5B) and their frequencies were assessed. G-CSF blockade effectively blocked expansion of all MPP subsets 24 hours after injury (Figure 5D). G-CSF blockade did not significantly alter the induction of CD41 on LT-HSC. These data demonstrate that G-CSF is a key driver of MPP expansion induced by trauma.
Prior studies have demonstrated that MyD88 signaling by Toll-like receptors including TLR4 and TLR2 is required to induce emergency hematopoiesis (8, 9, 27, 28). In contrast, studies by others have demonstrated MyD88-independent EH in the setting of experimental cecal-ligation and puncture induced sepsis (7). Others have demonstrated that sterile injury can induce G-CSF production in a MyD88-dependent manner in response to sterile chemical inflammation (29). To determine the role of MyD88 in the induction of G-CSF and EH by polytrauma, we first measured G-CSF levels in WT and MyD88−/− mice 24 hours after trauma. WT and MyD88−/− were subjected to polytrauma and both plasma G-CSF levels and progenitor populations were assayed by flow cytometry (representative FACS plots are shown in Figure 6A). We noted that the injury-induced G-CSF induction was completely abolished in MyD88−/− animals (Figure 6B).
Figure 6.

MyD88 Drives G-CSF Production and Hematopoietic Progenitor Expansion after Injury. WT (C57BL/6) and MyD88−/− mice were subjected to polytrauma. 24 hours after injury bone marrow and plasma was harvested and G-CSF measured by cytometric bead array (CBA). A. Representative FACS plots showing progenitors in WT and MyD88−/− mice 24 hours after injury. B. Plasma G-CSF from WT and MyD88−/− mice 24 hours after injury C. HSPC frequency was assayed by flow cytometry; *= p<0.05 by Mann-Whitney U test. D. 104 bone marrow cells were cultured in methylcellulose media. The number of cell-colony-forming units was counted at day 7 of culture; (mean+/−SEM, n=3/group, *=p<0.05 by t-test). B,C: Data are pooled from 3 independent experiments. Individual values shown, horizontal bar/whisker represent mean+/−SD). C: Data are pooled from 2 independent experiments.
We then evaluated the role of MyD88 on trauma-induced EH by measuring progenitor populations in WT and MyD88−/− mice 24 hours after polytrauma. Consistent with our G-CSF blockade experiments (Figure 5) we found that in the absence of MyD88 signaling, trauma had no effect on the frequency of LT-HSC or ST-HSC populations (Figure 6C). These data demonstrate that MyD88 signaling is required for both G-CSF production and expansion of LT-HSC and ST-HSC after polytrauma. Inflammation can be associated with changes in the progenitor cell-surface immunophenotype. To determine if the increased frequency of progenitor cells represented a true change in functional immature progenitor frequency, we measured the number of cell colony forming units (CFU-C) in the bone marrow of naïve and injured mice. Bone marrow cells from naïve and injured mice were cultured in methylcellulose media containing growth factors, and the number of colonies formed was enumerated after 7 days. Consistent with the increase in the frequency of LT-HSC as assayed by flow cytometry, we found that the bone marrow from injured WT mice produced higher numbers of colonies as compared to that isolated from naïve animals (Figure 6D). In contrast, injury had no effect on the number of colonies produced by bone marrow from MyD88−/− animals. We did note a trend toward increase in the number of CFU in the bone-marrow of the MyD88−/− mice as compared to WT animals. These data are consistent with our past studies demonstrating TLR-mediated signaling in the HSC leads to HSC quiescence, and that progenitors from untreated MyD88−/− or TLR4−/− mice have a repopulation advantage as compared to WT cells (27).
Given that we observed MyD88 dependent production of G-CSF in the setting of sterile injury, we hypothesized that this reflected activation of MyD88-dependent IL-1 signaling. To test this hypothesis, we treated mice with recombinant IL-1 receptor antagonists (IL-1RA, Anikinra, Sobi pharmaceuticals) 1 hour before polytrauma. Plasma was harvested 6 hours after injury and plasma G-CSF level was measured by cytometric bead array (CBA). We found that IL-1 blockade nearly completely abrogated injury-induced elaboration of G-CSF (Figure 7A). To determine the role of the IL-1/G-CSF axis in driving progenitor expansion, we measured the frequency of hematopoietic progenitor populations 24 hours after injury in mice treated with IL-1RA. Mice were treated with 3 doses of IL-1 RA, one given 16 hours before injury, a second at the time of injury and a 3rd dose 8 hours after injury. Bone marrow was harvested 24 hours after injury and progenitor populations measured by flow cytometry. Progenitor populations were identified as before (Figure 7B). Consistent with our G-CSF blockade experiments, we found that IL-1 blockade resulted in decreased frequencies of LT-HSC and ST-HSC but not the bulk MPP populations (Figure 7C). Again consistent with G-CSF blockade, IL-1RA treatment block the injury induced increase in MPP2 subpopulations, but there was only a modest (not statistically significant) trend toward a decrease in MPP3 (Figure 7C). IL-1 blockade did not have a significant effect on the MPP4 populations (Figure 7C).
Figure 7.
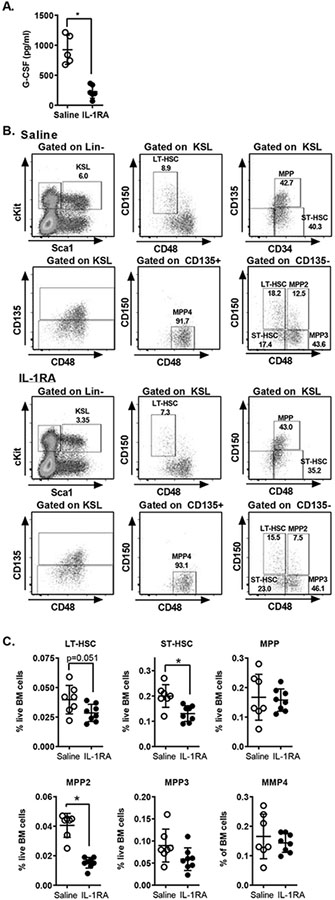
IL-1 Drives G-CSF Production and Progenitor Expansion after Injury. A. WT mice were treated with recombinant IL-1RA 1 hour before polytrauma; plasma G-CSF was measured by CBA. B, C. WT mice were treated with 3 doses of IL-1 RA (16 hours before injury, at the time of injury and 8 hours after injury). Bone marrow was harvested 24 hours after injury and analyzed by flow cytometry. B. Representative FACS plots showing progenitors in mice with and without IL-1RA. C. Frequency of LT-HSC, ST-HSC and MPP populations. *=p<0.05 by Mann-Whitney U Test. Data are pooled from 2–3 independent experiments. Individual values shown, horizontal bar/whisker represent mean+/−SD)
IL-1RA blocks the activity of the IL-1R1 receptor in response to both IL-1α and IL-1β. To determine whether IL-1α or IL-1β was driving G-CSF production, we measured the effect of specific cytokine blockade on G-CSF production in response to injury. Mice were treated with blocking antibodies against IL-1α, IL-1β or both in combination; control animals were treated with isotype control antibody. 6 hours after injury, plasma was harvested and G-CSF levels measured by cytometric bead array. We found that anti-IL-1α significantly blocked injury-induced G-CSF production (Figure 8A). There was a trend towards decreased G-CSF in the presence of antibody-mediated blocking of IL-1β, although this was not statistically significant. Although these data and data in Figure 7 suggest that IL-1 is driving G-CSF production and emergency hematopoiesis after injury, we did not detect any significant levels of IL-1α or IL-1β in the plasma of injured mice. To determine the cellular source of IL-1, we measured IL-1 protein levels in liver homogenates, as well as in cell lysates prepared from bone marrow and peritoneal lavage from naïve animals, as well as animals at 6 hours after injury. IL-1α and IL-1β were measured by CBA and the amount of IL-1 relative to total protein was calculated to account for differences in tissue mass and/or cellular composition. We found very low levels of IL-1 within bone marrow cells; injury had no effect on the levels of IL-1α in this tissue (Figure 8B), and was associated with a decrease in IL-1β (Figure 8C). In contrast we found that the peritoneal exudate cells from injured mice contained significantly higher levels of both IL-1α and IL-1β as compared to those from naïve animals (Figure 8B,C). There was no difference in IL-1 α levels in injured vs. naïve liver and minimal levels of IL-1β in injured liver tissue (Figure 8B,C).
Figure 8.
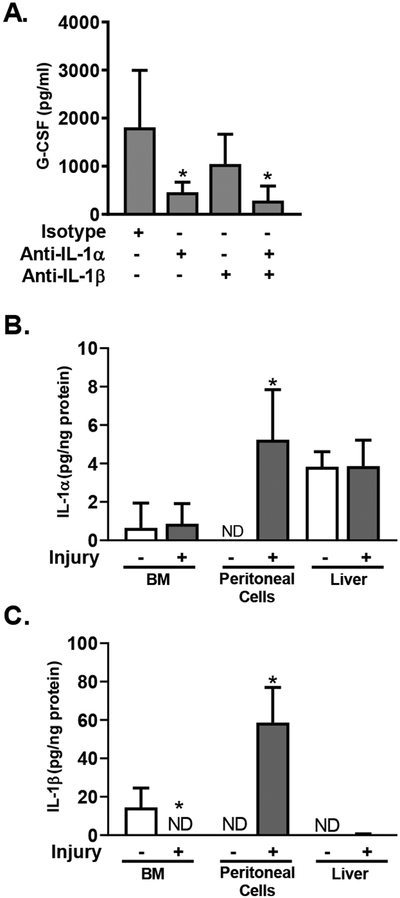
IL-1 Drives G-CSF Expression. A. WT mice were treated with antibodies against IL-1α and IL-1β alone or in combination as indicated, or with isotype control antibody, 1 hour before polytrauma. 6 hours after injury plasma G-CSF was measured. Data represented as mean+/−SD, n=4–5/group, data are pooled from 2 independent experiments. *=p<0.05 by Mann-Whitney U Test. B/C. IL-1 analysis in tissues from mice at 6h after injury vs. naive mice. IL-1α (B) and IL-1β (C) were measured by CBA in cell lysates from BM, peritoneal exudate, and blood, and in liver homogenates. Cytokine levels were normalized to total protein content. Data represented as mean+/−SD, n=4/group, data are pooled from 2 independent experiments. *=p<0.05 by Mann-Whitney U Test.
Discussion:
We report that sterile tissue injury alone results in expansion of immature hematopoietic progenitors including LT-HSC and MPP populations through IL-1/MyD88-dependent production of G-CSF, leading to a sustained increase in myeloid-biased progenitors at 10 days after insult. These data demonstrate that emergency hematopoiesis can be induced independent of exogenous pathogen-associated molecular patterns and describe a novel IL-1/G-CSF access in inducing emergency hematopoiesis. These data suggest a potential mechanism for the persistent changes in immune function seen in severely injured patients (30–32).
Previous work has demonstrated that exogenous G-CSF can increase the number of phenotypic HSC in the bone-marrow (27). Our data define a biologically relevant role for this pathway, with IL-1 and G-CSF driving expansion of pluripotent hematopoietic progenitors in a clinically relevant model of moderate, nonlethal polytrauma. We find that blocking injury-induced G-CSF almost completely abrogates the expansion of HSC induced by injury. These effects of G-CSF also extend to the MPP compartment, and we find that G-CSF similarly blocks the effect of injury on the MPP2, MPP3 and MPP4 subsets. Although we didn’t find a statistically significant decrease in the bulk MPP population, we did observe a trend toward decreased MPP frequency in the setting of G-CSF blockade after injury. Consistent with past literature, we have defined MPP based on expression of CD34 and FLT3 (CD135) (33) whereas the MPP subsets are defined based on expression of a combination of CD135/CD48 and CD150. There is incomplete overlap between the MPP2/3/4 populations and the ST-HSC and MPP populations as defined by these markers. In addition inflammation can affect the expression of cell surface markers on the progenitor populations and changes in cell surface marker expression may also contribute decreased magnitude of the effect of G-CSF on the MPP population (as compared to the MPP2/3/4 populations). Interestingly, G-CSF blockade had no effect on the frequency of the committed GMP and MEP populations, despite the well-defined role of G-CSF in granulopoiesis (4, 34, 35).
Consistent with myeloid biasing of the progenitors for up to 10 days, we do report an increase in myeloid cell populations in the bone-marrow and spleen up to 3 weeks after injury. We acknowledge that the differences were modest, and the functional consequences of these small differences in neutrophil and monocyte populations are unclear. Further, although we hypothesize that these data could be an expression of myeloid biasing of the progenitors by emergency hematopoiesis, there may be ongoing inflammation related to the injury that has incompletely resolved at this time. Future studies will be required to determine if the injury-induce expansion of progenitors has significant functional consequences.
Our results also define a new role for IL-1/MyD88 in emergency hematopoiesis after injury. Scumpia et al.(7) demonstrated that in the setting of endotoxemia or bacterial sepsis LT-HSC expansion is independent of MyD88 and IL-1 activity. In contrast, we find a complete block in both G-CSF production and expansion of the LT-HSC after trauma in the MyD88 knockout mice, and that IL-1 blockade with recombinant IL-1RA also almost completely blocked G-CSF production, largely recapitulating the effects of G-CSF blockade on the changes in hematopoietic progenitors induced by injury. These data are consistent with previous work demonstrating that G-CSF production in response to chemical peritonitis in driven by IL-1α and that IL-1α/IL1R signaling is required to initiate the inflammatory response to necrotic cell death (36, 37). Although we find that abrogating IL-1/MyD88 signalling effectively prevented both G-CSF production and emergency hematopoiesis, we did not detect elevated levels of IL-1α in the plasma at 3, 6 or 24 hours after injury (data not shown). However, we were able to detect a significant increase in IL-1 in the peritoneal exudate cells after injury. Liver injury is associated with a significant peritoneal infiltrate, and the increased IL-1 we detect likely reflects infiltrating neutrophils and the activation of peritoneal resident macrophages and neutrophils. These data are consistent with prior work demonstrating that in the setting of chemical peritonitis, IL-1α is produced by peritoneal neutrophils and drives G-CSF production, regulated by NADPH oxidase(29). Alternatively, Boettcher et al have demonstrated that in the setting of endotoxemia, G-CSF is produced by endothelial cells in a MyD88-dependent pathway(34). These authors presume that MyD88 was acting downstream of the Toll-like receptors in response to LPS. IL-1 is well known to be produced in response to LPS(38), and our results suggest that endothelial-cell G-CSF production in the setting of LPS may be mediated by IL-1 acting as a secondary messenger produced in response to LPS. Future studies will be required to parse the sources of G-CSF and IL-1.
Prior studies have evaluated changes in early hematopoietic progenitors after sterile injury (14, 17, 18). Schneider et al. reported that hemorrhagic shock caused an increase in the frequency of mixed population of progenitors, whereas Silva et al.(14) and Nacionales et al. (18) found no change in pluripotent progenitor populations in young animals after burn injury or polytrauma, although Silva et al. did report an increase in myeloid-committed progenitors based on ex-vivo culture assays. These studies utilize a heterogenous collection of cell-surface phenotypes to define the progenitor population. We describe a complete differential of progenitors in the bone marrow, and using rigorous definitions, we find that sterile injury results in expansion of both pluripotent progenitor populations and myeloid committed progenitors. We further find that the effects of injury on the hematopoietic compartment are sustained for at least 10 days after injury. The persistent changes in the bone-marrow progenitor cohort suggest that changes in progenitor function could underlie the persistent immune function defects observed in critically injured patients. Injured patients develop nosocomial infections at 5–10 days after their trauma (39). Most myeloid cells have a circulation half-life of 8–24 hours under baseline conditions (40, 41), and systemic insults such as critical illness are associated with margination of circulating cells with rapid replacement of those cells by new bone marrow emigrants (42). The leukocytes in circulation by 48 hours after injury are likely derived from progenitor cells in the bone marrow after the insult, and therefore any injury-induced changes in function may be encoded by the progenitor cells during differentiation. This hypothesis is consistent with recent data demonstrating that IL-1 mediated “immunologic training” of hematopoietic progenitors can lead to persistent changes in innate immune function (43, 44).
In conclusion, we find that sterile polytrauma injury induces expansion of the pluripotent hematopoietic stem and progenitor cell populations. Progenitor expansion is mediated by G-CSF, produced in response to IL-1 through a MyD88 dependent pathway. Progenitor expansion is associated with an increase in myeloid biased progenitors, including CD41+ LT-HSC and MPP2/MPP3 subpopulations, with the increase in myeloid-biased progenitors persisting for up to 21 days after injury. These results suggest that the persistent defects in immune function observed after injury may result from changes progenitor cohort and suggest that modulation of progenitor function may be a potential pathway to modulate injury-induced immune dysfunction.
Supplementary Material
Key Points:
Traumatic injury alone without infection can induce emergency hematopoiesis.
Trauma induces emergency hematopoiesis though IL-1 dependent production of G-CSF.
Footnotes
This manuscript supported in part by the Surgical Infection Society Junior Faculty Fellowship Award (IRT) and by NIH/NHLBI 1R01 HL134896–01 (LS)
References:
- 1.Cullen SM, Mayle A, Rossi L, and Goodell MA. 2014. Hematopoietic stem cell development: an epigenetic journey. Current topics in developmental biology 107: 39–75. [DOI] [PubMed] [Google Scholar]
- 2.Challen GA, Boles N, Lin KK, and Goodell MA. 2009. Mouse hematopoietic stem cell identification and analysis. Cytometry. Part A : the journal of the International Society for Analytical Cytology 75: 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi L, Challen GA, Sirin O, Lin KK, and Goodell MA. 2011. Hematopoietic stem cell characterization and isolation. Methods in molecular biology (Clifton, N.J.) 750: 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manz MG, and Boettcher S. 2014. Emergency granulopoiesis. Nature reviews. Immunology 14: 302–314. [DOI] [PubMed] [Google Scholar]
- 5.Takizawa H, Boettcher S, and Manz MG. 2012. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 119: 2991–3002. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez S, Chora A, Goumnerov B, Mumaw C, Goebel WS, Fernandez L, Baydoun H, HogenEsch H, Dombkowski DM, Karlewicz CA, Rice S, Rahme LG, and Carlesso N. 2009. Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood 114: 4064–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinstein JS, Cuenca AG, Al-Quran S, Bovio I, Akira S, Kumagai Y, and Moldawer LL. 2010. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. Journal of immunology (Baltimore, Md. : 1950) 184: 2247–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, Heikenwalder M, and Manz MG. 2012. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. Journal of immunology (Baltimore, Md. : 1950) 188: 5824–5828. [DOI] [PubMed] [Google Scholar]
- 9.Herman AC, Monlish DA, Romine MP, Bhatt ST, Zippel S, and Schuettpelz LG. 2016. Systemic TLR2 agonist exposure regulates hematopoietic stem cells via cell-autonomous and cell-non-autonomous mechanisms. Blood cancer journal 6: e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, Pless R, Gelman AE, Krupnick AS, and Miller MJ. 2010. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proceedings of the National Academy of Sciences of the United States of America 107: 18073–18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald B, and Kubes P. 2016. Innate Immune Cell Trafficking and Function During Sterile Inflammation of the Liver. Gastroenterology 151: 1087–1095. [DOI] [PubMed] [Google Scholar]
- 12.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, and Kubes P. 2010. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330: 362–366. [DOI] [PubMed] [Google Scholar]
- 13.Santangelo S, Gamelli RL, and Shankar R. 2001. Myeloid commitment shifts toward monocytopoiesis after thermal injury and sepsis. Annals of surgery 233: 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva KD, Gamelli RL, and Shankar R. 2001. Bone marrow stem cell and progenitor response to injury. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 9: 495–500. [DOI] [PubMed] [Google Scholar]
- 15.Livingston DH, Anjaria D, Wu J, Hauser CJ, Chang V, Deitch EA, and Rameshwar P. 2003. Bone marrow failure following severe injury in humans. Annals of surgery 238: 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livingston DH, Gentile PS, and Malangoni MA. 1990. Bone marrow failure after hemorrhagic shock. Circulatory shock 30: 255–263. [PubMed] [Google Scholar]
- 17.Schneider CP, Schwacha MG, and Chaudry IH. 2007. Impact of sex and age on bone marrow immune responses in a murine model of trauma-hemorrhage. Journal of applied physiology (Bethesda, Md. : 1985) 102: 113–121. [DOI] [PubMed] [Google Scholar]
- 18.Nacionales DC, Szpila B, Ungaro R, Lopez MC, Zhang J, Gentile LF, Cuenca AL, Vanzant E, Mathias B, Jyot J, Westerveld D, Bihorac A, Joseph A, Mohr A, Duckworth LV, Moore FA, Baker HV, Leeuwenburgh C, Moldawer LL, Brakenridge S, and Efron PA. 2015. A Detailed Characterization of the Dysfunctional Immunity and Abnormal Myelopoiesis Induced by Severe Shock and Trauma in the Aged. Journal of immunology (Baltimore, Md. : 1950) 195: 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnbull IR, Ghosh S, Fuchs A, Hilliard J, Davis CG, Bochicchio GV, and Southard RE. 2016. Polytrauma Increases Susceptibility to Pseudomonas Pneumonia in Mature Mice. Shock (Augusta, Ga.) 45: 555–563. [DOI] [PubMed] [Google Scholar]
- 20.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Brownstein BH, Mason PH, Baker HV, Finnerty CC, Jeschke MG, Lopez MC, Klein MB, Gamelli RL, Gibran NS, Arnoldo B, Xu W, Zhang Y, Calvano SE, McDonald-Smith GP, Schoenfeld DA, Storey JD, Cobb JP, Warren HS, Moldawer LL, Herndon DN, Lowry SF, Maier RV, Davis RW, and Tompkins RG. 2011. A genomic storm in critically injured humans. The Journal of experimental medicine 208: 2581–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, and Passegue E. 2015. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell stem cell 17: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Gottgens B. 2016. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128: e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu VW, Yusuf RZ, Oki T, Wu J, Saez B, Wang X, Cook C, Baryawno N, Ziller MJ, Lee E, Gu H, Meissner A, Lin CP, Kharchenko PV, and Scadden DT. 2016. Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 167: 1310–1322.e1317. [DOI] [PubMed] [Google Scholar]
- 24.Gekas C, and Graf T. 2013. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 121: 4463–4472. [DOI] [PubMed] [Google Scholar]
- 25.Gardner JC, Noel JG, Nikolaidis NM, Karns R, Aronow BJ, Ogle CK, and McCormack FX. 2014. G-CSF drives a posttraumatic immune program that protects the host from infection. Journal of immunology (Baltimore, Md. : 1950) 192: 2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka H, Ishikawa K, Nishino M, Shimazu T, and Yoshioka T. 1996. Changes in granulocyte colony-stimulating factor concentration in patients with trauma and sepsis. The Journal of trauma 40: 718–725; discussion 725–716. [DOI] [PubMed] [Google Scholar]
- 27.Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, Woloszynek JR, Greenbaum AM, and Link DC. 2014. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia 28: 1851–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Lieshout MH, Anas AA, Florquin S, Hou B, van’t Veer C, de Vos AF, and van der Poll T. 2014. Hematopoietic but not endothelial cell MyD88 contributes to host defense during gram-negative pneumonia derived sepsis. PLoS pathogens 10: e1004368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bagaitkar J, Pech NK, Ivanov S, Austin A, Zeng MY, Pallat S, Huang G, Randolph GJ, and Dinauer MC. 2015. NADPH oxidase controls neutrophilic response to sterile inflammation in mice by regulating the IL-1alpha/G-CSF axis. Blood 126: 2724–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenthal MD, and Moore FA. 2016. Persistent Inflammation, Immunosuppression, and Catabolism: Evolution of Multiple Organ Dysfunction. Surgical infections 17: 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanzant EL, Lopez CM, Ozrazgat-Baslanti T, Ungaro R, Davis R, Cuenca AG, Gentile LF, Nacionales DC, Cuenca AL, Bihorac A, Leeuwenburgh C, Lanz J, Baker HV, McKinley B, Moldawer LL, Moore FA, and Efron PA. 2014. Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. The journal of trauma and acute care surgery 76: 21–29; discussion 29–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, and Moore FA. 2012. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. The journal of trauma and acute care surgery 72: 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buza-Vidas N, Woll P, Hultquist A, Duarte S, Lutteropp M, Bouriez-Jones T, Ferry H, Luc S, and Jacobsen SE. 2011. FLT3 expression initiates in fully multipotent mouse hematopoietic progenitor cells. Blood 118: 1544–1548. [DOI] [PubMed] [Google Scholar]
- 34.Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, Kopf M, and Manz MG. 2014. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 124: 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panopoulos AD, and Watowich SS. 2008. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine 42: 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, and Rock KL. 2007. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nature medicine 13: 851–856. [DOI] [PubMed] [Google Scholar]
- 37.Kono H, Karmarkar D, Iwakura Y, and Rock KL. 2010. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. Journal of immunology (Baltimore, Md. : 1950) 184: 4470–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hesse DG, Tracey KJ, Fong Y, Manogue KR, Palladino MA Jr., Cerami A, Shires GT, and Lowry SF. 1988. Cytokine appearance in human endotoxemia and primate bacteremia. Surgery, gynecology & obstetrics 166: 147–153. [PubMed] [Google Scholar]
- 39.Sobrino J, and Shafi S. 2013. Timing and causes of death after injuries. Proceedings (Baylor University. Medical Center) 26: 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varol C, Yona S, and Jung S. 2009. Origins and tissue-context-dependent fates of blood monocytes. Immunology and cell biology 87: 30–38. [DOI] [PubMed] [Google Scholar]
- 41.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, and Jung S. 2013. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, Bigley V, Flavell RA, Gilroy DW, Asquith B, Macallan D, and Yona S. 2017. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. The Journal of experimental medicine 214: 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song WM, and Colonna M. 2018. Immune Training Unlocks Innate Potential. Cell 172: 3–5. [DOI] [PubMed] [Google Scholar]
- 44.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, Chatzigeorgiou A, Schlitzer A, Beyer M, Joosten LAB, Isermann B, Lesche M, Petzold A, Simons K, Henry I, Dahl A, Schultze JL, Wielockx B, Zamboni N, Mirtschink P, Coskun U, Hajishengallis G, Netea MG, and Chavakis T. 2018. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172: 147–161.e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.