

Abstract
It is predicted that pancreatic ductal adenocarcinoma (PDAC) will become the second most lethal cancer in the US by 2030. PDAC includes a fibrous-like stroma, desmoplasia, encompassing most of the tumor mass, which is produced by cancer-associated fibroblasts (CAFs) and includes their cell-derived extracellular matrices (CDMs). Since elimination of desmoplasia has proven detrimental to patients, CDM reprogramming, as opposed to stromal ablation, is therapeutically desirable. Hence, efforts are being made to harness desmoplasia’s anti-tumor functions. We conducted biomechanical manipulations, using variations of pathological and physiological substrates in vitro, to culture patient-harvested CAFs and generate CDMs that restrict PDAC growth and spread. We posited that extrinsic modulation of the environment, via substrate rigidity, influences CAF’s cell-intrinsic forces affecting CDM production. Substrates used were polyacrylamide gels of physiological (~1.5 kPa) or pathological (~7 kPa) stiffnesses. Results showed that physiological substrates influenced CAFs to generate CDMs similar to normal/control fibroblasts. We found CDMs to be softer than the corresponding underlying substrates, and CDM fiber anisotropy (i.e., alignment) to be biphasic and informed via substrate-imparted morphological CAF aspect ratios. The biphasic nature of CDM fiber anisotropy was mathematically modeled and proposed a correlation between CAF aspect ratios and CDM alignment; regulated by extrinsic and intrinsic forces to conserve minimal free energy. Biomechanical manipulation of CDMs, generated on physiologically soft substrates, lead to reduction in nuclear translocation of pERK1/2 in KRAS mutated pancreatic cells. ERK2 was found essential for CDM-regulated tumor cell spread. In vitro findings correlated with in vivo observations; nuclear pERK1/2 is significantly high in human PDAC samples. The study suggests that altering underlying substrates enables CAFs to remodel CDMs and restrict pancreatic cancer cell spread in an ERK2 dependent manner.
Keywords: cancer associated fibroblasts, cell-derived extracellular matrix, desmoplasia, fiber alignment, matrix-induced responses, pancreatic cancer, underlying substrate stiffness
Introduction
Fibroblastic stromal extracellular matrices (ECMs) modulate essential cellular behaviors such as differentiation, migration, proliferation, and survival [1]. In epithelial cancers such as pancreatic ductal adenocarcinoma (PDAC), loss of the homeostatic equilibrium of normal stroma induces mechanical and biochemical changes, resulting in dynamic activation of fibroblastic pancreatic stellate cells (PSC). In turn, the resultant activated cancer-associated fibroblasts (CAFs) remodel and deposit CAF-derived ECMs (CDMs), generating a dynamically altered fibrotic tumor microenvironment known as desmoplasia [2, 3]. Biomechanical characteristics of desmoplasia, such as ECM fiber anisotropy (i.e., parallel organized CDM fibers), have been correlated with poor cancer survival in numerous epithelial cancers, including PDAC [4–6], yet the mechanisms by which desmoplastic stroma promotes tumor progression remain unclear.
Along with stromal alterations, over 90% of all PDACs encompass Kirsten retrovirus associated to sarcoma (KRAS) mutations [7–9], which are evident during early phases of PDAC manifestation [10] and essential for both tumor initiation and progression [11, 12]. Despite the high frequency of activating KRAS mutations, even the most common PDAC predisposing point mutation, KRASG12D, known to initiate the precancerous benign lesion named pancreatic intraepithelial neoplasia, necessitates stromal reciprocity enhancement [13–15]. Notwithstanding the recognition of the key role of KRAS in PDAC, targeting this mutant GTPase has been difficult as preclinical promising drugs failed to show clinical efficacy [16, 17]. As an alternative approach, targeting the stromal reciprocity that maintains KRAS active [13] may assist in treating this disease.
While CAFs are responsible for producing and remodeling CDMs as well as maintaining many tumorigenic aspects of desmoplasia [18–20], ablation of CAFs is detrimental to patients [21]. Therefore, CAF reprogramming, i.e., harnessing the innate tumor-restrictive properties of the “normal” microenvironment, as opposed to stromal ablation, is an attractive therapeutic approach [22–26]. Many studies have suggested that pancreatic tumor stiffness is significantly greater than the physiological pancreas [27]. Tissue stiffness is a major factor that regulates naïve-to-CAF activation as well as CAFs’ ability to remodel desmoplastic ECMs (i.e., CDMs) [28]. The contractility of adherent cells (e.g., myofibroblastic CAFs), in concert with the extracellular physical properties of the substrate, constitute, respectively, the intrinsic and extrinsic forces needed to regulate tissue architecture (e.g., ECM isotropy) [25, 28–31]. Nonetheless, the biomechanical mechanisms that enable a stromal ECM production with tumor-restrictive, as opposed to tumor-permissive, capabilities remain unclear.
This study tests the hypothesis that manipulations altering ECM architecture or intracellular CAF myofibroblastic contractility could result in a “normalized” tumor-restrictive microenvironment. In testing our hypothesis, we investigated whether modulating substrate rigidity affects the ability of CAFs to adjust CDM fiber anisotropy and/or rigidity. We also questioned whether CDMs could be modified to restrict, rather than promote, tumorigenicity of PDAC cells. While our previous studies presented a mathematical model to explain the correlation between stiffness, cell polarization, and matrix alignment [32], we have modified this model to explain how physiological substrate stiffness triggers isotropic normalization of CDMs. Our results suggest that physiological stiffness can affect CAFs to produce CDMs that restrict tumor cell growth and spread by preventing nuclear localization of activated extracellular signal-regulated kinase (ERK) in an ERK2 dependent manner.
Results
To assess stiffness-modulated CAF morphology, patient-harvested CAFs [33] were cultured on collagen-I conjugated polyacrylamide gels of pancreatic physiological (physio-gel; ~1.5 kPa), or pathological (patho-gel; ~7 kPa) stiffnesses [27] using glass coverslips as a rigid-substrate control. As seen in Figure 1, the cell aspect ratio (i.e., length over width) of CAFs indicated a biphasic distribution, with a peak of about 3 times length over width for CAFs cultured on patho-gels, compared to CAF aspect ratios of 1.8 and 2.3, respectively, measured on physio-gels and glass coverslips. A similar, but discrete, trend was observed when control (i.e., inactive) fibroblasts were used (Supplemental Figure 1A). These results indicated that physiologically soft substrates can limit the aspect ratio of CAFs rendering round, as opposed to spindled, morphologies.
Figure 1: Changes in substrate stiffness dictate a biphasic distribution of CAF aspect ratios.

Images correspond to epi-fluorescent microscopy acquired CAFs labeled to depict cell morphology (CalceinAM; green) and nuclei (blue) cultured onto physio (~1.5 kPa), patho (~7 kPa) gels or glass coverslips. Graph includes measured CAF aspect ratios (length/breadth) calculated using MetaMorph software (below). Data is presented as median ± interquartile range. Asterisks correspond to * p<0.05 and ** p<0.01, compared to ratios attained by CAFs onto patho-gels.
We next questioned if the patterns of fiber alignment in CDMs correlate with the observed substrate stiffness-induced biphasic cell morphologies. For this, CAFs were cultured at high density on physio-gels, patho-gels or glass coverslips and allowed to produce CDMs. To assess the level of anisotropy at the cell-substrate interface, CDM fiber alignment was calculated from reconstructed quantitative confocal scanning images using the OrientationJ plugin for ImageJ software. For each condition, fiber alignment was calculated as the percentage of fibers oriented within 15 degrees from the measured mode orientation angle [33, 34]. Results revealed a biphasic fiber anisotropy: on physio-gels only ~30% of the fibers were aligned, while patho-gels induced a greater alignment (> 60%), yielding considerable fiber anisotropy. Using increasingly stiffer gels (e.g., ~20 kPa) CDMs produced were similar to the ones seen on patho-gels (not shown). Therefore, a decision was made to only utilize patho-gels (~7 kPa) in the study. Then again, on glass coverslips, only ~40% of the fibers were aligned (measuring at the cell-substrate interphase). These data suggest a positive correlation between the underlying surface-instructed cell morphology and CDM fiber anisotropy (Figure 2A). A similar trend, albeit to a lesser degree, was observed using control fibroblasts (Supplemental Figure 1B).
Figure 2: Changes in substrate stiffness dictate a biphasic distribution of CDM alignment and a linear increase in CDM stiffness.

(A) Reconstituted maximum projections of confocal images obtained from gel or glass to CDM interphases, as indicated. Fibronectin is shown in green merged with CAF cell nuclei (blue; top images) or as a monochromatic image (white; bottom images) containing a colored insert, indicative of fiber angle distributions, obtained with the ‘OrientationJ’ plugin of Image-J software and that was normalized using the hue function of Photoshop to show the cyan color as indication of fibers oriented on the mode measured angles. Color gradient bar on the right provides a color palate indicative of angle distributions. Graph includes the measured percentages of fibers distributed at 15˚ from the mode angle. Note that the CDM fiber anisotropy is at peak levels when CDMs are produced onto patho-gels. (B) Indentation moduli of decellularized ECMs, produced by control fibroblasts ECMs or CAF-generated CDMs cultured onto the indicated substrates. Results are presented as mean ± standard deviation. Asterisks denote the following significance compared to measurements obtained using the patho-gel condition in A or as indicated in B: * p<0.05, ** p<0.01, *** p<0.005 and **** p<0.001. For a comprehensive list of P values, please refer to Supplemental Table 1.
Interestingly, either on physio- or patho-gels, CAFs required only 3–5 days of culture to generate homogenous CDMs of >15 microns in thickness. This is less than half of the 8 days required to obtain CDMs on glass coverslips [33–35] with a CDM thickness of >5 microns and an overall combined fiber anisotropy of >55% (Supplemental Figure 2A). Similar results were obtained using additional patient-harvested CAFs (Supplemental Figure 2B). Taken together, our results suggest that both the intrinsic mechano-chemical coupling parameter of cells (i.e., CAFs and, to a lesser extent, in control fibroblasts) and the extrinsic effects imparted by the underlying substrate stiffness may modulate the level of CDM anisotropy.
Using atomic force microscopy (AFM) to gauge CDM vs. underlying gel stiffnesses, we first confirmed the indentation moduli of the engineered gels. As designed, gels exhibited the appropriate physiological and pathological stiffnesses of ~1.5 kPa and ~7 kPa, respectively. Interestingly, CDMs presented with numbers of magnitude lower stiffnesses (i.e., indentation moduli), compared to the corresponding underlying substrates (Figure 2B). Nonetheless, when control fibroblasts as opposed to CAFs were used to produce cell-derived ECMs, indentation moduli were ~1 kPa in all cases, regardless of substrate stiffness (Figure 2B). These results suggest that, in terms of the indentation moduli, the extrinsic effects imparted by the various substrates mostly affect matrices produced by CAFs. Furthermore, the substrate-directed CDM stiffness is independent of both fiber anisotropy and cell-aspect ratios. Altogether these results suggest that, by using substrates of pancreatic physiological stiffness (i.e., physio-gels of ~1.5 kPa), it is possible to modulate the CDMs production, with regards to both fiber alignment and stiffness, to phenotypically resemble normal ECMs.
In order to explain the biphasic underlying stiffness-dependence of both CAF cell aspect ratios and corresponding CDM fiber alignments, we applied and modified an existing mechanochemical mathematical “contractile cell model” that was previously used to explain the correlation between substrate stiffness and cell polarization [32]. This model described how cells assume energy-favorable morphologies by “sensing” underlying surfaces of variating stiffness via intrinsic chemical energy, arising from myosin motors, and extrinsic mechanical energy imparted by substrate stiffness. Here we used a similar approach to include the cell-substrate interfacial energy that is directly associated with cell shape to determine the aspect ratios needed for cells to attain a given shape by minimizing their total free energy. In line with our experimental results, this modified model predicted a biphasic distribution of cell aspect ratios as a function of increase in substrate stiffnesses. Figure 3A depicts a theoretical cell that is cultured on a surface (blue), where the cell’s aspect ratio (f) is defined as f = a/c and in which (a) is the cell’s length and (c) is its breadth (i.e., width). The accompanying graph depicts the hypothetical biphasic changes in cell-aspect ratios predicting CAF shape changes with a maximum aspect ratio found at a stiffness that is above the physiological stiffness of the normal pancreas. These theoretical calculations predict and validate the experimental observations suggesting: a) that to minimize their total free energy, CAFs can dynamically alter their acquired aspect ratios in response to changes in the stiffness of their underlying substrates, and b) that these alterations involve both intrinsic/chemical and extrinsic/mechanical energies.
Figure 3: Biphasic CDM alignment is explained in a minimum free energy model informed by substrate stiffness induced CAF aspect ratio.

(A) Depiction of the mathematical model conducted to explain the experimentally observed biphasic distribution in CAF aspect ratios. (A; left) Schematic representation of a cell (green) spread onto a 2D substrate (blue) used to calculate interface energy contribution to total energy. Both cell-substrate interface (red) and free cell surface (gold) are modeled as isotropic surfaces with surface energy γ_CM and γ_C (γ_C>γ_CM), respectively. (A; right) The minimum shape energies show biphasic response to changes in substrate modulus. K is the effective passive stiffness of the theoretical cellular actin network. (B) Depiction of mathematical model designed to explain the observed biphasic distribution in CDMs produced onto surfaces of increased stiffnesses, based on the observed/measured CAF aspect ratios. (B; left) Schematic depiction of the model suggesting that fiber alignment induced by single cell contraction: Light red area shows the spread of ECM fibers that are affected by a single cell, shown in the center. Red cones indicate the predicted local fiber orientations based on the center cell’s aspect ratio. (B; right) Graph presenting the predicted CDM fiber alignment expected when informed via the measured CAF aspect ratios obtained in Figure 1. Please refer to Supplemental Table 2 for additional data.
We next simulated the active crosstalk between CAFs and the underlying substrate to predict the effects of cell contraction on the initial fiber alignment. This simulation was informed by the measured cell aspect ratios and conducted by integrating a previously published model for fibrous ECM alignment [36] and the above depicted contractile cell models. Figure 3B shows that cell contraction prompts fiber alignment in a relative large region (gray), which is approximately 300 times that of the area of a cell. The red cones in Figure 3B indicate the predicted fiber orientation based on the cells’ measured aspect ratio. Both model predictions and experimental results show that cells become elongated when cultured on surfaces of intermediate stiffness, indicating that contraction is mostly uniaxial when the cells are cultured on the patho-gels, with CAFs (as opposed to control fibroblasts) exhibiting the highest aspect ratios (Figures 1, 2 and Supplemental 1). These results suggest that the observed biphasic aspect ratios, informed by the extrinsic stiffness of underlying substrates combined with the intrinsic contractility (i.e., chemical energy) of CAFs, dictate the observed biphasic anisotropy of CDMs via maintenance of low free energy.
Our studies, as well as studies by other groups, have demonstrated that, while CDMs (i.e., produced by CAFs) are tumor-permissive, e.g., by supporting cancer cell growth and invasion, ECMs derived from normal fibroblasts are tumor-restrictive, e.g., regulate transcription, limit cell motility and metastatic invasion as well as alter the manner in which tumor cells transmit integrin-dependent biochemical signals [37–40]. Hence, we asked whether CDMs produced by CAFs growing on physio-gels, but not on patho-gels, could functionally restrict cell growth and invasive spread. For this, CDMs [34] produced onto physio- vs. patho-gels were decellularized and used as substrates on which we cultured two human pancreatic ductal epithelial cells known as human pancreatic nestin expressing epithelial cells (HPNE): 1) Benign HPNEs immortalized using human telomerase reverse transcriptase (hTERT), and 2) hTERT immortalized and E6/E7/KRasG12D/small T antigen mutated HPNEs (K-HPNEs), which were transformed with oncogenic KRASG12D, concomitant with inactivation of tumor suppressors Rb and p53 [41]). CDM produced onto physio-gels limited cell proliferation, assessed by nuclear detection of Ki67 levels in K-HPNE cells, by ~2 fold compared to levels attained on CDMs produced on patho-gels (Figure 4A). Used as controls, syngeneic HPNE cells presented with similar proliferative trends, albeit to a lesser extent (Supplemental Figure 3A). When both cell types were tested using CDMs made on coverslips and compared to matrices made onto gels, proliferation levels correlated with measured anisotropic fiber levels, yet HPNE cells presented with reduced levels compared to K-HPNE cells (Supplemental Figure 3A). Bare gels (i.e., lacking CDMs) of pathological stiffness induced Ki67 incorporation/proliferation to levels akin to the ones observed on both cell types on glass, while physiological bare gels significantly restricted Ki67 incorporation in all cases (Supplemental Figure 3A).
Figure 4: CDMs generated on physiological substrates restrict K-HPNE cell growth and spheroid spreading by excluding pERK1/2 from the cell nucleus.
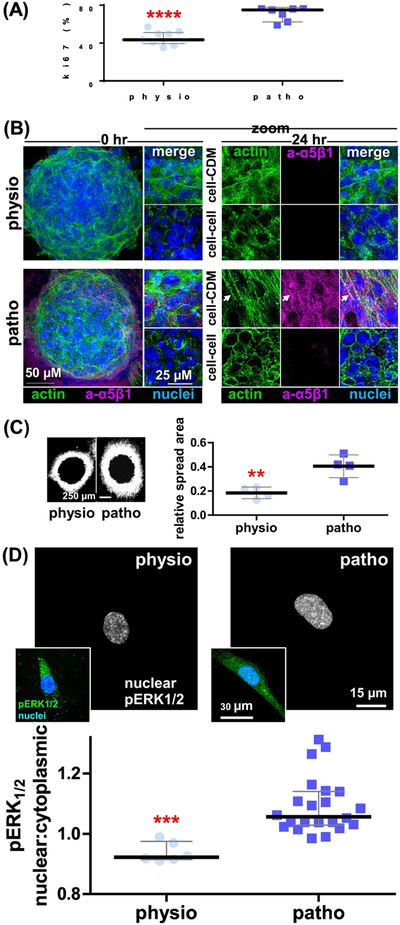
K-HPNE cells [41] were cultured in CDMs produced onto physio- vs. patho-gels. (A) Graph depicts measured percentages of nuclear Ki67 positive cells, showing median ± interquartile ranges. (B) Confocal images of K-HPNE spheroids at 0 and 24 hr cultured onto CDMs formed on Physio- and patho-gels highlighting nuclei (blue), cortical vs. stress fiber actin (green; phalloidin) and active fibronectin receptor levels and localizations (magenta; active α5β1-integrin using SNAKA51[43]) prompted by the assorted CDMs. Note how spheroids spreading onto CDMs made on patho-gerls are the only ones making 3D-matrix adhesions (arrow) [4 4] and that these are made only at the cell-CDM intersection and not at the center of the spheroids were cell-cell interactions are prevalent (i.e., evident via enrichment of cortical as opposed to F-actin stress fibers). Please refer to Movies 1 to 8 for additional details. (C) Epifluorescence microscopy images were used to generate area mask thresholds at times 0 and 24 hr of spread. Images show the corresponding net area spreads of RFP expressing K-HPNE cell spheroids at 24 hr (dark centers correspond to the original core areas of the spheroids at time 0). (C-Graph) area spreads corresponding to 95 percent of RFP-spheroid measured intensities. The y-axis depicts relative area spread, which was calculated by dividing the final area spread by the area measured at time 0 hr. (D) Indirect immunofluorescence depicting monochromatic images of nuclear pixels (selected using the nuclear channel) of pERK1/2 staining, generated using SMIA-CUKIE 2.1.0. https://github.com/cukie/SMIA. Inserts include merged image of pERK1/2 (green) and nuclei (blue) channels. (C-Graph) SMIA-CUKIE 2.1.0. https://github.com/cukie/SMIA generated pERK1/2 nuclear localization intensity levels divided by cytosol localized pERK1/2 levels. Asterisks denote significances of: ** p<0.01, *** p<0.005, and **** p<0.001 compared to nuclei:cytosol ratios obtained when using CDMs produced on patho-gels. Please refer to Supplemental Table 2 for additional data.
Since anisotropic ECMs have also been shown to promote tumor cell invasion in vitro and in vivo [38–40, 42], we tested if CDMs generated on physio-gels could also restrict the cell spread of K-HPNE cells [41]. For this, we cultured pre-made K-HPNE cell spheroids (Figure 4B and movies 1–8), for 4 hours (i.e., time 0), recorded the spheroid size and incubated for an additional 24 hours to allow K-HPNE cell migration into the assorted CDMs. Confocal spheroid phenotypic analyses, at 0 and 24 hours, were conducted using F-actin, active α5β1-integrin [43] and nuclei staining. Results, especially the ones obtained at 0 hours, served as architectural proof of effective spheroid formation; cortical actin was evident in cells at the middle of the spheres where cell-cell interactions are evident, while stress fibers were prevalent in cells at ventral spheroid locations for which cell-matrix interactions are predominant. Interestingly, 3D-adhesions [44], evident via active α5β1-integrin staining, were evident at cell-CDM adhesion sites in K-HPNEs in contact with CDMs formed onto patho-gels (Figure 4B and movies 1–8). In line with our hypothesis, we observed that areas of cell spread decreased by ~2 fold when the spheroids were cultured on CDMs produced on physio-gels, compared to areas of cells spreading into CDMs that were produced onto patho-gels (Figure 4C). As controls, the same spheroids were cultured using all assorted matrices and 2D substrates. As seen in Supplemental Figure 3B, control fibroblastic-derived ECMs played a restrictive role in all cases; limiting spreading areas similarly to the ones attained by K-HPNE cells cultured in CDMs produced onto physio-gels. These data suggest the possibility that ECMs produced by control “normal” fibroblasts are inherently restrictive regardless of the substrate used to produce them. Importantly, similar results to the ones obtained with K-HPNE cells, regarding both Ki67 and spheroid cell spreads, were also seen using the well-established KRASG12D mutant human PDAC cell line, Panc1 (Supplemental Figure 3C). Taken together, the data suggest that biomechanical manipulations of CDMs, which restore a physiological stiffness-induced isotropic CDM topology, can effectively restrain tumorigenic cell growth and spheroid cell spread to levels like the ones observed when normal (e.g., tumor-restrictive) fibroblastic-derived ECMs were used.
Nuclear accumulation of phosphorylated ERK1/2 (pERK1/2) is regarded as a downstream effect to constitutive KRAS signaling. Recent studies indicate that ERK2, rather than ERK1, is predominantly associated with the regulation of tumor cell invasion in 3D [45–50]. Hence, we questioned the ability of CDMs, produced onto physio- vs. patho-gels, to manipulate the K-HPNE cells and direct pERK1/2 localization (e.g., nuclear pERK1/2). Western blotting revealed no difference in pERK1/2 levels in KHPNE cells cultured on CDM produced on either physio- or patho-gels, yet there was a modest increase in pERK1/2 levels when K-HPNE cells were cultured on CDM produced on glass (supplemental Figure 4A). Importantly, compared to CDMs produced on patho-gels, the nuclear localization of pERK1/2 was modestly, yet significantly (p=0.0003), reduced by 18 % in K-HPNE cells cultured on CDMs produced on physio-gels (Figure 4D). ECM controls, testing all experimental matrices and bare gels, showed a similar trend (supplemental Figure 4B–C and supplemental Table 2). This data suggests that in tumorigenic/invasive cells, nuclear localization of pERK1/2 is controlled via alterations in the CDM that, in turn, are affected by fine-tuning the underlying substrate stiffnesses. To elucidate the role of the two forms of ERK, we compared the effects of U0126, an inhibitor that indirectly blocks both ERK1 and ERK2, to the effects obtained by specifically knocking down ERK1 vs. ERK2 expression (Figure 5). As seen in Figure 5A, U0126 reduced CDM-induced spheroid cell spreading of K-HPNE cells by ~40%, compared to the spheroid cell spreading measured using vehicle control. To transiently knock down either ERK1 and/or ERK2, we used specific siRNAs and, as controls, equal amounts of a scrambled (e.g., non-specific) siRNA. As seen in Figure 5B, knocking down ERK2, but not ERK1, decreased anisotropic CDM-induced spheroid cell spreading by ~60%. Interestingly, spreading of K-HPNE cells on CDMs produced on patho-gels under ERK1/2 or ERK2 blockage was similar to the cell spreading observed on both restrictive CDMs produced on physio-gels and by all the control fibroblast-derived ECMs (compare results in Figures 4, 5 and Supplemental Figure 3). To assure that the CDM-induced effects were indeed ERK2 dependent, we verified that the phosphorylation levels of p90RSK, one of several downstream targets of ERK2, was more significantly altered under pERK2 down-regulation and to a lesser extent under pERK1 siRNA knockdown (Figure 5). Taken together, our results suggest that ERK2 is essential for matrix-induced spheroid cell spread of human pancreatic tumor cells and that ERK2 blockage reduces anisotropic CDM-induced spheroid spreading.
Figure 5: CDM-induced K-HPNE spheroid cell spreading rates are regulated by tumoral pERK2.

(A) K-HPNE cell spheroids spreading through CDMs produced onto patho-gels were treated with 20 μM of U0126 to inhibit MEK1/2 upstream to pERK1/2. Spheroid area spreads were measured (graph) as before as well as in comparison to untreated and vehicle treated (DMSO) controls. Blots depict representative levels of pERK1/2 and phospho-p90RSK, downstream to pERK2, in total cell lysates (left) and in nuclear fractions (right). Representative measured optical densitometry (O.D.) are provided. (B) Same experiments as in A, but this time K-HPNE cells were transfected with lipofectamine (lipo) or scrambled controls as well as siRNAs to ERK1 and/or ERK2 as labeled on the figure. Note how effects are seen when ERK2 is downregulated. Significance was calculated in comparison to untreated controls and asterisks denote * p<0.05 and *** p<0.005.
Lastly but importantly, to further validate both mathematical predictions and the in vitro findings, we assessed the nuclear localization pERK1/2 in 8 matching normal (e.g., physiological) and tumor (e.g., pathological) human pancreas samples. For this, we used our recently published simultaneous multiplex immunofluorescent approach and accompanying software needed for quantitative digital imaging analysis [33]. Results, shown in Figure 6A, demonstrated a ~3-fold increase in levels of nuclear localization of pERK1/2 in human PDAC samples, compared to nuclear pERK1/2 measured in matching normal/non-pathological pancreatic epithelial cells.
Figure 6: Human normal stroma maintains pERK1/2 away from nuclei of pancreatic epithelial.
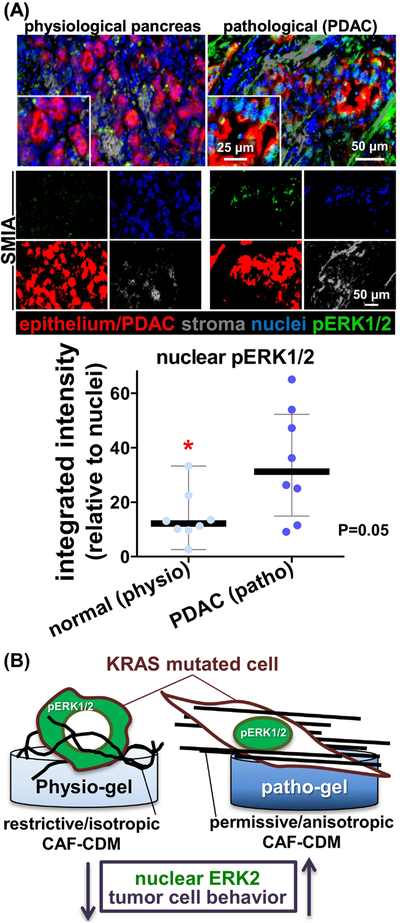
(A) Representative examples of pancreatic normal (physiological pancreas) and matched pathological PDAC formalin-fixed paraffin-embedded, FFPE, tissue samples stained and analyzed using the SMI approach and accompanying SMIA software [33]. Top panels correspond to merged images of a representative matching normal and PDAC sample showing epithelium/tumoral cells (red), nuclei (blue), stromal cells (grey) and pERK1/2 (green). The monochromatic panels shown below indicate “area masks” generated by the SMIA-CUKIE software (SMIA) and depict the levels of pERK1/2 (green) located solely at epithelial or tumoral (red) nuclei areas. Grey masks represent stromal areas. Graph summarizes the measured integrated intensity levels (obtained using SMIA-CUKIE) of nuclear tumoral (PDAC; patho) or normal epithelial (normal; physio) localized pERK1/2 levels. P value is indicated. (B) Summary cartoon to highlight that isotropic CDMs, obtained on a physiologically soft substrate, direct pERK1/2 (green) out of the nucleus; restricting pancreatic cancer cell invasive spread, akin to in vivo PDAC.
Taken together, results showing that increased occurrences of nuclear PDAC cell pERK1/2 levels correlate with our in vitro observations. The collected data suggest that point-mutated KRAS cells tend to maintain pERK1/2 in the nucleus under pathological conditions (Figure 6B), such as in the presence of significant stromal activation, as it is typically seen in human PDAC.
Discussion
Pioneering work by the late Dr. Patricia Keely demonstrated that tumor-associated stromal ECM architecture is altered and that stromal ECM anisotropy in vivo serves as a prognostic indicator of poor cancer patient outcomes [51]. ECM-imparted physical cues dictate directional migration, as migrating cells often use the ECM fibers as attachment points during invasion [42, 52]. Hence, aligned (anisotropic) ECM fibers are a characteristic signature for PDAC-associated desmoplasia and indicative of poor patient prognosis [4, 5, 53]. Ablation of PDAC-associated desmoplasia was tumor-promoting pre-clinically and detrimental to patients in clinical trials [21, 54, 55]. Hence, approaches that can modulate the pro-tumorigenic aspects of desmoplasia (e.g., desmoplastic ECM anisotropy) to harness the naturally tumor-restrictive features of a normal microenvironment are highly sought by the field. As such, genetic and biomechanical regulation of CAFs seeks to alter their ability to contract and produce an anisotropic ECM. For example, increased levels of stromal caveolin-1 in fibroblasts were shown to impact stromal ECM anisotropy, via p190RhoGAP activation, which increases intrinsic cell contractile forces and, in turn, facilitates in vivo metastatic escape [39]. The presence of syndecan-1 in stroma alters the ECM architecture, in vivo and in vitro, and induces the production of an anisotropic ECM that promotes cell invasion [56]. Further, overexpression of fibroblast activating protein (FAP) in naive fibroblasts prompts the formation of anisotropic ECM, akin to pancreatic CDM, and also induces increased PDAC cell invasion [40]. Alternatively, physical approaches have been used to modulate ECM anisotropy, however most systems rely on collagen gels or synthetic scaffolds which lack in vivo biochemical complexity. Thus, despite reports suggesting that ECM anisotropy is a key regulator of PDAC tumorigenesis [4, 5, 53], the particular biomechanical cues that enable production of tumor-restrictive ECM remain mostly elusive.
Substrate stiffness strongly influences a wide range of cell functions, including some with ECM-modifying properties, such as cell spreading, survival, proliferation, differentiation, and migration [57]. Substrate stiffness also enables ECM remodeling by modulating the expression of genes for ECM proteins and ECM-altering enzymes [58]. Fibronectin fibrillogenesis, which is necessary for collagen fibrillogenesis in vivo [59], is greatly affected by the underlying substrate stiffness [60]. Fibrillogenesis per se is necessary for altering cell-intrinsic forces [61]. Substrate stiffness can also drive collective cell migration, which is critical for some types of cancer invasion [62]. Nonetheless, not many studies have highlighted the role of substrate stiffness on CAF-derived matrix production, which is one of the most important factors believed to influence cancer progression [63]. To correctly interpret the role of biomechanical matrix properties that affect cell behavior (i.e., cell growth), it is important to select a system that accurately mimics the relevant in vivo physiological and pathophysiological microenvironments. In the present study, we adapted our previously described, cell-derived ECM system [34, 44] to identify means to alter desmoplastic ECM anisotropy and question the role that anisotropic ECMs could play in dictating pancreatic cancer cell tumorigenesis, such as cell proliferation and/or spheroid cell spread. The advantage of this well-characterized system is that it is comprised of natural, cell-derived materials and that it offers an in vivo-like biochemical complexity [44]. However, one known technical limitation of using CDMs produced on glass coverslips is a gradient-like heterogeneity in fiber anisotropy, apparent between the bottom (at the glass ECM interphase) and top ECM fiber layers (Movie 9). Remarkably, using the physio and patho-gels as shock absorbing/buffering materials not only permitted fine-tuning of the underlying substrate stiffness and direct the anisotropy of CDM fibers (Figure 2), but it allowed doing so in a homogenous way (Supplemental Figure 2).
We developed a mathematical model using the experimentally measured cell aspect ratios (Figure 1 and Supplemental 1A) to predict the anisotropy levels of matrix fibers being produced at the cell-substrate interphase. Substrate stiffness affects cytoskeletal actin organization, which, in turn, regulates numerous cellular functions, such as migration, which results in cell spreading [64]. Because of the organization of the actin network, cells contract along their long axes and generate aligned ECMs [36]. This effect is increasingly pronounced as the cell becomes more elliptical [30, 32]. Interestingly, intermediate stiffness, akin to pathological substrates (e.g. 7 kPa), is associated with induction of maximum elongation of cell and migration, whereas softer stiffness (e.g. 1.5 kPa) is associated with dramatic reduction in cell spreading [65, 66]. This prompted us to hypothesize that CAFs cultured on soft substrates may exhibit “normalization” of ECM anisotropy. Our published model, also referred to as “constitutive material model” [36], is a continuum model that addresses fiber alignment and long-range force transmission (i.e., the ability of cells to “sense” each other at long distances within an ECM-like mesh within matrices generated on physiological versus pathological environments). The model also captures the nonlinear elastic behavior of matrices in response to cellular contraction. Based on our experimental data (Figures 1 and 2) and supported by mathematical modeling (Figure 3), we suggest that extrinsic (e.g., substrate stiffness) and intrinsic forces (e.g., ECM producing cell’s contraction ability) collectively affect the aspect ratios of CAFs, which in turn control CAF-derived ECM anisotropy. Therefore, we propose changing substrate stiffness as novel means for manipulating extrinsic cues, which may influence the ability of human PDAC-associated CAFs to biomechanically remodel CDMs by altering their key biomechanical properties: anisotropy and indentation moduli. Our data (Figures 3 and 4) further demonstrated that tumor restrictive responses, particularly limiting spheroid cell spreading, are highly correlated with matrix anisotropy. This study is in line with the previous data that ECM anisotropy is a major predictor of tumor responses, particularly spheroid cell spreading [30, 67, 68].
Despite harboring “permanent” KRAS mutations, PDAC cells require stromal cues to effectively trigger and maintain constitutive KRAS activity in vivo [13]. High, epithelial/tumoral levels of ERK1/2 activity can be regarded as indicative of in vivo KRAS activity, since ERK1/2 is downstream to KRAS [69]. ERK1/2-regulated epithelial to mesenchymal transition has been linked to poor PDAC survival [70]. Activation of key mechano-transducing molecules such as YAP, which depend on ERK1/2 phosphorylation, is also a trademark of PDAC [71]. In this study, we observed sustained high levels of nuclear pERK1/2, as opposed to merely increased cytosolic levels, in KRAS-driven tumorigenic/invasive human PDAC cells (e.g., K-HPNE and Panc1) cultured on anisotropic matrices (i.e., CDMs produced onto patho-gels). Yet, pERK1/2 was effectively excluded from the nucleus when the cells were cultured on isotropic matrices (i.e., CDMs produced onto physio-gels). Additionally, ERK2, and not ERK1, was responsible for regulating the observed ECM-induced PDAC cell responses (e.g., spheroid cell spread areas in Figure 5). These data are in line with our own early studies and studies by others which have highlighted a role for ERK2 in tumorigenic responses to extracellular-imparted cues [45–50]. Interestingly, high phosphorylated ERK2 levels have been correlated with poor survival [72]. Specific nuclear localization of ERK2 induces epithelial mesenchymal transition [71] and contributes to drug resistance [73]. Further, increase in p90RSK phosphorylation levels are potentially indicative of an escalation in ERK2 activity since this molecule is one of several known ERK2 downstream effectors and because ERK2 is known to signal through its nuclear localization [74]. Our data, therefore, suggest the possibility that pERK2 is implicated in ECM-induced oncogenic KRAS-supported PDAC spheroid cell spreading (Figure 5). This observation also agrees with reports showing synthetic lethality between ERK1/2 inhibition and p90RSK or its downstream effector CDC25C [75]. Further, it has been reported that integrin activity is needed for ERK1/2 nuclear translocation [76]. Interestingly, levels and localization of the active conformation of the main fibronectin receptor, α5β1-integrin, were greatly affected by the substrate altered CDMs (Figure 4B). Future work will focus on assessing why, by remodeling the CDM architecture, integrin signaling is altered.
Lastly, this study demonstrated that human PDAC tissues present with enriched tumoral nuclear pERK1/2 while normal human tissues exhibit lower levels of nuclear pERK1/2 in the pancreatic epithelium (Figure 6). While increased overall levels of pERK1/2 have been reported in human PDAC [77], only few studies have looked at epithelial/tumoral nuclear pERK1/2 localization [78]. Therefore, these findings may have important implications regarding the therapeutic use of drugs that could exclude pERK1/2 from tumoral nuclei. Future efforts will be directed towards further evaluating mechanisms on how ERK2, enabled via anisotropic CDMs, may control pERK1/2 translocalization to PDAC cell’s nuclei.
Conclusions:
In this study, we have demonstrated that CAFs can be enticed to generate pancreatic cancer-restrictive CDMs, provided the underlying substrate rigidity matches that of a physiological pancreas. We propose that the in vitro measured results can be modeled mathematically, informed by the substrate stiffness’ extrinsic forces combined with CAFs’ intrinsic contractility, which jointly directs a biphasic matrix fiber anisotropy by maintenance of a low free energy. We found that CDMs generated by CAFs onto physiologically soft gels are tumor-restrictive and limit Ki67 incorporation, indicative of reduced rates of proliferation and spheroid cell spread of oncogenic (e.g., KRAS driven) pancreatic cancer cells. Our results also suggest that loss of nuclear pERK1/2 in cells cultured on isotropic CDMs is probably regulated by restricting ERK2 activity. The observed in vitro results correlated with in vivo measured epithelial nuclear pERK1/2 levels, supporting the validity of our tunable pathophysiological 3D CDM system. Hence, the therapeutic reprogramming of stromal ECM and/or targeting tumoral ERK2 may provide future means to contain PDAC, and possibly other KRAS-driven neoplasias.
Material and Methods
Contact for Reagent and Resource Sharing
All information (manufacturer/source) regarding the chemical and biological reagents has been compiled in the Reagents Table. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Drs. Edna Cukierman (Edna.Cukierman@fccc.edu) and Peter I. Lelkes (pilelkes@temple.edu).
Experimental Model and Subject Details
Cell Lines
Human pancreatic CAFs were isolated using an Institutional Review Board approved protocol. Cells were characterized, immortalized and authenticated as previously described [33, 34]. Since cultured NIH-3T3 fibroblasts (from ATCC, CRL-1658™) do not undergo spontaneous myofibroblastic activation [35], these cells were used as inactive fibroblastic cell controls as before [38]. Note that the study included the use of two independent, published, immortalized human CAF lines; while most of the results were conducted using a single line, key experiments were confirmed using an additional cell line harvested from another patient and previously reported [33]. All cells were maintained in a humidified incubator at 37°C and 5% CO2. Control fibroblasts and CAFs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; from Mediatech (Manassas, VA) supplemented with 10% FBS, 100 U/mL Penicillin, 100 mg/mL Streptomycin and 2 mM L-Glutamine. Syngeneic pancreatic ductal epithelial cells, HPNE and K-HPNE [41], from ATCC (CRL-4038™), were cultured in growth medium containing four parts of low glucose DMEM and one part M3 supplemented with 5% FBS containing 100 U/mL Penicillin and 100 mg/mL Streptomycin.
Method Details
Preparation of Polyacrylamide gels
Circular glass coverslips, 18 mm in diameter (Carolina Biological Supply Company; Burlington, NC), were activated using 3-Aminopropyl triethoxysilane (APTES) for 10 minutes and washed extensively with distilled water followed by treatment with 0.5% glutaraldehyde for 1 hr. To prepare the gel solutions, acrylamide and N,N′-Methylenebisacrylamide solution were mixed together in distilled water in the desired ratios to generate gel precursor solutions for predicted Young’s moduli of ~1.5 (physio-gels) and ~7.5 kPa (patho-gels) [79]. The final percentage of gel solutions for a ~1.5 kPa gels was 3% acrylamide and 0.15% bisacrylamide and 10% acrylamide and 0.1% bisacrylamide for a ~ 7.5 kPa gel. Gel stiffness were validated using atomic force microscopy. Gel polymerization was initiated by addition of crosslinkers 10% w/v APS and N,N,N′,N′-Tetramethylethylenediamine accelerator (TEMED) at dilution 1:000 and 1:10,000 respectively from their stock solutions. After gentle mixing, 120 μl of the gel solution were pipetted onto the activated coverslips and a dichlorodimethylsilane -treated coverslip was carefully placed on top of the gel solution. Gels were allowed to polymerize at room temperature for ~10–15 min. The top glutaraldehyde and dichlorodimethylsilane (DCDMS)-treated coverslip was gently lifted and gels were washed with Milli-Q water and sterilized under a UV lamp (365 nm) for 15 min. Covalent conjugation of gels with 50 μg/ml collagen-I was performed in 50 mM HEPES buffer, 8.5 pH. Collagen-I was crosslinked to the gels using Sulfo-SANPAH for 15 min under the UV lamp, as above. Collagen-coated gels were washed extensively with PBS and stored in PBS at 4°C for up to two weeks. The Collagen-conjugated gels were equilibrated for 30 min with culture media at 37°C prior to seeding with the various fibroblasts.
Preparation of CDMs onto polyacrylamide gels
Glass coverslips containing gels coated with collagen were placed inside wells of a 24 well cell culture plate (gel side up). Pyrex® cloning cylinders (Sigma Aldrich, St. Louis, MO), 8 mm (height) x 8 mm (diameter), were carefully placed at the center of gels and 100 μl culture medium containing 4×104 CAFs (or control fibroblasts) was carefully pipetted inside the cylinders. Cylinders were removed after ~1 hr. to allow the cells to attach. Next, the cells were covered with 1 ml of fibroblast culture medium, listed above, supplemented with 50 μg/ml ascorbic acid. A similar procedure, omitting the use of cloning cylinders, was employed for producing CDMs by seeding the cells (CAFs and normal controls) directly on collagen coated glass coverslips, as previously published [34]. For this, 1.25×105 cells per 12 mm coverslips were plated. In both cases, media including freshly weighted and diluted ascorbic acid (50 μg/ml) was added every day except on the last day (e.g., before extraction).
Following matrix production (3 days for CDMs on gels or 8 days for CDMs on glass), decellularized matrices were obtained using an alkaline detergent (0.5% Triton X-100 and 20 mM NH4OH in PBS), followed by DNase I (50 U per mL) treatment [34]. The resulting decellularized matrices were washed three times with PBS and stored at 4°C for up to 2 months. All decellularized matrix batches, of control fibroblast ECMs and CAF CDMs, underwent rigorous quality control as published [34].
Indirect immunofluorescence and image analysis
Indirect immunofluorescence was as previously described [34]. Briefly, samples were fixed/permeabilized, for 3 min, with 4% (w/v) paraformaldehyde (EM-grade from Electron Microscopy Sciences), 0.5 % (v/v) Triton X-100, and 50 mg/ml sucrose in Dulbecco’s phosphate-buffered saline and continued fixing, in the absence of triton, for 20 min. Samples were blocked for 2 hr with Odyssey blocking buffer (TBS) (LI-COR Biotechnology, NE, Cat. 927–50100). For matrix assessments, samples were incubated, for 1 hr with a rabbit anti-mouse fibronectin antibody (25 μg/ml, Abcam, UK Catalog no: ab2413). After incubation with primary antibody, the matrices were washed three times, for 10 min each, with tris buffer saline containing 0.5 % v/v Tween 20 (TBS-T buffer) and incubated at room temperature for 45 min with donkey anti-rabbit Cy5 conjugated secondary antibody (15 μg/ml, Jackson ImmunoResearch, PA Catalog no: 711–175-152). Samples were washed with TBS-T, three times. Nuclei were stained with SYBR green (1:50,000 dilution, Thermo Fisher Scientific, Waltham, MA) and samples were mounted as previously detailed [34]. Images were captured using a spinning disk confocal microscope (Ultraview, Perkin-Elmer Life Sciences, Boston, MA) equipped with a 60X (1.45 PlanApo TIRF) oil immersion objective. For each condition, three independent experiments were conducted and a minimum of 7 images per sample were obtained.
CDM (or control ECM) alignment measurements were conducted using ImageJ OrientationJ plug analyses as published [33, 34]. Isotropic CDMs were identified as matrices containing < 55% alignment, while anisotropy was identified as > 55% of fibers distributed at 15 degrees from the mode angle [33, 34].
For ERK1/2 subcellular localization quantification, K-HPNE [41] cells were incubated with rabbit anti-human phospho- p44/42 ERK1/2 (Thr202/Tyr204) (Cell Signaling Technology, Danvers, MA, Cat no. 4370,) followed by Cy5-coupled secondary antibody and SYBR green, as above. Subcellular localization of p-ERK1/2 (cytosolic vs. nuclear) was quantified using our publically available software, SMIA-CUKIE 2.1.0 https://github.com/cukie/SMIA [33]. Images corresponding to the same experimental conditions and the same staining procedures were processed identically; 16 to 8 bit level conversions were conducted using identical parameters. To find suitable thresholds, to inform the SMIA-CUKIE software, and distinguish between signal and noise, we applied intensity histogram distributions obtained from Photoshop. Selected threshold values (between 1 and 254) for each staining (pERK1/2 or nuclei) were consistently used through the study. Images were sorted into experimental folder batches, using the “make a batch” software in https://github.com/cukie/SMIA, to include monochromatic matching images of nuclei and pERK1/2 per simultaneously acquired images, which served as inputs for the SMIA-CUKIE 2.1.0 software. Mean intensity levels of pERK1/2 in nuclei vs. cytoplasmic fractions were measured and outputs were plotted as nuclei:cytoplasmic ratios. Shown in the figures are representative monochromatic image outputs, displaying positive pixels indicative of “nuclear” only pERK1/2 levels, accompanied by total pERK1/2 nuclei color overlays.
Human tissue samples,this protocol is based on the Franco-Barraza et. al., 2017 publication [33]. Eight pairs of matched sections of human pancreatic tissue of normal (physio) and PDAC (patho) tissues were obtained with approval by Fox Chase Cancer Center’s Institutional Review Board. The patients had consented to donate their samples for research purposes without restriction and in a decoded manner. Samples were pre-vetted by pathologist collaborating with Fox Chase’s Bio-Sample Repository Facility, which distributed the samples to the investigators. Tissues were fixed with formalin and embedded in paraffin. Staining: Slices containing samples were de-paraffinized and rehydrated as well as antigen retrieval following classic procedures for IHC. Odyssey blocking buffer was used to block samples prior to staining. First primary rabbit antibody against pERK1/2 antibody, (dilution 1:100) purchased from Cell Signaling Technologies (Cat. No. 4370S), was incubated overnight at 4°C. Samples were rinsed with PBS containing tween 20 at a concentration of 0.02% v/v at (PBS-T) three times for 5 min each before secondary incubations with anti-rabbit Cy5 conjugated antibodies (Cat no. 711–175-152 Jackson Immuno Research Inc. West Grove, PA). Samples were again rinsed and blocked with IgG fragments to avoid additional secondary antibody detection. To distinguish between epithelial and stromal regions, tissue samples were stained with primary antibodies against pan-cytokeratin (1:40 dilution) and vimentin (1:200) using primary anti-Mouse pan-cytokeratin (AE1/AE3) antibodies (1:40) from DAKO (Cat no. M3515) and Rabbit monoclonal to Vimentin antibody (1:200) from Abcam (ab92547). Incubations were performed for 90 min at room temperature. The samples were rinsed as above and incubated with corresponding, fluorophore pre-labeled secondary antibodies (Anti-mouse-Cy3 conjugated antibody (Cat no. 715–166-151) and Anti-rabbit Cy2 conjugated antibody (Cat no. 711–175-152) from Jackson Immuno Research Inc. West Grove, PA). Finally, nuclear staining was performed using Hoechst 33342 (1:50,000 dilution) for 15 min at room temperature. Sections were dehydrated and cleared with Toluene. Slides were kept overnight in dark at RT before imaging. Imaging was performed using the VECTRA SYSTEM microscope at 40X magnification.
Monochromatic images were separated using the Vectra software and 16 to 8 bit levels were conducted in an identical manner per channel. Images were feed to SMIA-CUKIE as before and analyses were conducted to provide graphs as well as image outputs on the main text. Details for image analyses were as above. The following filters were used DAPI (440–680), FITC (500–680), TRITC (570–690), and CY5 (680–720).
Atomic force microscopy (AFM)
The stiffnesses of the various (gel and cell-derived matrices) substrates were assessed by AFM-nanoindentation, carried out on a Dimension Icon AFM (BrukerNano, Santa Barbara, CA), using a custom-made microspherical tip. The colloidal probe used was generated by attaching a 5 μm-radius polystyrene microsphere (PolySciences, Warrington, PA) to the end of a tipples cantilever (Arrow-TL1Au, NanoAndMore USA, Watsonville, CA) using M-bond 610 epoxy (Structure Probe Inc., West Chester, PA). All tests were conducted using filtered 1× PBS to simulate a physiological fluid environment. The probe tip was programmed to indent into the sample at a constant z-piezo displacement rate of 5 μm/s, up to a maximum indentation depth ~ 1 μm. All CDMs (and control ECMs) used for this study were at least 8 μm thick, i.e. exceeding the minimum thickness of 7 μm required for CDM quality control [34]. Each sample was tested at a minimum of 10 randomly selected locations to ensure consistency and/or to account for spatial heterogeneity. The indentation modulus Eind was calculated by fitting the loading portion of each indentation force-depth curve to the Hertz model,
| (1) |
where F is the indentation force, D is the indentation depth, ν is the poison’s ratio (0.49 for highly swollen hydrogels) (57), and Rtip is the radius of the probe tip (≈ 5μm). Since the thickness of the gels (>200μm) is more than 2 orders of magnitude greater than the maximum indentation depth, the substrate constraint effect was minimal, and thus, finite thickness correction was not needed. The comparison between CDMs and adjacent bare gels was done by probing regions with or without the ECM on the same gel for consistency. This was possible because CDMs were constrained to the cloning cylinder areas, with the adjacent bare gel areas serving as internal controls. The mechanical properties of the latter were indistinguishable from those of intact bare gels that had never been coated with CDMs.
Mathematical model for predicting cell shape
Cells change their shapes in accordance with the physicochemical properties of the underlying substrate [80]. To mathematically understand how substrate stiffness influences cell morphology, we consider a cell cultured on a 2D substrate. We use an energy criterion to determine the cell shape, i.e., we hypothesized that a cell adjusts its shape in order to minimize the total free energy of the cell-substrate system. The total free energy can be written as,
| (2) |
where Ecell is the cell energy, Esubst is the elastic energy of the underlying substrate and Eint is the interface energy (including the basolateral cell-substrate interface and the apical free cell surface). The cell energy is a function of the elastic energy (accounting for cell deformation) and the motor density (accounting for contractility). Based on the model for contractile cells [32], Ecell can be written as,
| (3) |
where Uc is the cell energy density, is the elastic deformation of the cell and Pij is the motor density. The interface energy consists of the basolateral cell-substrate interface energy and the apical free cell surface energy.
| (4) |
where γcs and γc are interface/surface energy density for cell-substrate interface and free cell surface respectively, Scs and Sc are the area for cell-substrate interface and free cell surface respectively.
We characterized the cell shape by defining the aspect ratio f = a/c, where a stands for length and c is the cell breadth. For a given substrate (a fixed stiffness), we computed the total free energy of the cell-substrate system for various aspect ratios, and chose the energy-minimized one as the preferred cell shape. Next, we varied the substrate stiffness and obtained the hypothetical cell aspect ratio as a function of stiffness.
Mathematical model for predicting CDM alignment
In terms of the stress-dependent regulation of cell contractility, the contractile stress of the actin network can be written [32] as,
| (5) |
where ρ is the density of force-dipoles (representing myosin motors/contractility) in the actin network, ɛ is the strain of the actin network and K is the effective passive stiffness of the actin network. The contractility itself depends on the mechano-chemical coupling through mechano-signaling pathways, such as Rho-Rock and myosin light chain kinase [32];
| (6) |
where ρ0 is the contractility in the absence of adhesions, α and β denote mechano-chemical coupling parameters. Additional details of this model have been described elsewhere [32].
Short Interfering RNA (siRNA) Transfections
Transient transfections were performed on the K-HPNE cells [41], using Lipofectamine® 2000, following the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). Non-targeting SMARTpool and siRNA targeting ERK1 or ERK2, each comprising four distinct siRNA species, were from Life Technologies-Dharmacon (Lafayette, CO). Transfections were carried out in K-HPNE cell growth media, without FBS or antibiotics. Cells were trypsinized, plated at a density of 1×105 per well, in a 6-well plate, and mixed with transfection medium, as per manufacturer’s instructions. The plate was placed in the incubator for 5 hrs. Following incubation, media was replaced with regular K-HPNE cell growth media (as above) and cells were cultured for an additional 48 hrs. For spheroid spread experiments, cells were trypsinized 24 hrs post transfection and used for spheroid formation, followed by spheroid spread assay (see below).
Western blotting
Cell lysates were obtained using a cell lysis buffer from Cell Signaling Technology (Catalog no. 9803, Danvers, MA) supplemented with Pierce™ Phosphatase and Protease Inhibitor Mini Tablets (Cat no. 88667 and 88665, respectively) from Thermo Fisher Scientific (Waltham, MA). Proteins were resolved by 4–20% SDS-PAGE gels (Bio-Rad, Hercules, CA) at 60 V and transferred to PVDF membranes using semi-dry transfer (Bio-Rad, Hercules, CA). Protein transfer was carried out at 20 V for 30 min. Blots were incubated with the following primary antibodies: Rabbit anti-human Phospho- p44/42 ERK1/2 (Thr202/Tyr204) (Cat no. 4370) and rabbit anti-human total- p44/42 ERK1/2 (Cat no. 9102) from Cell Signaling Technology (Danvers, MA). Anti-phospho-p90RSK1 (Ser380) (Cat no. 04–418) and anti-human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cat no. MAB374) from Millipore (Billerica, MA). Horseradish peroxidase- conjugated, anti-species matched, secondary antibodies were from Sigma Aldrich (St. Louis, MO). Protein bands were visualized using the Protein Simple FluorChemE System, (San Jose, CA). For biochemical nuclear level assessments of pERK1/2 we used the Subcellular Protein Fractionation Kit (Thermo Scientific, Waltham, MA), according to the manufacturer’s instructions.
Cell proliferation assay (Ki67)
Pancreatic human epithelial cells HPNE and K-HPNE [41] were plated at a density of 2×104 cells/ml, per sample, and cultured for or 24 hrs. Cells were fixed as stated in Indirect immunofluorescence and digital imaging analyses section above, prior to staining with anti-Ki67 antibody (Cat no. ab15580, Abcam, Cambridge, UK), using the same protocols as above. The fraction of proliferating cells was determined by counting the number of cells stained positive for Ki67 divided by total number of nuclei, stained using Hoechst 33342 solution (Calbiochem, Billerica, MA). At least 5 images were taken per condition, a minimum of two samples was used for each experiment and experiments were performed independently a minimum of three times. Data from all three experiments was pooled and plotted.
Lentiviral infection of K-HPNE cells
Target cells were seeded at ~40% confluence in a 6 well plate and allowed to attach overnight. The next day, target cells were infected in the presence of media containing mCherry lentivirus (Plv-CMV-Puro vector) and 10 ug/ml polybrene (Santa Cruz). After 24 hrs, media was replaced with complete K-HPNE media and allowed to grow for an additional 48 hours. 72 hrs after the initial infection, cells were screened for the presence of mCherry, using the EVOS microscope system. After confirmation of red color, media was replaced with K-HPNE media containing puromycin (12 ug/mL) and selection of mCherry positive cells occurred over the next 7–10 days. The resulting cells that survived selection were then used for subsequent experiments.
Spheroid cell spreading assay
Red fluorescence protein (RFP)-expressing K-HPNE cells were trypsinized and resuspended in spheroid formation media (Irvine Scientific, Santa Ana, CA, Catalog ID: 91130) overnight; 30 μl drops containing 2.5×103 cells were carefully placed on a lid of a sterile 100 mm Petri dish. The dish was filled with 5 mL media, to avoid condensation or drying, and the lid with the “hanging drops” was carefully placed, drops facing down, and incubated overnight. Spheroids were carefully removed from the lids and placed, one by one, onto the assorted matrices, gels and glass substrates and allowed to adhere, for 4 hrs. Subsequently, the spheroid formation media replaced by regular pancreatic cancer growth media. Spheroid cell spreading assays lasted 24 hrs. Areas of cell spreading were visualized in an inverted microscope equipped with epifluorescent image acquisition capabilities, using a 10X objective (note that for images shown in Figure 4B and movies 1–8, images were acquired using a NIKON A1 confocal microscope using 20X and 60X objectives). Data were normalized to the initial size of each spheroid, as measured at time 0 (4 hrs after initial incubation). When indicated, spheroids were treated overnight with 20 μM of the MEK1/2 inhibitor U0126 from Calbiochem or the equivalent volume of DMSO, or pre-transfected 24 hrs prior to spheroid formation with the assorted siRNAs. Images were processed using MetaMorph 7.8.1.0 software (Molecular Devices, Downingtown, PA). A minimum of 5 spheres per condition were analyzed in at least three independent experiments. Similar experiments were conducted using Panc1 cells.
Quantification and Statistical Analysis
All experiments included a minimum of duplicate samples and repeated independently at least three times. Data was plotted using GraphPad Prism and analyzed using unpaired Student’s t-test analyzing unpaired conditions each time. Values are presented as median ± interquartile range or mean ± standard deviation, as indicated in the figure legends. Asterisks depicting statistical significance are indicated, when relevant.
Reagents Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Rabbit anti-mouse fibronectin antibody | Abcam | Cat # ab2413, RRID:AB_2262874 | |
| Donkey anti-rabbit Cy5 conjugated antibody | Jackson ImmunoResearch | Cat # 711–175-152, RRID:AB_2340 607 | |
| Rabbit anti-human phospho- p44/42 ERK1/2 (Thr202/Tyr204) | Cell Signaling Technology | Cat # 4370, RRID:AB_2315112 | |
| Rabbit anti-human total- p44/42 ERK1/2 | Cell Signaling Technology | Cat # 9102, RRID:AB_330744 | |
| Phospho-p90RSK1 (Ser380) antibody | Millipore | Cat # 04–418, RRID:AB_673094 | |
| Ki67 antibody | Abcam | Cat # ab15580, RRID:AB_443209 | |
| Histone H3 antibody | Cell Signaling Technology | Cat # 4499 | |
| Pan-cytokeratin (AE1/AE3) antibody | DAKO | Cat # M3515 | |
| Vimentin antibody | Abcam | Cat #ab92547 | |
| GAPDH antibody | Millipore | Cat # MAB374, RRID:AB_2107445 | |
| Biological Samples | |||
| Rat Tail Collagen-I | Thermo Fisher Scientific | Cat# A1048301 | |
| Surgical samples of patient harvest pancreatic tissue (normal and cancer tissue) | Fox Chase Cancer Center Biological sample Repository | NA | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| U0126 | Calbiochem | Cat # 662005 | |
| Sulfo-SANPAH | Thermo Fisher Scientific | Cat # 22589 | |
| APTES | Sigma-Aldrich | Cat # A3648 | |
| Glutaraldehyde | Sigma-Aldrich | Cat # G6257 | |
| Acrylamide | Sigma-Aldrich | Cat # A4058 | |
| N,N’ -Methylenebisacrylamide solution | Sigma-Aldrich | Cat # M1533 | |
| TEMED | Sigma-Aldrich | CAS Number 110–18-9 | |
| APS | Sigma-Aldrich | Cat # A3678 | |
| DCDMS | Sigma-Aldrich | Cat no. 440272 | |
| Paraformaldehyde | Electron Microscopy Sciences | CAS #30525–89-4 | |
| Odyssey blocking buffer (TBS) | LI-COR Biotechnology | Cat # 927–50100 | |
| SYBR green | Thermo Fisher Scientific | Cat # S7563 | |
| Lipofectamine® 2000 | Thermo Fisher Scientific | Cat # 11668027 | |
| Cell lysis buffer | Cell Signaling Technology | Cat # 9803 | |
| PierceTM Phosphatase | Thermo Fisher Scientific | Cat # 88667 | |
| Protease Inhibitor Mini Tablets | Thermo Fisher Scientific | Cat # 88665 | |
| Spheroid formation media | Irvine Scientific | Cat # 91130 | |
| Polybrene | Santa Cruz | Cat # sc-134220 | |
| Critical Commercial Assays | |||
| Subcellular Protein Fractionation Kit | Thermo Scientific | Cat # 78840 | |
| Experimental Models: Cell Lines | |||
| Human pancreatic CAFs | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5283834/ | NA | |
| NIH-3T3 fibroblasts | ATCC | Cat # CRL-1658™ | |
| Panc-1 cells | ATCC | RRID:CVCL_0480 | |
| hTERT-HPNE (HPNE) | ATCC https://www.ncbi.nlm.nih.gov/pubmed/17332339 | RRID:CVCL_C466 | |
| hTERT-HPNE E6/E7/KRasG12D (K-HPNE) | ATCC https://www.ncbi.nlm.nih.gov/pubmed/17332339 | RRID:CVCL_C469 | |
| Oligonucleotides | |||
| Non-targeting control siRNA | Life Technologies- Dharmacon | Cat # D-001810–01-05 | |
| siRNA targeting ERK1 | Life Technologies- Dharmacon | Cat # L-003592–00-0005 | |
| siRNA targeting ERK2 | Life Technologies- Dharmacon | Cat # L-003555–00-0005 | |
| Software and Algorithms | |||
| ImageJ OrientationJ plugin software | http://bigwww.epfl.ch/demo/orientation/ | RRID:SCR_014796 | |
| SMIA-CUKIE 2.1.0 | https://github.com/cukie/SMIA | RRID:SCR_014795 | |
| MetaMorph 7.8.1.0 software | Molecular Devices | RRID:SCR_002368 | |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | RRID:SCR_002798 | |
Supplementary Material
HIGHLIGHTS:
Substrate stiffness dictates a biphasic distribution of fibroblastic aspect ratios
Cancer-associated fibroblastic aspect ratios dictate extracellular matrix fiber alignment
Biphasic matrix fiber alignment is explained by a minimum free energy model
Physiologically soft substrates prompt formation of an isotropic matrix that restricts tumor cell growth and spheroid cell spread via nuclear pERK1/2 exclusion
Anisotropic matrix-induced pancreatic cancer cell spread is regulated by pERK2
Human normal stroma maintains pERK1/2 out of the nucleus of pancreatic epithelium
Acknowledgements:
The authors would like to dedicate this work to the memory of Dr. Patricia Keely whose work inspired this study. We thank Dr. E. Golemis for her input in revising and discussing this work, C. O’Donnell for proofing as well as S. Karamil, J. So, and S. Eble for technical help. We thank Dr. R. Francescone for molecular biology consulting.
Financial support was from the Commonwealth of Pennsylvania, Temple-FCCC’s Nodal Grant (EC, PIL, RM), NIH/NCI’s R01 CA113451 (EC), DOD W81XH-15–1-0170 (EC), funds from the Martin and Concetta Greenberg Pancreatic Cancer Institute (EC), and NIH/NCI’s CA006927 Core Grant Supported Facilities: Talbot Library, Biorepository, Translational, Histopathology, Biostatistics, Cell Imaging, Cell Culture, Instrument Shop, and Glass Washing.
Abbreviations:
- 2D
Two dimensional
- 3D
Three dimensional
- AFM
Atomic force microscopy
- APTES 3
Aminopropyl triethoxysilane
- CAF
Cancer-associated fibroblast
- CDM
Cancer-associated fibroblast-derived extracellular matrix
- DCDMS
dichlorodimethylsilane
- ECM
Extracellular matrix
- ERK
Extracellular signal-regulated kinase
- ERK1
Extracellular signal-regulated kinase 1
- ERK2
Extracellular signal-regulated kinase 2
- FAP
Fibroblast activating protein
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- hTERT
Human telomerase reverse transcriptase
- K-HPNE
hTERT immortalized and E6/E7/KRasG12D/small T antigen mutated, human pancreatic nestin expressing cell
- KRas
Kirsten retrovirus associated to sarcoma
- Patho-gel
Pathological stiffness matching acrylamide gel
- PDAC
Pancreatic ductal adenocarcinoma
- pERK
Phosphorylated extracellular signal-regulated kinase
- Physio-gel
Physiological stiffness matching acrylamide gel
- RFP
Red fluorescence protein
- siRNA
Short Interfering RNA
- TBS
T Tris buffer saline containing 0.5 % v/v Tween 20
- TEMED
N,N,N′,N′-Tetramethylethylenediamine accelerator
- UV
Ultraviolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Humphrey JD, Dufresne ER, Schwartz MA, Mechanotransduction and extracellular matrix homeostasis, Nature reviews. Molecular cell biology 15(12) (2014) 802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cukierman E, Stromagenesis, in: Schwab M (Ed.), Encyclopedia of Cancer, Springer; Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 4376–4379. [Google Scholar]
- [3].Sasser AK, Hall BM, Desmoplasia, in: Schwab M (Ed.), Encyclopedia of Cancer, Springer; Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 1344–1347. [Google Scholar]
- [4].Drifka CR, Loeffler AG, Mathewson K, Keikhosravi A, Eickhoff JC, Liu Y, Weber SM, Kao WJ, Eliceiri KW, Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection, Oncotarget 7(46) (2016) 76197–76213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Drifka CR, Tod J, Loeffler AG, Liu Y, Thomas GJ, Eliceiri KW, Kao WJ, Periductal stromal collagen topology of pancreatic ductal adenocarcinoma differs from that of normal and chronic pancreatitis, Mod Pathol 28(11) (2015) 1470–80. [DOI] [PubMed] [Google Scholar]
- [6].Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ, Aligned Collagen Is a Prognostic Signature for Survival in Human Breast Carcinoma, The American Journal of Pathology 178(3) (2011) 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M, Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes, Cell 53(4) (1988) 549–54. [DOI] [PubMed] [Google Scholar]
- [8].Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grutzmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA, Australian I Pancreatic Cancer Genome, Gill AJ, Eshleman JR Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV, Grimmond SM, Whole genomes redefine the mutational landscape of pancreatic cancer, Nature 518(7540) (2015) 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA, Genetics and biology of pancreatic ductal adenocarcinoma, Genes Dev 30(4) (2016) 355–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, Goggins M, Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia, Gastroenterology 142(4) (2012) 730–733 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M, Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice, J Clin Invest 122(2) (2012) 639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA, Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse, Cancer Cell 4(6) (2003) 437–50. [DOI] [PubMed] [Google Scholar]
- [13].Tape CJ, Ling S, Dimitriadi M, Kelly M McMahon, Jonathan D. Worboys, Hui S. Leong, Ida C. Norrie, Crispin J. Miller, G. Poulogiannis, Douglas A. Lauffenburger, C. Jørgensen, Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation, Cell 165(4) (2016) 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM, Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression, Nat Med 22(5) (2016) 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Logsdon CD, Lu W, The Significance of Ras Activity in Pancreatic Cancer Initiation, Int J Biol Sci 12(3) (2016) 338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H, Safran H, Humblet Y, Perez Ruixo J, Ma Y, Von Hoff D, Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer, J Clin Oncol 22(8) (2004) 1430–8. [DOI] [PubMed] [Google Scholar]
- [17].Baines AT, Xu D, Der CJ, Inhibition of Ras for cancer treatment: the search continues, Future medicinal chemistry 3(14) (2011) 1787–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kalluri R, The biology and function of fibroblasts in cancer, Nat Rev Cancer 16(9) (2016) 582–98. [DOI] [PubMed] [Google Scholar]
- [19].Mezawa Y, Orimo A, The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas, Cell Tissue Res 365(3) (2016) 675–89. [DOI] [PubMed] [Google Scholar]
- [20].von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A, The role of stromal cancer-associated fibroblasts in pancreatic cancer, Journal of hematology & oncology 10(1) (2017) 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, Khan GN, Blau JL, Craig R, Balis UG, Zalupski MM, Simeone DM, Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma, Clin Cancer Res 20(23) (2014) 5937–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, Attwood S, Garcia R, Ghassemi S, Fabry B, Del Rio Hernandez A, ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion, Nat Commun 7 (2016) 12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM, Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy, Cell 159(1) (2014) 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Whatcott CJ, Han H, Von Hoff DD, Orchestrating the Tumor Microenvironment to Improve Survival for Patients With Pancreatic Cancer: Normalization, Not Destruction, Cancer J 21(4) (2015) 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Alexander J, Cukierman E, Stromal dynamic reciprocity in cancer: intricacies of fibroblastic-ECM interactions, Current opinion in cell biology 42 (2016) 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR, Stromal reengineering to treat pancreas cancer, Carcinogenesis 35(7) (2014) 1451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Itoh Y, Takehara Y, Kawase T, Terashima K, Ohkawa Y, Hirose Y, Koda A, Hyodo N, Ushio T, Hirai Y, Yoshizawa N, Yamashita S, Nasu H, Ohishi N, Sakahara H, Feasibility of magnetic resonance elastography for the pancreas at 3T, J Magn Reson Imaging 43(2) (2016) 384–90. [DOI] [PubMed] [Google Scholar]
- [28].Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E, Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts, Nature cell biology 15(6) (2013) 637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, Janmey PA, Furth EE, Wells RG, Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis, Am J Physiol Gastrointest Liver Physiol 293(6) (2007) G1147–54. [DOI] [PubMed] [Google Scholar]
- [30].Ahmadzadeh H, Webster MR, Behera R, Jimenez Valencia AM, Wirtz D, Weeraratna AT, Shenoy VB, Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion, Proceedings of the National Academy of Sciences (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu R, Boudreau A, Bissell MJ, Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices, Cancer metastasis reviews 28(1–2) (2009) 167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shenoy VB, Wang H, Wang X, A chemo-mechanical free-energy-based approach to model durotaxis and extracellular stiffness-dependent contraction and polarization of cells, Interface focus 6(1) (2016) 20150067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Franco-Barraza J, Francescone R, Luong T, Shah N, Madhani R, Cukierman G, Dulaimi E, Devarajan K, Egleston BL, Nicolas E, Alpaugh KR, Malik R, Uzzo RG, Hoffman JP, Golemis EA, Cukierman E, Matrix-regulated integrin αvβ5 maintains α5β1-dependent desmoplastic traits prognostic of neoplastic recurrence, eLife 6 (2017) e20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Franco-Barraza J, Beacham DA, Amatangelo MD, Cukierman E, Preparation of extracellular matrices produced by cultured and primary fibroblasts, Curr Protoc Cell Biol Chapter 10(71) (2016) 10.9.1–10.9.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Amatangelo MD, Bassi DE, Klein-Szanto AJ, Cukierman E, Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts, Am J Pathol 167(2) (2005) 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang H, Abhilash AS, Chen CS, Wells RG, Shenoy VB, Long-range force transmission in fibrous matrices enabled by tension-driven alignment of fibers, Biophys J 107(11) (2014) 2592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kaukonen R, Mai A, Georgiadou M, Saari M, De Franceschi N, Betz T, Sihto H, Ventela S, Elo L, Jokitalo E, Westermarck J, Kellokumpu-Lehtinen PL, Joensuu H, Grenman R, Ivaska J, Normal stroma suppresses cancer cell proliferation via mechanosensitive regulation of JMJD1a-mediated transcription, Nat Commun 7 (2016) 12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Castello-Cros R, Khan DR, Simons J, Valianou M, Cukierman E, Staged stromal extracellular 3D matrices differentially regulate breast cancer cell responses through PI3K and beta1-integrins, BMC Cancer 9 (2009) 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibanez T, Pellinen T, Echarri A, Cerezo A, Klein-Szanto AJ, Garcia R, Keely PJ, Sanchez-Mateos P, Cukierman E, Del Pozo MA, Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis, Cell 146(1) (2011) 148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD, FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells, BMC Cancer 11 (2011) 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ, K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling, Cancer Res 67(5) (2007) 2098–106. [DOI] [PubMed] [Google Scholar]
- [42].Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ, Collagen reorganization at the tumor-stromal interface facilitates local invasion, BMC Med 4(1) (2006) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Clark K, Pankov R, Travis MA, Askari JA, Mould AP, Craig SE, Newham P, Yamada KM, Humphries MJ, A specific {alpha}5{beta}1-integrin conformation promotes directional integrin translocation and fibronectin matrix formation, J Cell Sci 118(2) (2005) 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cukierman E, Pankov R, Stevens DR, Yamada KM, Taking cell-matrix adhesions to the third dimension, Science 294(5547) (2001) 1708–12. [DOI] [PubMed] [Google Scholar]
- [45].Botta GP, Reginato MJ, Reichert M, Rustgi AK, Lelkes PI, Constitutive K-RasG12D activation of ERK2 specifically regulates 3D invasion of human pancreatic cancer cells via MMP-1, Mol Cancer Res 10(2) (2012) 183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Botta GP, Reichert M, Reginato MJ, Heeg S, Rustgi AK, Lelkes PI, ERK2-regulated TIMP1 induces hyperproliferation of K-Ras(G12D)-transformed pancreatic ductal cells, Neoplasia 15(4) (2013) 359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Radtke S, Milanovic M, Rosse C, De Rycker M, Lachmann S, Hibbert A, Kermorgant S, Parker PJ, ERK2 but not ERK1 mediates HGF-induced motility in non-small cell lung carcinoma cell lines, J Cell Sci 126(Pt 11) (2013) 2381–91. [DOI] [PubMed] [Google Scholar]
- [48].Bessard A, Fremin C, Ezan F, Fautrel A, Gailhouste L, Baffet G, RNAi-mediated ERK2 knockdown inhibits growth of tumor cells in vitro and in vivo, Oncogene 27(40) (2008) 5315–25. [DOI] [PubMed] [Google Scholar]
- [49].von Thun A, Birtwistle M, Kalna G, Grindlay J, Strachan D, Kolch W, von Kriegsheim A, Norman JC, ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-beta2, J Cell Sci 125(Pt 6) (2012) 1465–77. [DOI] [PubMed] [Google Scholar]
- [50].Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R, ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially, J Biol 5(5) (2006) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ, Aligned collagen is a prognostic signature for survival in human breast carcinoma, Am J Pathol 178(3) (2011) 1221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, Washington MK, Shi C, Franco OE, Weaver AM, Hayward SW, Li D, Webb DJ, Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin, The Journal of cell biology 216(11) (2017) 3799–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Conklin MW, Gangnon RE, Sprague BL, Van Gemert L, Hampton JM, Eliceiri KW, Bredfeldt JS, Liu Y, Surachaicharn N, Newcomb PA, Friedl A, Keely PJ, Trentham-Dietz A, Collagen Alignment as a Predictor of Recurrence after Ductal Carcinoma In Situ, Cancer Epidemiol Biomarkers Prev 27(2) (2018) 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rhim Andrew D., Oberstein Paul E., Thomas Dafydd H., Mirek Emily T., Palermo Carmine F., Sastra Stephen A., Dekleva Erin N., Saunders T, Becerra Claudia P., Tattersall Ian W., Westphalen CB, Kitajewski J, Fernandez-Barrena Maite G., Fernandez-Zapico Martin E., Iacobuzio-Donahue C, Olive Kenneth P., Stanger Ben Z., Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma, Cancer Cell 25(6) (2014) 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Özdemir Berna C., Pentcheva-Hoang T, Carstens Julienne L., Zheng X, Wu C-C, Simpson Tyler R., Laklai H, Sugimoto H, Kahlert C, Novitskiy Sergey V., De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses Harold L., Weaver Valerie M., Maitra A, Allison James P., LeBleu Valerie S., Kalluri R, Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival, Cancer Cell 25(6) (2014) 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yang N, Mosher R, Seo S, Beebe D, Friedl A, Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility, Am J Pathol 178(1) (2011) 325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wells RG, The role of matrix stiffness in regulating cell behavior, Hepatology 47(4) (2008) 1394–400. [DOI] [PubMed] [Google Scholar]
- [58].Haage A, Schneider IC, Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells, FASEB J 28(8) (2014) 3589–99. [DOI] [PubMed] [Google Scholar]
- [59].McDonald JA, Kelley DG, Broekelmann TJ, Role of fibronectin in collagen deposition: Fab’ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix, J Cell Biol 92(2) (1982) 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Carraher CL, Schwarzbauer JE, Regulation of matrix assembly through rigidity-dependent fibronectin conformational changes, J Biol Chem 288(21) (2013) 14805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Scott LE, Mair DB, Narang JD, Feleke K, Lemmon CA, Fibronectin fibrillogenesis facilitates mechano-dependent cell spreading, force generation, and nuclear size in human embryonic fibroblasts, Integr Biol (Camb) 7(11) (2015) 1454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ng MR, Besser A, Danuser G, Brugge JS, Substrate stiffness regulates cadherin-dependent collective migration through myosin-II contractility, J Cell Biol 199(3) (2012) 545–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Broders-Bondon F, Nguyen Ho-Bouldoires TH, Fernandez-Sanchez M-E, Farge E, Mechanotransduction in tumor progression: The dark side of the force, The Journal of Cell Biology (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Asparuhova MB, Gelman L, Chiquet M, Role of the actin cytoskeleton in tuning cellular responses to external mechanical stress, Scand J Med Sci Sports 19(4) (2009) 490–9. [DOI] [PubMed] [Google Scholar]
- [65].Lang NR, Skodzek K, Hurst S, Mainka A, Steinwachs J, Schneider J, Aifantis KE, Fabry B, Biphasic response of cell invasion to matrix stiffness in three-dimensional biopolymer networks, Acta Biomater 13 (2015) 61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Peyton SR, Putnam AJ, Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion, J Cell Physiol 204(1) (2005) 198–209. [DOI] [PubMed] [Google Scholar]
- [67].Drifka CR, Loeffler AG, Esquibel CR, Weber SM, Eliceiri KW, Kao WJ, Human pancreatic stellate cells modulate 3D collagen alignment to promote the migration of pancreatic ductal adenocarcinoma cells, Biomed Microdevices 18(6) (2016) 105. [DOI] [PubMed] [Google Scholar]
- [68].Fraley SI, Wu PH, He L, Feng Y, Krisnamurthy R, Longmore GD, Wirtz D, Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions, Scientific reports 5 (2015) 14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Roberts PJ, Der CJ, Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer, Oncogene 26(22) (2007) 3291–310. [DOI] [PubMed] [Google Scholar]
- [70].Javle MM, Gibbs JF, Iwata KK, Pak Y, Rutledge P, Yu J, Black JD, Tan D, Khoury T, Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer, Ann Surg Oncol 14(12) (2007) 3527–33. [DOI] [PubMed] [Google Scholar]
- [71].Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, Chetram M, Joshi M, Wang F, Kallakury B, Toretsky J, Wellstein A, Yi C, Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma, Science signaling 7(324) (2014) ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bartholomeusz C, Gonzalez-Angulo AM, Liu P, Hayashi N, Lluch A, Ferrer-Lozano J, Hortobagyi GN, High ERK protein expression levels correlate with shorter survival in triple-negative breast cancer patients, Oncologist 17(6) (2012) 766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lee S, Yoon S, Kim DH, A high nuclear basal level of ERK2 phosphorylation contributes to the resistance of cisplatin-resistant human ovarian cancer cells, Gynecol Oncol 104(2) (2007) 338–44. [DOI] [PubMed] [Google Scholar]
- [74].Bar-Gill AB, Efergan A, Seger R, Fukuda M, Sagi-Eisenberg R, The extra-cellular signal regulated kinases ERK1 and ERK2 segregate displaying distinct spatiotemporal characteristics in activated mast cells, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1833(9) (2013) 2070–2082. [DOI] [PubMed] [Google Scholar]
- [75].Chatterjee S, Huang EH, Christie I, Burns TF, Reactivation of the p90RSK-CDC25C pathway leads to bypass of the ganetespib induced G2/M arrest and mediates acquired resistance to ganetespib in KRAS mutant NSCLC, Mol Cancer Ther 16(8) (2017) 1658–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Aplin AE, Stewart SA, Assoian RK, Juliano RL, Integrin-mediated adhesion regulates ERK nuclear translocation and phosphorylation of Elk-1, The Journal of cell biology 153(2) (2001) 273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kong B, Michalski CW, Hong X, Valkovskaya N, Rieder S, Abiatari I, Streit S, Erkan M, Esposito I, Friess H, Kleeff J, AZGP1 is a tumor suppressor in pancreatic cancer inducing mesenchymal-to-epithelial transdifferentiation by inhibiting TGF-beta-mediated ERK signaling, Oncogene 29(37) (2010) 5146–58. [DOI] [PubMed] [Google Scholar]
- [78].Parikh N, Shuck RL, Nguyen TA, Herron A, Donehower LA, Mouse tissues that undergo neoplastic progression after K-Ras activation are distinguished by nuclear translocation of phospho-Erk1/2 and robust tumor suppressor responses, Mol Cancer Res 10(6) (2012) 845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tse JR, Engler AJ, Preparation of hydrogel substrates with tunable mechanical properties, Curr Protoc Cell Biol Chapter 10 (2010) Unit 10 16. [DOI] [PubMed] [Google Scholar]
- [80].Pelham RJ Jr., Y.-l. Wang, Cell locomotion and focal adhesions are regulated by substrate flexibility, PNAS 94(25) (1997) 13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.