

Abstract
BRAF kinase plays an important role in mitogen activated protein kinase (MAPK) signaling and harbors activating mutations in about half of melanomas and in a smaller percentage in many other cancers. Despite its importance, few in vitro studies have been performed to characterize the biochemical properties of full-length BRAF. Here we describe a strategy to generate an active, intact form of BRAF protein suitable for in vitro enzyme kinetics. We show that purified intact BRAF protein auto-phosphorylates its kinase activation loop and this can be enhanced by binding its MEK protein substrate through an allosteric mechanism. Our studies provide in vitro evidence that BRAF selectively binds to active RAS and that the BRAF/CRAF heterodimer is the most active form relative to their respective homodimers. Full-length BRAF analysis with small molecule BRAF inhibitors shows that two drugs, dabrafenib and vemurafenib, can modestly enhance BRAF's kinase activity at low concentration. Taken together, our characterization of intact BRAF contributes to a framework for understanding its role in cell signaling.
Keywords: BRAF, CRAF, RAS, RAF Kinase, MAPK Signaling, MEK, ATP-competitive Inhibitor, Auto-phosphorylation, Paradoxical Activation
Graphical Abstract
Here we report a strategy to obtain full-length BRAF that is significantly more active than its truncated counterpart. To gain a better understanding of its regulation, we conducted a detailed kinetic analysis of purified full-length BRAF to investigate the impact of BRAF inhibitors (BRAFi), MEK binding, RAS binding, and heterodimerization with CRAF on kinase activity in vitro.
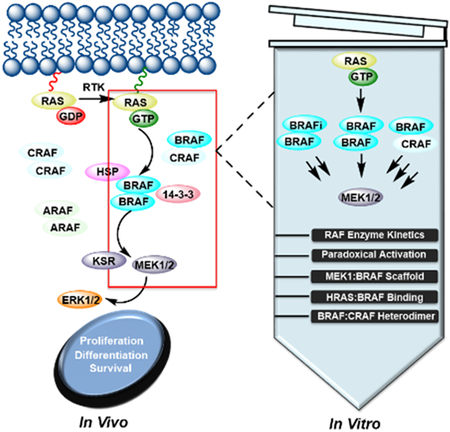
Introduction
BRAF is the most frequently mutated serine/threonine kinase in human cancers[1]. BRAF, together with its two isoforms ARAF and CRAF belong to the RAF family, which is a core component of the RAS-RAF-MEK-ERK pathway (MAPK pathway)[2]. Under physiological conditions, the RAF family is activated by binding to growth factor-stimulated RAS and signals to the downstream kinases MEK and ERK through a series of sequential phosphorylation and activation events[2]. Activated ERK then phosphorylates a diverse set of cytoplasmic and nuclear proteins to trigger cell proliferation, differentiation, and survival[2].
Multiple factors are involved in RAF activation such as dimerization, phosphorylation/dephosphorylation, membrane recruitment, and interactions with various accessory proteins. The RAF kinase family shares three conserved regions: CR1, CR2, and CR3[1b]. It is believed that the N-terminal CR1 and CR2 domains maintain autoinhibitory interactions with the C-terminal catalytic CR3 domain under resting conditions so that full-length RAF resides as an inactive monomer in the cytosol[3], although direct experimental evidence for this model has been lacking. CR1 contains a RAS binding domain (RBD) followed by a cysteine-rich domain/CRD (also noted as zinc finger motif) whose exact function is less defined. The serine/threonine-rich CR2 domain is believed to stabilize an inactive conformation of RAF kinase through binding to 14–3-3 proteins in a phosphorylation-dependent manner[4]. The current RAF activation model proposes that binding of active GTP-bound RAS to the RBD and dephosphorylation of CR2 relieve the inhibitory interactions among these domains to induce active homo- and/or heterodimers of the catalytic CR3 domain. In addition, membrane localization and phosphorylation of key residues are necessary steps for full activation of RAF, although their order and amplitude remain elusive. Site-directed mutagenesis and structural analyses of the catalytic CR3 domain demonstrate that allosteric interactions within the catalytic domain dimer contribute to kinase activation[5]. Due to the lack of structural information of the full-length RAF kinase, it is unclear how the N-terminal regulatory domains and the C-terminal catalytic domain coordinate to assemble the active conformation of the RAF kinase in the context of the full-length protein.
Several oncogenic BRAF mutations have been identified to promote constitutive MAPK pathway activity and subsequent tumorigenesis[1]. Targeting BRAF with small molecule inhibitors is now an established strategy for BRAF mutant cancers and particularly for melanoma. Two BRAF inhibitors, vemurafenib and dabrafenib, are FDA approved for clinical use[6]. Notably, BRAF inhibitors affect MAPK signaling in a mutation-specific manner. While they inhibit the catalytic activity of the BRAF V600E mutant and MAPK signaling to cause tumor regression[7], they also stimulate the same pathway in cells containing wild-type BRAF and oncogenic RAS to induce secondary malignancies, a phenomenon known as ‘paradoxical activation’[8]. These concerns surrounding current BRAF inhibitor therapy underscore the urgent need for further investigation into the unusual regulation mechanism of BRAF.
The paradoxical activation seen in the clinic has spurred intensive investigation on the underlying molecular mechanism. It is widely believed that BRAF inhibitor binding to BRAF promotes BRAF/CRAF heterodimerization and CRAF priming as a result of the formation of GTP-RAS/RAF complex [8–9]. However, relative to other BRAF inhibitors, vemurafenib and dabrafenib promote RAF dimerization to a much less extent but induce much higher level of paradoxical activation. The mechanism by which vemurafenib induces paradoxical activation has been proposed to work through the binding of vemurafenib to one protomer of a RAF dimer in the αC-helix OUT conformation and forcing the second protomer of the dimer to adopt a αC-helix IN active kinase conformation that is unable to bind vemurafenib[9–10]. An alternative but not mutually exclusive theory suggests that paradoxical activation is caused by relieving inhibitory autophosphorylation of the ATP-binding loop (P-loop)[11]. This mechanism predicts that all RAF inhibitors are likely to cause paradoxical activation. The other unexpected side effect of current BRAF therapies is intrinsic drug resistance, which is caused by the low potency of BRAF inhibitors against RAF that signals as a dimer in vivo[12]. In either scheme, binding of BRAF inhibitors to one RAF molecule decreases their binding affinity to the unbound RAF molecule within the dimer, referred to as ‘negative allostery’. Unfortunately, none of the two undesired side effects are captured by in vitro assays using truncated BRAF catalytic domain. Instead, BRAF inhibitors potently inhibit truncated BRAF catalytic domain in vitro. This discrepancy led us to speculate that purified full-length BRAF might recapitulate the actual in vivo mechanism more precisely.
Traditional approaches for studying RAF kinases have involved cell-based assays, immunoprecipitation kinase assays, and functional and structural characterization of the RAF catalytic domain. Results obtained from cell-based assays provide valuable information about the biology of RAF kinases, but typically depend on stimuli, growth cycle, cell type, and complex cellular contexts. Local concentrations and cellular stoichiometry of RAF proteins, MEK, and ERK are key determinants of MAPK signaling output and could vary significantly in different cells and tissues. Immunoprecipitation kinase assays are limited by sample heterogeneity and quantitative reliability. Although RAF proteins have been studied for over 30 years, much of the in vitro studies have been carried out on the truncated catalytic domain. While a wealth of X-ray structures of the BRAF catalytic domain in complex with various inhibitors exist, all these structures represent truncated, dephosphorylated, and inactive proteins. Here, we carry out quantitative enzymological studies on intact BRAF in a cell-free system to yield more physiologically relevant and controlled experiments.
Results and Discussion
Purification of Full-Length BRAF Protein from HEK293F Mammalian Cells.
Given the tendency of BRAF to become insoluble and unstable in the absence of chaperones and appropriate post-translational modifications, it is difficult to isolate intact RAFs suitable for in vitro analysis. Several commonly used expression systems, such as Escherichia coli strains and insect cells, have failed to produce active full-length BRAF protein. We carried out the expression in suspension-adapted mammalian HEK293F cells, which have a significantly higher potential for generating near native-like proteins[13]. We purified full-length BRAF from transiently transfected mammalian HEK293F cells. The size-exclusion chromatography (SEC) elution profile of full-length BRAF shows two peaks (Fig. 1A). Coomassie staining results demonstrate that the first peak (peak #1) contains both BRAF and a 70-kDa protein, which was identified as HSP70 protein by mass spectrometry (Supplementary Fig. 1). The majority of BRAF protein (~70% of total BRAF protein) elutes as peak #2 with minimal amount of chaperone proteins (Fig. 1B), suggesting that purified full-length BRAF adopts a predominantly dimer state in vitro. We concentrated the second peak for further enzymological characterization. As demonstrated by the coomassie staining (Fig. 1B), the purified BRAF enzyme is with good yield (~300 μg/L) and homogeneity, ideal for enzyme kinetics. To characterize the oligomerization state of purified full-length BRAF, we analyzed its native size using chemical crosslinking and SDS-PAGE. As shown in Fig. 1C, crosslinked BRAF displayed a distinct band at a size of ~ 200 kDa, corresponding to a dimer while only monomeric BRAF was detected when BRAF was electrophoresed without crosslinking. Together, gel filtration analysis of native BRAF and electrophoretic analysis of crosslinked BRAF suggest that purified full-length BRAF is predominantly dimeric in solution.
Figure 1.

(A) Size-exclusion chromatography of purified full-length BRAF. The elution volumes of protein standards (690, 158, 44, and 17 kDa) are indicated by arrows. (B) Coomassie blue–stained SDS–PAGE analysis of the purified full-length BRAF. FL-BRAF and HSP70 bands are indicated by arrows. Molecular weight markers (M) are indicated. (C) One microgram of purified full-length BRAF was incubated with various concentrations of BS3 (0, 7.8, 15.6, 31.25, 62.5, 125, and 250 μM) for 30 min at room temperature. The samples were separated with SDS-PAGE and stained with coomassie blue staining. BRAF dimers and BRAF monomers are indicated by arrows. (D) Quantification of the activity of 5 nM BRAF with or without the indicated amount (1, 5, and 25 nM) of HSP70 protein. Reaction mixtures were subjected to western blot analysis with anti-pMEK (top) and anti-HSP70 (bottom) antibodies. Representative images were shown from one of three independent experiments. (E) Specific enzyme activity of full-length BRAF (FL-BRAF) and kinase domain of BRAF (CD-BRAF). The kinase activity of BRAF was quantified via western blotting for MEK phosphorylation. The levels of phospho-MEK were analyzed using ImageJ software and three independent replicates were included.
HSP70 Has No Effect on the Kinase Activity of BRAF.
It is apparent that HSP70 causes formation of higher-order multimerization of BRAF, as demonstrated by the SEC chromatography. We purified kinase-dead MEK1 from E. coli for use as the RAF substrate and compared the specific activity of peak#1 and peak#2. Phosphorylated MEK1 was probed with phospho-specific antibody pMEK1 (Ser217/221) via immunoblotting. We did not observe any difference in catalytic capability between higher-order oligomeric BRAF (peak#1) and the apparent dimeric BRAF (peak#2). This is consistent with fluorescence imaging microscopy studies on BRAF in live cells that revealed both multimerization and dimerization[14]. A similar phenomenon was observed for purified full-length epidermal growth factor receptor[15] (unpublished data), suggesting that multimerization might represent a general mechanism of activation for protein kinases, in addition to dimerization. The purified full-length BRAF concentrated from peak#2 contains a trace amount of HSP70, which might interfere with kinetic studies. To address this possibility, we titrated various amount of purified HSP70 into BRAF and quantified the kinase activity of BRAF. As shown in Fig. 1D, the presence of HSP70 does not change the catalytic activity of BRAF, suggesting that the HSP70 that co-purified with BRAF unlikely effects interpretation of the follow-up kinase experiments as described below.
The Catalytic Activity of Full-length BRAF is Significantly Higher Compared with the Truncated Catalytic Domain.
Previous cellular studies have suggested that the N-terminal non-catalytic domain of RAF plays a negative regulatory role by preventing RAF dimerization[3]. One concern regarding purified full-length RAF proteins is that they might adopt an autoinhibited state with no or reduced catalytic activity. In light of this, for comparison we also expressed and purified the catalytic domain (CR3, residues 442–724) of BRAF from Sf9 cells, as previously reported[16]. Consistent with previous studies[17], the catalytic domain of BRAF is present predominantly as a monomer (Supplementary Fig. 2A&B), in contrast to full-length BRAF, which migrates on gel filtration as an apparent dimer. Consistent with its oligomeric state, the specific activity of the full-length BRAF is ~20 fold higher than that of catalytic domain (Fig. 1E). We did not detect 14–3-3 proteins in our purified protein using 14–3-3 (pan) antibody. The lack of 14–3-3 protein detection demonstrates that this protein is removed during our protein purification procedure. We propose that the absence of 14–3-3 proteins abrogates the autoinhibitory function of the N-terminal region.
It is possible that different expression systems could generate proteins with distinct post-translational modification patterns, thus influencing the different kinetic profiles of full-length BRAF vs. the isolated catalytic domain. Phosphorylation of the activation loop (Thr599/Ser602) has been well established as one critical step to achieve maximal activity of BRAF throughout the activation cycle[2, 18]. Phosphorylation of Thr599/Ser602 is believed to disrupt the closed inactive conformation of the kinase domain and initiate subsequent catalysis. We compared the phosphorylation status of the activation loop between catalytic domain and full-length proteins using phospho-Thr599 antibody. As shown in Supplementary Fig. 2, both catalytic domain and full-length BRAF have very low basal levels of activation loop phosphorylation that are comparable to each other (0 min time point). However, full-length BRAF is observed to auto-phosphorylate its activation loop in the presence of ATP while the monomeric catalytic domain was not able to phosphorylate its activation loop under the same conditions (Supplementary Fig. 2), further supporting our conclusion that full-length BRAF adopts a dimeric configuration capable of trans-phosphorylation. Regardless, the higher enzyme activity from our approach highlights the significance of using full-length BRAF in characterizing its biochemical parameters.
MEK1/BRAF Complex Primes Auto-phosphorylation of BRAF Activation Loop.
Similar to other kinases, RAF kinases undergo phosphorylation as a key mechanism of regulation. As expected, dephosphorylated BRAF upon treatment with calf intestine phosphatase (CIP) was inactive (Fig. 2A). We found that full-length BRAF auto-phosphorylates its own activation loop and that auto-phosphorylation reaches a plateau within 10 min (Fig. 2B). Therefore, 10 min of reaction time was chosen for the kinase assays conducted in this study, unless specified otherwise.
Figure 2.

(A) Comparison of the enzymatic activity of dephosphorylated versus native BRAF. The dephosphorylated BRAF was obtained after treatment of CIP protein phosphatase. BRAF protein purified from HEK293F cells is defined as native protein. The kinase activity of BRAF was quantified via western blotting for MEK phosphorylation. (B) Time-course of auto-phosphorylation of the activation loop. Purified BRAF was incubated with ATP for the indicated time 2.5, 5.0, 7.5, 10, 15, 20, and 30 min. (C) Auto-phosphorylation of the BRAF activation loop was increased by the presence of kinase-dead MEK substrate. BRAF was incubated with or without ATP and kinase-dead MEK for 5 min at 30 °C and probed with phospho-Thr599 BRAF antibody. (D) Autoradiograph showing incorporation of radioactive phosphate (32P) into BRAF and MEK simultaneously. BRAF and 32P-labeled ATP were incubated with or without kinase-dead MEK for 30 min at 30 °C. The top band represents auto-phosphorylated BRAF (p-BRAF). The bottom band represents phosphorylated MEK (p-MEK).
Surprisingly, the presence of kinase-dead MEK1 triggered a dramatic increase in activation loop auto-phosphorylation (Fig. 2C), although overall auto-phosphorylation of BRAF was decreased, as quantified using P-32 autoradiograph (Fig. 2D). To further investigate this, we did quantitative proteomics on two micrograms of purified BRAF pre-incubated with either ATP and Mg2+ or ATP, Mg2+ and kinase-dead MEK1 for 30 min at 25 °C. BRAF and BRAF in a mixture with MEK1 were separated by SDS-PAGE. The protein bands corresponding to BRAF were cut out, digested with trypsin, and enriched for phospho-peptides. The mass spectrometry data demonstrate that auto-phosphorylation of Thr599 was increased 2.3-fold in the presence of kinase-dead MEK1 (Supplementary Table 1), confirming that binding of MEK1 to BRAF primes the activation loop for stimulatory auto-phosphorylation. As the purified MEK1 in our study is inactive, our studies reveal a scaffolding function of MEK1 independent of its kinase activity, providing an additional layer of complexity regarding RAF regulation.
Steady-State Kinetics of Full-Length BRAF.
As little enzymatic characterization of intact BRAF has been conducted, we developed a quantitative radiometric assay to measure the turnover number of BRAF by monitoring total ATP consumption. In these assays, protein kinase activity is measured by quantifying the incorporation of radiolabeled phosphate from γ−32P-ATP into BRAF and BRAF substrate MEK1. As shown in Fig. 3A&B, the reaction rate is linear versus reaction time and enzyme concentration, supporting that the reactions catalyzed by BRAF under the conditions of our assay meet the prerequisite for steady-state kinetics. Subsequently, we chose BRAF enzyme concentration of 50 nM, MEK1 substrate concentration of 200 nM, and reaction time of 10 min for the following steady-state kinetics. The Km value for ATP was measured to be 6.6 ±1.5 μM (Fig. 3C), close to the previously reported Km value of 5 μM[8a]. The measured turnover number of BRAF was 0.44 min−1, which is a relatively slow rate for a human kinase (which typically range from 3 to 60 min−1). A plausible explanation for this low apparent kcat is that we used a low concentration of purified MEK1 protein (200 nM) as the substrate for RAF kinase assays, due to the limited solubility of MEK protein. Alternatively, the low turnover number could reflect that BRAF partnering with another protein such as CRAF is required to achieve maximal RAF activity[4].
Figure 3:

Enzymatic characterization of BRAF. (A&B) The kinase activity of BRAF is linear versus enzyme concentration (25, 50, 100, and 200 nM) (A) as well as reaction time (5.0, 7.5, 10, and 12.5 min) (B). (C) Steady-state kinetic analysis of BRAF. The values of Km for ATP and kcat were obtained with varying concentrations of ATP (3.13, 6.25, 12.5, 25.0, 50.0 μM) and fixed kinase-dead MEK concentration (200 nM) and BRAF enzyme concentration (50 nM).
Purified Full-length BRAF Recapitulates ‘Paradoxical Activation’ Induced by Dabrafenib and Vemurafenib.
Dabrafenib and vemurafenib are ATP-competitive BRAF inhibitors that have been approved to treat metastatic melanoma patients carrying the V600E mutation[6, 19]. It has been an enigma that the notorious paradoxical activation of BRAF by inhibitors in cancer cell lines and clinics has not been observed with in vitro kinase assays[1b], which led us to speculate that intact BRAF might display actual in vivo mechanisms more precisely. We applied an enzyme-linked immunosorbent assay (ELISA)[20] to quantify the potency of RAF inhibitors against BRAF. The ELISA readout is phosphorylated MEK1, which was probed with phospho-specific antibody pMEK1 (Ser217/221). As shown in Fig. 4A, dabrafenib modestly stimulates full-length BRAF at sub-saturating concentrations (< 1 nM) and inhibits BRAF only at higher concentrations, with an IC50 value of 50 nM. A similar trend was observed with vemurafenib, another ATP-competitive inhibitor approved as anti-BRAF drug (Fig. 4B). By comparing the different inhibition profiles between full-length BRAF and the catalytic domain of BRAF (Supplementary Fig. 2C), it appears that dimerization is highly relevant for paradoxical activation. Therefore, our preparation of full-length BRAF may correspond more faithfully to cellular pharmacological properties lacking from truncated BRAF.
Figure 4.

Dose-responsive inhibition curves of ATP-competitive inhibitors. (A&B) Effects of different concentrations of dabrafenib and vemurafenib on the activity of full-length BRAF. (C) Dose-response curves of various ATP-competitive inhibitors against full-length BRAF. Summary of percent of activity (± s.d.) calculated from three independent experiments is shown.
Evaluation of Other BRAF Inhibitors Using Full-Length BRAF.
Given the limitations of current BRAF drugs, a new generation of inhibitors with distinct structural and biochemical properties has emerged[9]. We sought to compare their inhibition potency towards full-length BRAF. To this end, we conducted in vitro analyses of other BRAF inhibitors: PLX-7904, TAK-632, and SB-590885 (Fig. 4C & Supplementary Fig. 2C). PLX-7904 is known as a paradox-breaker, which has been shown to overcome paradoxical MAPK pathway activation both in vivo and in cancer cell lines[21]. The chemical structure of PLX-7904 is only subtly different from that of vemurafenib, however, our purified full-length protein appears to reflect their distinct biological outcomes, emphasizing the biological relevance of evaluating RAF inhibitors using the full-length BRAF protein. Different from vemurafenib and dabrafenib that are less potent against dimeric BRAF due to negative allostery, TAK-632 and SB-590885 are representatives of RAF inhibitors that potently inhibit dimeric BRAF[22]. Both TAK-632 and SB-590885 belong to type I inhibitors that stabilizes the αC-helix IN position, thus are able to occupy both promoters within a dimer to effectively inhibit dimeric RAF[9]. The measured IC50 values of TAK-632 (0.70 nM) and SB-590885 (0.11 nM) are ~30–200 fold lower than that of vemurafenib, consistent with our observation that purified full-length BRAF exists in the dimeric form. TAK-632 and SB-590885 did not exhibit paradoxical activation in vitro. However, this does not conflict with previous cell-based assays demonstrating that TAK-632 and SB-590885 display paradoxical activation in cells[22], as they lead to the interaction of RAF with GTP-loaded RAS, followed by transactivation of RAF kinases[9, 23]. Meanwhile, TAK-632 was demonstrated to promote dimerization of purified RAF catalytic domains in vitro[24]. Full-length BRAF molecules under our assay conditions already form stable dimers, therefore TAK-632 and SB-590885 did not activate purified full-length BRAF. Our studies further establish that type I inhibitors require active RAS for paradoxical activation of full-length BRAF.
HRAS Interacts with Intact BRAF In Vitro.
As a result of RAS binding, autoinhibition interactions between the catalytic domain and the regulatory domains of RAF are relieved and RAF proteins are recruited to the plasma membrane for further activation. Binding of RAS with the isolated RBD of RAF has been studied in detail[25]. ATP-competitive inhibitors have been reported to allosterically enhance RAS-RAF association[23]. However, little is known about whether the RAS-RBD interaction exerts long-range, allosteric conformational activation on the catalytic site of RAF kinases. Purified full-length BRAF contains the RBD, which makes it suitable to investigate how RAS binding affects the biochemical properties of BRAF. We expressed and purified full-length HRAS from E. coli with a yield of ~ 1 mg/L (Fig. 5A). As RAS is active only in its dimeric form, we generated GST-HRAS protein to ensure that the purified HRAS adopts a dimeric configuration. Purified GST-HRAS was loaded with the non-hydrolyzable GTP analog GppNHp, as reported previously[26]. SEC demonstrates that the GppNHp-bound HRAS runs as an apparent dimer, with a size of ~90 kDa (Fig. 5B). We reconstituted the GppNHp-RAS/ BRAF complex, which was validated by a pull-down experiment (Fig. 5C). In a negative control experiment, GDP-bound HRAS did not bind BRAF (Fig. 5C), suggesting that the protein-protein interaction we observed between GppNHp-bound HRAS and full-length BRAF is physiologically relevant.
Figure 5:

(A) Coomassie blue–stained SDS-PAGE analysis of the purified HRAS, as indicated by the arrow. (B) Top: Size-exclusion chromatography of the purified HRAS. The elution volumes of protein standards (690, 158, 44, and 17 kDa) are indicated by arrows; Bottom: Individual fractions were analyzed by SDS-PAGE and stained with coomassie blue. (C) In vitro affinity pull-down experiments. After Flag-tagged BRAF proteins were bound to Flag-M2 antibody conjugated agarose beads, the samples were incubated with either GDP-HRAS or GppNHp-HRAS. After washing, resin-bound proteins were probed with anti-GST antibody (upper panel). The corresponding two lower panels demonstrate comparable protein loading of the lanes. (D) Comparison of the activity of 5 nM BRAF with or without the indicated amounts (0, 2.5, 5.0, 10, 20, 50, 75, 100, and 200 nM) of GppNHp-HRAS.
By quantifying MEK1 phosphorylation, we did not observe an enhancement of BRAF activity by the formation of the BRAF/HRAS complex (Fig. 5D). These data support that BRAF activation events subsequent to BRAF dimerization are independent of RAS binding. This observation is in accordance with the occurrence of several oncogenic BRAF mutations that bypass their reliance on interactions with RAS by promoting constitutive BRAF dimerization. In the context of such BRAF mutations, constitutive dimerization is sufficient to fully stimulate its kinase activity. Furthermore, our in vitro system replicates the RAS-BRAF interaction and could serve as a valid model to study in vitro binding of RAS to functional full-length BRAF.
Similarly, we monitored the kinase activity of BRAF in the presence and absence of Zn2+ to investigate the role of the zinc finger domain (cysteine rich domain, CRD), which has remained controversial. In these assays, Zn2+ had no effect on the overall kinase catalysis in vitro (data now shown). These results are not surprising, since it has been reported that CRD might regulate the association of BRAF with membrane lipids and multiprotein complexes[27]. A combination of in vitro and in vivo approaches is necessary to unveil the complex regulation of BRAF.
The BRAF/CRAF Heterodimer is the most active RAF dimer in Vitro.
The low kcat value of BRAF led us to hypothesize that the BRAF/CRAF heterodimer is the major effector of RAS. To investigate the catalytic activity of RAF heterodimers, we immuoprecipitated full-length CRAF from HEK293F cells. Consistent with the notion that CRAF by itself has very low basal activity[28], the activity of purified CRAF is undetectable under our reaction conditions. By mixing separately purified CRAF and BRAF, we found that the mixture of BRAF and CRAF has an activity up to ~20 fold higher than that of BRAF alone (Fig. 6A). Since the activity of CRAF alone is below the detection limit, we deduce that the increased activity is due to the formation of the BRAF/CRAF heterodimer, which was verified by pull-down experiments (Fig. 6B).
Figure 6:

(A) Comparison of the specific activities of 20 nM BRAF with and without 100 nM CRAF. The activity was measured by radiometric kinase assay and normalized to the activity of 20 nM BRAF. The activity of 100 nM CRAF by itself is below the detection limit. (B) A pull-down assay shows the formation of a BRAF/CRAF complex. (C) The kinase activity of the BRAF/CRAF mixture is non-linear versus reaction time. (D) Autoradiograph showing incorporation of radioactive phosphate (32P) into BRAF and MEK, simultaneously. The top band represents phosphorylated BRAF (p-BRAF). The bottom band represents phosphorylated MEK (p-MEK). No phosphorylated CRAF was detected. (E) The mixture of BRAF/CRAF is more resistant to dabrafenib. Autoradiograph showing incorporation of radioactive phosphate (32P) into BRAF and MEK simultaneously. The results are representative of three independent tests. Error bars indicate standard deviation.
Intriguingly, the mixture of BRAF/CRAF behaves differently from BRAF. First, the reaction rate of BRAF/CRAF was not linear with reaction time but rather had a distinct lag phase (Fig. 6C), suggesting that stimulatory phosphorylation is involved in activating the BRAF/CRAF heterodimer. By monitoring phosphorylation of BRAF and CRAF via P-32 autoradiograph, we identified that only BRAF was phosphorylated under the reaction conditions (Fig. 6D). Since phosphorylation of Ser338, Thr491, and Ser494 in CRAF is essential for full activation of the kinase[2], we attempted to quantify phospho-Ser338/Thr491 of CRAF in the presence of BRAF and MEK1, however, we did not detect any signal. Apparently, full activation of CRAF requires other proteins in addition to BRAF. Second, the BRAF/CRAF mixture was more resistant to dabrafenib, compared with BRAF alone (Fig. 6E). This finding is consistent with a previous report that elevated CRAF protein levels associate with drug resistance to RAF inhibition in melanoma[29]. Our data not only confirm that the BRAF/CRAF heterodimer is more active than the respective homodimers, but also demonstrate that the purified full-length BRAF and CRAF self-associate to form heterodimers. Although it is not clear which RAF isoform directly phosphorylates MEK1, we believe that one RAF isoform functions as an allosteric activator of the other RAF isoform in the reconstituted BRAF/CRAF heterodimer.
Conclusions
Regulation of BRAF activity is a highly complex process involving association with scaffold proteins and RAS, membrane recruitment, dimerization, and phosphorylation events[2]. Despite a well-established role for BRAF in tumorigenesis, its mechanism of activation remains incompletely understood. Due to the high plasticity of the BRAF kinase domain, it is critical to study intact BRAF instead of the truncated catalytic domain to investigate its regulation mechanism. A detailed kinetic analysis of functional full-length BRAF is still missing. In this study, we report a strategy to obtain intact BRAF that is significantly more active than its truncated counterpart. To gain a better understanding of its regulation, we conducted a detailed kinetic analysis to investigate the impact of RAF inhibitors, auto-phosphorylation of activation loop, RAS binding, and heterodimerization with CRAF on kinase activity in vitro.
An intramolecular interaction between the kinase domain and the N-terminal regulatory domains is believed to enforce RAF auto-inhibition in quiescent cells and is disrupted upon RAS binding[30], although high resolution structural information for this is currently not available. Our kinetic data demonstrate that the kinase domain of BRAF, lacking the N-terminal regulatory domains, has very low activity and that full-length BRAF is ~20-fold more active than the isolated catalytic domain. We believe that this phenomenon excludes the possibility of an auto-inhibited state of purified BRAF, which is further supported by the fact that the kinase activity of full-length BRAF is not affected by RAS binding. We conclude that removing accessory proteins, such as 14–3-3, during the protein purification procedure abrogates BRAF auto-inhibition and leads to RAS-independent dimerization of the kinase domain. Our conclusion that full-length BRAF forms stable dimers is supported by our crosslinking analysis, gel filtration data as well as our ability to observe paradoxical activation with dabrafenib and vemurafenib inhibitors and the observed kinetics with BRAF/CRAF heterodimers.
A crystal structure of MEK1 in complex with the catalytic domain of BRAF was previously reported and the MEK1-BRAF complex was shown to dissociate in response to MEK1 phosphorylation[31]. Although the high affinity BRAF/MEK1 complex is independent of BRAF dimerization, BRAF adopts an active conformation in the complex[31]. In addition, MEK has been proposed to interact with kinase suppressor of RAS (KSR) to drive BRAF-KSR heterodimerization, which allosterically activates BRAF[32]. Our work further clarifies another step in BRAF activation by showing that association with MEK1 facilitates auto-phosphorylation of the BRAF activation loop that is necessary for full activation of BRAF kinase. In such a model, MEK1 acts as a scaffolding protein to stabilize the active conformation of BRAF and aligns the activation loop for auto-phosphorylation. Subsequent to auto-phosphorylation, fully activated BRAF phosphorylates MEK1 to relay downstream signal. This model is consistent with our observation that BRAF cannot phosphorylate peptide substrates. Different from versatile kinases that have a broad spectrum of protein substrates, BRAF specifically phosphorylates Ser217/221 of MEK1/2, the only currently identified RAF substrates. Our findings suggest a novel function of MEK in regulating the MAPK pathway and provide a rationale for the strict substrate selectivity towards MEK1/2.
Our enzyme kinetic data reveal a weak kinase activity of BRAF homodimers and CRAF homodimers. Although it is possible that the purified RAF homodimers were not fully activated in the absence of accessory proteins, we suspect that the measured lower level of activity in our assays could be sufficient for physiological functions of RAF. RAF is capable of serving as a kinase activator of other RAF family members beyond its direct role in catalysis, as catalytic activity in RAF family members is dependent on a side-to-side dimer in which one protomer (receiver) is switched on by an allosteric interaction with the second protomer (activator)[5a]. Our in vitro data identified the BRAF/CRAF heterodimer as the most active form. The physiological relevance of the BRAF/CRAF heterodimerization is further demonstrated by the occurrence of several oncogenic kinase-dead BRAF mutants that partner with CRAF to develop lung adenocarcinoma[33]. Thus, it is reasonable to conclude that RAF does in fact have a low level of kinase activity through homodimerization and RAF is activated much more efficiently by heterodimerization. RAF is one of several examples of a protein kinase that can serve as a scaffolding protein. It is currently not clear under which circumstances RAF prefers to form heterodimers instead of homodimers.
Our data also shed light on the paradoxical activation of small molecule BRAF inhibitors observed in the clinic[8]. Previous cellular studies and structural characterization of truncated catalytic domain of BRAF suggest that RAF inhibitors activate the MAPK pathway through promoting RAF dimerization in a RAS-dependent manner[23]. In this model, ATP-competitive inhibitor binding events in the active site relieve intracellular auto-inhibition to trigger RAS association and subsequent RAF dimerization. Although the structural basis underlying the positive cooperativity between RAF inhibitors and RAS association remains elusive, different approaches agree that the inhibitor-bound RAF molecule trans-activates the inhibitor-free RAF molecule through allosteric interaction within a dimer, hence leading to downstream ERK signaling[8]. However, it is unclear whether enhanced RAF dimerization per se is the sole factor to induce paradoxical activation. Our purified full-length BRAF protein already adopts a dimeric configuration, but still was activated by dabrafenib and vemurafenib under sub-saturating concentrations. Our data provide clear in vitro evidence that certain ATP-competitive inhibitors have an intrinsic ability to activate BRAF kinase independent of other proteins, such as RAS and CRAF, most likely through modifying the kinase domain conformation. In this scenario, binding of inhibitors such as dabrafenib and vemurafenib to the BRAF catalytic domain not only lowers the binding affinity of inhibitors to the other protomer, but also makes the inhibitor-bound protomer a more efficient allosteric activator of the other protomer of the dimer. As a result, BRAF proteins that are not occupied by inhibitors are less sensitive to inhibitors and are more allosterically activated so that they compensate for the inhibited activity related to inhibition of the other protomer. With higher concentrations of inhibitor, both protomers are bound to the inhibitor and kinase activity is abrogated. In brief, the measured BRAF activity under our assay conditions is the composite of inhibition, allosteric activation, and negative cooperativity. We believe that the models of paradoxical activation are dependent on the biochemical properties of various RAF inhibitors.
Our findings have important therapeutic implications as well. Apparently, BRAF is a highly dynamic protein with various conformations and thus can be activated via distinct mechanisms. BRAF inhibitors with diverse chemical structures can elicit different effects on both kinase activity as well as protein-protein interactions. In contrast to the extant kinase domain based model, our full-length BRAF appears to better reflect RAF biological function in vivo, and thus provides a useful tool to evaluate potential RAF inhibitors in the design of new therapies targeting BRAF in disease.
Experimental Section
Compounds and Reagents
Radioactive ATP γ-P32 (# NEG002Z250UC) was purchased from Perkin Elmer. Vemurafenib, Dabrafenib, PB-PLX7904, TAK-632, AZ-628, GDC-0879, SB-590885 were purchased from Selleckchem. Inhibitors were dissolved in DMSO to produce 10 mM stocks and stored at −20°C. Non-hydrolyzable GTP analog GppNHp (G0635) was purchased from Sigma. Alkaline Phosphatase, Calf Intestinal (CIP) (M0290) was purchased from NEB. Gel filtration standard (#151–1901) was purchased from Bio-Rad. All other reagents were purchased without further purification.
Antibodies
Anti-p-MEK1/2 Ser217/221 (#9121) and anti-HSP70 (#4873S) were purchased from Cell Signaling. Anti-p-BRAF T599 (#PA5–37497) was purchased from Invitrogen. Anti-FLAG M2 (#F1804), anti-FLAG M2 agarose resin (#A2220), and anti-FLAG M2 magnetic resin (#M8823) were purchased from Sigma Aldrich. Anti-GST (#SC-138) was purchased from Santa Cruz. Talon metal affinity resin (#635501) was purchased from Takara. Profinity IMAC Ni charged resin (#156–0131) was purchased from Bio-Rad.
Plasmids
BRAF-WT/6x-HIS/FLAG and CRAF-WT/6x-HIS/HA were prepared using standard cloning procedures with pCDNA™ 4/TO (Invitrogen) as the vector. The plasmids #40775 (human BRAF) and #51124 (human CRAF) were purchased from Addgene and were used as the templates to amplify BRAF and CRAF, respectively. The following primers were designed for full-length BRAF cloning: ACCATGGGG CATCATCATCATCATCATATGGCGGCGCTGAGCGGTG and AGCGGCCGCTCACTTGTCATCGTCATCCTTGTAATCGTGGACAGGAAACGCAC. The following primers were designed for full-length CRAF cloning: TCCACCATGGGGCACCACCACCACCACCACATGGAGCACATACAGGGAG and GAGCGGCCGCCTAAGCGTAATCTGGAACATCGTATGGGTAGAAGACAGGCAGCCTCGG. HRAS-expressing plasmid was purchased from Addgene (plasmid#55653).
Full-Length RAF Purification
Full-length BRAF (Uniprot ID# P15056) and CRAF (Uniprot ID# P04049) proteins were expressed in HEK293F cells, following the protocol previously developed for EGFR[13, 34]. The cell pellet was thawed on ice and resuspended in lysis buffer (150 mM NaCl, 20 mM HEPES pH 7.4, 1 mM Na3VO4, 1 mM PMSF, 10 % glycerol and a CompleteTM protease inhibitor table Sigma Aldrich (product #11836153001)). The homogenous suspension was sonicated at 30% amplitude for 20 seconds on and 1 minute off on ice (until the solution became viscous and cells were completely lysed) using a Branson 250 Digital Sonifier Ultrasonic Cell Disruptor with a microtip attached (model #102C). The lysed cell solution was centrifuged at 20,000 rpm for 40 minutes at 4°C. Once the centrifugation is completed, the supernatant was filtered with a 0.45 μm filter and the supernatant was applied to the pre-equilibrated resin in low salt buffer (20 mM HEPES pH 7.4, 150 mM NaCl and 10% glycerol) with slow rotation for 2–4 hrs at 4°C. The resin was washed 3 times with 1 mL of low salt buffer (5 min incubations) and 2 times with high salt buffer (500 mM NaCl, 20 mM HEPES pH 7.4 and 10% Glycerol) alternating low, high, low, etc. For removal of HSP70, an ATP wash (5 mM ATP, 20 mM MgCl2 in low salt buffer) was used to remove HSP proteins. This ATP wash was performed 3–5 times with 10 min incubations with rotation. After the last ATP wash, a final low salt wash was performed before elution of FL-BRAF with 1x FLAG peptide (200 μg/mL diluted in low salt buffer) for 2–12 hours. Elutions were analyzed by SDS-Page and pooled for further purification on size exclusion chromatography on a Superdex 200 10/300 GL column (Cat#28–9909-44). Appropriate protein fractions were concentrated with a millipore concentrator cut off 30 kDa (Cat#UFC803024) and flash frozen in liquid nitrogen before storage at −80°C.
BS3 Crosslinking
The crosslinking reagent, bis(sulfosuccinimidyl)suberate BS3, was prepared according to the manufacturers protocol (thermos scientific cat# 21585). In brief, 2 mg of BS3 was dissolved and diluted to 25 mM with ddH2O. BS3 was then diluted to final working concentrations of 0–1000 μM. BRAFWT (~ 1 μg) and BS3 were incubated for 30 minutes at room temperature to allow the crosslinking to occur and 4x loading dye (Tris-HCl, β-mercaptoethanol, EDTA, SDS, bromophenol blue and glycerol) was used to quench the reaction. The samples were then boiled for 10 minutes before loading on a 10% SDS-PAGE gel. After electrophoresis, the gel was stained with coomassie staining buffer (coomassie brilliant blue G250, acetic acid, methanol, and ddH2O) and destained with destaining buffer (acetic acid, methanol, and ddH2O).
Kinase-Dead MEK (Uniprot ID#Q02750) Purification
Kinase-dead MEK1–6xHIS protein was purified from BL21 codon plus E. coli modified from a previous publication[35]. In brief, culture flasks were incubated at 37°C until an OD of 0.6–0.8 was achieved then induced with 0.5 mM IPTG and incubated at 16°C with 200 rpm rotation overnight. The cells were harvested and washed with PBS before lysing in lysis buffer (Sigma Aldrich Complete™ EDTA-Free protease inhibitor tablets (product#11836170001), 20 mM HEPES pH 7.4, 150 mM NaCl, 10 mM βME, 5 mM Imidazole, and 5% Glycerol) with sonication. The lysed cells were pelleted and the supernatant was added to 1 mL of Nickel resin and incubated for 2 hours. The protein bound resin was washed with low (150 mM NaCl, 20 mM HEPES pH 7.4 and 5% Glycerol) and high (500 mM NaCl, 20 mM HEPES pH 7.4 and 5% Glycerol) salt washes to remove non-specific binding. MEK1 was eluted with elution buffer (200 mM imidazole, 150 mM NaCl, 20 mM HEPES pH 7.4 and 5% Glycerol) and further purified, concentrated and stored as described above.
GppNHp-HRAS (Uniprot ID#Q61411) Purification
GST-tagged full-length HRAS was purified from BL21 codon plus E. coli, modified as previously described[26]. In brief, cell pellet was incubated with lysis buffer (50 mM HEPES pH8.0, 150 mM NaCl, 1 mM EDTA, 5% Glycerol, 1 mg/mL lysozyme, and protease inhibitor cocktail) for 1hr at room temperature and followed by brief sonication. Soluble cell lysate was incubated with pre-equilibrated glutathione resin for 4 h at 4°C. After extensive washing, HRAS protein was eluted off the resin with elution buffer (50 mM HEPES pH8.0, 150 mM NaCl, 1 mM EDTA, 5% Glycerol, and 10 mM reduced glutathione). Purified HRAS was incubated with CIP and GppNHp for 2h at room temperature. After adding 20 mM MgCl2, GppNHp-loaded HRAS was further purified on a Superdex 200 size exclusion chromatography column.
Western blotting-Based Kinase Assay
Kinases were diluted in 2x dilution buffer (25 mM HEPES pH 7.4, 0.125 mg/mL BSA and 300 mM NaCl) to a final 2x concentration and mixed with 2x cocktail (HEPES pH 7.4, MgCl2, DTT, β-glycerolphosphate, MEK and ATP) for 5–10 min at 30°C. The reaction was quenched with 4x loading dye and subjected to SDS-PAGE immunoblotting. The PVDF membranes were probed with the appropriate primary and HRP-tagged secondary antibodies followed by a chemiluminescent incubation. Quantitative analyses of the immunoblots were executed with ImageJ software.
Radioactive Spin-Column Assay
The kinase assay was developed as previously reported[15]. RAF was diluted in 2x dilution buffer (25 mM HEPES pH 7.4, 0.125 mg/mL BSA and 300 mM NaCl) to a final 2x concentration and mixed with 2x cocktail (25 mM HEPES pH 7.4, 10 mM MgCl2, 1 mM DTT, 50 mM β-glycerolphosphate, 10% glycerol, 200 nM MEK, 1–200 uM non-radioactive ATP and 1μCi/reaction buffer radioactive ATP γ-P32) for 5–15 min at 30°C. The reaction was quenched with 100 mM EDTA. The samples were loaded onto a 30 kDa spin column Omega (part #OD030C35) and washed with phosphate buffer (25 mM Mono- /475 mM di-basic phosphate and 250 mM NaCl). These columns were added to scintillation fluid (reference contained just scintillation fluid and the reaction buffer). The total radioactivity in the reference and sample vials were measured with a scintillation counter and calculated for Product/time, product/enzyme or V/E.
BRAF ELISA Inhibition Assays
ELISA assays were performed as previously described[36]. Pierce glutathione coated plates (cat.#15240) were washed with 1x THBS (25 mM HEPES pH 7.4, 140 mM NaCl, 0.05 % Tween-20) followed by a GST-MEK (0.0025 mg/mL final) incubation for >2hrs at RT with shaking. During the MEK incubation step, 100x inhibitor was added to 2x RAF. After a one-hour incubation, 2x cocktail buffer containing ATP was added to the glutathione-coated plate followed by 2x RAF/inhibitor and incubated at 30°C for 15 min. All wells were washed with 1x THBS and incubated for 5 minutes at RT (2x), then primary anti-pMEK (1:5,000) was added to the plate and allowed to incubate with shaking at RT for one hour. This plate was then washed with 1x THBS twice (5 minutes per wash) and secondary anti-rabbit HRP (1:5,000) was added to the plate and allowed to incubate for one hour at RT. The plate was washed three times with wash buffer (5 min each wash). On the final wash, the Pierce super signal pico chemiluminescent substrate reagents were mixed together (cat.#37070). The luminescence was measured on Biotek Synergy 2.
In-Gel Radioactive Assay
A 2x cocktail buffer (HEPES pH 7.4, MgCl2, β- glycerolphosphate, DTT, 200 uM ATP, 4 mM Na3VO4, and 1 µL radioactive ATP) was mixed with 2x wild-type FL-BRAF (25 mM HEPES pH 7.4, 0.125 mg/mL, and 300 mM NaCl). This was incubated for 30 minutes at RT before being transferred to SDS-Page, dried, and imaged on a STORM imager.
Supplementary Material
Acknowledgements
We thank Philip A. Cole for helpful discussion. The study was funded by grants from the W.W. Smith Charitable Fund to Z. Wang and National Institutes of Health (P01 CA114046 to R. Marmorstein).
References
- [1] a).Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA, Nature 2002, 417, 949–954 [DOI] [PubMed] [Google Scholar]; b) Holderfield M, Deuker MM, McCormick F, McMahon M, Nat Rev Cancer 2014, 14, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lavoie H, Therrien M, Nat Rev Mol Cell Biol 2015, 16, 281–298. [DOI] [PubMed] [Google Scholar]
- [3].Muslin AJ, Tanner JW, Allen PM, Shaw AS, Cell 1996, 84, 889–897. [DOI] [PubMed] [Google Scholar]
- [4].Rushworth LK, Hindley AD, O’Neill E, Kolch W, Mol Cell Biol 2006, 26, 2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] a).Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS, Cell 2013, 154, 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M, Nature 2009, 461, 542–545. [DOI] [PubMed] [Google Scholar]
- [6] a).Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Group B-S, N Engl J Med 2011, 364, 2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr., Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB, Lancet 2012, 380, 358–365. [DOI] [PubMed] [Google Scholar]
- [7].Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K, Nature 2010, 467, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8] a).Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S, Nature 2010, 464, 431–435 [DOI] [PubMed] [Google Scholar]; b) Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N, Nature 2010, 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, Wu X, Zhang C, Bollag G, Herlyn M, Fagin JA, Lujambio A, Gavathiotis E, Poulikakos PI, Cancer cell 2016, 30, 501–503. [DOI] [PubMed] [Google Scholar]
- [10].Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, Wolchok JD, de Stanchina E, Chandarlapaty S, Poulikakos PI, Fagin JA, Rosen N, Cancer cell 2012, 22, 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, Tellew J, Hardy S, Hekmat-Nejad M, Stuart DD, McCormick F, Nagel TE, Cancer cell 2013, 23, 594–602. [DOI] [PubMed] [Google Scholar]
- [12].Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, Misteli T, Chapman PB, Sosman JA, Graeber TG, Ribas A, Lo RS, Rosen N, Solit DB, Nature 2011, 480, 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Qiu C, Tarrant MK, Boronina T, Longo PA, Kavran JM, Cole RN, Cole PA, Leahy DJ, Biochemistry 2009, 48, 6624–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nan X, Collisson EA, Lewis S, Huang J, Tamguney TM, Liphardt JT, McCormick F, Gray JW, Chu S, Proc Natl Acad Sci U S A 2013, 110, 18519–18524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang Z, Longo PA, Tarrant MK, Kim K, Head S, Leahy DJ, Cole PA, Nat Struct Mol Biol 2011, 18, 1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xie P, Streu C, Qin J, Bregman H, Pagano N, Meggers E, Marmorstein R, Biochemistry 2009, 48, 5187–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DY, Bouvier M, Sicheri F, Therrien M, Nat Chem Biol 2013, 9, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang BH, Guan KL, EMBO J 2000, 19, 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J, N Engl J Med 2012, 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Grasso M, Estrada MA, Ventocilla C, Samanta M, Maksimoska J, Villanueva J, Winkler JD, Marmorstein R, ACS Chem Biol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, Tsang G, Marimuthu A, Cho H, Wu G, Wang W, Fong D, Nguyen H, Shi S, Womack P, Nespi M, Shellooe R, Carias H, Powell B, Light E, Sanftner L, Walters J, Tsai J, West BL, Visor G, Rezaei H, Lin PS, Nolop K, Ibrahim PN, Hirth P, Bollag G, Nature 2015, 526, 583–586. [DOI] [PubMed] [Google Scholar]
- [22]a).Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, Van Horn RD, Yin T, Zhang X, Yadav V, Chen SH, Gong X, Ma X, Webster Y, Buchanan S, Mochalkin I, Huber L, Kays L, Donoho GP, Walgren J, McCann D, Patel P, Conti I, Plowman GD, Starling JJ, Flynn DL, Cancer cell 2015, 28, 384–398 [DOI] [PubMed] [Google Scholar]; b) Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S, Cancer Res 2013, 73, 7043–7055. [DOI] [PubMed] [Google Scholar]
- [23].Jin T, Lavoie H, Sahmi M, David M, Hilt C, Hammell A, Therrien M, Nat Commun 2017, 8, 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Grasso M, Estrada MA, Berrios KN, Winkler JD, Marmorstein R, J Med Chem 2018, 61, 5034–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, Mattos C, Structure 2015, 23, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]; Tran NH, Wu X, Frost JA, The Journal of biological chemistry 2005, 280, 16244–16253. [DOI] [PubMed] [Google Scholar]
- [26].Fischer A, Hekman M, Kuhlmann J, Rubio I, Wiese S, Rapp UR, The Journal of biological chemistry 2007, 282, 26503–26516. [DOI] [PubMed] [Google Scholar]
- [27].Okada T, Hu CD, Jin TG, Kariya K, Yamawaki-Kataoka Y, Kataoka T, Mol Cell Biol 1999, 19, 6057–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]; Mott HR, Carpenter JW, Zhong S, Ghosh S, Bell RM, Campbell SL, Proc Natl Acad Sci U S A 1996, 93, 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Garnett MJ, Rana S, Paterson H, Barford D, Marais R, Mol Cell 2005, 20, 963–969. [DOI] [PubMed] [Google Scholar]
- [29].Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J, Cancer Res 2008, 68, 4853–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stanton VP Jr., Cooper GM, Mol Cell Biol 1987, 7, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, Morales T, Smith D, Brandhuber BJ, Hymowitz SG, Malek S, Cancer cell 2014, 26, 402–413. [DOI] [PubMed] [Google Scholar]
- [32].Lavoie H, Sahmi M, Maisonneuve P, Marullo SA, Thevakumaran N, Jin T, Kurinov I, Sicheri F, Therrien M, Nature 2018, 554, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]a).Nieto P, Ambrogio C, Esteban-Burgos L, Gomez-Lopez G, Blasco MT, Yao Z, Marais R, Rosen N, Chiarle R, Pisano DG, Barbacid M, Santamaria D, Nature 2017, 548, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R, Cell 2010, 140, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang Z, Candelora C, Methods Mol Biol 2017, 1487, 23–33. [DOI] [PubMed] [Google Scholar]
- [35].Luo C, Xie P, Marmorstein R, J Med Chem 2008, 51, 6121–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Qin J, Xie P, Ventocilla C, Zhou G, Vultur A, Chen Q, Liu Q, Herlyn M, Winkler J, Marmorstein R, J Med Chem 2012, 55, 5220–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.