

Abstract
Background
In non-small cell lung cancer (NSCLC), mesenchyme to epithelial transition (MET) protein abundance increases with disease stage and is implicated in resistance to tyrosine kinase inhibitors. To better clarify the impact of MET overexpression on tumor behavior, we investigated a large cohort of patients who underwent curative surgical resection to determine whether MET gene amplification or protein abundance was prognostic.
Methods
Tissue microarrays (TMAs) were constructed using triplicate 1 mm cores of FFPE primary NSCLC specimens. TMAs underwent immunohistochemical (IHC) staining with the SP44 clone (Ventana) and cores were considered positive if >50% of tumor exhibited 2+ staining. The highest of triplicate values was used. MET gene amplification was detected using either SISH using Ventana’s MET DNP probe or FISH using the D7S486/CEP 7 Abbott Probe. DNA was subjected to mutational profiling using Sequenom’s LungCarta panel.
Results
Data from two institutions comprising 763 patients (516; 68%) male were generated, including 360 stage I, 226 stage II, 160 stage III and 18 resected stage IV. High MET protein expression was detected in 25% (193/763), and was significantly more common in adenocarcinomas than squamous cell carcinoma (P<0.01). MET gene copy number (GCN) correlated with high MET protein expression by IHC (P=0.01). Increased MET protein expression was associated with EGFR and KRAS mutations (P<0.01 for both). Once polysomy was excluded, true MET gene amplification was detected in only 8/763 (1%) of samples. In multivariate analysis, neither MET protein abundance nor GCN were correlated to overall patient survival.
Conclusions
MET expression by IHC and GCN amplification was not prognostic in this large Caucasian surgical series. MET’s primary role remains as a therapeutic target.
Keywords: Immunohistochemistry, in situ hybridisation, mesenchyme to epithelial transition receptor (MET receptor), mesenchyme to epithelial transition amplification (MET amplification), non-small cell lung cancer (NSCLC)
Introduction
The discovery of oncogenic driver mutations in non-small cell lung cancer (NSCLC) and the ability to abrogate their signalling pathways using small molecule inhibitors has transformed treatment paradigms. Large international studies have shown that mutations or gene rearrangements in NSCLC can be found in up to 50% of cases (1). Many such alterations, such as mutations in epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK), have prognostic significance due their association with response to therapy, whilst other biomarkers, such as alterations p53, are independently prognostic (2). The mesenchyme to epithelial transition (MET) receptor plays a critical role in embryonic tissue and organ development (3). It plays a key role in proliferation and migration, and is oncogenic in many malignancies, including NSCLC (4,5). MET can be aberrantly activated by overexpression of the gene itself, by over-expression of its sole known ligand, hepatocyte growth factor (HGF), increased DNA copy number, or through activating point mutations, particularly in exon 14 (6,7). Both MET amplification and exon 14 skipping mutations have been associated with response to MET inhibitors (8-10).
Several approaches to targeting dysregulated MET have been investigated, including multikinase inhibitors, such as crizotinib and cabozantinib, selective MET inhibitors such tivantinib, anti-MET monoclonal antibodies (MAbs) and anti-HGF antibodies (4,11). A phase II study of onartuzumab, an anti-MET MAb designed to block HGF binding, with erlotinib was negative overall, but in patients with increased MET protein expression (determined by an immunohistochemical (IHC) assay called MET Dx), progression-free survival (PFS) and overall survival (OS) were both improved (12). However, a subsequent phase III trial of MET Dx positive patients failed to confirm these findings (13). Other trials have suggested that MET amplification, as determined by increased MET gene copy number (GCN), could be a more reliable biomarker. Camidge and colleagues presented data from a group of patients with true MET amplified tumors treated with the MET/ALK inhibitor crizotinib. They reported responses in most patients with medium (>2.2 but <5 fold) to high (>5 fold) amplification of the gene relative to the chromosome 7 centromere (CEP7) (14). These data support the clinical potential of effectively targeting MET.
Critical to understanding MET amplification’s role in the pathogenesis of NSCLC is distinguishing its impact on survival from its role as a predictive biomarker. Several studies have reported conflicting results for the role of MET as a prognostic marker. Cappuzzo et al. showed that MET GCN was associated with poorer clinical outcome (15), in contrast to Dziadziuszko et al. who, using a different methodology, did not find any association between GCN or protein expression and survival (16). Tran et al. found that MET overexpression by IHC and GCN was associated with favorable prognosis (17), and despite the fact that two meta-analyses concluded MET GCN was associated with poor prognosis, these were limited by comparisons of vastly different assays (18,19). Thus, the impact of MET on survival remains unclear.
To further clarify the role of MET IHC and GCN, we investigated GCN and performed IHC for MET using the SP44 clone from Ventana used in the MET Dx assay in 763 surgically resected cases from two tertiary oncology centres. We profiled for common mutations in a subset of 426 cases.
Methods
Patients included
Under a Human Research Ethics approved protocol, clinicopathological data for patients undergoing surgical resection for NSCLC were prospectively collected. Two patient cohorts were used from large academic centres, the Austin Hospital (AH) and The Peter MacCallum Cancer Centre (PMC) Melbourne, Australia. All patients were treated with curative intent surgery followed by either adjuvant chemotherapy or observation. Cancer specific and overall survival were captured and updated from local cancer registry services.
Tissue microarray (TMA) protein expression and GCN evaluation
TMAs were constructed using 1 mm cores of archival formalin fixed paraffin embedded NSCLC tissue in triplicate (Beecher Instruments, Sun Prairie, WI). In brief, three distinct but highly cellular areas were cored for each patient. TMAs were stained for MET expression using the SP44 clone rabbit monoclonal antibody (Ventana Medical Systems, Tucson, AZ) as described previously (16).
Two independent pathologists (K Asadi and AL Morey) assessed staining and documented staining intensity (0–3+) as well as percentage of tumor cells stained (0–100%). Patients were divided according to the criteria used in the MET Dx assay (20) where >50% of cells staining 2+ or more was considered positive.
MET GCN was analyzed using bright-field microscopy and dual silver in situ hybridisation (SISH) employing MET and CEP7 specific probes according to protocols provided by the manufacturer (Ventana Medical System). The assessment of GCN was performed by a pathologist (AL Morey) blinded to the IHC results. Analysis was performed on each core in the TMA; any discordance (heterogeneity) between cores in the same patient was noted. Clustered signals were estimated based on size of single gene signals in tumor nuclei. Cases were scored as non-amplified (diploid) if mean MET copy number was <2.5, non-amplified (polysomic) if MET copies were 2.5–4, non-amplified (high polysomic) if average MET copies were >4 (but MET/CEP7 ratio <2), low level amplified if MET copies were >4 (but <10) and ratio >2, high level amplified if MET copies were >10 and ratio >2. All MET amplified cases showed formation of signal clusters rather than having dispersed gene signals.
For the PMC cohort MET FISH was employed, utilising dual colour fluorescent in situ hybridisation (FISH). Slides were deparaffinized and then placed in Heat Pretreatment Solution (Invitrogen Corporation, Camarillo, CA) in a Pascal pressure cooker (DakoCytomation, Carpinteria, CA) at 124 °C for 2 minutes. The slides were then washed in several changes of distilled water prior to the addition of enough Enzyme Reagent (Invitrogen Coporation, Camarillo, CA) to cover the tissue, and incubated at room temperature for 30 minutes. Slides were washed in distilled water then dehydrated in graded alcohols (70%, 85% and 100%) and air dried at room temperature. Ten microlitres of D7S486/CEP 7 FISH Probe Kit (MET) (Abbott Molecular, Des Plaines, IL) was placed onto the tissue sections, which were then coverslipped and sealed. Slides were denatured at 85 °C for 5 minutes and hybridized at 37 °C for a minimum of 14 hours on a Dako Hybridiser (Dako, Fort Collins, CO). Following hybridisation, slides were placed in 2× SSC/0.1% NP40 at room temperature for removal of coverslips and then transferred to 2× SSC/0.3% NP40 at 73 °C for 2 minutes. Slides were mounted using Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). Fifty tumor nuclei were evaluated as per the scoring criteria indicated: cases were considered as non-amplified (diploid) if mean MET copy number was <2.5, non-amplified (polysomic) if MET copies were 2.5–4, non-amplified (high polysomic) if average MET copies were >4 (but MET/CEP7 ratio <2), low level amplified if MET copies were >4 (but <10) and ratio >2, high level amplified if MET copies were >10 and ratio >2.
Mutational profiling
Mutational profiling was only performed for the AH dataset. DNA was isolated from FFPE blocks and profiled using Sequenom’s Oncocarta Panel v.1.0 as previously described (21). Only mutations with a frequency >10% and able to be validated by Sanger sequencing were included in the final analysis.
Statistical analysis
Differences in patient demographics were assessed with χ2 tests. Survival was analysed using the Kaplan-Meier method. Univariate and multivariate Cox proportional hazard ratio modelling analyses were used to calculate survival hazard ratios. The proportional hazards assumption was satisfied for all variables. All analyses were performed in SPSS, version 24.0.0.0 (IBM Corporation, Armonk, NY). A P value of <0.05 was considered significant throughout.
Results
Patients
Clinicopathologic data were available for a total of 763 patients who underwent surgical resection for NSCLC. Table 1 summarizes the combined cohort, stratified by MET protein expression. All pathological stage IV patients had resection of synchronous brain metastases at diagnosis. Increased MET expression by IHC was seen in significantly more patients with adenocarcinoma [MET high, 130 patients (31%) vs. MET low, 283 (69%)] than in squamous cell tumors [MET high, 31 patients (13%) vs. MET low, 209 (87%); P<0.01] (Table 1). Other histologies also had significantly higher proportion of increased MET expression than squamous cell carcinoma, but similar to adenocarcinoma [MET high, 32 (29%) vs. MET low, 78 (71%)]. High MET expression was present in statistically indistinguishable proportions of early- and late-stage tumors on chi-square analysis (P=0.09); however, numerically a greater proportion of early-stage cancers had increased MET protein abundance, compared with late-stage cancers. A significantly higher proportion of those with high MET IHC also had an elevated GCN (based on the median), but these did not necessarily reach the pre-specified criteria for amplification.
Table 1. Clinicopathologic characteristics of combined cohort divided by MET IHC.
| Characteristics | Combined cohort, n [%] | ||
|---|---|---|---|
| Low MET (n=570) | High MET (n=193) | P | |
| Age (years), median (range) | 67.2 (29.0–87.0) | 67.1 (29.0–85.0) | |
| Sex | 0.16 | ||
| Male | 393 [76] | 122 [24] | |
| Female | 177 [71] | 71 [29] | |
| Histology | <0.01 | ||
| Adenocarcinoma | 283 [50] | 130 [67] | |
| Squamous | 209 [37] | 31 [16] | |
| Other | 78 [14] | 32 [17] | |
| Stage | 0.09 | ||
| IA | 117 [21] | 57 [30] | |
| IB | 139 [24] | 46 [24] | |
| IIA | 94 [16] | 23 [12] | |
| IIB | 89 [16] | 20 [10] | |
| IIIA | 110 [19] | 39 [20] | |
| IIIB | 9 [2] | 2 [1] | |
| IV | 12 [2] | 6 [3] | |
| MET GCN (based on median) | 0.01 | ||
| Low | 308 [54] | 84 [44] | |
| High | 262 [46] | 109 [56] | |
Median GCN was 2.0. MET, mesenchyme to epithelial transition; IHC, immunohistochemical; GCN, gene copy number.
Association of MET expression with mutations
For 426 cases from one centre, somatic point mutations were characterized using a MassArray platform (Table 2). Activating EGFR mutations were seen in 26 patients (6%) and KRAS mutations in 90 (21%). Both KRAS (P<0.001) and EGFR mutations (P<0.001) were associated with increased MET expression, although the effect sizes were small (P=0.22 and 0.17, respectively). Other mutations were less frequent and were not associated with statistically significant changes in MET protein abundance.
Table 2. Mutational characteristics of Austin Hospital cohort divided by MET IHC.
| Characteristics | Austin Hospital cohort, n [%] | ||
|---|---|---|---|
| Low MET (n=307) | High MET (n=119) | P | |
| KRAS mutation (yes) | 42 [14] | 48 [40] | <0.001 |
| EGFR mutation (yes) | 10 [3] | 16 [13] | <0.001 |
| EGFR SISH copy number, median (range) | 2.3 (1.2–25.1) | 2.45 (0.0–13.6) | 0.85 |
| EGFR IHC score, median (range) | 174 (0.0–300.0) | 165 (0.0–300.0) | 1.0 |
MET, mesenchyme to epithelial transition; IHC, immunohistochemical; SISH, silver-enhanced in situ hybridisation.
MET protein overexpression was not prognostic
Given that MET Dx positive patients have poorer survival (20) and conflicting data from recent studies, we correlated MET protein abundance with survival in our dataset. Increased MET expression by IHC was associated with improved survival of borderline significance on univariate analysis (HR 0.81, 95% CI: 0.65–1.01; P=0.06) (Figure 1), but not in multivariate analysis, where stage accounted for the differences seen in univariate analysis (HR 0.87; 95% CI: 0.70–1.09; P=0.24) (Table 3). The cohort was divided into resectable (stage I–IIIA) and unresectable/metastatic (stage IIIB/IV) groups. The stage I–IIIA group with increased MET expression also showed improved OS on univariate analysis (HR 0.79, 95% CI: 0.63–0.99, P=0.04). However, as with the analysis of the entire cohort, the increased MET expression seen in early stage disease accounted for this difference. In the stage IIIB/IV there was no difference in survival (HR 1.02, 95% CI: 0.41–2.53, P=0.97). Thus, MET expression was associated with pathologic stage at diagnosis, but was not an independent prognostic factor above established clinical variables.
Figure 1.
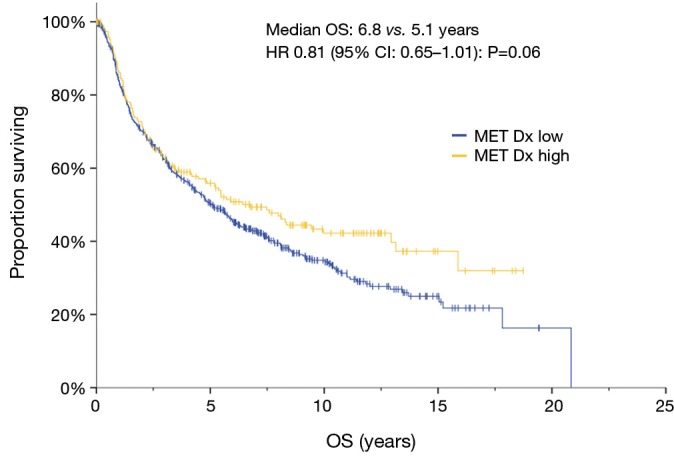
Kaplan-Meier curve of overall survival MET IHC of high expression vs. low expression. MET Dx: >50% of cells staining 2+ or more considered positive. MET, mesenchyme to epithelial transition; IHC, immunohistochemical; OS, overall survival.
Table 3. Univariate and multivariable analysis of OS (n=465).
| Variables | Univariate analysis | Multivariable analysis | |||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | P (Wald test) | HR | 95% CI | P (Wald test) | ||
| MET++ score (>50) | 0.81 | 0.65–1.01 | 0.06 | 0.87 | 0.70–1.09 | 0.24 | |
| MET GCN | 1.02 | 0.95–1.10 | 0.55 | – | – | – | |
| Sex (male) | 0.85 | 0.77–0.95 | <0.01 | 0.89 | 0.80–0.99 | 0.03 | |
| Histology (SQ vs. ADC) | 0.96 | 0.78–1.19 | 0.74 | – | – | – | |
| Histology (other vs. ADC) | 1.06 | 0.80–1.40 | 0.70 | – | – | – | |
| Stage (IB vs. IA) | 1.35 | 0.98–1.84 | 0.06 | 1.33 | 0.77–2.21 | 0.07 | |
| Stage (IIA vs. IA) | 1.92 | 1.38–2.67 | <0.01 | 1.83 | 1.00–3.20 | <0.01 | |
| Stage (IIB vs. IA) | 2.37 | 1.70–3.29 | <0.01 | 2.23 | 1.13–3.76 | <0.01 | |
| Stage (IIIA vs. IA) | 4.10 | 3.03–5.56 | <0.01 | 4.01 | 2.29–6.70 | <0.01 | |
| Stage (IIIB vs. IA) | 5.93 | 2.71–12.97 | <0.01 | 5.81 | 1.05–11.7 | <0.01 | |
| Stage (IV vs. IA) | 4.73 | 2.65–8.42 | <0.01 | 4.50 | 1.64–8.87 | <0.01 | |
OS, overall survival; MET, mesenchyme to epithelial transition; IHC, immunohistochemical; SQ, squamous cell carcinoma; ADC, adenocarcinoma.
MET GCN was not prognostic
To understand to what extent MET protein abundance reflects genomic copy number change, we used SISH (in the AH cohort) or FISH (in the PMC cohort) to evaluate MET gene amplification, normalized to the CEP7 probe to account for polysomy (Figure 2). True MET amplification was seen in only 8 of 763 (1.0%) cases all of which were in the AH cohort. Importantly, one of the amplified cases showed heterogeneity of amplification. No cases of GCN amplification were seen in the PMC cohort. We also dichotomized GCN at the median for the combined cohort, and neither elevated GCN nor MET amplification were significantly associated with survival on univariate or multivariable analysis.
Figure 2.

SISH analysis of MET amplification. Three examples of MET amplification assessed by SISH. (A) Uniform high-level MET amplification with large signal clusters; (B) heterogenous MET amplification: junction between a region of amplification (left) and polysomy (right); (C) cep7 probe used to confirm polysomy rather than amplification, with equal numbers of cep7 red and MET black signals. MET, mesenchyme to epithelial transition; SISH, silver-enhanced in situ hybridisation.
Discussion
MET expression has been employed as both a prognostic and predictive factor in NSCLC, though the results have been inconsistent. Increased MET expression by IHC and increased GCN have been associated with improved survival in one study (17), no association with survival in others (16,22), and worse overall survival in two meta-analyses (18,23). As a predictive marker, MET amplification through increased GCN or exon 14 mutations have both been used to select for, and are associated with, response to MET targeted therapy (24-26). Given this background, and in a large cohort, we found that MET expression by IHC was not associated with prognosis. Tumors with mutations in EGFR and KRAS had elevated MET protein abundance. MET gene amplification, as defined by increased GCN without polysomy, was only observed in 1% cases and was not associated with prognosis.
The contradictory results in these studies may have resulted from the specific antibodies used, differing patient populations or the proportion of each histology. In our dataset, increased MET expression was seen more commonly in adenocarcinomas than squamous cell carcinomas. In the study by Dziadziuszko et al., there were a high proportion of patients with squamous cell cancers (103/189) and expression was increased in squamous cell carcinomas (16). However, our data concur with those from other studies, including the randomized Phase II trial using onartuzumab, in which the majority of MET Dx positive patients were adenocarcinomas (17,20).
A clear understanding of the effect of MET expression by IHC on prognosis remains elusive. Our data suggest no effect on prognosis in early stage disease, in keeping with Li et al. (22) who found no difference in survival in a cohort with advanced disease. Furthermore, the meta-analysis of 13 studies concluded a poorer prognosis in resected patients, predominantly of Asian origin, with MET positive tumors by IHC (18). Of note, 12/13 studies included in this meta-analysis originated from China or Japan and the largest of the included studies used the same antibody (SP44) on 883 cases, predominantly adenocarcinomas (27). Those studies that have shown an association between MET IHC and survival have used either rabbit polyclonal or monoclonal antibodies, suggesting that MET may only be prognostic in the context of specific IHC protocols. Certainly, it does not appear to exhibit a consistent and reliable association with survival.
GCN is an area of renewed interest, given that data suggest it may be a predictive marker for response to the MET inhibitors. In the study from Cappuzzo and colleagues, and two subsequent meta-analyses, GCN was also reported to be an independent poor prognostic marker, although in multivariate analysis the effect was reduced (15,18,19). Again, the majority of studies included in the meta-analyses were derived from Asian populations. Differing methodologies were also employed from SISH and FISH to bright-field in situ hybridisation (BISH) in the largest study (27). The definition of positivity is variable with some studies excluding polysomy, while others were more permissive. Our data excluded polysomy and did not find an association with survival in the small minority of samples with true gene amplification.
The discovery of exon 14 skipping mutations has complicated this picture, as it can lead to MET overexpression detectable by IHC (11). In addition, these mutations have also been associated with copy number gain (28). Awad et al. found MET mutations in 28/933 (3%) non-squamous NSCLC by next-generation sequencing, of which 6/28 had “high-level” GCN (>3 MET/CEP7 ratio), and another 8/28 had “low-level” GCN (1–3 MET/CEP7 ratio). Interestingly, despite the presence of the exon 14 mutations IHC expression ranged from faint to maximal (as assessed with an H-score identical to ours). Recently these mutations were reported to be more frequent in older smokers with sarcomatoid NSCLC (28). This phenotype was very uncommon in our cohort with sarcomatoid NSCLC found in only five patients, suggesting that the expected mutation frequency in this dataset would be low. The independent contribution of each of MET overexpression by IHC, increased GCN and exon 14 mutations to prognosis is as yet unknown, and the utility of each as a predictive biomarker remains an area of ongoing exploration in current clinical trials.
MET has been identified as a promising therapeutic target in a number of reported trials. Data from a Phase I trial using the multikinase inhibitor crizotinib in patients with exon 14 skipping mutations demonstrated anti-tumor activity in 10/15 patients with five confirmed partial responses and durable efficacy (24). In a parallel study, the investigators screened archival samples and found true gene amplification (>2.2 MET/CEP7 copies) occurred in 30/800 (3.8%) samples, with high MET amplification (≥5) occurring in only 6 (0.8%) cases. Interestingly crizotinib was associated with partial responses in 4/6 patients with high MET amplification and 3/6 patients with intermediate MET amplification (14). Similarly, MET protein overexpression was associated with increased overall response rate (ORR) in the phase I trial of the highly-potent, selective MET inhibitor, capmatinib. Of patients with increased GCN (≥5) 5/8 had PRs, and in those patients with MET IHC 3+ the ORR was 29% (5/17) (26).
These data highlight the validity of MET as a target in tumors reliant on the MET signalling pathway. Certainly, in these cases, MET GCN appears to be a useful predictive marker, although its role as a prognostic marker may not be as straightforward to elucidate. Acknowledging differences in assay, the correlation between GCN and IHC has not been uniform across studies, although in our dataset all eight MET amplified cases also had a MET H-score >140.
To our knowledge, this is one of the largest studies to comprehensively profile a group of predominantly Caucasian patients for MET expression, GCN and co-existent mutations. However, our study has several limitations. Most importantly, exon 14 skipping mutations were not assessed. This may underplay the effect these mutations have on both MET overexpression and prognosis. Secondly, both datasets were investigated using TMAs. While three cores were interrogated from each tumor sample, tumor heterogeneity needs to be considered when using this approach. Thirdly, assessing H-scores are subjective and may have underestimated heterogeneity. To combat these differences, two pathologists scored the TMA slides independently and tumors with discrepant scores were reviewed and consensus reached. In addition, though an association between MET expression and pathologic stage, we did not have complete clinical staging data to examine whether this relationship held true in both cases. Lastly the GCN data were derived also using a TMA and SISH/FISH. Assessing GCN ratios using SISH can be difficult in archival tissues where signal is generally weaker than evident by FISH. While dual staining was tried for some, most of the cases were done with parallel CEP7 and MET on contiguous slices of the TMA. Technically this could cause minor discrepancies in the MET:CEP7 ratio, however with three cores per tumor, such discrepancies were considered unlikely to affect classification as amplified.
Conclusions
Our data demonstrate that in a Caucasian population, MET expression was associated with adenocarcinoma histology, EGFR and KRAS mutations but not with prognosis. Similar to other studies, MET GCN amplification only occurred in a small subset of patients. Prognostic markers in advanced NSCLC may not necessarily translate into earlier stage disease, however MET remains an important target for not only oncogene-addicted tumors but also in TKI resistant tumors. Defining the optimal predictive biomarker remains a critical hurdle to overcome.
Acknowledgements
None.
Ethical Statement: The study was approved by institutional/regional/national ethics committee/ethics board of Austin Hospital (No. H2006/02394). This study will not affect outcomes or management of subjects included. No consent was deemed necessary for inclusion but patient data has remained secure through the course of the study.
Footnotes
Conflicts of Interest: G Rivalland reports personal fees from Astra-Zeneca, non-financial support from Roche, outside the submitted work. P Mitchell reports personal fees and other from Merck, personal fees and non-financial support from Roche, from Boehringer Ingelheim, non-financial support from Bristol Myers-Squibb, from Celgene, non-financial support and other from Astra-Zeneca, outside the submitted work. AL Morey reports speaking fees from Pfizer. B Solomon reports advisory Boards and Honoraria from Pfizer, Novartis, Roche-Genetech, AstraZeneca, BMS, and Merck. T John reports personal fees from Pfizer, personal fees from Roche, personal fees from AstraZeneca, personal fees from BMS, personal fees from Novartis, outside the submitted work. The other authors have no conflicts of interest to declare.
References
- 1.Sholl LM, Aisner DL, Varella-Garcia M, et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol 2015;10:768-77. 10.1097/JTO.0000000000000516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitsudomi T, Hamajima N, Ogawa M, et al. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: a meta-analysis. Clin Cancer Res 2000;6:4055-63. [PubMed] [Google Scholar]
- 3.Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984;311:29-33. 10.1038/311029a0 [DOI] [PubMed] [Google Scholar]
- 4.Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol 2013;31:1089-96. 10.1200/JCO.2012.43.9422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olivero M, Rizzo M, Madeddu R, et al. Overexpression and activation of hepatocyte growth factor/scatter factor in human non-small-cell lung carcinomas. Br J Cancer 1996;74:1862-8. 10.1038/bjc.1996.646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cipriani NA, Abidoye OO, Vokes E, et al. MET as a target for treatment of chest tumors. Lung Cancer 2009;63:169-79. 10.1016/j.lungcan.2008.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnaswamy S, Kanteti R, Duke-Cohan JS, et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res 2009;15:5714-23. 10.1158/1078-0432.CCR-09-0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol 2011;6:942-6. 10.1097/JTO.0b013e31821528d3 [DOI] [PubMed] [Google Scholar]
- 9.Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842-9. 10.1158/2159-8290.CD-14-1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015;5:850-9. 10.1158/2159-8290.CD-15-0285 [DOI] [PubMed] [Google Scholar]
- 11.Drilon A, Cappuzzo F, Ou SI, et al. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol 2017;12:15-26. 10.1016/j.jtho.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spigel DR, Burris HA, 3rd, Greco FA, et al. Randomized, double-blind, placebo-controlled, phase II trial of sorafenib and erlotinib or erlotinib alone in previously treated advanced non-small-cell lung cancer. J Clin Oncol 2011;29:2582-9. 10.1200/JCO.2010.30.7678 [DOI] [PubMed] [Google Scholar]
- 13.Spigel DR, Edelman MJ, O'Byrne K, et al. Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J Clin Oncol 2017;35:412-20. 10.1200/JCO.2016.69.2160 [DOI] [PubMed] [Google Scholar]
- 14.Camidge DR, Ou SHI, Shapiro G, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol 2014;32:8001 10.1200/jco.2014.32.15_suppl.8001 [DOI] [Google Scholar]
- 15.Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009;27:1667-74. 10.1200/JCO.2008.19.1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dziadziuszko R, Wynes MW, Singh S, et al. Correlation between MET gene copy number by silver in situ hybridization and protein expression by immunohistochemistry in non-small cell lung cancer. J Thorac Oncol 2012;7:340-7. 10.1097/JTO.0b013e318240ca0d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran TN, Selinger CI, Kohonen-Corish MR, et al. Alterations of MET Gene Copy Number and Protein Expression in Primary Non-Small-Cell Lung Cancer and Corresponding Nodal Metastases. Clin Lung Cancer 2016;17:30-8.e1. 10.1016/j.cllc.2015.08.002 [DOI] [PubMed] [Google Scholar]
- 18.Guo B, Cen H, Tan X, et al. Prognostic value of MET gene copy number and protein expression in patients with surgically resected non-small cell lung cancer: a meta-analysis of published literatures. PLoS One 2014;9:e99399. 10.1371/journal.pone.0099399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimou A, Non L, Chae YK, et al. MET gene copy number predicts worse overall survival in patients with non-small cell lung cancer (NSCLC); a systematic review and meta-analysis. PLoS One 2014;9:e107677. 10.1371/journal.pone.0107677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2013;31:4105-14. 10.1200/JCO.2012.47.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John T, Starmans MH, Chen YT, et al. The role of Cancer-Testis antigens as predictive and prognostic markers in non-small cell lung cancer. PLoS One 2013;8:e67876. 10.1371/journal.pone.0067876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li A, Niu FY, Han JF, et al. Predictive and prognostic value of de novo MET expression in patients with advanced non-small-cell lung cancer. Lung Cancer 2015;90:375-80. 10.1016/j.lungcan.2015.10.021 [DOI] [PubMed] [Google Scholar]
- 23.Kim JH, Kim HS, Kim BJ. Prognostic value of MET copy number gain in non-small-cell lung cancer: an updated meta-analysis. J Cancer 2018;9:1836-45. 10.7150/jca.24980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drilon AE, Camidge DR, Ou SHI, et al. Efficacy and safety of crizotinib in patients (pts) with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J Clin Oncol 2016;34:abstr 108.
- 25.Wu YL, Kim DW, Felip E, et al. Phase (Ph) II safety and efficacy results of a single-arm ph ib/II study of capmatinib (INC280) þ gefitinib in patients (pts) with EGFR mutated (mut), cMET-positive (cMETþ) non-small cell lung cancer (NSCLC). J Clin Oncol 2016;34:abstr 9020.
- 26.Schuler MH, Berardi R, Lim WT, et al. Phase (Ph) I study of the safety and efficacy of the cMET inhibitor capmatinib (INC280) in patients (pts) with advanced cMET+ non-small cell lung cancer (NSCLC). J Clin Oncol 2016;34:abstr 9067.
- 27.Tsuta K, Kozu Y, Mimae T, et al. c-MET/phospho-MET protein expression and MET gene copy number in non-small cell lung carcinomas. J Thorac Oncol 2012;7:331-9. 10.1097/JTO.0b013e318241655f [DOI] [PubMed] [Google Scholar]
- 28.Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 2016;34:721-30. 10.1200/JCO.2015.63.4600 [DOI] [PubMed] [Google Scholar]