

Abstract
Autoimmune bullous diseases with ocular involvement consist of a group of systemic entities that are characterized by formation of autoantibodies against the proteins of the epithelial basement membrane zone of the conjunctiva. Mostly, the elderly are affected by these diseases. The characteristic patterns of mucocutaneous involvement and the specific tissue components targeted by these autoantibodies are differentiating features of these diseases. Ocular pemphigus vulgaris exhibits intraepithelial activity, whereas the autoimmune activity in linear immunoglobulin A disease, mucous membrane pemphigoid, and epidermolysis bullosa acquisita occurs at a subepithelial location. Given the increased risk for blindness with delays in diagnosis and management, early detection of ocular manifestations in these diseases is vital. The precise diagnosis of these autoimmune blistering diseases, which is essential for proper treatment, is based on clinical, histological, and immunological evaluation. Management usually includes anti-inflammatory and immunosuppressive medications. Inappropriate treatment results in high morbidity and even potential mortality.
Keywords: Eye, Autoimmune Bullous Diseases, Epidermolysis Bullosa Acquisita, Linear Immunoglobulin A Disease, Mucous Membrane Pemphigoid, Ocular Pemphigus Vulgaris, Paraneoplastic Pemphigus
INTRODUCTION
The autoimmune bullous diseases are caused by an immune response to proteins of the desmosomes or the basement membrane zone (BMZ).[1] They include mucous membrane pemphigoid, pemphigus vulgaris, paraneoplastic pemphigus, linear immunoglobulin A disease, lichen planus, pemphigus foliaceus, dermatitis herpetiformis, and epidermolysis bullosa acquisita [Table 1]. These autoimmune bullous diseases can be differentiated on the basis of characteristic patterns of mucocutaneous involvement and the specific tissue components targeted by the autoantibodies. Ocular pemphigus vulgaris exhibits intraepithelial activity, whereas the autoimmune activity in linear immunoglobulin A disease, mucous membrane pemphigoid, and epidermolysis bullosa acquisita occurs at a subepithelial location.[2,3,4,5] Both inflammatory cells and complement are necessary for the formation of a blister.[6,7] Autoantibodies in pemphigus vulgaris cause acantholysis and blister formation.[8,9,10] Linear immunoglobulin A disease, mucous membrane pemphigoid, and epidermolysis bullosa acquisita are the subepidermal autoimmune bullous diseases that result from autoantibodies against one or more components of the BMZ.[11,12] These autoantibodies activate complement, resulting in a cellular inflammatory infiltrate that disrupts the BMZ.[13,14] Given the increased risk for blindness with a delay in diagnosis and management, early detection of ocular manifestations in these diseases is vital. The precise diagnosis of these autoimmune bullous diseases, which is essential for proper treatment, is based on clinical, histological, and immunological evaluation. Management usually includes anti-inflammatory and immunosuppressive medications. Elderly individuals are particularly vulnerable to the side effects of these medications. Herein, we describe the etiologies and clinical manifestations of autoimmune bullous diseases with eye involvement and summarize the available treatment as well as recent developments in the management of these diseases.
Table 1.
Summary of clinical characteristics, etiopathogenesis and paraclinical findings in different autoimmune bullous diseases.
| Disease | Ocular Findings | Systemic Findings | Etiopathogenesis | Histology/laboratory |
|---|---|---|---|---|
| MMP | Chronic cicatrizing conjunctivitis, subconjunctival bullae, conjunctival ulceration, pseudomembrane formation, fornix shortening, symblepharon, trichiasis, dry eye, corneal epithelial defect, ulceration and vascularization, ocular surface keratinization | Oral (100%), pharynx (43%), nasal mucosa (38%), larynx (30%), genital mucosa (20-35%), rectum (11%), esophagus (7%), skin (10-43%) | Type-2 hypersensitivity reaction, autoantibodies against BMZ antigens (BP180, BPAG 2) | Subepithelial bullae, Linear immunoglobulin and complement deposition along BMZ detected by DFA or immunoperoxidase assay, circulating anti-BM autoantibodies |
| PV | Non-cicatrizing conjunctivitis, conjunctival erosions, lid margin erosions (especially medial aspect of the lower eyelid) | Skin, oral mucosa, larynx, esophagus, anus, cervix, vulva | Autoantibodies against desmosomal antigens desmoglein 1 and 3 | Suprabasal (intra-epithelial) blister, intercellular immunoglobulin and complement deposits detected by DFA, anti-desmoglein 1 and 3 autoantibodies detected by ELISA |
| PNP | Similar to MMP. Bilateral cicatrizing conjunctivitis, subconjunctival bullae, conjunctival ulceration, pseudomembrane formation, fornix shortening, symblepharon, dry eye, corneal epithelial defect, ulceration and melting | Oral mucosa (severe intractable stomatitis), nasopharynx, oropharynx, nasal septum, hypopharynx, larynx, tracheobronchial mucosa, esophagus, epiglottis, skin (latent) | Autoantibodies with cross-reactivitiy against epidermal (desmosomal) antigens | Suprabasal acantholysis, necrosis of epidermal keratinocytes, intercellular and/or BMZ deposition of IgG and complement |
| LAD | May resemble MMP, may be asymptomatic. Conjunctival scarring, subconjunctival fibrosis, trichiasis, entropion, inferior fornix shortening and symblepharon with secondary corneal scarring | Annular tense bullae in perioral region, perineum, and extremities | Autoantibodies against LAD-1/collagen type VII Drug-related: vancomycin, captopril, amiodarone, phenytoin. Secondary to preceding illness, malignancy, or other autoimmune disorder | Subepidermal bullae, linear deposition of IgA and complement C3 along the BMZ detected by DFA, circulating anti-BMZ autoantibodies |
| EBA | Chronic conjunctivitis, symblepharon, keratitis, subepithelial corneal vesiculation, perforation, scarring | Milia and scarring at sites of trauma (e.g. elbows, knees, buttock, dorsa of feet and hands) and nasal, pharyngeal, esophageal, and genital mucosa | IgG autoantibodies against collagen type VII, disruption of lamina densa of the EBM | Subepidermal blister with intact epidermis, linear thick band IgG deposit along BMZ detected by DFA, circulating IgG autoantibodies against type VII collagen using ELISA or IFA |
BMZ, basement membrane zone; BM, basement membrane; EBM, epithelial basement membrane; MMP, mucus membrane pemphigoid; PV, pemphigus vulgaris; PNP, paraneoplastic pemphigus; LAD, linear IgA disease; EBA, epidermolysis bullosa acquisita; DFA, direct immunofluorescent assay; IFA, indirect immunofluorescent assay.
REVIEW METHOD
A PubMed review was performed, analyzing all publications from 1970 to 2017 concerning the topic “autoimmune bullous diseases with ocular involvement” (keywords: autoimmune bullous diseases, eye, mucous membrane pemphigoid, ocular pemphigus vulgaris, paraneoplastic pemphigus, linear immunoglobulin A disease, epidermolysis bullosa acquisita). Animal and human studies published in English (full text) were included for this review.
MUCOUS MEMBRANE PEMPHIGOID
Mucous membrane pemphigoid (MMP, formerly called ocular cicatricial pemphigoid) is a chronic vesiculobullous disease of the mucous membranes that manifests on the eyes primarily through progressive scarring of the conjunctiva. This condition is rare, with estimates in the ophthalmology patients ranging from 1 in 8,000 to 1 in 46,000.[15,16] Its annual incidence in France and Germany is estimated to be 1.3 and 2.0 per 1 million people per year, respectively.[17] Involved patients present typically with bilateral ocular and mucocutaneous lesions in the fifth or sixth decade of life, although the disease can manifest at any age. There is no apparent geographic or racial predilection and women are more frequently affected, with a female to male ratio of 1.5:1.[18] Several human leukocyte antigens (HLAs) such as HLA-DR2, HLADR4, and HLA-DQw7 (DQB1*0301) have been associated with increased susceptibility to MMP, but this association is not observed in all patients; hence, HLA typing is not useful.[19,20,21]
Any mucous membrane lined by stratified squamous epithelium may be involved in MMP. Almost all patients develop oral involvement and 61% to 80% of patients present with ocular involvement.[18,22] The disease is confined to the eye (ocular pemphigoid) in approximately 20% of patients.[23,24] Other involved mucous membranes include the pharynx (43%), nasal mucosa (38%), larynx (30%), genital mucosa (20%–35%), rectum (11%), and esophagus (7%).[23,24] The disease can involve any oral structure, although the lips and tongue are less affected than the gingival, buccal mucosal, and palatal surfaces. Difficulty in swallowing may be an important early symptom. There is no correlation between the severity of involvement of one mucous membrane and the severity or presence of involvement in other mucous membranes. Skin involvement occurs in 10% to 43% of patients and has a variable course, including generalized bullous eruption similar to that of bullous pemphigoid (rare) and isolated lesions in the head and neck area that may heal with scarring (more frequent).[25] MMP has a variable course, ranging from a limited course with mild ocular scarring that remits following treatment, to an intermittent course, and to a chronic, progressive disease with only partial response to treatment, necessitating lifelong follow-up.[26,27,28] The latter form is observed in approximately one third of patients.[26]
Pathogenesis
The exact mechanism of MMP remains unknown, although it may represent an antibody tissue-specific antigen-mediated (type-2 hypersensitivity) reaction in which cell injury results from autoantibodies against cell surface antigens in the BMZ. Various autoantibody targets have been described. The most common targets of autoimmunity are 180 kDa bullous pemphigoid antigen (BP180, BPAG 2), a protein that spans the basal keratinocyte cell membrane into the lamina lucida of the dermoepidermal junction, and laminin 332 (also known as laminin 5 or epiligrin), a component of the lamina lucida, suggesting that the clinical diagnosis of MMP encompasses various pathophysiologic disease entities.[29] The antibodies activate complement, with a subsequent breakdown of the conjunctival membrane. Additionally, a number of proinflammatory cytokines, including interleukin 1 (IL-l) and tumor necrosis factor α (TNF-α), are overexpressed.[29] The increased level of TNF-α in the conjunctival tissues of patients with MMP induces the expression of migration inhibition factors. Additionally, macrophage colony-stimulating factor has also been shown to have elevated levels in the conjunctival tissue of patients with active MMP. Therefore, cellular immunity may also play a role.[29]
Ocular Manifestations
MMP is a chronic cicatrizing conjunctivitis that is usually bilateral; however, asymmetrical presentation is not uncommon. The diagnosis of unilateral MMP should be made with caution, because other diseases may masquerade as MMP (see below). Remissions and exacerbations are common. Affected patients frequently present with recurrent episodes of mild and nonspecific conjunctivitis with an occasional mucopurulent discharge. They complain of red eyes, itching, burning, foreign body sensation, dryness or tearing, all of which are common symptoms and findings of chronic conjunctivitis.
In the early phase, slit-lamp examination may reveal papillary conjunctivitis associated with diffuse conjunctival hyperemia and edema, and formation of subconjunctival bullae, which may rupture, leading to ulceration and pseudomembrane formation.[15,16] Transient bullae of the conjunctiva rupture, leading to subepithelial fibrosis (stage I). Fine gray white linear opacities are best seen with an intense but thin slit beam. As the disease progresses, further scarring is manifested that results in inferior and superior fornix shortening (stage II) and bulbar and palpebral conjunctival fusion (symblepharon, stage III) [Figure 1].[30,31,32] More severe fibrosis may lead to restricted ocular motility due to extensive symblephara, or even the fusion of the superior and inferior palpebral conjunctiva leading to ankyloblepharon (stage IV).[33] This staging system (Foster stages I-IV) has been used widely as a disease progression marker in ocular MMP.[34] The conjunctival contraction leads to entropion (inversion of the eyelid margins) and lagophthalmos (incomplete eyelid closure). Lash metaplasia including distichiasis (eyelash growth arising from meibomian glands) and trichiasis (misdirected eyelash growth, usually toward the globe) may follow.[15] Recurrent attacks of conjunctival inflammation can lead to dry eye that is caused by the destruction of conjunctival goblet cells (mucin deficiency) and the obstruction of meibomian gland orifices (lipid deficiency) and lacrimal gland ductules (aqueous deficiency). In addition, depletion of limbal stem cells (perilimbal basal epithelial cells that repopulate the corneal epithelial cell layers through continuous proliferation) plays a recognized role in the development of vision threatening keratopathy, presenting initially as small epithelial defects and ultimately as large corneal ulcerations with resultant total corneal vascularization.[35,36] The combination of tear deficiency, lid abnormalities, and limbal stem cell deficiency leads to keratinization of the already thickened conjunctiva, corneal abrasions, and vascularization, and further scarring, ulceration, and epidermalization of the ocular surface with eventual blindness [Figure 1].
Figure 1.
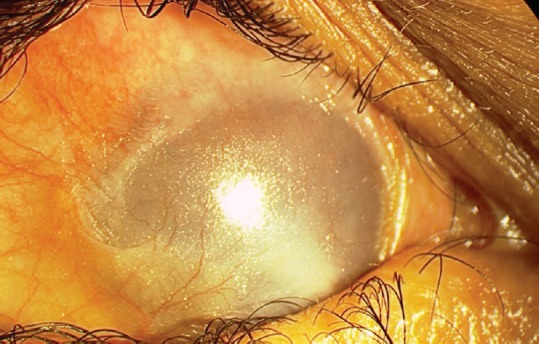
Epidermalization of the ocular surface in mucous membrane pemphigoid. The combination of tear deficiency, lid abnormalities, and limbal stem cell deficiency leads to keratinization of the conjunctiva, and to corneal scarring and vascularization.
Blistering, erosive, scarring conjunctivitis, with or without other associated mucocutaneous involvement, should suggest the diagnosis of MMP. To diagnose this condition in its early stages, ophthalmologists should watch for subtle inferior forniceal contraction and symblephara, which can be detected when the lower eyelid is pulled down while the patient looks up [Figure 2]. Oral mucosal lesions may be a clue that can lead to early diagnosis.
Figure 2.
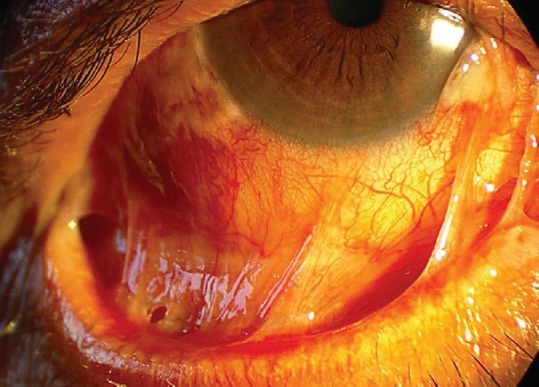
Inferior fornix shortening and bulbar and palpebral conjunctival fusion (symblepharon) in mucous membrane pemphigoid. The lesions are best evident when the lower eyelid is pulled down while the patient looks up.
Activity of ocular inflammation has been classified as absent, mild (conjunctival hyperemia, mild stromal edema), moderate (extensive or marked conjunctival hyperemia with stromal edema, significant tissue thickening), or severe (inflammation in all 4 quadrants, severe mucosal edema, limbitis, conjunctival and corneal ulceration).[37] In addition to the Foster stages, several sub-classifications have been described for the detailed monitoring of the disease progression.[38,39,40,41] For instance, Tauber stages subdivide Foster stages II and III based on the extent of vertical and horizontal involvement of the inferior fornix, respectively. Tauber stages a, b, c, and d correspond to loss of inferior fornix depth (stage II) or horizontal involvement by symblephara (stage III) in magnitudes of less than 25%, 25% to 50%, 50% to 75%, and 75% to 100%, respectively. Foster stage III may be further described by the number of symblephara.[39] Disease activity and severity have been used in guidelines for starting systemic therapy for ocular MMP, which is recommended in active stage II disease.[42] Since progression is often slow, photographic documentation in differing gaze positions is useful for clinical staging and for evaluating the disease course and response to therapy.
The differential diagnosis of MMP includes postinfectious conditions (trachoma, adenoviral conjunctivitis, or streptococcal conjunctivitis), other autoimmune or autoreactive conditions (sarcoidosis, scleroderma, ocular pemphigus vulgaris, dermatitis herpetiformis, linear immunoglobulin A dermatosis, epidermolysis bullosa acquisita, lichen planus, Stevens-Johnson syndrome, epidermolysis bullosa, atopic keratoconjunctivitis, or graft-vs-host disease), or prior conjunctival trauma. The clinician should also consider drug-induced MMP or pseudopemphigoid, which has a clinical picture similar to that of MMP and occurs as a side effect of the long-term use of certain topical ophthalmic medications including timolol, demecarium bromide, pilocarpine, epinephrine, idoxuridine, and echothiophate iodide.[43] Pseudopemphigoid has several differentiating features, such as the lack of mucocutaneous involvement in the topical drug induced form, and disease that does not progress after the discontinuation of the offending agent.[44]
Diagnostic Studies
Pathologic support for a diagnosis of MMP can be obtained from a conjunctival biopsy sent for histopathologic examination and direct immunofluorescent or immunoperoxidase staining. Biopsy specimens should be obtained from an actively affected area of the conjunctiva or, if involvement is diffuse, from the inferior conjunctival fornix. Oral mucosal biopsies are also useful, especially in the presence of an active lesion. Early specimens from conjunctival lesions reveal a subepithelial vesicle with underlying mixed inflammatory infiltrate including neutrophils, plasma cells, lymphocytes, and eosinophils.[45] In chronic cases, histologic examination of the conjunctiva in MMP shows epithelial metaplasia as conjunctival squamous keratinization, parakeratosis, a scarcity of goblet cells, and increased fibroblast and mast cell activity and proliferation.[46] The ensuing cicatrization yields an end result of conjunctival fibrosis. Electron microscopic evaluation reveals a cleft in the lamina lucida similar to that in bullous pemphigoid.[47] Disruption of the BMZ includes redundancy and variation of basal laminar thickness, increased desmosome concentration, and disorganized collagen fibrils.[47]
A diagnosis of MMP is confirmed through immunofluorescent and immunoperoxidase laboratory techniques that reveal characteristic linear immunoglobulin and complement deposition along conjunctival, buccal, or cutaneous tissue basement membranes in most cases.[48] However, false negative results are not uncommon. The sensitivity of detecting immune deposits increases from between 50% and 52% with immunofluorescence alone to 83% when labeled immunoperoxidase is used.[49] IgG and C3 are the most common immune deposits in both the skin and mucosal basement membranes. However, immunohistochemical staining techniques can demonstrate IgM and/or IgA localized in the BMZ of the conjunctiva.[49] Purely ocular MMP exhibits immune deposition in the upper lamina lucida, whereas mucocutaneous involvement is characterized by deposition in the lower lamina lucida and lamina densa.[50] End stage disease may produce negative results because of the destruction of the basement membranes.
Some patients with MMP have circulating anti-basement membrane antibody that demonstrates regularly clustered lower lamina lucida and lamina densa immunostaining.[51] The levels of these circulating autoantibodies may correspond to clinical disease improvement with intravenous immunoglobulin (IVIG) treatment.[52,53] In pseudopemphigoid, conjunctival biopsies may or may not be positive for immunoreactants.
Management
A multidisciplinary approach is often necessary in the management of MMP, with the involvement of primary care physicians, otolaryngologists, oral surgeons, ophthalmologists, dentists, dermatologists, gastroenterologists, and gynecologists. Chronic blepharitis and meibomian gland dysfunction require a regular regimen of warm compresses, lid scrubs, and erythromycin ophthalmic ointment at bedtime. Oral doxycycline 100 mg once or twice daily can also be added to this therapy. Topical corticosteroids such as triamcinolone acetonide may be used for limited acute and early-stage cases that present with ocular lesions, although topical treatment alone is not adequately effective in stopping MMP progression.[54] Posterior lid margin conjunctival keratinization may respond to topical retinoid treatment. When progressive conjunctival cicatrization is present, subconjunctival mitomycin C or steroid injection may be efficacious as a temporizing measure to slow disease progression.[55]
In most cases, systemic treatment is required for acute exacerbations and chronic disease. Classifying patients into low-risk and high-risk groups is valuable when determining the proper regimen.[23] The low-risk group consists of patients who have only oral mucosa or oral mucosa and skin involvement. These patients can be treated more conservatively since they have a much lower incidence of scarring. Patients with MMP involving nasopharyngeal, laryngeal, genital, ocular, and esophageal mucosae, as well as patients with rapidly progressing ocular disease, should be treated using the high-risk algorithm. Low-risk patients may be successfully treated with dapsone, while high-risk patients respond best to treatment with cyclophosphamide.[56] Dapsone, a drug previously used to treat leprosy (Hansen disease) and dermatitis herpetiformis, must be avoided in patients with sulfa allergy or glucose-6-phosphate dehydrogenase (G6PD) deficiency. Therefore, it is advisable to test for G6PD deficiency before treatment is initiated. However, even those without this enzymatic deficiency may develop hemolytic anemia.[26,27] In contraindicated cases or in those who do not have adequate response to dapsone, cytotoxic agents such as azathioprine, mycophenolate mofetil, sulfasalazine, or methotrexate are used for suppression of conjunctival inflammatory activity and prevention of progressive cicatrization.[26,27] Mycophenolate mofetil and azathioprine are generally less effective than cyclophosphamide. Persistent inflammation should prompt a transition to treatment with cyclophosphamide, which may be used along with a rapid systemic prednisone taper.[26,27] Monotherapy with systemic steroids should be avoided because they are not as effective as other immunosuppressive therapies and are associated with significant systemic toxicity when used at required doses.[26,27] Additionally, the disease may reactivate with the tapering of steroid treatment, supporting the use of systemic steroids as adjunct rather than sole therapy in patients who are refractory to immunosuppressive agents. The usual therapeutic dose of cyclophosphamide is 1.5–2.0 mg/kg per day in divided doses. The therapeutic target is a reduction in white blood cell count to the range of 2,000–3,000 cells per microliter. This treatment should be continued for at least 1 year after the improvement of the symptoms, and patients should be routinely monitored for known adverse effects while on this medication.[26,27] Consultation with an internist, dermatologist, or oncologist experienced in cytotoxic therapy is recommended when administering immunosuppressive agents such as cyclophosphamide. Patients who fail to respond to conventional therapies may be treated with IVIG or biologic agents such as rituximab, infliximab, daclizumab, or etanercept.[57,58] Although IVIG therapy is used in refractory cases or treatment-intolerant patients, it is also playing an increasing role as a primary systemic treatment of MMP due to its ability to avoid the limiting side effects of other agents. Increasing evidence shows that IVIG therapy can result in a decrease in circulating autoantibody against B4 through the treatment course.[28,57,58,59,60,61,62] IVIG is usually administered in dosages of approximately 2–3 g/kg per cycle, with each cycle lasting 3 to 5 consecutive days each month. Clinical improvement and disease remission have been shown to continue with increased time between cycles and a gradual tapering of IVIG.[52] On average, patients who had a successful halting of disease progression had received an average of 32 cycles over 35 months.[63]
Ocular surface reconstruction, including surgical correction of eyelid deformities or eyelash ablation for trichiasis, may be required to achieve ocular surface quiescence. Removing aberrant lashes, which is important in preventing corneal erosions or ulcers, can be performed using mechanical epilation, cryotherapy, bipolar electrolysis, radiofrequency ablation, or laser ablation. Punctal occlusion, which may already have resulted from cicatrization, can be helpful in managing dry eye associated with MMP. Permanent punctal occlusion with cautery is often required because patients with cicatrizing conjunctivitis have a higher rate of spontaneous extrusion of silicone punctal plugs. Buccal mucosal and hard palate grafting can be useful techniques in fornix reconstruction in severe cases. Ocular surgery can cause severe flares of the disease; therefore, surgical intervention should be avoided when the disease is active.[15,64] In cases of advanced corneal keratinization or ulceration, one may perform amniotic membrane transplantation or penetrating keratoplasty (PK). Standard PK is generally associated with a very guarded prognosis in patients who develop severe corneal disease in MMP. As limbal stem cell deficiency is thought to be present in blistering disease, limbal stem cell transplantation (LSCT) is playing an increasing role in reconstructing ocular surface and restoring useful vision to a functionally blind eye, although surrounding host tissue disease can the limit long-term success of limbal stem cell transplants.[65,66] When PK and LSCT are unsuccessful, keratoprostheses offer another option for the rehabilitation of vision with some success.[15,16,64,65,66,67,68,69] However, the prognosis of Boston keratoprosthesis type I is guarded due to severe dry eye and other ocular surface abnormalities in these cases.[70] Other types of keratoprostheses such as Boston keratoprosthesis type II, osteo-odonto-keratoprosthesis and Dacron keratoprosthesis had more favorable visual outcome in these cases.[71,72]
PEMPHIGUS VULGARIS
Pemphigus vulgaris (PV) is an autoimmune disorder affecting the skin and mucous membranes. The incidence of pemphigus is not clear, but it has been estimated to be 0.76 to 5 new cases per million per year. Those of Jewish ancestry have a much higher incidence (16–32 cases per million per year).[73] Different HLA haplotypes have been reported in different populations; HLA-DRB1 * 0403 and HLA-DQB1 * 0503 are major histocompatibility loci associated with increased genetic predisposition in Ashkenazi Jews and non-Jewish patients, respectively.[74,75] HLA-DRB*1507, HLA-DRB1 * 04, and HLA-DRB1 * 14 have been reported in Japanese patients with PV.[76,77] Pemphigus vulgaris can affect patients of any age but it most commonly arises between 30 and 50 years of age.[78]
The skin lesions can be found on the scalp, face, trunk, axilla, groin, and other pressure points. Oral and mucosal lesions are seen in 80% to 90% of cases and may be the initial presentation [Figure 3]. Compared to oral mucosa involvement, the eyes are less frequently involved. Other affected mucosal surfaces include the larynx, esophagus, anus, cervix, and vulva.[79,80] In contrast to tense blisters seen in bullous pemphigoid, patients with PV present with flaccid blisters on an erythematous base.[80] The blisters may present with Nikolsky's sign (describing the separation of the epithelium with tangential pressure on the skin surface), which offers a moderately sensitive but highly specific sign for the diagnosis of PV.[81]
Figure 3.
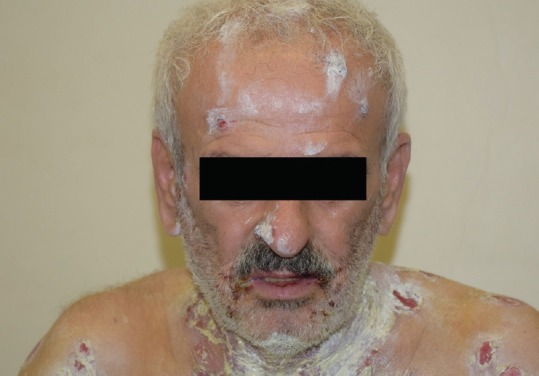
Pemphigus vulgaris. The skin lesions are present on the scalp, face, and perioral region. The oral mucosa is also involved.
Pathogenesis
In ocular PV (OPV), autoantibodies are secreted against antigens that are expressed in the basal cells of the conjunctival epithelium, fading in the suprabasal layers.[82] These pemphigus antigens include the desmosomal cadherins desmoglein 1 and 3.[83,84] Having a molecular weight of 160 kDa, desmoglein 1 is expressed throughout the epidermis but is most concentrated within the superficial layers. In contrast, desmoglein 3, a member of the cadherin family of cell adhesion molecules with a molecular weight of 130 kDa, is a component of the keratinocyte cell membrane and is primarily expressed in basal and directly suprabasal layers of the epidermis. Binding of the autoantibodies to these antigens results in destruction of adhesion between keratinocytes (acantholysis), manifested by blister formation just above the basal layer of the epidermis.[83,84] It has been found that patients with only desmoglein 1 autoantibodies typically have only skin lesions (as in pemphigus foliaceus). Patients with both desmoglein 1 and 3 autoantibodies can have both skin and mucosal involvement, and patients having only desmoglein 3 autoantibodies have only oral involvement. In most patients with PV, other non-desmoglein desmosomal proteins may compensate for the desmoglein 3 function loss in the conjunctiva, which explains why ocular involvement occurs in a minority of patients.[85]
Other immune system components may also play a role in PV. Early conjunctival inflammation is presumably evoked by the deposition of autoantibodies against intercellular adhesion molecules.[86] Interleukin 6 (IL-6) and TNF-α have been found to be increased in serum and in blister fluid in patients who have OPV.[83] Along with IL-6 and TNF-α, IL-1 is an important proinflammatory cytokine that mediates the blistering processes of PV through activating innate and acquired immune responses.
Ocular Manifestations
The incidence of ocular involvement in OPV is probably underestimated owing to a lack of large-scale studies focusing on ocular manifestations of PV.[87,88,89,90] A recent study reported that ocular involvement is present in 16.5% of patients with PV.[91] Ocular involvement in PV is limited to the eyelids and conjunctiva and does not appear to affect visual acuity; patients have a full recovery without sequelae.[87] The most commonly reported symptoms are redness, photophobia, pain, and ocular irritation.[87,91,92,93] Mild and moderate noncicatrizing bilateral conjunctivitis is the most frequent type of ocular involvement [Figure 4].[91] More severe ocular involvement appearing as palpebral conjunctival erosion is also commonly observed and is more prevalent than previously reported. Erosion of the bulbar conjunctiva is rarely detected.[91] Lid margin erosions can develop in lower and upper lids, and lid margin erosions in the medial aspect of the lower eyelid may be pathognomonic of this disease [Figure 4].[88] Unlike in MMP, severe dry eye due to lipid, mucin, and aqueous layer deficiency is not present in OPV.[86,88]
Figure 4.
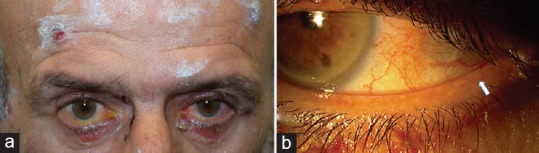
Ocular involvement in pemphigus vulgaris. a) The patient has eyelid erosions and noncicatrizing bilateral conjunctivitis. b) Lid margin blisters are visible in the lateral aspect of the lower eyelid (arrow).
The ocular manifestations tend to occur simultaneously or after mucocutaneous presentations in the majority of cases. A significant minority show ocular lesions as the first manifestation of PV.[91] Ocular involvement is not an indicator of more severe disease and can occur in partial remission, or in minor or major relapses of PV. For this reason, and because the ocular symptoms of PV are likely to be mild and not associated with discomfort, ophthalmologists should be familiar with pemphigus-associated ocular lesions and a complete ocular examination is often required to detect mild abnormalities.[88,91]
Diagnostic Studies
Histopathology reveals an intraepidermal blister, with changes consisting of intercellular edema with loss of intercellular attachments in the basal layer. The blisters are formed from suprabasal epidermal cells that subsequently separate from the basal cells that remain attached to the basement membrane.[94] No dermal cellular infiltration is evident. Direct and indirect immunofluorescence confirm the diagnosis of OPV.[80] Direct immunofluorescence demonstrates in vivo intercellular deposits of antibodies, primarily IgG, on the keratinocyte surface in and around lesions throughout the epidermis, with IgG1 and IgG4 as the most frequent subclasses.[95] Other immunoreactants, including complement C3 and IgM, can be deposited less frequently than IgG.[89] This pattern of intracellular deposition throughout the dermis is not specific for OPV and can be seen in pemphigus erythematosus, pemphigus foliaceus, and pemphigus vegetans. Indirect immunofluorescence demonstrates the presence of circulating IgG autoantibodies that bind to the epidermis in 80% to 90% of affected patients who have OPV and that correlate with disease course.[83] Laboratory use of ELISA to reveal desmoglein 1 and 3 antibodies provides reproducible, objective, and quantitative data that allow differentiation of OPV from pemphigus foliaceus. Because of these advantages, ELISA is likely to become a routine test for the diagnosis of PV.[92]
Management
Although the mainstay of treatment for OPV is systemic corticosteroids, various side effects limit its use. Steroid-sparing agents including cyclosporine, azathioprine, cyclophosphamide, dapsone, and mycophenolate mofetil are often used to avoid these side effects. Immunosuppressive agents and glucocorticoids can induce remission in the majority of PV patients; however, the associated mortality remains at 5% to 15% as a consequence of adverse effects.[96,97] Rituximab has been reported to induce a prolonged clinical remission following a single course of four treatments in patients who have OPV in addition to pemphigus foliaceus. The response to rituximab suggests that this medication may be a valuable therapeutic option for OPV patients who are refractory to standard immunosuppressive therapy.[83] Etanercept has also proved to be efficacious in OPV and allows for easy tapering and discontinuation of treatment with prednisolone and azathioprine.[82] In patients who do not respond to these immunosuppressive agents, IVIG can be used as an effective treatment alternative.[88] Its early use is of significant benefit in patients who may experience life threatening complications from immunosuppression.
Ocular lesions caused by PV respond well to systemic corticosteroids and/or immunosuppressive therapy. Local therapy consists of topical ophthalmic corticosteroids and the frequent use of artificial tears and lubricating ointment.[87,93] It has been reported that subconjunctival injection of triamcinolone acetonide 20 mg, 2 weeks apart, is successful in the management of chronic conjunctival ulcerations at the lid margin that were previously unresponsive to lubrication, topical steroids, and systemic treatment.[98]
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus (PNP) is associated with neoplasms, which can be benign or malignant. The neoplasm is usually a chronic lymphocytic leukemia, lymphoma (non-Hodgkin's type), Castleman's disease, or thymoma. Other hematologic disorders associated with PNP are monoclonal gammopathy, Waldenstrom macroglobulinemia, and Hodgkin lymphoma.[99,100] Both genders are equally affected and the disease has an average age of onset of 51 years.[101] Children and adolescents can also present with PNP, which is usually associated with Castleman's disease.[102]
Patients present with erosive and blistering skin and mucosal lesions. Mucous membrane involvement occurs in almost all patients with PNP. Mucosal involvement is characterized by persistent, painful blisters or erosions in the lips, gingival, buccal, and lingual mucosa, nasopharynx, oropharynx, nasal septum, hypopharynx, larynx, tracheobronchial mucosa, esophagus and epiglottis.[103,104,105] Severe intractable stomatitis is one of the most striking clinical features of PNP in which the lesions characteristically cross the vermillion border and extend from the cutaneous lip onto the vermillion lip, leading to thickened hemorrhagic mucosal erosions and slough. Stomatitis may be resistant to therapy. The lungs and gastrointestinal tract as well as involvement of the genital and ocular mucosa may also occur. Cutaneous lesions typically develop after mucous membrane lesions, with an interval between days and months.[106] Cutaneous findings may be quite polymorphic and can present as tense or flaccid blisters, erosions, lichenoid-appearing eruptions, erythema multiforme like lesions, and erythematous macules. Involvement of the palms and soles, including erythema multiforme like lesions and keratotic papules, can also be seen and distinguishes PNP from PV, which typically spares the palms and soles.[99] The lichenoid eruption, which is most commonly seen on the trunk and extremities, varies from isolated, scattered, pruritic, violaceous papules to more widespread lichenoid-appearing papules.[107] Given the different cutaneous presentations, the differential diagnosis of PNP may include graft-versus-host disease, erythema multiforme, lichenoid dermatitis, Stevens-Johnson syndrome, toxic epidermolytic necrolysis, bullous pemphigoid, and morbilliform drug eruption.[103]
The neoplasm is diagnosed before or shortly after the development of PNP which is associated with an existing neoplasm in approximately two thirds of patients and is a marker for an occult neoplasm in a third of cases.[108] The mortality rate is high (approximately 93%) when the disease is associated with a malignant neoplasm.[108] Death is due to the progression of malignancy or complications of immunosuppressive therapy. Approximately one third of the deaths in paraneoplastic pemphigus are due to pulmonary insufficiency (bronchiolitis obliterans).
Pathogenesis
The relationship between PNP and neoplasms is not well known. Antibodies against multiple desmosomal antigens have been reported, including those against desmogleins 1 and 3 and members of the plakin protein family (envoplakin, plectin, periplakin, desmoplakin I, desmoplakin II, and bullous pemphigoid antigen I).[103,109,110] It is proposed that the tumor produces proteins that activate autoimmune responses through cross-reactivity with epidermal antigens.[101,108]
Ocular Manifestations
Ocular symptoms are similar to those in MMP and include redness, pain, ocular irritation, mucous discharge, and worsening of vision. Patients may present with a bilateral cicatrizing conjunctivitis with shortening of the fornices and symblepharon formation. Other clinical signs are eyelid margin thickening, conjunctival erosion, pseudomembranous conjunctivitis, and corneal epithelial defects. A Schirmer tear production test may show a decrease in baseline tear secretion.[103,105,111] Other ocular examinations are usually unremarkable. Bilateral corneal melting has been reported in severe cases.[112,113]
Diagnostic Studies
The histological findings are polymorphous and can have marked variability, reflecting the rather heterogeneous cutaneous presentation of PNP. Suprabasal acantholysis similar to that seen in pemphigus vulgaris is present in most biopsy specimens.[103,104] It is common to observe vacuolar degeneration of basal cells and necrosis of epidermal keratinocytes. Some of these findings share features with erythema multiforme. As expected with lichenoid dermatitis, dense lymphocytic infiltrate in the papillary dermis is evident in lichenoid lesions.[108] IgG or C3 can be detected intercellularly and/or at the basement membrane zone using direct immunofluorescence microscopy. Deposition of C3 at the BMZ is analogous to what is seen in erythema multiforme, and deposition of IgG around epidermal cells is identical to that in PV. A conjunctival biopsy can be helpful for the diagnosis of PNP when biopsy of other mucocutaneous sites is misleading.[114]
Management
Treatment of the underlying pathology and suppression of the immune response to prevent production of the pathogenic autoantibodies are the main treatments.[108,115,116] Benign tumors including Castleman's disease and thymoma can be surgically excised, resulting in serological and clinical improvement. Management of the underlying malignant neoplasm may not lead to improvement in PNP, necessitating immunosuppressive therapy. Most of the immunosuppressive medications used in the management of PNP are those used in the management of other autoimmune blistering diseases, including PV. Compared to mucosal lesions, skin lesions better respond to treatment.
Prednisone at the dose of 1–2 mg/kg/day generally leads to partial improvement (especially of the skin lesions). Prednisone (1 mg/kg/day) cleared up PNP limited to the skin in a patient with non-Hodgkin's lymphoma (in remission), and the patient remained disease free for 18 months after diagnosis.[117] A combination of prednisone (1–2 mg/kg/day) and cyclosporine (5 mg/kg/day) could control symptoms in a subset of patients with PNP and chronic lymphocytic leukemia.[101,117,118,119] These patients had also received intermittent pulse cyclophosphamide for their underlying leukemia. The skin and mucosal lesions were effectively reduced in two patients using the combination of prednisone and azathioprine (100 mg/day).[101,120] Very high-dose cyclophosphamide (50 mg/kg/day intravenously for 4 consecutive days) significantly improved symptoms in some patients with PNP, but the recurrence rate was high.[119] The combination of mycophenolate mofetil, prednisone, and azathioprine, followed by mycophenolate mofetil alone after the discontinuation of the other two drugs has been found to be effective for the management of PNP [116]. Rituximab may be promising in patients who have underlying B-cell lymphomas such as CD20-positive follicular lymphoma and follicular non-Hodgkin lymphoma.[121,122,123]
Similar to cases of MMP, vigorous and early treatment may help prevent vision loss and blindness in PNP cases.[111,114] In addition to treatment of the underlying malignancy and systemic immunosuppressive therapy, ocular therapy may include the use of frequent preservative-free artificial tears, sodium hyaluronate, 10% N-acetylcysteine, and topical corticosteroid drops.[105,111,112,114,124] A surgical approach such as amniotic membrane transplantation may be necessary in cases of extensive forniceal contraction and conjunctival scarring.[88,114]
LINEAR IMMUNOGLOBULIN A DISEASE
Linear immunoglobulin A disease (LAD) is a rare disease characterized by circulating autoimmune antibodies to the epithelial BMZ, with the prevalence estimated to be 0.6 cases per 100,000 adults in Utah[125] and 0.13 cases per 250,000 individuals in France.[126] An estimated incidence of 1 case per 250,000 individuals per year has been reported in southern England.[126] Association with HLA-B8 has been noted.[127] Both genders are equally affected. LAD patients present in a bimodal age distribution, with pediatric disease presenting at a mean age of 3.3 to 4.5 years and adult cases at a mean age of 52 years.[128,129,130]
Affected patients typically present with a lengthened prodrome of a cutaneous burning sensation or pruritus before the emergence of lesions. The skin lesions classically present as annular tense bullae, resembling a “string of pearls” or “cluster of jewels” surrounding a patch of erythematous skin.[125] Lesions typically involve the perioral region, perineum, and extremities. In addition, mucous membrane involvement is seen in adults, with oral, genital, and conjunctival lesions that can result in scarring. These lesions may appear gradually or, in drug-associated cases, more acutely.[125,127,128] LAD typically follows an exacerbating and remitting disease course, with the possibility of progression to blindness. Overall, remission takes place in 64% of pediatric patients, frequently within 2 years of symptomatic onset.[125] Adult patients experience less frequent remission (48%) and a more prolonged disease course lasting an average of 5 to 6 years.[125,126,127] Although ocular and oral lesions may leave persistent scarring as in the case with MMP, cutaneous lesions usually heal without scarring.[131]
Pathogenesis
Associations between LAD and various suspected etiologies include drug-induced cases that are most commonly caused by vancomycin hydrochloride.[132,133] Other suspected drugs associated with one or several cases of LAD include captopril, amiodarone, and phenytoin.[130,134,135,136] Thus, the clinician should examine the presenting patient's medication regimen for possible iatrogenic causes. Other associations have been considered, including preceding illness, malignancy, and coexistence with other autoimmune disorders.[125,128,133]
Ocular Manifestations
LAD patients commonly complain of ocular symptoms including eye pain, dry eyes, foreign-body sensation, burning, or ocular discharge. Slit-lamp examination may disclose ocular abnormalities even in the absence of ocular complaints. These abnormalities include fine conjunctival scarring, subconjunctival fibrosis and subsequent inner canthal architecture loss, trichiasis, entropion, inferior forniceal foreshortening, and symblepharon formation with secondary corneal scarring. The ocular changes may be clinically indistinguishable from those of MMP. Because ocular involvement is common (approximately 50% of LAD patients) and may be asymptomatic, all LAD patients should undergo ophthalmologic examination.[130]
Diagnostic Studies
As in MMP, biopsy and immunofluorescent study are the predominant modalities used to confirm the diagnosis of LAD. Histopathologic examination of cutaneous blisters in LAD reveals characteristic findings, including neutrophil infiltration along the BMZ and subepidermal cleavage with inflammatory cells in the upper dermis. In some cases, neutrophilic microabscesses may form within the papillary ridges. Mature lesions demonstrate subepidermal blistering with a predominantly neutrophilic infiltrate, although eosinophils and monocytes may be present. In patients with atypical presentations, additional evaluation such as a Tzanck smear to rule out herpes virus infection and a Gram stain and bacterial culture of blister fluid to rule out bullous impetigo may be diagnostically informative.[125,128,130] A direct immunofluorescent assay of involved and spared areas of skin shows corresponding linear IgA (and less frequently IgG or IgM) and complement C3 deposition along the basement membrane.[130,137] Similar deposition is observed with analysis of conjunctival and oral mucosa in affected patients.[130,137] The addition of indirect immunofluorescence or direct immunoelectron microscopy techniques increases sensitivity, with the former identifying circulating anti-BMZ autoantibodies in approximately one half of LAD-affected patients.[130,137] Circulating autoantibody titers are characteristically higher in children than in adults but are generally a low-frequency finding in both age groups. The use of immunoperoxidase in direct and indirect immunoelectron microscopy localizes immunoglobulin deposition to the lamina lucida and to the target antigen LAD-1, secreted by keratinocytes as a component of the anchoring filament.[130,137,138] Immunoblotting and immunoelectron microscopy studies show reactivity between LAD sera and several different antigenic targets including type VII collagen.[139,140] Prediction of a particular patient's disease course or clinical response to treatment should take this intra-disease variability into account because patients are less treatment responsive in cases in which type VII collagen is the molecular target antigen.[128] This would explain the variation in the clinical picture and therapeutic responses seen with LAD.[139,140]
Management
Drug-induced cases typically resolve quickly with identification and withdrawal of the offending medication.[131,132,133,134,135,136] In cases of LAD induced by vancomycin, cessation of the drug prevents the formation of the new lesions within approximately 2 weeks.[131,141] In persistent cases of drug-induced LAD, a short oral steroid course may be beneficial.[125,130] Localized treatment addresses the mucosal and cutaneous lesions, although bullae do not usually require specific care beyond hygienic dressings, with topical antibacterial agents such as mupirocin used for infected lesions. The use of dapsone or sulfapyridine is successful in the majority of cases, resulting in noticeable response to treatment within several days.[125,126,127,128] Treated disease lasting longer than a few weeks may benefit from adjunct therapy including prednisone, dicloxacillin, sulfamethoxypyridazine, and colchicine.[142,143] As in MMP, treatment of refractory cases with IVIG has shown efficacy.[144,145,146]
EPIDERMOLYSIS BULLOSA ACQUISITA
Epidermolysis bullosa acquisita (EBA) is an autoimmune blistering disease of the mucous membranes and skin.[147] There are three different phenotypes of this disease: mechanobullous, inflammatory, and cicatricial pemphigoid. The disease is rare, with an estimated annual incidence of 0.25 per 1 million in Western Europe.[148] It has been noted that patients of Korean descent have a higher predisposition to EBA.[149] The male-to-female ratio is 1:1.4.[149] An association with HLA-DR2 has been found in most black patients from the southeastern part of the United States. HLA-DR2 depicts hyperimmunity that is observed in EBA, indicating an autoimmune etiology for this entity. However, no significant HLA allelic associations with EBA have been found in white patients.[150] EBA typically manifests in the third to fifth decade, with patients exhibiting milia and scarring blisters at sites of trauma, including elbows, knees, buttocks, and the dorsa of the hands and feet. The scarring nature of EBA can result in hair loss and nail destruction.[148,151] EBA is most commonly non- or mildly inflammatory (mechanobullous form), manifested as tense bullae and vesicles. The blisters may be hemorrhagic and rupture easily, with subsequent erosion.[152] The blisters in the classic mechanobullous noninflammatory EBA resemble those in dystrophic epidermolysis bullosa because both disease processes involve type VII collagen.[153] These lesions can also mimic bullous lupus erythematosus and porphyria cutanea tarda in the elderly. Mucosal involvement similar to that in MMP may develop in some patients, and others may present with inflammatory blisters that are tense and widespread, resembling bullous pemphigoid.[154] EBA can involve the pharyngeal, nasal, ocular, and esophageal mucosa or, in children, the oral mucosa, genitals, and ocular mucosa, causing symblepharon formation.[155,156]
Pathogenesis
EBA is mediated by IgG autoantibodies against type VII collagen (a dermal protein of 290 kDa), which is a major component of the anchoring fibrils in the lamina densa of the epithelial basement membrane.[147] A small subset of EBA is known to be due to IgA rather than to IgG.[157] Although the exact role of these autoantibodies is unknown, it has been proposed that they disrupt the assembly of type VII collagen into anchoring fibrils and interfere with their interactions with other extracellular matrix molecules.[158] Autoantibodies to type VII collagen have also been found to activate complement in both in vivo and in vitro studies. The complement components C3, C5b, and the membrane attack complex have been found in patients who have EBA. Passive transfer of IgG into mice from a patient who had EBA was shown to induce dermal edema and a granulocytic infiltrate in the superficial dermis, with immune deposits at the BMZ; however, no clinical disease was observed.[159]
Ocular Manifestations
The patients present with various aspects of chronic conjunctivitis, symblepharon formation, keratitis, subepithelial corneal vesiculation, perforation, and possibly opacification. In very severe cases (most notably cases mediated by IgA), total blindness has been reported.[155,156]
Diagnostic Studies
Various tests are used to diagnose EBA, including histopathology obtained from the border of a fresh blister, direct immunofluorescence on normal-looking perilesional skin, indirect immunofluorescence with the patient's serum on salt-split normal human skin substrate, ELISA, fluorescent overlay antigen mapping, immunoelectron microscopy, and Western immunoblotting.[144,160] Lesional skin histology initially reveals edema within the papilla in addition to edema and vacuolar alteration along the dermal-epidermal junction. A subepidermal blister forms as the disease progresses, with the epidermis remaining intact. Neutrophilic inflammation, which is minimal in the classic form of EBA and more abundant in the inflammatory form, along with eosinophils and monocytes within the dermis can be seen histologically.[156,161] The infiltrate is evident perivascularly, in the interstitium, and around follicles, and can lead to fibrosis in older lesions. Direct immunofluorescence reveals the immune-mediated disease process, with a thick band of IgG and, to a lesser extent, C3 deposited linearly at the BMZ.[154] Other immunoreactants such as IgM or IgA may also be seen. Indirect immunofluorescence demonstrates the presence of IgG antibodies directed toward type VII collagen and is used to differentiate EBA from bullous pemphigoid.[162] The IgG autoantibodies in patients who have bullous pemphigoid bind to the epidermal roof of salt-split skin, whereas the dermal floor pattern of indirect immunofluorescence on salt-split skin substrate is also found in the sera of patients who have bullous systemic lupus erythematosus and antiepiligrin cicatricial pemphigoid.[160] In immunoblotting, circulating autoantibodies bind type VII collagen, whereas detection of anti-type VII collagen antibodies by ELISA uses fusion proteins corresponding to different portions of the non-collagenous domain of type VII collagen.[163] Urine porphyrin levels and antinuclear antibody should be investigated to exclude porphyria cutanea tarda and to search for evidence of bullous systemic lupus erythematosus, which is also characterized by autoantibodies directed against type VII collagen.
Management
EBA has a protracted course that lasts several years in most patients and is usually resistant to treatment. The likelihood of disease remission is unpredictable. The primary therapeutic choices include immunosuppressive or anti-inflammatory agents. Systemic immunosuppressive drugs including prednisone, azathioprine, cyclophosphamide, methotrexate, and cyclosporine are used in various dosages. A group of anti-inflammatory drugs including dapsone, sulfapyridine, and colchicine appear to be effective and may decrease inflammation because of their antineutrophil effects.[164,165] Children who have EBA respond better with dapsone and prednisone, whereas patients who have bullous systemic lupus erythematosus usually respond rapidly and completely to dapsone.[166] Photochemotherapy was reported to be effective in one study but resulted in only partial improvement in two of three patients in another study.[167] IVIG has also recently been found to be effective as a sole therapeutic agent and when used in combination with other immunomodulatory agents such as rituxamab.[156,158] The basis for treatment with rituximab is to deplete pre-, immature, and mature B lymphocytes; cases of dramatically improved symptoms within a few weeks and a decrease in steroid requirements have been noted.[158] Of note, the patients who respond best to rituximab are those who have pemphigoid-type EBA.[144]
CONCLUSION
Management of the ocular involvement in autoimmune blistering diseases including OPV, EBA, MMP, and LAD usually requires collaborative efforts among the immunologist, the dermatologist, the ophthalmologist, and other specialists. Although there is significant overlap in the underlying pathophysiology and mucocutaneous lesions among this group of diseases, certain characteristics help differentiate them: (1) primarily mucous membrane involvement, with scarring in MMP and without scarring in LAD; and (2) tense cutaneous blisters in areas of trauma in EBA versus flaccid blisters and acantholysis in OPV. Further, the commonalities in clinical presentation strongly emphasize the importance of tissue biopsy for diagnostic confirmation. To guide appropriate therapy, a biopsy should preferably be performed early in the disease course after a treatment-resistant chronic conjunctivitis has been identified. Treatment with monoclonal antibodies such as rituximab, daclizumab, and infliximab and treatment with IVIG play an increasing role for these diseases, with early initiation providing cessation of disease progression. Future directions in the management of these blistering diseases consist of further characterization of the target antigens stimulating the autoimmune process and the development and commercialization of additional ELISAs and other tests. A focus on early initiation of therapy with existing immunoregulatory agents and the development of new agents will aid in avoiding the morbidities of these chronic diseases.
Declaration of Patient Consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial Support and Sponsorship
Nil.
Conflicts of Interest
There are no conflicts of interest.
REFERENCES
- 1.Burgeson RE, Christiano AM. The dermal-epidermal junction. Curr Opin Cell Biol. 1997;9:651–658. doi: 10.1016/s0955-0674(97)80118-4. [DOI] [PubMed] [Google Scholar]
- 2.Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–877. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- 3.Rappersberger K, Roos N, Stanley JR. Immunomorphological and biochemical identification of the pemphigus foliaceus autoantigen within desmosomes. J Invest Dermatol. 1992;99:323–330. doi: 10.1111/1523-1747.ep12616659. [DOI] [PubMed] [Google Scholar]
- 4.Kárpáti S, Amagai M, Prussick R, Cehrs K, Stanley JR. Pemphigus vulgaris antigen, a desmoglein type of cadherin, is localized within keratinocyte desmosomes. J Cell Biol. 1993;122:409–415. doi: 10.1083/jcb.122.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanley JR. Cell adhesion molecules as targets of autoantibodies in pemphigus and pemphigoid, bullous diseases due to defective epidermal cell adhesion. Adv Immunol. 1993;53:291–325. doi: 10.1016/s0065-2776(08)60503-9. [DOI] [PubMed] [Google Scholar]
- 6.Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. 2005;115:870–878. doi: 10.1172/JCI21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sitaru C, Zillikens D. Mechanisms of blister induction by autoantibodies. Exp Dermatol. 2005;14:861–875. doi: 10.1111/j.1600-0625.2005.00367.x. [DOI] [PubMed] [Google Scholar]
- 8.Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, et al. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol. 1985;85:538–541. doi: 10.1111/1523-1747.ep12277362. [DOI] [PubMed] [Google Scholar]
- 9.Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. 1982;306:1189–1196. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 10.Hu CH, Michel B, Schiltz JR. Epidermal acantholysis induced in vitro by pemphigus autoantibody. An ultrastructural study. Am J Pathol. 1978;90:345–362. [PMC free article] [PubMed] [Google Scholar]
- 11.Diaz LA, Giudice GJ. End of the century overview of skin blisters. Arch Dermatol. 2000;136:106–112. doi: 10.1001/archderm.136.1.106. [DOI] [PubMed] [Google Scholar]
- 12.Stanley JR. Pemphigus and pemphigoid as paradigms of organ-specific, autoantibody-mediated diseases. J Clin Invest. 1989;83:1443–1448. doi: 10.1172/JCI114036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. 1997;100:1256–1263. doi: 10.1172/JCI119639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP 180. J Clin Invest. 1993;92:2480–2488. doi: 10.1172/JCI116856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen QD, Foster CS. Cicatricial pemphigoid: Diagnosis and treatment. Int Ophthalmol Clin. 1996;36:41–60. doi: 10.1097/00004397-199603610-00007. [DOI] [PubMed] [Google Scholar]
- 16.Foster CS, Neumann R, Tauber J. Long-term results of systemic chemotherapy for ocular cicatricial pemphigoid. Doc Ophthalmol. 1992;82:223–229. doi: 10.1007/BF00160769. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320–332. doi: 10.1016/S0140-6736(12)61140-4. [DOI] [PubMed] [Google Scholar]
- 18.Laskaris G, Sklavounou A, Stratigos J. Bullous pemphigoid, cicatricial pemphigoid, and pemphigus vulgaris. A comparative clinical survey of 278 cases. Oral Surg Oral Med Oral Pathol. 1982;54:656–662. doi: 10.1016/0030-4220(82)90080-9. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed AR, Foster S, Zaltas M, Notani G, Awdeh Z, Alper CA, et al. Association of DQw7 (DQB1 * 0301) with ocular cicatricial pemphigoid. Proc Natl Acad Sci U S A. 1991;88:11579–11582. doi: 10.1073/pnas.88.24.11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhol K, Udell I, Haider N, Yunis JJ, Mohimen A, Neuman R, et al. Ocular cicatricial pemphigoid. A case report of monozygotic twins discordant for the disease. Arch Ophthalmol. 1995;113:202–207. doi: 10.1001/archopht.1995.01100020086034. [DOI] [PubMed] [Google Scholar]
- 21.Chan LS, Wang T, Wang XS, Hammerberg C, Cooper KD. High frequency of HLA-DQB1 * 0301 allele in patients with pure ocular cicatricial pemphigoid. Dermatology. 1994;189(Suppl 1):99–101. doi: 10.1159/000246943. [DOI] [PubMed] [Google Scholar]
- 22.Foster ME, Nally FF. Benign mucous membrane pemphigoid (cicatricial mucosal pemphigoid): A reconsideration. Oral Surg Oral Med Oral Pathol. 1977;44:697–705. doi: 10.1016/0030-4220(77)90379-6. [DOI] [PubMed] [Google Scholar]
- 23.Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: Definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370–379. doi: 10.1001/archderm.138.3.370. [DOI] [PubMed] [Google Scholar]
- 24.Thorne JE, Anhalt GJ, Jabs DA. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology. 2004;111:45–52. doi: 10.1016/j.ophtha.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Bruch-Gerharz D, Hertl M, Ruzicka T. Mucous membrane pemphigoid: Clinical aspects, immunopathological features and therapy. Eur J Dermatol. 2007;17:191–200. doi: 10.1684/ejd.2007.0148. [DOI] [PubMed] [Google Scholar]
- 26.Foster CS, Wilson LA, Ekins MB. Immunosuppressive therapy for progressive ocular cicatricial pemphigoid. Ophthalmology. 1982;89:340–353. doi: 10.1016/s0161-6420(82)34791-0. [DOI] [PubMed] [Google Scholar]
- 27.Foster CS, Ahmed AR. Intravenous immunoglobulin therapy for ocular cicatricial pemphigoid: A preliminary study. Ophthalmology. 1999;106:2136–2143. doi: 10.1016/S0161-6420(99)90496-7. [DOI] [PubMed] [Google Scholar]
- 28.Heiligenhaus A, Shore JW, Rubin PA, Foster CS. Long-term results of mucous membrane grafting in ocular cicatricial pemphigoid. Implications for patient selection and surgical considerations. Ophthalmology. 1993;100:1283–1288. doi: 10.1016/s0161-6420(93)31487-9. [DOI] [PubMed] [Google Scholar]
- 29.Rashid KA, Gürcan HM, Ahmed AR. Antigen specificity in subsets of mucous membrane pemphigoid. J Invest Dermatol. 2006;126:2631–2636. doi: 10.1038/sj.jid.5700465. [DOI] [PubMed] [Google Scholar]
- 30.Leonard JN, Wright P, Williams DM, Gilkes JJ, Haffenden GP, McMinn RM, et al. The relationship between linear IgA disease and benign mucous membrane pemphigoid. Br J Dermatol. 1984;110:307–314. doi: 10.1111/j.1365-2133.1984.tb04636.x. [DOI] [PubMed] [Google Scholar]
- 31.Frith PA, Venning VA, Wojnarowska F, Millard PR, Bron AJ. Conjunctival involvement in cicatricial and bullous pemphigoid: A clinical and immunopathological study. Br J Dermatol. 1989;73:52–56. doi: 10.1136/bjo.73.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venning VA, Frith PA, Bron AJ, Millard PR, Wojnarowska F. Mucosal involvement in bullous and cicatricial pemphigoid. A clinical and immunopathological study. Br J Dermatol. 1988;118:7–15. doi: 10.1111/j.1365-2133.1988.tb01744.x. [DOI] [PubMed] [Google Scholar]
- 33.Mutasim DF, Pelc NJ, Anhalt GJ. Cicatricial pemphigoid. Dermatol Clin. 1993;11:499–510. [PubMed] [Google Scholar]
- 34.Foster CS. Cicatricial pemphigoid. Trans Am Ophthalmol Soc. 1986;84:527–663. [PMC free article] [PubMed] [Google Scholar]
- 35.Schwab IR. Cultured corneal epithelia for ocular surface disease. Trans Am Ophthalmol Soc. 1999;97:891–986. [PMC free article] [PubMed] [Google Scholar]
- 36.Liesegang TJ. Conjunctival changes associated with glaucoma therapy: Implications for the external disease consultant and the treatment of glaucoma. Cornea. 1998;17:574–583. doi: 10.1097/00003226-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Elder MJ, Bernauer W. Monitoring of activity and progression in cicatrising conjunctivitis. Dev Ophthalmol. 1997;28:111–122. doi: 10.1159/000060709. [DOI] [PubMed] [Google Scholar]
- 38.Mondino BJ, Brown SI. Ocular cicatricial pemphigoid. Ophthalmology. 1981;88:95–100. doi: 10.1016/s0161-6420(81)35069-6. [DOI] [PubMed] [Google Scholar]
- 39.Tauber J, Jabbur N, Foster CS. Improved detection of disease progression in ocular cicatricial pemphigoid. Cornea. 1992;11:446–451. doi: 10.1097/00003226-199209000-00015. [DOI] [PubMed] [Google Scholar]
- 40.Rowsey JJ, Macias-Rodriguez Y, Cukrowski C. A new method for measuring progression in patients with ocular cicatricial pemphigoid. Arch Ophthalmol. 2004;122:179–184. doi: 10.1001/archopht.122.2.179. [DOI] [PubMed] [Google Scholar]
- 41.Reeves GM, Lloyd M, Rajlawat BP, Barker GL, Field EA, Kaye SB. Ocular and oral grading of mucous membrane pemphigoid. Graefes Arch Clin Exp Ophthalmol. 2012;250:611–618. doi: 10.1007/s00417-011-1853-z. [DOI] [PubMed] [Google Scholar]
- 42.Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid. Ocul Surf. 2013;11:259–266. doi: 10.1016/j.jtos.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Butt Z, Kaufman D, McNab A, McKelvie P. Drug-induced ocular cicatricial pemphigoid: A series of clinico-pathological reports. Eye (Lond) 1998;12:285–290. doi: 10.1038/eye.1998.66. [DOI] [PubMed] [Google Scholar]
- 44.Bernauer W, Broadway DC, Wright P. Chronic progressive conjunctival cicatrisation. Eye (Lond) 1993;7:371–378. doi: 10.1038/eye.1993.75. [DOI] [PubMed] [Google Scholar]
- 45.Fine JD. Cicatricial pemphigoid. In: Wojnarowska F, Briggaman RA, editors. Management of Blistering Diseases. New York: Raven Press; 1990. pp. 83–92. [Google Scholar]
- 46.Bernauer W, Wright P, Dart JK, Leonard JN, Lightman S. The conjunctiva in acute and chronic mucous membrane pemphigoid. An immunohistochemical analysis. Ophthalmology. 1993;100:339–346. doi: 10.1016/s0161-6420(93)31644-1. [DOI] [PubMed] [Google Scholar]
- 47.Brauner GJ, Jimbow K. Benign mucous membrane pemphigoid. An unusual case with electron microscopic findings. Arch Dermatol. 1972;106:535–540. doi: 10.1001/archderm.106.4.535. [DOI] [PubMed] [Google Scholar]
- 48.Setterfield J, Bhogal B, Morgan P, et al. Clinical and immunopathological correlation in cicatricial pemphigoid: A study of 36 cases. British Society for Investigative Dermatology Annual Meeting, Oxford, April 6–7, 1995. Br J Dermatol. 1995;132:631–660. [Google Scholar]
- 49.Power WJ, Neves RA, Rodriguez A, Dutt JE, Foster CS. Increasing the diagnostic yield of conjunctival biopsy in patients with suspected ocular cicatricial pemphigoid. Ophthalmology. 1995;102:1158–1163. doi: 10.1016/s0161-6420(95)30896-2. [DOI] [PubMed] [Google Scholar]
- 50.Prost C, Robin H, Caux F, et al. Direct immunoelectron microscopy on the conjunctiva in ocular cicatricial pemphigoid versus subepidermal autoimmune bullous dermatoses with ocular involvement: Report of 10 cases. Abstracts for the 1996 Annual Meeting Society for Investigative Dermatology. J Invest Dermatol. 1996;106:805–950. [Google Scholar]
- 51.Bédane C, Prost C, Bernard P, Catanzano G, Bonnetblanc JM, Dubertret L. Cicatricial pemphigoid antigen differs from bullous pemphigoid antigen by its exclusive extracellular localization: A study by indirect immunoelectron microscopy. J Invest Dermatol. 1991;97:3–9. doi: 10.1111/1523-1747.ep12477198. [DOI] [PubMed] [Google Scholar]
- 52.Letko E, Bhol K, Foster SC, Ahmed RA. Influence of intravenous immunoglobulin therapy on serum levels of anti-beta 4 antibodies in ocular cicatricial pemphigoid. A correlation with disease activity. A preliminary study. Curr Eye Res. 2000;21:646–654. [PubMed] [Google Scholar]
- 53.Leverkus M, Bhol K, Hirako Y, Pas H, Sitaru C, Baier G, et al. Cicatricial pemphigoid with circulating autoantibodies to beta4 integrin, bullous pemphigoid 180, and bullous pemphigoid 230. Br J Dermatol. 2001;145:998–1004. doi: 10.1046/j.1365-2133.2001.04543.x. [DOI] [PubMed] [Google Scholar]
- 54.Silverman S, Jr, Gorsky M, Lozada-Nur F, Liu A. Oral mucous membrane pemphigoid. A study of sixty-five patients. Oral Surg Oral Med Oral Pathol. 1986;61:233–237. doi: 10.1016/0030-4220(86)90367-1. [DOI] [PubMed] [Google Scholar]
- 55.Celis Sánchez J, López Ferrando N, García Largacha M, González del Valle F, Ortiz de la Torre MJ. Subconjunctival mitomycin C for the treatment of ocular cicatricial pemphigoid. Arch Soc Esp Oftalmol. 2002;77:501–506. [PubMed] [Google Scholar]
- 56.Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003:CD004056. doi: 10.1002/14651858.CD004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heffernan MP, Bentley DD. Successful treatment of mucous membrane pemphigoid with infliximab. Arch Dermatol. 2006;142:1268–1270. doi: 10.1001/archderm.142.10.1268. [DOI] [PubMed] [Google Scholar]
- 58.Papaliodis GN, Chu D, Foster CS. Treatment of ocular inflammatory disorders with daclizumab. Ophthalmology. 2003;110:786–789. doi: 10.1016/S0161-6420(02)01932-2. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt E, Seitz CS, Benoit S, Bröcker EB, Goebeler M. Rituximab in autoimmune bullous diseases: Mixed responses and adverse effects. Br J Dermatol. 2007;156:352–356. doi: 10.1111/j.1365-2133.2006.07646.x. [DOI] [PubMed] [Google Scholar]
- 60.Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355:1772–1779. doi: 10.1056/NEJMoa062930. [DOI] [PubMed] [Google Scholar]
- 61.Gürcan HM, Ahmed AR. Frequency of adverse events associated with intravenous immunoglobulin therapy in patients with pemphigus or pemphigoid. Ann Pharmacother. 2007;41:1604–1610. doi: 10.1345/aph.1K198. [DOI] [PubMed] [Google Scholar]
- 62.Ahmed AR. Use of intravenous immunoglobulin therapy in autoimmune blistering diseases. Int Immunopharmacol. 2006;6:557–578. doi: 10.1016/j.intimp.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 63.Sami N, Letko E, Androudi S, Daoud Y, Foster CS, Ahmed AR. Intravenous immunoglobulin therapy in patients with ocular-cicatricial pemphigoid: A long-term follow-up. Ophthalmology. 2004;111:1380–1382. doi: 10.1016/j.ophtha.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 64.Schwab IR, Reyes M, Isseroff RR. Successful transplantation of bioengineered tissue replacements in patients with ocular surface disease. Cornea. 2000;19:421–426. doi: 10.1097/00003226-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 65.Tsubota K, Satake Y, Ohyama M, Toda I, Takano Y, Ono M, et al. Surgical reconstruction of the ocular surface in advanced ocular cicatricial pemphigoid and Stevens-Johnson syndrome. Am J Ophthalmol. 1996;122:38–52. doi: 10.1016/s0002-9394(14)71962-2. [DOI] [PubMed] [Google Scholar]
- 66.Samson CM, Nduaguba C, Baltatzis S, Foster CS. Limbal stem cell transplantation in chronic inflammatory eye disease. Ophthalmology. 2002;109:862–868. doi: 10.1016/s0161-6420(02)00994-6. [DOI] [PubMed] [Google Scholar]
- 67.Koizumi N, Inatomi T, Suzuki T, Sotozono C, Kinoshita S. Cultivated corneal epithelial stem cell transplantation in ocular surface disorders. Ophthalmology. 2001;108:1569–1574. doi: 10.1016/s0161-6420(01)00694-7. [DOI] [PubMed] [Google Scholar]
- 68.Daya SM, Ilari FA. Living related conjunctival limbal allograft for the treatment of stem cell deficiency. Ophthalmology. 2001;108:126–133. doi: 10.1016/s0161-6420(00)00475-9. [DOI] [PubMed] [Google Scholar]
- 69.Tsai RJ, Li LM, Chen JK. Reconstruction of damaged corneas by transplantation of autologous limbal epithelial cells. N Engl J Med. 2000;343:86–93. doi: 10.1056/NEJM200007133430202. [DOI] [PubMed] [Google Scholar]
- 70.Palioura S, Kim B, Dohlman CH, Chodosh J. The Boston keratoprosthesis type I in mucous membrane pemphigoid. Cornea. 2013;32:956–961. doi: 10.1097/ICO.0b013e318286fd73. [DOI] [PubMed] [Google Scholar]
- 71.Lee R, Khoueir Z, Tsikata E, Chodosh J, Dohlman CH, Chen TC. Long-term visual outcomes and complications of Boston keratoprosthesis type II implantation. Ophthalmology. 2017;124:27–35. doi: 10.1016/j.ophtha.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 72.Falcinelli G, Falsini B, Taloni M, Colliardo P, Falcinelli G. Modified osteo-odonto-keratoprosthesis for treatment of corneal blindness: Long-term anatomical and functional outcomes in 181 cases. Arch Ophthalmol. 2005;123:1319–1329. doi: 10.1001/archopht.123.10.1319. [DOI] [PubMed] [Google Scholar]
- 73.Amagai M. Pemphigus. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. St. Louis, MO: Elsevier; 2012. p. 461. [Google Scholar]
- 74.Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, Awdeh Z, et al. Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A. 1990;87:7658–7662. doi: 10.1073/pnas.87.19.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, Alper CA, et al. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A. 1991;88:5056–5060. doi: 10.1073/pnas.88.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miyagawa S, Niizeki H, Yamashina Y, Kaneshige T. Genotyping for HLA-A, B and C alleles in Japanese patients with pemphigus: Prevalence of Asian alleles of the HLA-B15 family. Br J Dermatol. 2002;146:52–58. doi: 10.1046/j.1365-2133.2002.04564.x. [DOI] [PubMed] [Google Scholar]
- 77.Miyagawa S, Higashimine I, Iida T, Yamashina Y, Fukumoto T, Shirai T. HLA-DRB1 * 04 and DRB1 * 14 alleles are associated with susceptibility to pemphigus among Japanese. J Invest Dermatol. 1997;109:615–618. doi: 10.1111/1523-1747.ep12337585. [DOI] [PubMed] [Google Scholar]
- 78.Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61–73. doi: 10.1016/S0140-6736(05)66829-8. [DOI] [PubMed] [Google Scholar]
- 79.Smith RJ, Manche EE, Mondino BJ. Ocular cicatricial pemphigoid and ocular manifestations of pemphigus vulgaris. Int Ophthalmol Clin. 1997;37:63–75. doi: 10.1097/00004397-199703720-00006. [DOI] [PubMed] [Google Scholar]
- 80.Bhol KC, Rojas AI, Khan IU, Ahmed AR. Presence of interleukin 10 in the serum and blister fluid of patients with pemphigus vulgaris and pemphigoid. Cytokine. 2000;12:1076–1083. doi: 10.1006/cyto.1999.0642. [DOI] [PubMed] [Google Scholar]
- 81.Challacombe SJ, Setterfield J, Shirlaw P, Harman K, Scully C, Black MM. Immunodiagnosis of pemphigus and mucous membrane pemphigoid. Acta Odontol Scand. 2001;59:226–234. doi: 10.1080/00016350152509256. [DOI] [PubMed] [Google Scholar]
- 82.Tenner E. Ocular involvement in pemphigus vulgaris. J Am Acad Dermatol. 2006;55:725. doi: 10.1016/j.jaad.2006.01.067. author reply 725–726. [DOI] [PubMed] [Google Scholar]
- 83.Michailidou EZ, Belazi MA, Markopoulos AK, Tsatsos MI, Mourellou ON, Antoniades DZ. Epidemiologic survey of pemphigus vulgaris with oral manifestations in northern Greece: Retrospective study of 129 patients. Int J Dermatol. 2007;46:356–361. doi: 10.1111/j.1365-4632.2006.03044.x. [DOI] [PubMed] [Google Scholar]
- 84.Bianciotto C, Herreras Cantalapiedra JM, Alvarez MA, Méndez Díaz MC. Conjunctival blistering associated with pemphigus vulgaris: Report of a case. Arch Soc Esp Oftalmol. 2005;80:365–368. doi: 10.4321/s0365-66912005000600011. [DOI] [PubMed] [Google Scholar]
- 85.Olszewska M, Komor M, Mazur M, Rogozinski T. Response of ocular pemphigus vulgaris to therapy. Case report and review of literature. J Dermatol Case Rep. 2008;2:1–3. doi: 10.3315/jdcr.2008.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baer JC, Hemady RK. Ocular manifestations of systemic immune disease. In: Bosniak S, editor. Principles and Practice of Ophthalmic Plastic and Reconstructive Surgery. Vol. 1. Philadelphia, PA: WB Saunders Company; 1996. pp. 94–119. [Google Scholar]
- 87.Daoud YJ, Cervantes R, Foster CS, Ahmed AR. Ocular pemphigus. J Am Acad Dermatol. 2005;53:585–590. doi: 10.1016/j.jaad.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 88.Laforest C, Huilgol SC, Casson R, Selva D, Leibovitch I. Autoimmune bullous diseases: Ocular manifestations and management. Drugs. 2005;65:1767–1779. doi: 10.2165/00003495-200565130-00003. [DOI] [PubMed] [Google Scholar]
- 89.Merchant S, Weinstein M. Pemphigus vulgaris: The eyes have it. Pediatrics. 2003;112:183–185. doi: 10.1542/peds.112.1.183. [DOI] [PubMed] [Google Scholar]
- 90.Lifshitz T, Levy J, Cagnano E, Halevy S. Severe conjunctival and eyelid involvement in pemphigus vulgaris. Int Ophthalmol. 2004;25:73–74. doi: 10.1023/b:inte.0000031736.69290.18. [DOI] [PubMed] [Google Scholar]
- 91.Akhyani M, Keshtkar-Jafari A, Chams-Davatchi C, Lajevardi V, Beigi S, Aghazadeh N, et al. Ocular involvement in pemphigus vulgaris. J Dermatol. 2014;41:618–621. doi: 10.1111/1346-8138.12447. [DOI] [PubMed] [Google Scholar]
- 92.Palleschi GM, Giomi B, Fabbri P. Ocular involvement in pemphigus. Am J Ophthalmol. 2007;144:149–152. doi: 10.1016/j.ajo.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 93.Elchahal S, Kavosh ER, Chu DS. Ocular manifestations of blistering diseases. Immunol Allergy Clin North Am. 2008;28:119–136. doi: 10.1016/j.iac.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 94.Vaillant L. Bullous autoimmune diseases of the oral mucosa. Rev Stomatol Chir Maxillofac. 1999;100:230–239. [PubMed] [Google Scholar]
- 95.Hall VC, Liesegang TJ, Kostick DA, Lookingbill DP. Ocular mucous membrane pemphigoid and ocular pemphigus vulgaris treated topically with tacrolimus ointment. Arch Dermatol. 2003;139:1083–1084. doi: 10.1001/archderm.139.8.1083. [DOI] [PubMed] [Google Scholar]
- 96.Bhol K, Tyagi S, Natarajan K, Nagarwalla N, Ahmed AR. Use of recombinant pemphigus vulgaris antigen in development of ELISA and IB assays to detect pemphigus vulgaris autoantibodies. J Eur Acad Dermatol Venereol. 1998;10:28–35. [PubMed] [Google Scholar]
- 97.Kavosh ER, Bielory L. Allergic disease of the eye. In: Mahmoudi M, editor. Allergy and Asthma: Practical Diagnosis and Management. 1st ed. New York: McGraw Hill; 2007. pp. 21–31. [Google Scholar]
- 98.Kozeis N, Tyradellis S, Dragiotis E, Eleftheriadis H. Triamcinolone acetonide for rare ocular manifestations of pemphigus vulgaris: A case report. Clin Ophthalmol. 2010;4:365–368. doi: 10.2147/opth.s8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu X, Zhang B. Paraneoplastic pemphigus. J Dermatol. 2007;34:503–511. doi: 10.1111/j.1346-8138.2007.00322.x. [DOI] [PubMed] [Google Scholar]
- 100.Joly P, Richard C, Gilbert D, Courville P, Chosidow O, Roujeau JC, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619–626. doi: 10.1067/mjd.2000.107488. [DOI] [PubMed] [Google Scholar]
- 101.Robinson ND, Hashimoto T, Amagai M, Chan LS. The new pemphigus variants. J Am Acad Dermatol. 1999;40:649–671. doi: 10.1016/s0190-9622(99)70145-3. [DOI] [PubMed] [Google Scholar]
- 102.Mimouni D, Anhalt GJ, Lazarova Z, Aho S, Kazerounian S, Kouba DJ, et al. Paraneoplastic pemphigus in children and adolescents. Br J Dermatol. 2002;147:725–732. doi: 10.1046/j.1365-2133.2002.04992.x. [DOI] [PubMed] [Google Scholar]
- 103.Anhalt GJ, Kim SC, Stanley JR, Korman NJ, Jabs DA, Kory M, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990;323:1729–1735. doi: 10.1056/NEJM199012203232503. [DOI] [PubMed] [Google Scholar]
- 104.Fullerton SH, Woodley DT, Smoller BR, Anhalt GJ. Paraneoplastic pemphigus with autoantibody deposition in bronchial epithelium after autologous bone marrow transplantation. J Am Med Assoc. 1992;267:1500–1502. [PubMed] [Google Scholar]
- 105.Lam S, Stone MS, Goeken JA, Massicotte SJ, Smith AC, Folberg R, et al. Paraneoplastic pemphigus, cicatricial conjunctivitis, and acanthosis nigricans with pachydermatoglyphy in a patient with bronchogenic squamous cell carcinoma. Ophthalmology. 1992;99:108–113. doi: 10.1016/s0161-6420(92)32030-5. [DOI] [PubMed] [Google Scholar]
- 106.Frew JW, Murrell DF. Current management strategies in paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome) Dermatol Clin. 2011;29:607–612. doi: 10.1016/j.det.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 107.Hsiao CJ, Hsu MM, Lee JY, Chen WC, Hsieh WC. Paraneoplastic pemphigus in association with a retroperitoneal Castleman's disease presenting with a lichen planus pemphigoides-like eruption. A case report and review of literature. Br J Dermatol. 2001;144:372–376. doi: 10.1046/j.1365-2133.2001.04030.x. [DOI] [PubMed] [Google Scholar]
- 108.Anhalt GJ. Paraneoplastic pemphigus. Adv Dermatol. 1997;12:77–96. discussion 97. [PubMed] [Google Scholar]
- 109.Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T. Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest. 1998;102:775–782. doi: 10.1172/JCI3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gili A, Ngan BY, Lester R. Castleman's disease associated with pemphigus vulgaris. J Am Acad Dermatol. 1991;25:955–959. doi: 10.1016/0190-9622(91)70293-b. [DOI] [PubMed] [Google Scholar]
- 111.Tam PM, Cheng LL, Young AL, Lam PT. Paraneoplastic pemphigus: An uncommon cause of chronic cicatrising conjunctivitis? BMJ Case Rep 2009. 2009 doi: 10.1136/bcr.12.2008.1306. doi: 10.1136/bcr.12.2008.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Beele H, Claerhout I, Kestelyn P, Dierckxens L, Naeyaert JM, De Laey JJ. Bilateral corneal melting in a patient with paraneoplastic pemphigus. Dermatology. 2001;202:147–150. doi: 10.1159/000051622. [DOI] [PubMed] [Google Scholar]
- 113.Anan T, Shimizu F, Hatano Y, Okamoto O, Katagiri K, Fujiwara S. Paraneoplastic pemphigus associated with corneal perforation and cutaneous alternariosis: A case report and review of cases treated with rituximab. J Dermatol. 2011;38:1084–1089. doi: 10.1111/j.1346-8138.2010.01192.x. [DOI] [PubMed] [Google Scholar]
- 114.Meyers SJ, Varley GA, Meisler DM, Camisa C, Wander AH. Conjunctival involvement in paraneoplastic pemphigus. Am J Ophthalmol. 1992;114:621–624. doi: 10.1016/s0002-9394(14)74494-0. [DOI] [PubMed] [Google Scholar]
- 115.Hertzberg MS, Schifter M, Sullivan J, Stapleton K. Paraneoplastic pemphigus in two patients with B-cell non-Hodgkin's lymphoma: Significant responses to cyclophosphamide and prednisolone. Am J Hematol. 2000;63:105–106. doi: 10.1002/(sici)1096-8652(200002)63:2<105::aid-ajh14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 116.Williams JV, Marks JG, Jr, Billingsley EM. Use of mycophenolate mofetil in the treatment of paraneoplastic pemphigus. Br J Dermatol. 2000;142:506–508. doi: 10.1046/j.1365-2133.2000.03365.x. [DOI] [PubMed] [Google Scholar]
- 117.Dega H, Laporte JL, Joly P, Gabarre J, André C, Delpech A, et al. Paraneoplastic pemphigus associated with Hodgkin's disease. Br J Dermatol. 1998;138:196–198. doi: 10.1046/j.1365-2133.1998.02056.x. [DOI] [PubMed] [Google Scholar]
- 118.Camisa C, Helm TN, Liu YC, Valenzuela R, Allen C, Bona S, et al. Paraneoplastic pemphigus: A report of three cases including one long-term survivor. J Am Acad Dermatol. 1992;27:547–553. doi: 10.1016/0190-9622(92)70220-a. [DOI] [PubMed] [Google Scholar]
- 119.Korman NJ. New and emerging therapies in the treatment of blistering diseases. Dermatol Clin. 2000;18:127–137. doi: 10.1016/s0733-8635(05)70153-4. [DOI] [PubMed] [Google Scholar]
- 120.Krunic AL, Kokai D, Bacetic B, Kesic V, Nikolic MM, Petkovic S, et al. Retroperitoneal round-cell liposarcoma associated with paraneoplastic pemphigus presenting as lichen planus pemphigoides-like eruption. Int J Dermatol. 1997;36:526–529. doi: 10.1111/j.1365-4362.1997.tb01154.x. [DOI] [PubMed] [Google Scholar]
- 121.Aoi J, Makino K, Sakai K, Masuguchi S, Fukushima S, Jinnin M, et al. Case of paraneoplastic pemphigus with follicular lymphoma treated with rituximab. J Dermatol. 2013;40:285–286. doi: 10.1111/1346-8138.12095. [DOI] [PubMed] [Google Scholar]
- 122.Heizmann M, Itin P, Wernli M, Borradori L, Bargetzi MJ. Successful treatment of paraneoplastic pemphigus in follicular NHL with rituximab: Report of a case and review of treatment for paraneoplastic pemphigus in NHL and CLL. Am J Hematol. 2001;66:142–144. doi: 10.1002/1096-8652(200102)66:2<142::AID-AJH1032>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 123.Schmidt E, Hunzelmann N, Zillikens D, Bröcker EB, Goebeler M. Rituximab in refractory autoimmune bullous diseases. Clinical and Exp Dermatol. 2006;31:503–508. doi: 10.1111/j.1365-2230.2006.02151.x. [DOI] [PubMed] [Google Scholar]
- 124.Ahuero AE, Jakobiec FA, Bhat P, Ciralsky JB, Papaliodis GN. Paraneoplastic conjunctival cicatrization: Two different pathogenic types. Ophthalmology. 2010;117:659–664. doi: 10.1016/j.ophtha.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 125.Chorzelski TP, Jabłońska S, Maciejowska E. Linear IgA bullous dermatosis of adults. Clin Dermatol. 1991;9:383–392. doi: 10.1016/0738-081x(91)90030-o. [DOI] [PubMed] [Google Scholar]
- 126.Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol. 1995;131:48–52. [PubMed] [Google Scholar]
- 127.Collier PM, Wojnarowska F, Welsh K, McGuire W, Black MM. Adult linear IgA disease and chronic bullous disease of childhood: The association with human lymphocyte antigens Cw7, B8, DR3 and tumour necrosis factor influences disease expression. Br J Dermatol. 1999;141:867–875. doi: 10.1046/j.1365-2133.1999.03110.x. [DOI] [PubMed] [Google Scholar]
- 128.Wojnarowska F, Allen J, Collier P. Linear IgA disease: A heterogeneous disease. Dermatology. 1994;189(Suppl 1):52–56. doi: 10.1159/000246930. [DOI] [PubMed] [Google Scholar]
- 129.Jabłońska S, Chorzelski TP, Rosinska D, Maciejowska E. Linear IgA bullous dermatosis of childhood (chronic bullous dermatosis of childhood) Clin Dermatol. 1991;9:393–401. doi: 10.1016/0738-081x(91)90031-f. [DOI] [PubMed] [Google Scholar]
- 130.Aultbrinker EA, Starr MB, Donnenfeld ED. Linear IgA disease. The ocular manifestations. Ophthalmology. 1988;95:340–343. doi: 10.1016/s0161-6420(88)33188-x. [DOI] [PubMed] [Google Scholar]
- 131.Klein PA, Callen JP. Drug-induced linear IgA bullous dermatosis after vancomycin discontinuance in a patient with renal insufficiency. J Am Acad Dermatol. 2000;42:316–323. doi: 10.1016/s0190-9622(00)90102-6. [DOI] [PubMed] [Google Scholar]
- 132.Onodera H, Mihm MC, Jr, Yoshida A, Akasaka T. Drug-induced linear IgA bullous dermatosis. J Dermatol. 2005;32:759–764. doi: 10.1111/j.1346-8138.2005.tb00839.x. [DOI] [PubMed] [Google Scholar]
- 133.Friedman IS, Rudikoff D, Phelps RG, Sapadin AN. Captopril-triggered linear IgA bullous dermatosis. Int J Dermatol. 1998;37:608–612. doi: 10.1046/j.1365-4362.1998.00544.x. [DOI] [PubMed] [Google Scholar]
- 134.Kuechle MK, Stegemeir E, Maynard B, Gibson LE, Leiferman KM, Peters MS. Drug-induced linear IgA bullous dermatosis: Report of six cases and review of the literature. J Am Acad Dermatol. 1994;30:187–192. doi: 10.1016/s0190-9622(94)70015-x. [DOI] [PubMed] [Google Scholar]
- 135.Primka EJ, 3rd, Liranzo MO, Bergfeld WF, Dijkstra JW. Amiodarone-induced linear IgA disease. J Am Acad Dermatol. 1994;31:809–811. doi: 10.1016/s0190-9622(09)80051-0. [DOI] [PubMed] [Google Scholar]
- 136.Acostamadiedo JM, Perniciaro C, Rogers RS., 3rd Phenytoin-induced linear IgA bullous disease. J Am Acad Dermatol. 1998;38:352–356. doi: 10.1016/s0190-9622(98)70582-1. [DOI] [PubMed] [Google Scholar]
- 137.Kelly SE, Frith PA, Millard PR, Wojnarowska F, Black MM. A clinicopathological study of mucosal involvement in linear IgA disease. Br J Dermatol. 1988;119:161–170. doi: 10.1111/j.1365-2133.1988.tb03197.x. [DOI] [PubMed] [Google Scholar]
- 138.Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults. A comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol. 1988;19:792–805. doi: 10.1016/s0190-9622(88)70236-4. [DOI] [PubMed] [Google Scholar]
- 139.Darling TN, Cardenas AA, Beard JS, Sau P, Yee CL, Zone JJ, et al. A child with antibodies targeting both linear IgA bullous dermatosis and bullous pemphigoid antigens. Arch Dermatol. 1995;131:1438–1442. [PubMed] [Google Scholar]
- 140.Zone JJ, Pazderka Smith E, Powell D, Taylor TB, Smith JB, Meyer LJ. Antigenic specificity of antibodies from patients with linear basement membrane deposition of IgA. Dermatology. 1994;189(Suppl 1):64–66. doi: 10.1159/000246933. [DOI] [PubMed] [Google Scholar]
- 141.Nousari HC, Kimyai-Asadi A, Caeiro JP, Anhalt GJ. Clinical, demographic, and immunohistologic features of vancomycin-induced linear IgA bullous disease of the skin. Report of 2 cases and review of the literature. Medicine (Baltimore) 1999;78:1–8. doi: 10.1097/00005792-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 142.Pulimood S, Ajithkumar K, Jacob M, George S, Chandi SM. Linear IgA bullous dermatosis of childhood: Treatment with dapsone and co-trimoxazole. Clin Exp Dermatol. 1997;22:90–91. [PubMed] [Google Scholar]
- 143.Ang P, Tay YK. Treatment of linear IgA bullous dermatosis of childhood with colchicine. Pediatr Dermatol. 1999;16:50–52. doi: 10.1046/j.1525-1470.1999.99015.x. [DOI] [PubMed] [Google Scholar]
- 144.Segura S, Iranzo P, Martinez-de Pablo I, Mascaró JM, Jr, Alsina M, Herrero J, et al. High-dose intravenous immunoglobulins for the treatment of autoimmune mucocutaneous blistering diseases: Evaluation of its use in 19 cases. J Am Acad Dermatol. 2007;56:960–967. doi: 10.1016/j.jaad.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 145.Letko E, Bhol K, Foster CS, Ahmed AR. Linear IgA bullous disease limited to the eye: A diagnostic dilemma: Response to intravenous immunoglobulin therapy. Ophthalmology. 2000;107:1524–1528. doi: 10.1016/s0161-6420(00)00245-1. [DOI] [PubMed] [Google Scholar]
- 146.Kroiss MM, Vogt T, Landthaler M, Stolz W. High-dose intravenous immune globulin is also effective in linear IgA disease. Br J Dermatol. 2000;142:582. doi: 10.1046/j.1365-2133.2000.03395.x. [DOI] [PubMed] [Google Scholar]
- 147.Schattenkirchner S, Eming S, Hunzelmann N, Krieg T, Smola H. Treatment of epidermolysis bullosa acquisita with mycophenolate mofetil and autologous keratinocyte grafting. Br J Dermatol. 1999;141:932–933. doi: 10.1046/j.1365-2133.1999.03176.x. [DOI] [PubMed] [Google Scholar]
- 148.Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397–399. doi: 10.1007/s10227-001-0008-y. [DOI] [PubMed] [Google Scholar]
- 149.Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. 2007;178:6514–6521. doi: 10.4049/jimmunol.178.10.6514. [DOI] [PubMed] [Google Scholar]
- 150.Liu Y, Shimizu H, Hashimoto T. Immunofluorescence studies using skin sections of recessive dystrophic epidermolysis bullosa patients indicated that the antigen of anti-p200 pemphigoid is not a fragment of type VII collagen. J Dermatol Sci. 2003;32:125–129. doi: 10.1016/s0923-1811(03)00092-6. [DOI] [PubMed] [Google Scholar]
- 151.Cox NH, Bearn MA, Herold J, Ainsworth G, Liu C. Blindness due to the IgA variant of epidermolysis bullosa acquisita, and treatment with osteo-odonto-keratoprosthesis. Br J Dermatol. 2007;156:775–777. doi: 10.1111/j.1365-2133.2006.07739.x. [DOI] [PubMed] [Google Scholar]
- 152.Yancey KB. The pathophysiology of autoimmune blistering diseases. J Clin Invest. 2005;115:825–828. doi: 10.1172/JCI24855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen M, Doostan A, Bandyopadhyay P, Remington J, Wang X, Hou Y, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. Am J Pathol. 2007;170:2009–2018. doi: 10.2353/ajpath.2007.061212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Trigo-Guzmán FX, Conti A, Aoki V, Maruta CW, Santi CG, Resende Silva CM, et al. Epidermolysis bullosa acquisita in childhood. J Dermatol. 2003;30:226–229. doi: 10.1111/j.1346-8138.2003.tb00376.x. [DOI] [PubMed] [Google Scholar]
- 155.Kasperkiewicz M, Zillikens D. Rituximab (anti-CD20) for the treatment of autoimmune bullous diseases. Der Hautarzt. 2007;58:115–116. doi: 10.1007/s00105-007-1284-2. 118–121. [DOI] [PubMed] [Google Scholar]
- 156.Letko E, Bhol K, Anzaar F, Perez VL, Ahmed AR, Foster CS. Chronic cicatrizing conjunctivitis in a patient with epidermolysis bullosa acquisita. Arch Ophthalmol. 2006;124:1615–1618. doi: 10.1001/archopht.124.11.1615. [DOI] [PubMed] [Google Scholar]
- 157.Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol. 2004;22:129–138. doi: 10.1016/j.clindermatol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 158.McCuin JB, Hanlon T, Mutasim DF. Autoimmune bullous diseases: Diagnosis and management. Dermatol Nurs. 2006;18:20–25. [PubMed] [Google Scholar]
- 159.Das JK, Sengupta S, Gangopadhyay AK. Epidermolysis bullosa acquisita. Indian J Dermatol Venereol Leprol. 2006;72:86. doi: 10.4103/0378-6323.19736. [DOI] [PubMed] [Google Scholar]
- 160.Sadler E, Schafleitner B, Lanschuetzer C, Laimer M, Pohla-Gubo G, Hametner R, et al. Treatment-resistant classical epidermolysis bullosa acquisita responding to rituximab. Br J Dermatol. 2007;157:417–419. doi: 10.1111/j.1365-2133.2007.08048.x. [DOI] [PubMed] [Google Scholar]
- 161.Hakki SS, Celenligil-Nazliel H, Karaduman A, Usubütün A, Ertoy D, Ayhan A, et al. Epidermolysis bullosa acquisita: Clinical manifestations, microscopic findings, and surgical periodontal therapy. A case report. J Periodontol. 2001;72:550–558. doi: 10.1902/jop.2001.72.4.550. [DOI] [PubMed] [Google Scholar]
- 162.Sitaru C. Experimental models of epidermolysis bullosa acquisita. Exp Dermatol. 2007;16:520–531. doi: 10.1111/j.1600-0625.2007.00564.x. [DOI] [PubMed] [Google Scholar]
- 163.Ishii N, Yoshida M, Hisamatsu Y, Ishida-Yamamoto A, Nakane H, Iizuka H, et al. Epidermolysis bullosa acquisita sera react with distinct epitopes on the NC1 and NC2 domains of type VII collagen: Study using immunoblotting of domain-specific recombinant proteins and postembedding immunoelectron microscopy. Br J Dermatol. 2004;150:843–851. doi: 10.1111/j.1365-2133.2004.05933.x. [DOI] [PubMed] [Google Scholar]
- 164.Haufs MG, Haneke E. Epidermolysis bullosa acquisita treated with basiliximab, an interleukin-2 receptor antibody. Acta Derm Venereol. 2001;81:72. doi: 10.1080/000155501750208353. [DOI] [PubMed] [Google Scholar]
- 165.Hallel-Halevy D, Nadelman C, Chen M, Woodley DT. Epidermolysis bullosa acquisita: Update and review. Clin Dermatol. 2001;19:712–718. doi: 10.1016/s0738-081x(00)00186-3. [DOI] [PubMed] [Google Scholar]
- 166.Stein JA, Mikkilineni R. Epidermolysis bullosa acquisita. Dermatol Online J. 2007;13:15. [PubMed] [Google Scholar]
- 167.Suchniak JM, Diaz LA, Lin MS, Fairley JA. IgM-mediated epidermolysis bullosa acquisita. Arch Dermatol. 2002;138:1385–1386. doi: 10.1001/archderm.138.10.1385. [DOI] [PubMed] [Google Scholar]