

Abstract
Despite much attention to the use of biomarkers for predicting Alzheimer’s disease, little information is available at the individual level. We used the population-based Mayo Clinic Study of Aging to estimate absolute risk of cognitive impairment by biomarker group. Risk increased with age and any biomarker abnormality. For example, a 75-year-old with abnormal amyloid and cortical thinning biomarkers has ≈20% chance of cognitive impairment by age 80 whereas with normal biomarkers the chance is <10%. Persons with only one abnormal biomarker had similar intermediate risks.
Keywords: Mild cognitive impairment, amyloid beta, neurodegeneration
Introduction
There is a great deal of interest in biomarker based staging of preclinical Alzheimer’s disease (AD), in part due to ongoing development of therapies for early intervention1, 2. In the National Institute on Aging-Alzheimer’s Association committees in 2011, two classes of AD biomarkers were identified: measures of amyloid (A) by CSF or amyloid PET and measures of neuronal injury (N) by CSF, FDG PET, or MRI3–5. When cognitively unimpaired (CU) individuals are categorized based on having normal (−) or abnormal (+) levels of A and N, the A+N+ group consistently has an elevated risk of progression to subsequent cognitive impairment6, 7. Whether A−N+, also termed suspected non-Alzheimer pathophysiology (SNAP)8, is a risk factor for cognitive impairment is an important open question. Further, little is known about the implications of these biomarkers in terms of absolute risk of cognitive impairment for the elderly patient in clinical practice. Data most relevant to clinical practice should come from community-based studies, rather than highly selected referral samples, and should provide predictive information at the individual patient level. To address both of these gaps in current knowledge, we evaluated the role of imaging based biomarkers of A and N in predicting progression from CU to MCI or dementia for individual patients in a population-based study of aging and cognition9.
Methods
Study Design and Participants
All participants in this study were enrolled in the Mayo Clinic Study of Aging (MCSA), a longitudinal, population-based study of residents of Olmsted County, Minnesota9, and participated in brain imaging. MCSA participants were evaluated clinically approximately every 15 months. Each evaluation included separate assessments by a study coordinator, physician and a psychometrist9. The final clinical diagnosis is determined by consensus using previously published criteria10. For the purpose of this study individuals were categorized as cognitively unimpaired (CU), having mild cognitive impairment (MCI), or having dementia.
This study was approved by the Mayo Clinic and the Olmsted Medical Center Institutional Review Boards. All participants provided written informed consent at the time of enrollment.
Imaging
Amyloid PET imaging was performed using Pittsburgh Compound-B. Late uptake images were acquired from 40–60 minutes post injection. A cortical composite meta ROI was referenced to uptake in the cerebellar crus to create a standardized uptake value ratio (SUVR) as previously published11. Structural MRI imaging was performed at 3T. Our N measure was a composite AD-characteristic cortical thickness measure averaging entorhinal, inferior temporal, middle temporal, and fusiform gyri thickness estimated using FreeSurfer 5.312. Based on a recent analysis, amyloid PET SUVR values greater than 1.42 (centiloid 19) and cortical thickness values less than 3.5mm were considered abnormal11. These cut-points were used to form four groups: A−N−, A+N−, A−N+, and A+N+.
Statistical Analysis
We investigated progression from CU to MCI or dementia, treating death prior to cognitive impairment as a competing event that precludes MCI or dementia. We defined baseline as a participant’s first MCSA visit with both amyloid PET and MRI and limited our analysis to those who were CU and aged ≥70 at this baseline and had some follow-up. To estimate rates of progression to MCI or dementia by biomarker group, we used Poisson regression including age, sex, education level, and A/N group as main effects. The absolute risk of observed cognitive impairment over time was computed using the competing risks approach described in Putter 13, i.e. accounting for the fact that some subjects will never progress to MCI/dementia because they die before doing so. We note that since MCSA visits occurred approximately 15 months apart while death could occur many years after the final clinical visit, only deaths that occurred within 15 months of a participant’s last visit were counted as events.
Results
Of 763 CU participants, 26% were A−N−, 15% were A+N−, 30% were A−N+, and 28% were A+N+ with demographic comparisons shown in Table 1. Men were more likely to be N+ (p=0.002) and both A+ and N+ were associated with older age (p<0.001). Over a median follow-up of four years, 159 (22%) individuals progressed to MCI (n=152) or dementia (n=7). About a quarter of our sample (24%) had one 15-month follow-up visit, 13% had two, and 63% had 3 or more. In our sample, progression rates expressed as events per 100 person years (95% CI) were 2.4 (1.8–3.2) at age 75 and 6.5 (5.5–7.6) at age 85. Averaging over men and women and assuming some college education, progression rates (95% CIs) at age 75 were 3.9 (2.7–5.7) among A+N+, 1.1 (0.7–1.9) among A−N−, 2.3 (1.4–3.7) among A+N−, and 2.3 (1.5–3.4) among A−N+. By age 85, the rates increased to 8.9 (6.8–11.5) among A+N+, 2.6 (1.5–4.3) among A−N−, 5.2 (3.3–8.1) among A+N− and 5.2 (3.3–7.0) among A−N+. Treating the A−N− group as the reference, the relative rate (95% CI) for A+N+ was 3.5 (2.1– 6.2), for A+N− was 2.0 (1.1–3.9), and for A−N+ was 2.0 (1.2–3.6). Within a biomarker group, rates are an estimated 20% higher in men (RR 1.2, 95% CI 0.9–1.7) and 30% higher in those with only a high school education (RR 1.3, 95% CI 1.0–1.9).
Table 1.
Baseline characteristics of the study participants by biomarker group
| All (N=763) |
A–N– (N=202) |
A+N– (N=118) |
A–N+ (N=229) |
A+N+ (N=214) |
|
|---|---|---|---|---|---|
| Age, y | |||||
| Median (IQR) | 77 (74, 82) | 75 (72, 78) | 76 (72, 81) | 78 (75, 84) | 80 (77, 84) |
| Range | 70 to 95 | 70 to 92 | 70 to 93 | 70 to 93 | 70 to 95 |
| Sex, n (%) | |||||
| Male | 414 (54%) | 91 (45%) | 61 (52%) | 136 (59%) | 126 (59%) |
| Female | 349 (46%) | 111 (55%) | 57 (48%) | 93 (41%) | 88 (41%) |
| Education, median (IQR), y | 14 (12, 16) | 14 (12, 17) | 14 (12, 17) | 14 (12, 17) | 14 (12, 16) |
| Education level, n (%) | |||||
| High school or less | 244 (32%) | 68 (34%) | 36 (31%) | 77 (34%) | 63 (29%) |
| Some college | 519 (68%) | 134 (66%) | 82 (69%) | 152 (66%) | 151 (71%) |
| APOE status, n (%) | |||||
| Carrier | 202 (26%) | 37 (18%) | 57 (48%) | 30 (13%) | 78 (36%) |
| Non-carrier | 561 (74%) | 165 (82%) | 61 (52%) | 199 (87%) | 136 (64%) |
| Cognitive z scores, median (IQR)* | |||||
| Global | 0.0 (−0.7, 0.7) | 0.3 (−0.4, 1.0) | 0.1 (−0.6, 0.8) | 0.0 (−0.6, 0.7) | −0.3 (−1.0, 0.4) |
| Memory | 0.0 (−0.8, 0.7) | 0.1 (−0.5, 0.9) | 0.1 (−0.5, 0.8) | 0.0 (−0.7, 0.7) | −0.3 (−1.1, 0.4) |
| Attention | 0.1 (−0.6, 0.7) | 0.4 (−0.2, 1.0) | 0.2 (−0.5, 0.7) | 0.1 (−0.7, 0.6) | −0.3 (−0.9, 0.5) |
| Language | 0.1 (−0.6, 0.7) | 0.3 (−0.4, 0.9) | 0.1 (−0.5, 0.8) | 0.1 (−0.5, 0.7) | −0.2 (−0.8, 0.4) |
| Visuospatial | 0.1 (−0.6, 0.7) | 0.2 (−0.6, 0.9) | 0.0 (−0.7, 0.7) | 0.2 (−0.5, 0.7) | −0.1 (−0.8, 0.5) |
| Follow-up, y† | |||||
| Median (IQR) | 4.4 (2.4, 6.5) | 5.1 (2.7, 6.6) | 5.1 (2.6, 6.7) | 5.1 (2.5, 6.5) | 3.8 (1.5, 6.2) |
| Range | 0.3 to 11.4 | 0.6 to 9.0 | 1.0 to 10.3 | 1.1 to 11.4 | 0.3 to 11.0 |
| No. progressed to MCI or dementia | 159 | 17 | 22 | 49 | 71 |
| No. died before MCI or dementia | 39 | 5 | 2 | 10 | 22 |
Cognitive domain z scores are averages 3 tests for memory, 2 tests for attention, 2 tests for language, and two tests for visuospatial normed to subjects in the study. The global domain score is an average of four domains normed to subjects in the study.
Follow-up calculated as time from baseline to MCI or death within 15 months of last visit. Some individuals died before a return visit.
Figure 1 shows the estimated probability of cognitive impairment accounting for the competing risk of death for a 75 year old and an 85 year old with some college education averaged among men and women to emphasize risk differences by biomarker group. The data are plotted both with age and time in study on the abscissa. Table 2 shows the 5- and 10-year risks of progressing to MCI/dementia by age and sex for two levels of education. Based on the estimated risks, a CU 75 year old A−N− woman with some college education has an estimated 6% chance of becoming cognitively impaired within 5 years. This increases to 19% assuming A+N+. Participants who were either A+N− or A−N+ had intermediate risks of progression. Table 2 also illustrates that risk clearly increases with age, is somewhat higher in men, and is somewhat lower for those with some college education.
Figure 1.
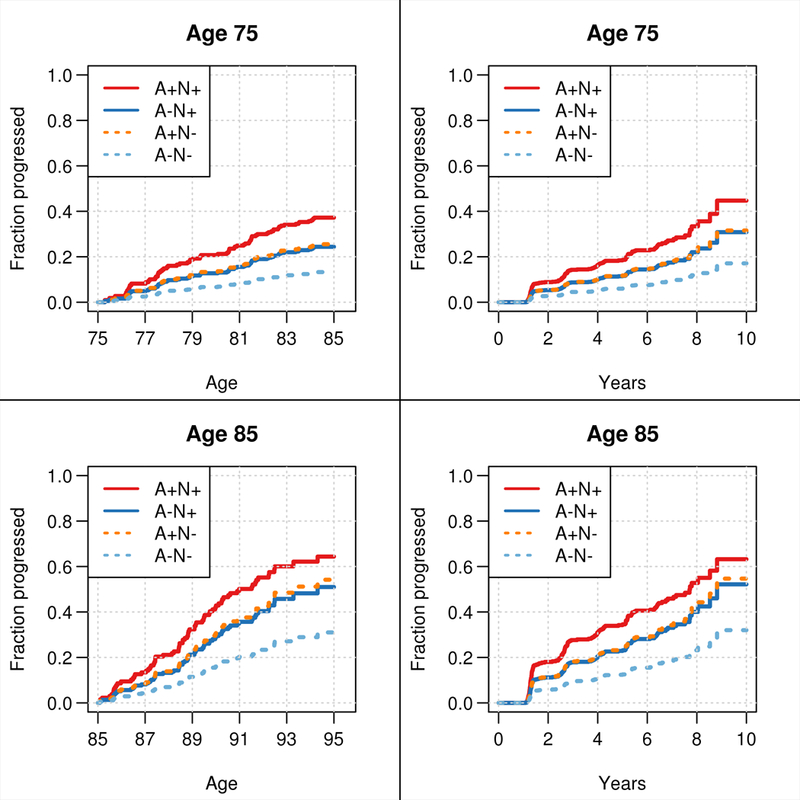
Probability of MCI or dementia over time accounting for the competing risk of death. Cause specific survival models were fit using age as the time scale (left column) and using years from imaging as the time scale (right column). Probabilities depend on age and ages 75 and 85 were chosen to illustrate this dependence. For simplification of presentation, estimates are shown averaged over sex and for an individual with some college education (i.e., ≥13 years).
Table 2.
Probability or risk of progression to MCI or dementia stratified by biomarker group over 5- and 10-year time horizons after adjusting for the competing risk of death. Estimates are shown separately for men and women and by education level.
| Time | A−N− | A+N− | A−N+ | A+N+ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Age | horizon (y) | Education* | Female | Male | Female | Male | Female | Male | Female | Male |
| 70 | 5 | High school | 5% | 6% | 10% | 12% | 10% | 12% | 16% | 19% |
| Some college | 4% | 5% | 8% | 9% | 7% | 9% | 12% | 15% | ||
| 10 | High school | 12% | 15% | 24% | 28% | 23% | 27% | 36% | 41% | |
| Some college | 10% | 12% | 19% | 22% | 18% | 21% | 29% | 33% | ||
| 75 | 5 | High school | 8% | 9% | 16% | 19% | 15% | 18% | 24% | 28% |
| Some college | 6% | 7% | 12% | 15% | 12% | 14% | 19% | 23% | ||
| 10 | High school | 16% | 18% | 29% | 34% | 28% | 33% | 43% | 48% | |
| Some college | 12% | 14% | 23% | 28% | 22% | 26% | 35% | 40% | ||
| 80 | 5 | High school | 8% | 10% | 17% | 20% | 16% | 19% | 25% | 29% |
| Some college | 7% | 8% | 13% | 16% | 12% | 15% | 20% | 23% | ||
| 10 | High school | 25% | 29% | 45% | 51% | 43% | 48% | 57% | 61% | |
| Some college | 20% | 23% | 37% | 42% | 35% | 40% | 48% | 52% | ||
| 85 | 5 | High school | 19% | 22% | 35% | 40% | 33% | 38% | 48% | 53% |
| Some college | 15% | 17% | 28% | 33% | 27% | 31% | 40% | 44% | ||
| 10 | High school | 36% | 41% | 61% | 67% | 57% | 63% | 71% | 74% | |
| Some college | 29% | 33% | 51% | 57% | 48% | 54% | 63% | 66% | ||
High school education is defined as 12 or fewer years of education while some college is defined as 13 or more
Discussion
These results extend our work examining effects of amyloid positivity on cognition14 to document the dual importance of amyloid positivity and neurodegeneration as substantial risk factors for cognitive impairment. Having both normal amyloid and normal neurodegeneration was protective. In contrast, individuals with both abnormal amyloid and abnormal neurodegeneration had much higher rates of progression to MCI or dementia over an average of four years of follow-up. Interestingly, in the general community setting, the presence of either abnormal amyloid or neurodegeneration constituted a similar risk for progression.
With the availability of imaging and/or CSF biomarker information in clinical practice it is uncertain how to convey the data to patients. Our results provide easily interpretable estimates of the chance of becoming cognitively impaired in 5- and 10-year windows stratified by biomarker group. Further, estimates are reported by three important risk factors: sex, education level, and age. Of these, risk varies to the greatest degree by age but male sex and lower education are contributing factors that remain important in an era of biomarkers15. We have previously shown that these risk factors also influence MCSA participation16 and therefore it is important account for them in regression analyses.
Our biomarker findings should be interpreted in the context of previous reports while recognizing that different definitions of amyloidosis and abnormal neurodegeneration using imaging based versus CSF based biomarkers make direct comparisons difficult. Using largely the same definition of A+ but an N+ based on abnormal hippocampal atrophy or abnormal FDG PET, an early report from the MCSA examining progression by 15 months found biomarker risk patterns that were quite similar to those reported here17. On the other hand, two somewhat comparable studies offer little evidence that A−N+ is a risk factor for progression. Vos and colleagues used CSF biomarkers7. Their A+N+ group had the highest cumulative incidence of progression followed by A+N−. However, the risk in their A−N+ group was comparable to that of the A−N− group. This disparate prognosis for SNAP individuals compared to our findings invites careful consideration of the processes underlying CSF-based or MRI-based definitions of N+. More recently, Burnham and colleagues reported that in the Australian Imaging, Biomarker and Lifestyle (AIBL) study, those with A+N+, compared to A−N−, had more than a five-fold increased hazard of progression to MCI; those with A+N− had a more than a two-fold increased hazard; and those with A−N+ had only a moderately elevated hazard (HR=1.3) 6. With brain atrophy on MRI a well-established risk factor for cognitive impairment, and given our current findings, we think it likely that neurodegeneration in the absence of amyloidosis is an important risk factor for MCI. Further, recent reports suggest amyloid and neurodegeneration are important complementary risk factors for dementia18, 19.
There is likely risk associated with AD biomarker levels that are just below those deemed abnormal. Further, the biomarker groups examined here do not address important neuropathological features such as cerebrovascular pathology, tau-based neurofibrillary tangles, TDP-43, and alpha synuclein. For a given individual, there are likely many factors contributing to cognitive function in aging 20. Still, our absolute risk estimates may be useful to clinicians in counseling patients with respect to the role of AD biomarkers in aging or those planning clinical trials. And because these data were from a population-based sample they are less subject to biases that may be encountered in referral settings.
Acknowledgements
This research was supported by the National Institutes of Health/National Institute on Aging (U01 AG006786, P50 AG016574, R01 AG011378, R01 AG041851) and made possible by the Rochester Epidemiology Project (R01 AG034676). We thank the Alzheimer’s Association, the GHR Foundation, the Elsie and Marvin Dekelboum Family Foundation, and the Mayo Foundation for Medical Education and Research.
Footnotes
Potential conflicts of interest
Dr. Petersen is a consultant for GE Healthcare which manufactured the MRI and PET scanners used in this study. Dr. Lowe receives research grants from GE Health Care which manufactured the MRI and PET scanners used in this study.
References
- 1.Thal LJ, Kantarci K, Reiman EM et al. The role of biomarkers in clinical trials for Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cummings J, Zhong K. Biomarker-driven therapeutic management of Alzheimer’s disease: establishing the foundations. Clin Pharmacol Ther. 2014;95:67–77 [DOI] [PubMed] [Google Scholar]
- 3.McKhann GM, Knopman DS, Chertkow H et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2011;7:263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert MS, DeKosky ST, Dickson D et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2011;7:270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, Aisen PS, Beckett LA et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnham SC, Bourgeat P, Dore V et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. Lancet Neurol. 2016;15:1044–1053 [DOI] [PubMed] [Google Scholar]
- 7.Vos SJ, Xiong C, Visser PJ et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jack CR Jr., Knopman DS, Chetelat G et al. Suspected non-Alzheimer disease pathophysiology--concept and controversy. Nat Rev Neurol. 2016;12:117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts RO, Geda YE, Knopman DS et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194 [DOI] [PubMed] [Google Scholar]
- 11.Jack CR Jr., Wiste HJ, Weigand SD et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017;13:205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Wiste HJ, Weigand SD et al. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain. 2015;138:3747–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Putter H, Fiocco M, Geskus RB. Tutorial in biostatistics: competing risks and multi-state models. Stat Med. 2007;26:2389–2430 [DOI] [PubMed] [Google Scholar]
- 14.Roberts RO, Aakre JA, Kremers WK et al. Prevalence and Outcomes of Amyloid Positivity Among Persons Without Dementia in a Longitudinal, Population-Based Setting. JAMA Neurol. 2018;75:970–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts RO, Geda YE, Knopman DS et al. The incidence of MCI differs by subtype and is higher in men: the Mayo Clinic Study of Aging. Neurology. 2012;78:342–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts RO, Knopman DS, Syrjanen JA et al. Weighting and standardization of frequencies to determine prevalence of AD imaging biomarkers. Neurology. 2017;89:2039–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knopman DS, Jack CR Jr., Wiste HJ et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012;78:1576–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez OL, Becker JT, Chang Y et al. Amyloid deposition and brain structure as long-term predictors of MCI, dementia, and mortality. Neurology. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolk DA, Sadowsky C, Safirstein B, et al. Use of flutemetamol f 18–labeled positron emission tomography and other biomarkers to assess risk of clinical progression in patients with amnestic mild cognitive impairment. JAMA Neurol. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petersen RC. How early can we diagnose Alzheimer disease (and is it sufficient)? The 2017 Wartenberg lecture. Neurology. 2018;91:395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]