

Summary
Neuronal activity-dependent transcription is tuned to ensure precise gene induction during periods of heightened synaptic activity, allowing for appropriate responses of activated neurons within neural circuits. The consequences of aberrant induction of activity-dependent genes on neuronal physiology are not yet clear. Here, we demonstrate that in the absence of synaptic excitation, the bHLH-PAS family transcription factor ARNT2 recruits the NCoR2 co-repressor complex to suppress neuronal activity-dependent regulatory elements and maintain low basal levels of inducible genes. This restricts inhibition of excitatory neurons, maintaining them in a state that is receptive to future sensory stimuli. By contrast, in response to heightened neuronal activity, ARNT2 recruits the neuronal-specific bHLH-PAS factor NPAS4 to activity-dependent regulatory elements to induce transcription and thereby increase somatic inhibitory input. Thus, the interplay of bHLH-PAS complexes at activity-dependent regulatory elements maintains temporal control of activity-dependent gene expression and scales somatic inhibition with circuit activity.
Graphcial Abstract
eTOC Blurb
Sharma et al. report context-specific ARNT2 transcription factor complexes that restrict activity-dependent transcription, and subsequent recruitment of somatic inhibition, to periods of increased membrane depolarization in pyramidal neurons. These mechanisms ensure that somatic inhibition scales appropriately with circuit activity.
Introduction
Sensory experience drives the development and maturation of the nervous system in part through the activation of an intricate program of gene transcription (Leslie and Nedivi, 2011; Yap and Greenberg, 2018). The sensory-dependent gene program in neurons is induced when action potentials promote sufficient calcium influx at the cell soma to activate signaling networks that transiently induce transcription at the loci of immediately early genes (IEGs), including those that encode FOS and JUN family members (AP-1) and the neuronal PAS domain protein 4 (NPAS4) (Greenberg and Ziff, 1984; Greenberg et al., 1986; Lin et al., 2008; Morgan and Curran, 1986). The AP-1 family and NPAS4 are transcription factors (TFs) that activate programs of gene transcription that are remarkably cell-type-specific (Mardinly et al., 2016; Spiegel et al., 2014; Vierbuchen et al., 2017), and thus tailored to the function of each neuronal subtype within the brain. Notably, mutations in specific components of this activity-regulated signaling network can contribute to cognitive disorders, including intellectual disability, autism spectrum disorders, and schizophrenia (De Rubeis et al., 2014; Ebert and Greenberg, 2013). These observations underscore the importance of the activity-dependent gene program for neuronal function and circuit plasticity.
An emerging view is that the spatial and temporal precision of gene expression in neurons is coordinated by the binding of select TFs to promoters and enhancers in the genome (Kim et al., 2010; Long et al., 2016; Nord et al., 2015; Tyssowski et al., 2018). Enhancers are DNA sequences that can act over a distance of several hundred kilobases to potentiate gene expression by delivering regulatory transcriptional complexes to gene promoters adjacent to the site of transcriptional initiation (Gray et al., 2015). Across a variety of cell types and species, a balance of activating and repressive factors bind these regulatory elements to ensure that gene transcription is spatiotemporally controlled (Koenecke et al., 2017; Nord et al., 2013; Pattabiraman et al., 2014). Additionally, it is now appreciated that in neurons, membrane depolarization leads to the activation of thousands of regulatory elements across the genome, a process driven by a series of regulated molecular events, including the remodeling of nucleosomes to facilitate the binding of sequence-specific transcription factors and the modification of histone octamers (e.g. acetylation, phosphorylation) to relax chromatin structure and establish platforms for the recruitment of additional transcriptional machinery (Gray et al., 2015; Heinz et al., 2015). While the mechanisms driving inducible gene transcription in neurons have been extensively studied (Joo et al., 2016; Kim et al., 2010; Malik et al., 2014), comparatively little is understood about whether and how repression of activity-dependent regulatory elements controls activity-dependent gene expression programs.
Given the narrow temporal window during which experience drives activity-dependent gene transcription to produce synaptic changes, neurons have likely evolved multiple strategies to restrict inducible transcription prior to sensory stimulation. Long-term silencing of regulatory elements and genes is thought to be mediated in part by the methylation of DNA via the coordinated action of the DNA methyltransferase DNMT3A and methyl-binding protein MeCP2 during embryonic and postnatal development (Feng et al., 2010; Kinde et al., 2015; Lister et al., 2013; Stroud et al., 2017). However, the transient transcriptional activation of inducible genes is thought to circumvent long-term epigenetic silencing, suggesting alternative mechanisms that might reversibly suppress these sites specifically during periods of low stimulation (Stroud et al., 2017). Previous studies that focused exclusively on the promoters of individual IEGs reported that specific protein complexes bind in the absence of nuclear calcium influx to maintain low levels of gene expression, possibly through recruitment of histone deacetylases (Carrion et al., 1999; Mellstrom et al., 2014; Panteleeva et al., 2004; Qiu and Ghosh, 2008). For example, in silenced neurons, the histone deacetylase HDAC4 transiently localizes to the nucleus, where it represses genes encoding synaptic proteins in concert with the MEF2 family of transcription factors (Chawla et al., 2003; Sando et al., 2012). Additional studies suggest that restriction of inducible gene expression to the appropriate context may occur primarily at the level of chromatin accessibility of enhancer elements (Ostuni et al., 2013). In dentate granule neurons (Su et al., 2017), fibroblasts (Vierbuchen et al., 2017), and immune cells (Ostuni et al., 2013), stimulus-responsive enhancers are occluded by histone octamers and thus nucleosomes limit binding of TF complexes in the absence of stimulation However, the full repertoire of repressive mechanisms that restrict activity-dependent regulatory elements across the neuronal genome is lacking and the functional consequences of de-repressing activity-dependent transcriptional programs on neural circuits remain unclear.
To address these gaps in knowledge, we first sought to identify and characterize the elements responsible for specifically regulating neuronal activity-dependent gene expression. We employed NPAS4 as a molecular marker of such loci, as NPAS4 is selectively induced in neurons specifically in response to membrane depolarization-induced calcium signaling (Lin et al., 2008; Ramamoorthi et al., 2011). Once activated, NPAS4 induces neuronal subtype-specific gene programs and cellular responses (Bloodgood et al., 2013; Spiegel et al., 2014). For example, in hippocampal CA1 pyramidal neurons, NPAS4 activates a program of gene expression that recruits inhibitory synapses to the pyramidal neuron soma, while in somatostatin-positive inhibitory neurons, NPAS4 induces a gene program that enhances the number of excitatory synapses that form on NPAS4-expressing somatostatin neurons. Defects in these NPAS4-dependent effects on synaptic architecture have been hypothesized to underlie developmental and behavioral deficits in mice harboring a deletion of the Npas4 gene, including defective ocular dominance column plasticity, altered social interaction, and impaired contextual fear and reward-related learning (Heslin and Coutellier, 2017; Mardinly et al., 2016; Ramamoorthi et al., 2011; Taniguchi et al., 2017). Despite the known effects of NPAS4 on synaptic physiology and behavior, it is not clear whether and how the NPAS4-bound subset of stimulus-responsive regulatory elements is kept off prior to sensory stimulation so that activity-dependent gene transcription occurs in a precise and temporally controlled manner.
In the present study, we characterized the molecular mechanisms that restrict the activity of NPAS4-bound regulatory elements prior to stimulation. We first generated a comprehensive atlas of NPAS4 binding sites in stimulated neurons to identify the enhancers and promoters that could then be examined prior to stimulation. We identified the bHLH-PAS transcription factor ARNT2 as a heterodimeric partner of NPAS4 that is expressed prior to neuronal activity. Unlike NPAS4, we find that ARNT2 binds to these same elements prior to stimulation, and perturbation of ARNT2 in unstimulated neurons results in the de-repression of activity-dependent transcription and an enhancement of somatic inhibition. ARNT2 functions in silent neurons by recruiting the NCoR2 repressor complex components (NCoR2, TBL1, HDAC3) to activity-dependent regulatory elements to suppress activity-dependent genes and restrict inhibitory synaptic plasticity. In response to neuronal activity, NPAS4 is induced and forms a complex with ARNT2 to induce gene expression, thus shifting the balance between repression and activation. Surprisingly, NCoR2 is also inducibly recruited to NPAS4-bound enhancers in stimulated neurons and in this context appears to augment the expression of activity-dependent genes. Thus, through the recruitment of distinct cofactors in the basal and stimulated states, ARNT2 exerts opposing effects on activity-regulated transcription to maintain an appropriate level of somatic inhibition as CA1 pyramidal neurons respond to changing levels of sensory input.
Results
Identification of NPAS4-bound inducible regulatory elements
We hypothesized that identifying the set of regulatory elements bound by NPAS4 would provide insight into mechanisms of activity-dependent transcription unique to neurons. We performed ChIP-seq for NPAS4 in primary cultured cortical neurons exposed to elevated levels of KCl (55 mM), which depolarizes the neuronal membrane and leads to robust induction of Npas4 mRNA and protein (Lin et al., 2008). We identified 11,199 NPAS4 binding sites across the genome, of which 76% were promoter-distal (Figures 1A, S1A). By contrast, in untreated neurons, where NPAS4 is not expressed, we observed minimal binding of NPAS4 across the genome (Figures 1A, S1B). We confirmed the specificity of the NPAS4 ChIP by demonstrating significantly reduced binding in neurons transduced with Npas4 shRNA (Figure S1B).
Figure 1. Identification of NPAS4-bound inducible enhancers.

A. ChIP-seq signal for NPAS4 genomic binding sites in unstimulated (left) and stimulated (right) cortical neuron cultures sorted by descending binding strength. Each NPAS4 binding site is represented as a single horizontal line (blue) centered at the peak summit with flanking 2kb. Color denotes ChIP-seq signal intensity (displayed in log2(counts per million + 1)).
B. Upper panels: Log2 fold change in ATAC-seq signal comparing 2hr membrane depolarization by addition of 55 mM KCl (stimulated) to samples not treated with KCl (unstimulated). Data plotted as a function of log2 normalized ATAC-seq signal (counts per million) in unstimulated cultured cortical neurons at enhancers (left) and promoters (right). Dark gray points denote activity-regulated sites (>1.5 fold increase in ATAC signal upon depolarization, FDR < .05). Lower panels: Box-whisker plots showing median and distribution of log2 normalized ATAC-seq signal in the unstimulated condition at non-NPAS4 bound regulatory elements (red), NPAS4-bound regulatory elements (blue), and non-regulatory sequences 5kb away from 10,000 randomly chosen ATAC-seq positive regions (yellow). *** p < .001, Wilcoxon signed-rank test.
C. Upper panels: Log2 fold change in H3K27ac-seq signal comparing 2hr membrane depolarization by addition of 55 mM KCl (stimulated) to samples not treated with KCl (unstimulated). Data plotted as a function of log2 normalized H3K27ac-seq signal (counts per million) in unstimulated cultured cortical neurons at enhancers (left) and promoters (right). Dark green points denote activity-regulated sites (>1.5 fold increase in H3K27ac signal upon depolarization, FDR < .05). Lower panels: Box-whisker plots showing median and distribution of log2 normalized H3K27ac-seq signal in the unstimulated condition at non-NPAS4 bound regulatory elements (red), presumptive NPAS4-bound regulatory elements (blue), and non-regulatory sequences 5kb away from 10,000 randomly chosen ATAC-seq positive regions (yellow). *** p < .001, Wilcoxon signed-rank test.
D. H3K27ac log2-fold change between depolarized Npas4fl/fl neurons infected with Cre compared to control GFP plotted as quantiles of NPAS4 binding strength at enhancer regions (1 < 4). − indicates sites not bound by NPAS4. *** indicates p-value <0.001 in a Wilcoxon rank sum test comparing the fold change distribution for a given quantile relative to non-bound sites.
E. H3K27ac log2-fold change between unstimulated neurons overexpressing NPAS4 compared to control GFP plotted as quantiles of NPAS4 binding strength at enhancer regions (1 < 4). − indicates sites not bound by NPAS4. *** indicates p-value <0.001 in a Wilcoxon rank sum test comparing the fold change distribution for a given quantile relative to non-bound sites.
F. UCSC genome browser ChIP-seq tracks displaying NPAS4-dependent effects on H3K27ac at a representative enhancer in cortical cultures stimulated with 55mM KCl. Each ChIP-seq track is labeled for the ChIP-ed factor, time point after membrane depolarization, and treatment.
G. Scatter plot of relative luciferase activity in 6hr KCl-depolarized neurons as a function of NPAS4 binding strength. Relative luciferase activity was calculated as the ratio of (luciferase/renilla in Npas4 shRNA-treated cells)/(luciferase/renilla in control shRNA-treated cells). Each point represents an independent biological replicate for a given luciferase construct (13 luciferase constructs plotted). Shaded lines indicate 95% confidence interval around line of regression. Pearson correlation: Npas4 shRNA#1 R= −.68 p-value = 2.2e-08; Npas4 shRNA#2 R= −.82; p-value = 4.1e-09). Spearman correlation: Npas4 shRNA#1 R= −.0.67 p-value = p=7.1e-08; Npas4 shRNA#2 R= −.74; p-value = 6.6e-07).
H. Heatmap of mean scRNA-seq bHLH-PAS TF expression by cell type in cultured cortical neurons. Columns denote cell type at the specified stimulation condition (unstimulated, 1 hour 55mM KCl, 6 hours 55mM KCl). Expression in units of mean transcripts per thousand (TPT).
I. Mean co-expression of Npas4 and bHLH-PAS transcription factors across Npas4-positive cells of indicated neuronal subtype. Fraction of co-expression indicated in each cell.
To focus on the functional NPAS4 binding sites, we established a list of active regulatory elements in the primary neuron culture system by performing ATAC-seq (assay for transposase-accessible chromatin by sequencing), which measures chromatin accessibility, and ChIP-seq for acetylation of histone 3 lysine 27 (H3K27ac), a chromatin modification whose abundance scales with regulatory element activity (Buenrostro et al., 2015; Creyghton et al., 2010; Rada-Iglesias et al., 2011) (Figure 1B, 1C, Methods). We defined active regulatory elements as regions where ATAC-seq and H3K27ac ChIP-seq signals intersect in at least one of the experimental conditions used in this study and identified 31,138 peaks by these criteria (8,469 TSS-proximal elements (promoters) and 22,669 TSS-distal elements (enhancers)) (Figure S1C). Intersecting these active regulatory elements with our NPAS4 ChIP-seq peaks yields 8,966 NPAS4-bound regulatory elements (2,614 promoters and 6,352 enhancers).
We next asked if NPAS4-bound activity-dependent regulatory elements require NPAS4 for their activation. We derived primary cortical neurons from Npas4fl/fl mice and expressed via lentivirus either GFP as a control or Cre and then stimulated the neurons with KCl to trigger activity-dependent transcription. When NPAS4 is deleted, we observe a statistically significant decrease in H3K27ac ChIP signal at the majority of NPAS4-bound activity-regulated enhancers (Figure 1D) and promoters (Figure S1D). Additionally, lentiviral overexpression of NPAS4 in wildtype, unstimulated cortical neurons resulted in increased H3K27ac at NPAS4-bound sites (Figures 1E, 1F, S1E). To further confirm NPAS4’s ability to activate enhancers and promoters, we cloned DNA sequences with strong and weak NPAS4 binding into luciferase reporters (Table S1) and examined their depolarization-dependent activation in neurons transfected with either a non-targeting shRNA or shRNAs targeting Npas4. Consistent with a role for NPAS4 in enhancer activation, knockdown of NPAS4 significantly reduced inducible reporter activity, particularly at high confidence NPAS4 binding sites (Figures 1G, S1F).
Single cell RNA-seq identifies ARNT2 as a candidate regulator of enhancers in silent neurons
Having identified the NPAS4-bound activity-dependent regulatory elements, we next examined these elements prior to stimulation. We refer to these elements as presumptive NPAS4 binding sites when they are analyzed in unstimulated neurons where NPAS4 is not expressed. We found that in unstimulated neurons, presumptive NPAS4 binding sites have higher levels of chromatin accessibility and H3K27ac acetylation than regulatory regions that do not bind NPAS4 (Figures 1B, 1C). Since we observe accessible chromatin and H3K27ac signal at presumptive NPAS4-bound sites prior to stimulation (Figures 1B, 1C) it seems unlikely that occlusion by histone octamers is the repressive mechanism restricting NPAS4-bound regulatory elements. Rather, we hypothesized that these accessible presumptive NPAS4 binding sites might be bound by an unidentified complex that is removed or inactivated in response to neuronal activity. As NPAS4 is a member of the bHLH-PAS family, whose members form obligate homo- or heterodimers to bind DNA (Ooe et al., 2004), we considered the possibility that in unstimulated neurons another bHLHPAS domain protein might be expressed which binds to the presumptive NPAS4 sites and functions as a repressor. To identify bHLH-PAS proteins that are expressed in neurons prior to stimulation, we performed single-cell RNA-sequencing (scRNA-seq) from primary neuron cultures using the inDrops platform (Klein et al., 2015). In this dataset, as well as previously published scRNA-seq data from the visual cortex, we found ARNT2 to be the most robustly expressed bHLH-PAS transcription factor across all neuronal cell clusters and activity states (Figures 1H, S1G-L) and confirmed that ARNT2 and NPAS4 are co-expressed in individual neurons (Figure 1I).
ARNT2 suppresses activity-dependent transcription in neurons prior to neuronal activity
As ARNT2 is the most highly expressed PAS domain protein in unstimulated neurons, we asked if ARNT2 binds to presumptive NPAS4 regulatory elements in the absence of membrane depolarization. We generated an anti-ARNT2 antibody (Figures S2A, S2B) and performed ChIP-seq in unstimulated cultured cortical neurons. We identified 17,490 ARNT2 binding sites (14,323 that overlap with our set of active elements) which display a similar genome-wide distribution to NPAS4 sites (Figures 2A, S2C). We observed a two-fold reduction in aggregated ARNT2 ChIP-seq signal in neuronal cultures transduced with two independent Arnt2 shRNAs, confirming ChIP-seq specificity (Figures S2D, S2E). We also determined ARNT2 binding in vivo using hippocampal tissue from wildtype mice raised in standard housing, where hippocampal neurons are relatively inactive (Lin et al., 2008) (Figure 2A). Notably, ARNT2 binds at a significant fraction of presumptive NPAS4 regulatory elements in both primary cultures and in adult hippocampus (Figure 2B). Consistent with a role for NPAS4 in regulatory element activation, twice as many NPAS4:ARNT2 co-bound sites display a significant 1.5-fold increase in H3K27ac in response to membrane depolarization compared sites bound by ARNT2 alone. These two classes of sites show no differences in their promoter/enhancer distribution, H3K27ac signal, or ATAC signal (Figure S2F).
Figure 2. ARNT2 represses activity-dependent transcription prior to neuronal activity.

A. ChIP-seq signal for ARNT2 at presumptive NPAS4 sites in unstimulated cortical neuron cultures (left) and CA1 hippocampal tissue derived from standard housed mice (right) sorted by descending binding strength. Each ARNT2 binding site is represented as a single horizontal line (orange) centered at the peak summit with flanking 2kb. Color denotes ChIP-seq signal intensity (displayed in log2(counts per million + 1)). Peaks are split between distal enhancers bound by NPAS4 (n= 6,352) and TSS-proximal promoters bound by NPAS4 (n = 2,614).
B. Venn diagram of overlap between NPAS4 binding sites in stimulated neurons (2 KCl) and ARNT2 binding sites in unstimulated cultured cortical neurons (0 KCl, left) and adult hippocampus (Strd Housing, right). Fisher’s exact test odds ratios (OR) and p-values for set intersection shown.
C. Left panel: KSM representations of top motif enriched when comparing the 5,000 best NPAS4-positive/ARNT2-positive enhancers against the 4,632 best NPAS4-negative/ARNT2-positive sites. Right panel: Reversed analysis showing top motif enriched in 4,632 best ARNT2-only bound sites KMAC output partial area under receiver operating characteristic curve (auc) values shown.
D. Upper panel: –log10(false discovery rate) (FDR) versus log2 fold change in gene expression between Arnt2 shRNA and control shRNA-infected hippocampal hemispheres in mice raised in standard housing conditions with low basal transcriptional activity. ARNT2-regulated genes are defined as fold change≥1.5 and FDR <0.05 when comparing Arnt2 shRNA-injected animals against control shRNA (demarcated by top left and top right sectors). Activity-regulated genes (plotted in orange) are defined as fold change≥1.5 and FDR <0.05 when comparing kainate-injected animals against saline-injected controls (Figure S2H). Lower panel: Box and whisker plots display the distribution of log2 fold changes (Arnt2 shRNA/control shRNA) of all genes (gray) and activity-dependent genes (orange). p = 1.4e-117 in the Wilcoxon rank sum test.
E. Gene expression in log2(RPKM+1) for all activity-regulated genes in control shRNA or Arnt2 shRNA-infected hippocampal hemispheres of standard housed animals. Each gene is connected by a line across its expression values between the two conditions. Orange colored lines indicate genes within 50kb of an ARNT2 ChIP-seq peak.
F. Normalized ATAC signal in CA1 pyramidal neurons isolated from Arnt2 shRNA and control shRNA-infected hippocampi in standard housed mice for NPAS4-bound enhancers (left) and promoters (right). Data plotted by quantiles of in vitro ARNT2 binding strength (3 > 1). − indicates sites not bound by ARNT2. *** p<.001, Wilcoxon signed-rank test comparing distributions of Arnt2 shRNA and control shRNA conditions by quantile.
Motif analysis of the strongest 5,000 NPAS4:ARNT2 co-bound and 4,632 ARNT2 only-bound enhancers revealed a previously described NPAS4:ARNT2 motif (Ooe et al., 2004) as the most significantly enriched hit (Figure 2C). However, NPAS4:ARNT2 co-bound sites are enriched for the sequence TCGTG-A, while ARNT2-only sites are more CG-rich (e.g. GNGNA-CGTG-C), suggesting a potential sequence preference for NPAS4 recruitment to a subset of ARNT2 bound sites (Figure 2C). Finally, we note enrichment for activity-dependent TF motifs (e.g. AP-1) in the NPAS4:ARNT2 co-bound set relative to ARNT2 only-bound sites, consistent with the hypothesis that ARNT2 enhancers are activity-dependent if they recruit NPAS4 (Figure S2G).
Given the low levels of activity-dependent transcription in unstimulated neurons, we considered whether ARNT2 might actively suppress activity-dependent gene expression prior to stimulation. Alternatively, ARNT2 may passively prime sites for NPAS4 recruitment but not directly repress gene expression. To distinguish between these potential mechanisms, we assessed changes to gene transcription upon ARNT2 depletion by injecting AAVs expressing an Arnt2 shRNA or a control, non-targeting shRNA into the left or right hippocampus respectively and performing RNA-seq on infected tissue (Figures S2H-J). We observed that in Arnt2 shRNA-treated samples, activity-dependent genes specifically increase in expression compared to the set of all genes (which show no change), suggesting that ARNT2 dampens activity-dependent gene expression in unstimulated neurons (Figure 2D). Furthermore, we observe that 99% of activity-dependent genes within 50kb of an ARNT2 binding site show up-regulation upon ARNT2 knockdown, consistent with direct ARNT2-mediated repression. However, the set of ARNT2-repressed genes also includes non-activity-inducible genes, suggesting that ARNT2 repression is not restricted to activity-dependent genes (Figures 2D, 2E). To exclude the possibility that the increased expression of activity-regulated genes is an indirect effect of ARNT2 depletion on neuronal excitability, we measured and observed no significant changes in the spiking responses to current injection or evoked excitatory currents (eEPSCs) between wildtype and Arnt2 shRNA-infected neurons (Figures S4D, S4F, Methods).
We next asked whether ARNT2 might repress transcription by promoting changes to chromatin at ARNT2 binding sites. We isolated CA1 pyramidal neurons infected with an Arnt2 or control shRNA using the INTACT approach (Mo et al., 2015) and performed ATAC-seq (Methods). Disrupting ARNT2 expression in unstimulated neurons increased chromatin accessibility across all quantiles of ARNT2 binding strength (Figure 2F). We also observed a similar trend towards increased H3K27ac signal at activity-dependent regulatory elements bound by ARNT2, but this increase did not correlate with ARNT2 binding strength, suggesting that ARNT2-mediated repression may modify chromatin independently of H3K27ac at the measured timepoints (Figure S2K). Indeed, knocking down ARNT2 expression in unstimulated neurons modestly increases luciferase reporter activity at a subset of ARNT2 sites (Figure S2L). Taken together, these findings suggest that ARNT2 is as a repressor of activity-regulated gene expression in unstimulated neurons.
Identification of ARNT2 cofactors in unstimulated neurons
As ARNT2 does not possess a repressor domain (Ooe et al., 2009; Teh et al., 2006), we sought to identify potential repressive ARNT2 cofactors by immunoprecipitating ARNT2 from neuronal lysates and performing mass spectrometry (IP-MS). In addition to the known ARNT2 heterodimer partners NPAS3, NPAS1, and NPAS4 (Wu et al., 2016) (Figures 3A, S3A, S3B, Table S2), the most abundant peptides identified were derived from NCoR2, the nucleating member of the NCoR2 co-repressor complex (Chen and Evans, 1995; Perissi et al., 2010). We also identified NCoR2 complex components TBLR1, TBL1, and HDAC3 as interactors of ARNT2 in neurons (Figures 3A, 3B). We confirmed by immunoblot analysis that NCoR2 and TBL1 co-immunoprecipitate with ARNT2 in unstimulated cortical neurons and adult hippocampus derived from animals raised in standard housing conditions (Figure 3C), thus validating the IP-MS observation that ARNT2 interacts with the NCoR2 complex during periods of low neuronal activity.
Figure 3. ARNT2 recruits the NCoR2 co-repressor complex to enhancers in unstimulated neurons.

A. −log10(FDR) versus log2 fold change in peptides obtained by immunoprecipitation followed by mass spectrometry (IP-MS) with an anti-ARNT2 antibody in stimulated primary cortical neurons relative to peptides immunoprecipitated with an IgG control. Blue points indicate proteins with FDR <0.1 and log2FC > 0. Purple points indicate members of the NCoR2 co-repressor complex.
B. Peptide counts for NCoR2 complex components identified by ARNT2 IP-MS across three replicates from cultured cortical neurons.
C. Immunoblot analysis of immunoprecipitations performed in cultured neurons and adult hippocampus confirms the interaction between ARNT2 and the NCoR2 complex components NCoR2 and TBL1. The known ARNT2 interactor NPAS3 serves as a positive control for the co-IP.
D. Heatmaps of normalized ChIP-seq signal for NCoR2, TBL1, and HDAC3 at presumptive NPAS4 enhancers (upper plots) and promoters (lower plots) in unstimulated (0 hr KCl) cortical neuron cultures. Each binding site is represented as a single horizontal line centered at the peak summit and the intensity of color correlates with sequencing signal. Peaks are ordered by decreasing NPAS4 peak intensity.
E. Representative UCSC genome browser ChIP-seq tracks demonstrating co-localization of the NCoR2 complex at a presumptive NPAS4 enhancer. Each ChIP-seq track is labeled by target, stimulus, and tissue.
F. Overlap of NCoR2 and ARNT2 binding in unstimulated neurons with presumptive NPAS4 binding sites for enhancers (left) and promoters (right). *** p=0, as determined by bootstrapped random sampling approach detailed in Methods.
G. Mean NCoR2 ChIP-seq signal in control or ARNT2-depleted neurons at presumptive NPAS4-bound enhancers (left) or enhancers not bound by NPAS4:ARNT2 (right).
ARNT2 recruits the NCoR2 complex to suppress activity-dependent genes in neurons
The NCoR2 complex has been suggested to repress transcription in a variety of cell types primarily through the action of its enzymatic component, the histone deacetylase HDAC3 (Guenther et al., 2000; Mottis et al., 2013). While NCoR2 function in mature neurons remains unclear, we wondered if one critical role of this complex is to modulate activity-dependent gene expression via interaction with ARNT2. We therefore performed NCoR2, TBL1, and HDAC3 ChIP-seq in unstimulated cortical neurons and observed binding of all three complex components at presumptive NPAS4 regulatory elements across the genome (Figures 3D–F, S3C, S3D). To reduce the detection of false-positive NCoR2 binding sites, we performed ChIP-seq with two independent antibodies generated against distinct NCoR2 domains and verified that shRNA-mediated depletion of NCoR2 in neuronal cultures significantly reduces NCoR2 ChIP-seq signal (Figures 3E, S3E, S3F, Methods).
We next asked whether ARNT2 is required to recruit NCoR2 to presumptive NPAS4 binding sites by performing NCoR2 ChIP-seq in unstimulated neurons transduced with control or Arnt2 shRNA. NCoR2 binding at these elements is significantly reduced in ARNT2-depleted neurons (Figure 3G), in contrast to elements not bound by NPAS4:ARNT2, indicating that ARNT2 is partially required for the specific targeting of NCoR2 to presumptive NPAS4 sites (Figure 3G).
Having shown that ARNT2 recruits NCoR2 to activity-dependent enhancers in silent neurons, we next examined whether NCoR2 works in concert with ARNT2 to repress activity-dependent genes. To this end, we injected AAVs expressing Cre-GFP and a catalytically inactive control (ΔCre-mCherry) into the left or right hippocampus of Ncor2fl/fl mice respectively and performed RNA-seq (Figure S4A). We found that activity-dependent genes were broadly upregulated in hippocampi depleted of NCoR2 compared to all genes, as observed in ARNT2-depleted neurons (Figures 2D, 4A, 4B, S4B, S4C). As for the ARNT2 loss of function experiments, we confirmed that spiking responses to current injection (input/output curve) and evoked excitatory currents (eEPSCs) were unchanged upon depletion of NCoR2 (Figures S4E, S4G). These experiments suggest that in the absence of neuronal activity, ARNT2 and the NCoR2 complex restrain activity-dependent gene transcription.
Figure 4. NCoR2 represses activity-dependent genes in neurons to modulate somatic inhibition.

A. Upper panel: –log10(FDR) versus log2 fold change in gene expression between Cre-GFP and ΔCre-mCherry-infected hippocampal hemispheres in Ncor2fl/fl mice raised in standard housing conditions. NCoR2-regulated genes are defined as fold change≥1.5 and FDR <0.05 when comparing Cre-injected animals against ΔCre (demarcated by top left and top right sectors). Activity-regulated genes (plotted in purple) are defined as fold change≥1.5 and FDR <0.05 when comparing kainate-injected animals against saline-injected controls. Lower panel: Box and whisker plots display the distribution of log2 fold changes (Cre/ΔCre) of all genes (gray) and activity-dependent genes (purple). p = 3.3e-61 in the Wilcoxon rank sum test.
B. Gene expression in log2(RPKM+1) for all activity-regulated genes in ΔCre-mCherry or Cre-GFP- infected hippocampal hemispheres of standard housed animals. Each gene is connected by a line across its expression values between the two conditions. Purple colored lines indicate genes within 50kb of an ARNT2 ChIP-seq peak.
C. eIPSC amplitude simultaneously measured from wildtype uninfected (black) and neighboring CA1 pyramidal neurons infected by AAVs expressing Arnt2 shRNA#1 or Arnt2 shRNA#2 (red) in response to stimulation of axons in stratum pyramidale (St. pyr). Slices were obtained from animals raised in standard housing conditions. Individual pairs of recorded neurons are represented by paired points, with bars showing mean ± standard error of the mean (SEM). Representative traces shown above all data. * p < 0.05, paired two-tailed t-test.
D. eIPSC amplitude simultaneously measured from wildtype uninfected (black) and neighboring CA1 pyramidal Ncor2fl/fl neurons infected by AAVs expressing Cre-GFP (Ncor2-KO, green) in response to stimulation of axons in stratum pyramidale (St. pyr). Slices were obtained from animals raised in standard housing conditions. Individual pairs of recorded neurons are represented by paired points, with bars showing mean ± SEM (bottom). Representative trace shown above all data. *p < 0.05, paired two-tailed t-test.
ARNT2:NCoR2 loss of function impairs recruitment of somatic inhibition
Since the NPAS4-dependent gene program has previously been reported to promote somatic inhibition onto CA1 pyramidal neurons, we investigated whether reducing expression of either ARNT2 (by shRNA) or NCoR2 (by Cre in NCoR2fl/fl mice) results in increased somatic inhibition under standard housing conditions where activity-dependent genes are not expressed (Bloodgood et al., 2013; Lin et al., 2008). We found that both ARNT2- and NCoR2-depleted neurons display an increase in eIPSC amplitude when compared to neighboring control neurons (Figures 4C, 4D). These observations support a model in which removal of ARNT2:NCoR2-mediated repression of the activity-dependent program is sufficient to trigger aberrant inhibition without affecting neuronal excitability, as measured by I/O curve and eEPSCs (Figures S4D-G).
ARNT2:NPAS4 facilitates activity-dependent gene induction
We next investigated how neurons overcome ARNT2:NCoR2-mediated repression upon stimulation to induce transcription and promote activity-dependent modifications to neural circuits. Prior work has shown that ARNT2 and NPAS4 directly interact (Ooe et al., 2004), suggesting that ARNT2 may mediate NPAS4-dependent gene activation. We confirmed the direct interaction of NPAS4 and ARNT2 by performing NPAS4 IP-MS in membrane-depolarized cortical neurons (Figure 5A, Table S2) and validated the results by 1) reciprocal co-immunoprecipitation of NPAS4 and ARNT2 from embryonic cortical neurons (Figures 5B, S5A) and adult brain extracts (Figure 5B) and 2) using 293T overexpression assays (Figure S5B). These data indicate that ARNT2 is the primary heterodimer partner of NPAS4 in neurons.
Figure 5. ARNT2 is required for NPAS4-dependent gene activation.

A.−log10(FDR) versus log2 fold change in peptides obtained by IP-MS with an anti-NPAS4 antibody in stimulated primary cortical neurons relative to peptides immunoprecipitated from control samples. Control samples include a non-specific IgG and immunoprecipitation with an anti-NPAS4 antibody from unstimulated neurons, where NPAS4 is not expressed. Blue points indicate proteins with FDR <0.1 and log2FC > 0.
B. Immunoblot analysis of anti-NPAS4 immunoprecipitates performed in cultured neurons or hippocampal tissue confirming the interaction between ARNT2 and NPAS4 both in vitro and in vivo.
C. Overlap between NPAS4 binding sites in stimulated cultured cortical neurons (2 KCl) and ARNT2 binding sites in both stimulated cultured neurons (2 KCl, top) and adult hippocampal tissue derived from mice treated with kainate (KA, bottom) to induce neuronal activity. Fisher’s exact test odds ratios (OR) and p-values for set intersections shown on figure.
D. Mean normalized NPAS4 ChIP-seq signal in control or ARNT2-depleted neurons across NPAS4-bound enhancers.
E. Normalized H3K27ac ChIP-seq signal in control shRNA (gray) and Arnt2 shRNA (green) transduced cultured neurons at NPAS4-bound enhancers (left) and promoters (right). Data plotted by quantiles of in vitro ARNT2 binding strength (3 > 1). − indicates sites not bound by ARNT2. *** p<.001, Wilcoxon signed-rank test comparing distributions of Arnt2 shRNA and control shRNA conditions by quantile.
F. UCSC genome browser ChIP-seq tracks displaying H3K27ac signal at a representative NPAS4-bound enhancer in cortical cultures stimulated with 55mM KCl. Each ChIP-seq track is labeled for the ChIP-ed factor, time point after membrane depolarization, and treatment.
G. −log10(FDR) versus log2 fold change in gene expression NPAS4Full-Length (left) or NPAS4ΔPAS (right) expressing neurons to GFP controls in unstimulated cortical neurons. NPAS4-regulated genes are indicated in blue. Npas4 and its known target gene Bdnf are labeled in both plots for reference.
We next asked if ARNT2, in addition to its repressive role in silent neurons, mediates NPAS4dependent activation in stimulated neurons. We confirmed that ARNT2 is bound at 89% of NPAS4bound regulatory elements in stimulated neurons (Figures 5C, S5C). To test the requirement of ARNT2 for targeting NPAS4 to the genome, we performed NPAS4 ChIP-seq in depolarized neurons transduced with an Arnt2 shRNA. NPAS4 protein levels were unchanged in ARNT2-depleted neurons (Figure S5D), but NPAS4 binding to activity-regulated enhancers was strongly reduced relative to control shRNA-treated neurons, indicating that NPAS4 requires ARNT2 to bind its target sites (Figure 5D).
Given its role in NPAS4 targeting, we tested whether depletion of ARNT2 decreases enhancer and promoter activation in stimulated neurons. We observed a significant decrease in H3K27ac ChIP-seq signal at NPAS4:ARNT2 co-bound regulatory elements (Figures 5E, 5F) and decreased luciferase reporter activity at the majority of NPAS4 sites tested (Figures S5E, S5F). These data indicate that, in contrast to its repressive function in unstimulated neurons, ARNT2 is required for the activation of NPAS4-bound regulatory elements in stimulated neurons. We therefore hypothesized that disrupting the NPAS4:ARNT2 interaction would also impair NPAS4-dependent gene transcription. Indeed, expression of a mutant NPAS4 incapable of binding ARNT2 (NPAS4ΔPAS) failed to increase gene expression (Figures 5G, S5G). Taken together, these data demonstrate that ARNT2 is required for NPAS4-dependent transcription in stimulated neurons.
Neuronal activity partially decouples ARNT2:NCoR2 from its repressive role
Given that an NPAS4:ARNT2 activator complex is recruited to activity-dependent regulatory elements upon membrane depolarization, we anticipated that neuronal activity might trigger the eviction of the repressive ARNT2:NCoR2 complex, thus contributing to induced transcription as previously suggested (Telese et al., 2015). However, we did not detect a reduction in binding of NCoR2, TBL1, or HDAC3 at two hours post-depolarization in cortical neurons (Figures 6A, 6B). Instead we observed no change in TBL1 and HDAC3 and, surprisingly, increased NCoR2 ChIP signal specifically at NPAS4bound regulatory elements (Figures 6A, 6B). Although we cannot exclude the possibility that this increase in NCoR2 ChIP-seq signal is due to differential epitope accessibility in stimulated neurons, we consistently observed this result with two distinct anti-NCoR2 antibodies (Figure S6A). We also found that increased NCoR2 binding to NPAS4-bound enhancers upon membrane depolarization fails to occur in ARNT2depleted neurons, suggesting that the activity-dependent recruitment of NCoR2 requires ARNT2 (Figures S6B, 6C). Notably, NPAS4 did not co-immunoprecipitate with either TBL1 or NCoR2 in adult hippocampal or in cultured cortical lysates (Methods), suggesting that ARNT2 forms separate complexes with NPAS4 and NCoR2 under these conditions (Figures 5A, S6C).
Figure 6. Activity partially decouples ARNT2:NCoR2 from its repressive role.

A. Heatmaps of normalized ChIP-seq signal for unstimulated (0 hr KCl) and depolarized (2 hr KCl) cortical neuron cultures (left eight columns) at presumptive NPAS4 enhancers. Rightmost two columns show ARNT2 binding in adult hippocampal tissue in standard housing (Strd) or following neuronal stimulation via the injection of kainate (KA). Each binding site is represented as a single horizontal line centered at the peak summit and intensity of color correlates with sequencing signal for the indicated factor. Peaks are ordered by decreasing NPAS4 peak intensity.
B. Mean normalized ChIP-seq signal for indicated factors. Light lines indicate signal at non-NPAS4 bound enhancers and dark lines indicate signal at NPAS4-bound enhancers.
C. Mean normalized NCoR2 ChIP-seq signal intensity at NPAS4-bound enhancers (upper panel) and promoters (lower panel) in unstimulated (gray) or stimulated neurons transduced with either a control (solid purple) or Arnt2 shRNA (dashed purple). The induction of NCoR2 upon depolarization observed in control samples is lost upon ARNT2 knockdown at enhancers but not promoters.
D. Upper panel: −log10(FDR) versus log2 fold change between Arnt2 shRNA and control shRNA infected hippocampal hemispheres in mice injected with the glutamatergic receptor agonist kainate (KA). Arnt2 regulated genes are defined as fold change ≥1.5 and FDR <0.05 when comparing Arnt2 shRNA-injected animals against controls (demarcated by top left and top right sectors). Activity-regulated genes (plotted in orange) are defined as fold change ≥1.5 and FDR <0.05 when comparing kainate-injected animals against saline-injected controls. Lower panel: Box and whisker plots display the distribution of log2 fold changes (Arnt2 shRNA/control shRNA) of all genes (gray) and activity-dependent genes (orange). p = 1.3e-45 in the Wilcoxon rank sum test.
E. Upper panel: −log10(FDR) versus log2 fold change between Cre-GFP and ΔCre-mCherry infected hippocampal hemispheres in Ncor2fl/fl mice injected with the glutamatergic receptor agonist kainate. NCoR2-regulated genes are defined as fold change ≥1.5 and FDR <0.05 when comparing Cre-GFP-injected animals against ΔCre-mCherry controls (demarcated by top left and top right sectors). Activity-regulated genes (plotted in purple) are defined as fold change ≥1.5 and FDR <0.05 when comparing kainate-injected Ncor2fl/fl animals against saline-injected controls. Lower panel: Box and whisker plots display the distribution of log2 fold changes (Cre-GFP/ΔCre-mCherry) of all genes (gray) and activity-dependent genes (orange). p = 7.8e-81 in the Wilcoxon rank sum test.
F. eIPSC amplitude simultaneously measured from control uninfected (wildtype, black) and neighboring CA1 pyramidal neurons infected by AAVs expressing Arnt2 shRNA#1 or Arnt2 shRNA#2 (red) in response to stimulation of axons in stratum pyramidale (St. pyr). Slices were obtained from animals injected with kainate 24 hours before sacrifice. Individual pairs of recorded neurons are represented by paired points, with bars showing mean ± SEM. Representative traces shown above all data. * p < 0.05, paired two-tailed t-test.
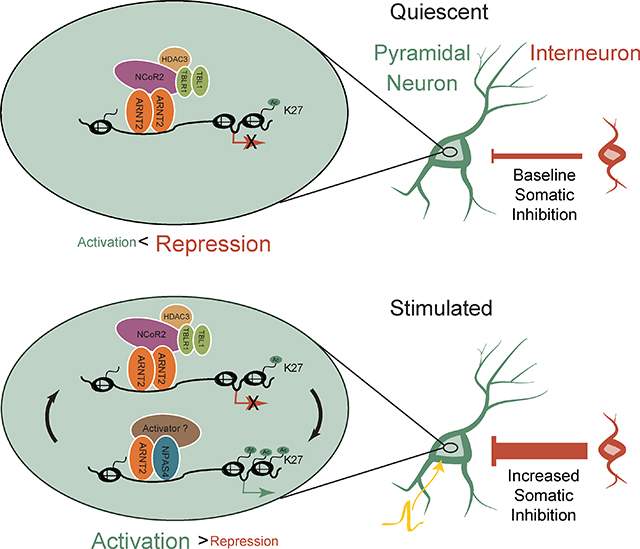
G. In unstimulated neurons, the ARNT2 recruits the NCoR2 complex to presumptive NPAS4-bound regulatory elements, suppressing activity-dependent gene transcription and preventing aberrant recruitment of somatic inhibition (top panel). Upon neuronal activation (yellow action potential), NPAS4:ARNT2 overcomes repression to activate enhancers and induce gene expression. NCoR2 is also inducibly recruited to these sites and may contribute to gene activation (not shown in model). By contrast, HDAC3 and TBL1 binding at these sites remains unchanged with stimulation and may act as a repressive brake that is transiently overcome by the recruitment of activating factors like NPAS4. This enables the precise tuning of gene expression and subsequent temporally-specific recruitment of somatic inhibition (bottom panel).
The recruitment of ARNT2 and NCoR2 to NPAS4-bound enhancers could represent an additional function for ARNT2:NCoR2 beyond the repression observed in silent neurons. To test this, we injected viruses delivering Arnt2 shRNA or Cre into the hippocampus (Figure S4A) to deplete either ARNT2 or NCoR2 in CA1 and collected RNA from these mice six hours after the injection of kainate, a glutamatergic agonist that robustly induces activity-dependent genes (Bloodgood et al., 2013). We found that the expression of the activity-dependent gene set is significantly reduced in kainate-stimulated hippocampi lacking ARNT2 or NCoR2 compared to the contralateral control hemisphere. By contrast, overall expression of the set of all genes is not affected by ARNT2 or NCoR2 depletion (Figures 6D, 6E, S6D). Depletion of NCoR2 also decreases the ability of NPAS4:ARNT2-bound regulatory sites to drive luciferase activity in stimulated neurons (Figure S6E). Together, these data provide evidence that ARNT2 and NCoR2 boost activity-dependent gene induction in membrane-depolarized neurons.
Since NPAS4:ARNT2 stimulates activity-dependent gene transcription in membrane-depolarized neurons, we hypothesized that depletion of ARNT2 in stimulated neurons might phenocopy the reduction in somatic inhibition observed when NPAS4 function is disrupted (Bloodgood et al., 2013). We injected AAVs expressing either of two distinct Arnt2 shRNAs into the hippocampus, stimulated the mice with kainate to induce NPAS4 and other activity-dependent genes, and compared eIPSC amplitude between ARNT2-depleted and neighboring control neurons (Figure 6F). We found that eIPSC amplitude is reduced in ARNT2-depleted neurons compared to neighboring control neurons, suggesting that in response to neuronal activity ARNT2 drives expression of activity-regulated genes that promote increased somatic inhibition onto CA1 pyramidal neurons. In contrast, in the absence of synaptic activity, ARNT2 represses activity-dependent gene expression and restricts somatic inhibition (Figures 2D, 4C).
Discussion
We have identified an ARNT2:NCoR2 repressor complex and demonstrated its importance for maintaining low levels of activity-dependent gene expression in unstimulated neurons. Disruption of this protein complex causes up-regulation of activity-dependent genes and dysregulation of somatic inhibition in silent pyramidal neurons. In response to neuronal activity, ARNT2 complexes with NPAS4 and activates NPAS4-dependent stimulus-responsive elements (Figure 6G). We also observe NCoR2 recruitment to NPAS4-bound sites upon depolarization and reveal a surprising role for NCoR2 in promoting stimulus-responsive gene expression. We propose that NPAS4:ARNT2 rapidly converts repressed regulatory regions to their activated state, but it remains to be further explored by what mechanism ARNT2:NCoR2 contributes to activity-dependent gene expression in membrane-depolarized neurons. Together, these data identify a molecular mechanism in neurons that restricts inducible gene expression to periods of time marked by increased neuronal activity, while maintaining transcriptional quiescence during inactivity.
Maintaining transcriptional inactivity during periods of low stimulation
Our study supports a growing body of evidence highlighting the importance of enhancer elements for coordinated control of stimulus-induced transcription. One strategy is to restrict enhancer activation to the appropriate temporal window by nucleosomal occlusion of DNA (Ostuni et al., 2013; Vierbuchen et al., 2017), which is then relieved by the stimulus-inducible transcription factors FOS and JUN via direct recruitment of chromatin remodeling complexes (e.g. BAF) (Vierbuchen et al., 2017). Alternatively, previous studies have identified a separate class of enhancers that display characteristic features of transcriptionally active sites in unstimulated cells, including open chromatin, RNAPII binding, or modest levels of H3K27ac, but paradoxically are not associated with increased transcription (Kim et al., 2010; Ostuni et al., 2013). In the present study, we have shown that at such activity-dependent regulatory elements, the primary control of inducibility is achieved through the tunable repressor complex ARNT2:NCoR2.
The presence of ARNT2:NCoR2 complexes at presumptive NPAS4-bound regulatory elements in silent neurons raises the intriguing possibility that this complex could select these enhancers during neuronal maturation, thereby establishing the landscape of NPAS4-dependent elements poised for use in the adult brain. Indeed, given that NPAS4 induces cell-type-specific gene expression programs, it is tempting to speculate a role for ARNT2 in selecting enhancers critical for generating neuronal sub-typespecific transcriptional responses to sensory experience (Mardinly et al., 2016; Spiegel et al., 2014). Determining the functions of ARNT2 in alternative cell types will be an exciting area for future investigation. As examples, the known ARNT2 interacting partners NPAS3 and NPAS1 (Figure 3A) are highly enriched in inhibitory interneurons (Figure 1D) where they may compete with NPAS4 for ARNT2, providing alternative mechanisms of cell-type-specific enhancer selection and gene repression (Sabatini et al., 2018).
Transcriptional Control by Multifunctional Complexes
The regulated recruitment of repressor or activator complexes to DNA by transcription factors, such as ARNT2, may be a general mechanism by which signal-responsive transcription is controlled in biologically distinct systems. For example, transforming growth factor beta (TGF-β) and nuclear hormone receptor transcription factor superfamilies regulate diverse cellular responses via interactions with both activator (p300, CBP) and repressor (NCoRs and HDACs) complexes (Hill, 2016). Our study provides a new variation on this theme in the nervous system by identifying distinct activating and repressing roles for ARNT2 in regulating inhibitory synaptic plasticity. Consistent with previous reports, we have identified a reduction in stimulus-dependent gene induction upon NCoR2 depletion in membrane-depolarized neurons, suggesting a possible activator role for the canonically repressive NCoR2 (Adikesavan et al., 2014; Berghagen et al., 2002; Peterson et al., 2007). How NCoR2 switches its role from repressive to activating is not yet clear. One possible mechanism is that ARNT2:NCoR2 decouples from its canonical partners TBL1 and HDAC3 and competitively inhibits the recruitment of the repressive ARNT2:NCoR2:HDAC3 complex to regulatory elements. Alternatively, post-translational modifications to NCoR2 may alter its binding partners (Guenther et al., 2000; Peterson et al., 2007; Zhang et al., 2002). Indeed, the NCoR2 complex components TBL1 and TBLR1 proteins have both repressive and activating functions due to their ability to promote cofactor exchange (Perissi et al., 2004). While it remains to be examined which of these mechanisms is involved in NCoR2’s switch to activator in membrane-depolarized neurons, our genome-wide results support the emerging view that that the level and duration of membrane depolarization ultimately determine the function of these recruited regulatory complexes.
Temporal specificity of activity-dependent transcription for circuit plasticity
Although we primarily present evidence for how ARNT2:NCoR2 inhibits activity-dependent gene expression prior to sensory stimulation, it will be interesting to explore if ARNT2:NCoR2:HDAC3 also functions post-depolarization to return active regulatory elements to transcriptional quiescence. Indeed, recent work demonstrates that the chromatin remodeler complex NuRD is important for dampening IEG expression post-stimulation (Yang et al., 2016). Future studies will be necessary to determine if the interplay of NuRD and ARNT2:NCoR2/ARNT2:NPAS4 influences the off-rate kinetics of activity-dependent transcription. It is also conceivable that ARNT2:NCoR2:HDAC3 recruitment following a stimulus may impose a refractory state on activation, wherein additional stimulation of the cell is insufficient to induce activity-dependent gene expression for some period of time. Such molecular mechanisms could impart additional modes of control to ensure that excitatory-inhibitory balance is maintained appropriately across wide variations in neuronal activity.
We speculate that the temporal precision of activity-dependent gene transcription is critical for circuit changes that occur in response to changes in behavioral state. For example, associative learning paradigms require consistent coincidence detection between a stimulus and context across time. While the molecular and circuit changes underlying these rapid associations remain unclear, activity-dependent changes in transcription have been shown to be critical for the consolidation of addictive behaviors in drug reward-related behavioral paradigms (Nestler, 2014; Renthal et al., 2007; Taniguchi et al., 2017). These findings highlight the importance of continued study of the mechanisms that regulate the temporal specificity of activity-dependent transcription in normal and pathological behaviors. Of note, mutations in components of the NCoR2 repressor complex are reported to contribute to intellectual disability and schizophrenia (Gulsuner et al., 2013; O’Roak et al., 2012; Pons et al., 2015; Tabet et al., 2014; Takata et al., 2016). Thus, our study identifies the ARNT2:NCoR2 complex as a possible mechanistic link between these disorders and activity-dependent gene expression, underscoring how stimulus-dependent control of inhibition may contribute to brain development and go awry in disease.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Michael Greenberg, meg@hms.harvard.edu
EXPERIMENTAL MODEL DETAILS
Mouse models
Animals were handled according to protocols approved by the Harvard University Standing Committee on Animal Care and were in accordance with federal guidelines. The following mouse lines were used: Npas4fl/fl (Lin et al., 2008), Ncor2fl/fl (Shimizu et al., 2015), B6;129-Gt(ROSA)26Sor<tm5(CAGSun1/sfGFP)Nat>/J (Mo et al., 2015), B6.Cg-Tg(Camk2a-cre)T29–1Stl/J (Tsien et al., 1996), wildtype C57/Bl6 (JAX 000664). Both male and female mice were used in equal proportions for all experiments conducted. For experiments in which seizures were induced, kainic acid (5–10 mg/kg (electrophysiology)) or 15–20 mg/kg (genomic analysis and immunoprecipitations)) (Sigma Aldrich, K0250) was injected intraperitoneally. Mice were sacrificed 2 or 6 hours after injection, as indicated, for genomic-level analyses based on prior work indicating that immediate early gene transcription factors and late-response genes are robustly induced at 2 and 6 hours post-stimulus onset, respectively. Mice were sacrificed 16–24 hours after injection for electrophysiological analysis to allow sufficient time for the expression of the activity-dependent gene program and the execution of potential synaptic regulation but far in advance of any measurable seizure related cellular toxicity.
Mouse neuron culture
Embryonic cortices from C57Bl/6 or transgenic (Npas4fl/fl) mice were dissected at ages E16.5-E17 and then dissociated with papain (Sigma Aldrich; 10108014001). Cultures were generated by combining multiple embryos (~ 5–6) of both males and females (mixed sex cultures). Papain digestion was terminated with the addition of ovomucoid (trypsin inhibitor; Worthington). Cells were gently triturated through a P1000 pipette and passed through a 40 micron filter. Neurons were plated onto cell cultures dishes precoated overnight with poly-D-lysine (20 μg/mL) and laminin (4 μg/mL). Neurons were grown in Neurobasal medium (Gibco) containing B27 supplement (2%), penicillin-streptomycin (50 U/mL penicillin and 50 U/mL streptomycin) and glutaMAX (1 mM). Neurons were grown in incubators maintained at 37°C and a CO2 concentration of 5%. Neurons were harvested in all experiments at 7DIV, and fresh media was added at 3–4DIV (25% total volume), unless otherwise indicated. In all experiments, independent replicates were generated by preparing primary cultures from pools of embyros dissected on different days and maintained in culture for 7DIV. For immunoprecipitation and mass spectrometry (IPMS) experiments or ChIP, cortical neurons were plated at a density of 25–30 × 106 / dish on 15 cm dishes. To prevent spurious activity, for IP-MS experiments and NCoR2 complex ChIPs, 7DIV neurons were silenced on 6DIV overnight by the addition of 1 μM TTX (Abcam ab120055) and 100 μM AP5 (Thermo Fisher 01–061-0).
METHOD DETAILS
Stereotaxically guided surgery
P13–15 mice of equal proportion male and female were anaesthetized by isoflurane inhalation (3–5% for induction, 1–3% maintenance) and positioned within a stereotaxic frame (Kopf) where animal temperature was maintained at ~37°C by a heat pad. All surgeries were performed according to protocols approved by the Harvard University Standing Committee on Animal care and were in accordance with federal guidelines. Fur around the scalp area was removed using a shaver and sterilized with three alternating washes with betadine and 100% ethanol. A burr hole was drilled through the skill above the CA1 region of hippocampus (medial/lateral +/− 2.9 mm; anterior/posterior −2.5 mm; dorsal/ventral: 2.8 mm) to allow for specific targeting of this region with a glass pipette pulled to a tip diameter of roughly 50 microns. 250300 nL of AAV virus was injected at 150 nL/min and the pipette was left in place upon completion of viral infusion to allow for viral spreading. All animals were given postoperative analgesic (flunixin, 2.5 mg per kg) as well as additional injections at 12 hour intervals for the 72 hours following surgery. For in vivo genomic datasets, mice were sacrificed and analyzed 10 days post surgery.
Virus production
For shRNA treatments in vitro, 21 bp targeted sequences from TRC (Sigma Aldrich) were cloned into the FUW vector downstream of the U6 promoter. The following sequences were used: Control shRNA (GCGCGATAGCGCTAATAATTT), Arnt2 shRNA#1 (TGTCGGACAAGGCAGTAAAT), Arnt2 shRNA#2 (CGCTATTATCATGCCATAGAT), Npas4 shRNA#1 (GGTTGACCCTGATAATTTA), Npas4 shRNA #2 (ACACAGAGAAAGAGCAAAA), Ncor2 shRNA#1 (CCTGTCTAAAGCCTTAACTAA), Ncor2 shRNA#2 (CCAGTGTAAGAACTTCTACTT), Ncor2 shRNA#3 (GAAAGGCACTCATGGGTAAAT). To examine the importance of NPAS4’s ability to dimerize with ARNT2 for stimulating gene expression GFP, NPAS4 (NPAS4Full-length), or a mutant NPAS4 lacking the PAS domain (NPAS4ΔPAS) were subcloned into the FUW backbone.
To produce lentivirus, 10 μg of lentiviral plasmid (either shRNA-expressing or NPAS4 overexpression) was transfected into HEK293T cells along with third generation packaging plasmids pMDL (5 μg), RSV (2.5 μg), and VSVG (2.5 μg). At 12–16 hours following transfection, 293T cells were switched to Neurobasal media containing B27 supplement, and supernatant containing virus was collected at 36 hours post transfection. To infect neurons, on 2DIV, one third of neurobasal media was replaced with viral supernatant. Cells were collected at 7DIV for either ChIP or RNA-seq. To test the specificity of NCoR2 ChIPs, cells were infected with a combination of Ncor2 shRNAs #1, #2, #3.
To deplete ARNT2 in CA1 neurons for electrophysiology, in vivo RNA-seq, and ATAC-seq, control shRNA (GCGCGATAGCGCTAATAATTT), Arnt2 shRNA#1 (TGTCGGACAAGGCAGTAAAT), Arnt2 shRNA#2 (CGCTATTATCATGCCATAGAT), were cloned downstream of the U6 promoter and mCherry or GFP florescence was driven off the CAG promoter. Construct names are denoted in the Key Resources table. All AAV backbones were generated by using standard cloning and molecular biology techniques. AAVs (serotype 2/1) were prepared by the Boston Children’s Hospital Viral Core.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3K27ac | Abcam | Cat# Ab4729, RRID: AB_2118291 |
| anti-ARNT2 | This paper | N/A |
| anti-NPAS4 | Lin et al., 2008 | N/A |
| anti-NPAS3 | Gift of Steven McKnight Lab | |
| anti-NCoR2 | Thermo Fisher | Cat# PA1–843, RRID:AB_2149135 |
| anti-NCoR2 | Millipore | Cat# 06–891, RRID: AB_310286 |
| anti-TBL1 | Abcam | Cat# Ab24528 |
| anti-TBL1 | Santa Cruz | Cat# sc-137006, RRID:AB_2199796 |
| anti-HDAC3 | Santa Cruz | Cat# sc11416X |
| anti-β-TUBULIN3 | Covance | Cat# MMS-435P |
| anti-GAPDH | Sigma | Cat# G9545, RRID:AB_796208 |
| anti-MYC | Millipore | Cat# 05–724, RRID:AB_309938 |
| anti-MYC, clone 9E10 | Developmental Studies Hybridoma Bank http://dshb.biology.uiowa.edu/ | NA |
| anti-FOS | Santa Cruz | Cat# sc-7202 X; RRID:AB_2106765 |
| anti-GFP | Invitrogen | G10362 |
| anti-Histone H3 | Abcam | Cat# 1791; RRID:AB_302613 |
| Rabbit IgG | Santa Cruz | Cat# sc-66931; RRID:AB_1125055 |
| anti-M2-FLAG | Sigma Aldrich | Cat# A2220 |
| Bacterial and Virus Strains | ||
| AAV2/1-CAG-Cre-GFP | Boston Children’s Hospital Viral Core | |
| AAV2/1-CAG-ΔCre-mCherry | Boston Children’s Hospital Viral Core | |
| AAV2/1-U6-shRNA-CAG-mCherry | Boston Children’s Hospital Viral Core | |
| AAV2/1-U6-non-targeting shRNA-CAG-GFP | Boston Children’s Hospital Viral Core | |
| FUW Lentivirus (pMDL/RSV/VSVG packaging) | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TTX | Abcam | Cat# ab120055 CAS: 18660–81-6 |
| Kainic acid monohydrate | Sigma Aldrich | Cat# K0250 CAS: 58002–62-3 |
| DL-AP5 | Thermo Fisher | Cat# 01–061-0 CAS: 76326–31-3 |
| CPP | Tocris | Cat# 0173 CAS: 126453–07-4 |
| NBQX | Tocris | Cat#0190 CAS:118876–58-7 |
| Gabazine | Tocris | Cat# SR 95531 CAS: 104104–50-9 |
| EGS (ethylene glycol bis(succinimidyl succinate) | Fisher | Cat# 21565 CAS: 70539–42-3 |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | E1910 |
| Nextera DNA Library Prep Kit | Illumina | FC-121–1030 |
| Ovation Ultralow System V2 | Nugen | 0344–32 |
| NEBNext® rRNA Depletion Kit | New England Biolabs | E6310X |
| NEBNext Ultra Directional RNA Library Prep Kit | New England Biolabs | E7420L |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE121660 |
| Raw and analyzed visual cortex scRNA-seq data | Hrvatin et al. 2018 | GEO: GSE102827 |
| Experimental Models: Organisms/Strains | ||
| Npas4fl/fl | Lin et al., 2008 | Greenberg Lab |
| NCoR2f/lfl | Shimizu et al., 2015 | Hollenberg Lab |
| C57BL6/J | The Jackson Laboratory | JAX 000664 |
| B6.Cg-Tg(Camk2a-cre)T29–1Stl/J | The Jackson Laboratory | JAX 005359 |
| B6;129-Gt(ROSA)26Sor<tm5(CAG-Sun1/sfGFP)Nat>/J https://www.jax.org/strain/021039 | The Jackson Laboratory | JAX 021039 |
| Oligonucleotides | ||
| Primers for Luciferase Assays (Table S1) | ||
| non-targeting GCGCGATAGCGCTAATAATTT | ||
| Arnt2 shRNA#1 TGTCGGACAAGGCAGTAAAT | ||
| Arnt2 shRNA#2 CGCTATTATCATGCCATAGAT | ||
| Ncor2 shRNA#1 CCTGTCTAAAGCCTTAACTAA | ||
| Ncor2 shRNA#2 CCAGTGTAAGAACTTCTACTT | ||
| NCoR2 shRNA#3 GAAAGGCACTCATGGGTAAAT | ||
| Npas4 shRNA#1 GGTTGACCCTGATAATTTA | ||
| Npas4 shRNA#2 ACACAGAGAAAGAGCAAAA | ||
| Recombinant DNA | ||
| FUW-U6-Npas4-shRNA#1 | This Paper | |
| FUW-U6-Npas4-shRNA#2 | This Paper | |
| FUW-U6-Arnt2-shRNA#1 | This Paper | |
| FUW-U6-Arnt2-shRNA#2 | This Paper | |
| FUW-U6-Ncor2-shRNA#1 | This Paper | |
| FUW-U6-Ncor2-shRNA#2 | This Paper | |
| FUG-U6-Ncor2-shRNA#3 | This Paper | |
| pGL4.11 Genomic Binding Sites; See Table S1 | This Paper | Addgene #59744 |
| FUW-Npas4-full-length | This Paper | |
| FUW-Npas4ΔPas | This Paper | |
| FUGW | Lois et al., 2002 | Addgene #14883 |
| pEF1a-Arnt-Myc | This Paper | |
| pEF1a-Arnt2-Myc | This Paper | |
| pEF1a-Ephexin5-Myc | This Paper | |
| pGEX-Arnt2–312:712 | This paper | |
| AAV-CAG-Cre-GFP | Gift from Matt During (Ohio State University) | |
| AAV-CAG-ΔCre-mCherry | This paper; modified from Matt During | |
| AAV-U6-shRNA-CAG-mCherry (see sequences above) | This paper; modified from Matt During | |
| AAV-U6-non-targeting shRNA-CAG-GFP | This paper; modified from Matt During | |
| Software and Algorithms | ||
| Subread v1.5.1 | Liao et al., 2013 | http://subread.sourceforge.net/ |
| Trimmomatic (v0.36) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| bedGraphToBigWig (UCSC-tools v363) | UCSC-tools | http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/ |
| featureCounts (Subread package, v1.5.1) | Liao et al., 2013 | http://subread.sourceforge.net/ |
| MACS2 (v 2.1.0) | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| IDR | Li, 2011 | https://drive.google.com/file/d/0B_ssVVyXv8ZSX3luT0xhV3ZQNWc/view?usp=sharing |
| EdgeR(v3.16.5) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Limma(v3.30.13) | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Bh-tsne (commit 36b169c) | van der Maaten, 2014 | https://github.com/lvdmaaten/bhtsne |
| GEM software (v3.4) | Guo et al., 2018 | http://groups.csail.mit.edu/cgs/gem/kmac/ |
| Other | ||
Acute slice preparation and electrophysiology
For electrophysiology, we first injected the CA1 of postnatal 13–15 day old mice with an AAV that drives the expression of Arnt2 shRNA under the U6 promoter and mCherry under the CAG promoter. Transverse hippocampal slices were prepared from P21-P28 mice of the indicated genotypes. Animals were anesthetized with ketamine/xylazine and transcardially perfused with ice-cold choline-based artificial cerebrospinal fluid (choline-ACSF) equilibrated with 95% O2/5% CO2 consisting of (in mM): 110 cholineCl, 25 NaHCO3, 1.25 Na2HPO4, 2.5 KCl, 7 MgCl2, 25 glucose, 0.5 CaCl2, 11.6 ascorbic acid, 3.1 pyruvic acid. Cerebral hemispheres were quickly removed and placed into ice-cold choline-ACSF. Tissue was rapidly blocked and transferred to a vibratome (Leica VT1000), where 300 micron slices were cut. Slices were subsequently transferred to a holding chamber containing ACSF consisting of (in MM): 127 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose, and saturated with 95% O2/5% CO2. Slices were incubated at 32°C for 20 min after cutting and then maintained at room temperature. All recordings were performed within 4–5 hours of slice preparation and showed AAV infection measured by fluorescence in ~10–30% of CA1 neurons. Slices showing >30% of CA1 neurons infected were discarded from further analysis.
Simultaneous paired whole-cell voltage-clamp recordings were obtained from morphologically identified CA1 pyramidal neurons visualized with infrared, differential interference contrast microscopy as well as fluorescence microscopy, where appropriate. In all recordings, neurons were held at −70 mV and recordings were made with borosilicate glass patch pipettes with open pipette resistance ranging from 3–5 M. Patch pipettes were filled with internal solution containing (in mM):147 CsCl, 5 Na2-phosphocreatine, 10 HEPES, 2 MgATP, 0.3 Na2GTP, 2 EGTA and 5 QX-314 for voltage clamp recordings (eIPSC), 135 Cs-Methanesulfonate, 15 HEPES, 0.5 EGTA, 5 mM TEA-cl, 1 mM MgCl2, 0.16 CaCl2, 2 mM Mg-ATP, 0.3 Na-GTP, 10 phosphocreatine, 2 QX-314 for eEPSC, and 126 K-Gluconate, 4 KCl, 10 HEPES, 4 MgATP, 10 phosphocreatine for current clamp recordings. Evoked inhibitory postsynaptic currents were pharmacologically isolated by adding 10 μM CPP (Tocris) and 10 μM NBQX (Tocris) to the ACSF perfusion. Similarly, 10 μM gabazine (Tocris) was used to pharmacologically isolated eEPSCs. Extracellular stimulation was delivered via theta glass stimulation electrode placed in the center of stratum pyramidale within 100–200 microns of the cell pair being recorded. The stimulus strength was the minimum required to generate an eIPSC in both ARNT2-depleted and NCoR2-depleted and wildtype neurons. Recording from neighboring neurons increased the probability that these neurons receive synaptic input from the same population of inhibitory axons while simultaneous recordings insured that the neighboring Knockdown/KO and wildtype neurons were exposed to an identical extracellular stimulus. Action potentials were measured by injecting 200ms steps of current in 50pA increments from 0 pA to 400 pA. All cells included in the analysis were held between −65 mV and −70 mV with <30pA of constant current injection.
Neuronal transfection and luciferase reporter assays
DNA sequences to test in luciferase reporter assays were chosen based on the strength of NPAS4 and ARNT2 binding, as assessed by normalized ChIP-seq read counts from ChIPs in primary cortical cultures. Sites included both strongly bound (i.e. selected from top 100 high-confidence NPAS4 binding sites determined by IDR peak calling and with a normalized read count > 30) and weakly bound sites (i.e. no consensus IDR peak and normalized count <25). Regions were PCR amplified from mouse genomic DNA and subcloned into the pGL4.11 vector using standard Gibson assembly. See Table S1 for genomic locations and primers.
To conduct the luciferase assays, 400,000 mouse cortical neurons were plated onto 24-well culture dishes and transfected with plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. At 5DIV, neurons were transfected with 1 μg of total DNA consisting of 450 ng of firefly luciferase reporter DNA in pGL4.11, 50 ng pGL4.74 renilla luciferase reporter DNA (Promega), and 500 ng of shRNA construct. 2 μl of Lipofectamine (Invitrogen) was used for each 1 μg of DNA. DNA-lipofectamine complexes were added drop wise to neurons and incubated for 2 hours, after which the transfection media was replaced with conditioned neuronal media. Neurons were silenced on 6DIV overnight by the addition of 1 μM TTX (Abcam ab120055) and 100 μM AP5 (Thermo Fisher 01–061-0). At 7DIV, neurons were depolarized by the addition of 55 mM KCl and lysates were collected 6 hours post KCl addition. Briefly, neurons were washed 2X with PBS and lysed by the addition of 500 μl of Passive Lysis Buffer (Dual-Luciferase Reporter Assay System, Promega, E1910). 20 μl of each lysate was added to one well of a costar white polystyrene 96-well assay plate (Corning). Luciferase Assay reagent II (LARII) and Stop & Glo reagent (Dual-luciferase assay system, Promega) were added to neuronal lysates and luciferase/renilla measurements made with a Synergy 4 Hybrid Microplate Reader (BioTek). To control for variations in transfection efficiency and cell lysate generation, all values are displayed as luciferase/renilla ratios. With each experiment, 2–4 independent wells were transfected, and luciferase/renilla ratios from independent wells transfected within an experiment were averaged for each condition. Biological replicates conducted on separate days were performed a minimum of three times for each enhancer construct and shRNA condition.
ChIP-seq library preparation
For ChIP of transcription factors and histone modifications, primary neurons were cross-linked via addition of 1% formaldehyde for 10 min at room temperature and quenched by the addition of 0.125 M glycine for 5 min at room temperature. To ChIP the NCoR2 complex, we found it necessary to perform dual crosslinking by incubating neurons for 30 min with 1.5 mM EGS (ethylene glycol bis(succinimidyl succinate) (Fisher, 21565)), followed by 10 min incubation in 1% formaldehyde and standard quenching for 5 minutes at room temperature with 0.125 M glycine. Nuclei were isolated from cell pellets by 10 minute incubation in lysis buffer 1 (in mM: 100 HEPES-NaOH pH 7.5, 280 NaCl, 2 EDTA, 2 EGTA, 0.5% Triton X-100, 1% NP-40, 20% Glycerol) followed by washing in buffer containing 10 mM Tris-HCl pH 8.0, 200 mM NaCl. Chromatin was sheared using a Bioruptor (Diagenode) on high power mode for 50 cycles with 30 sec pulses in sonication buffer (in mM: 10 Tris-HCl pH 8.0, 100 NaCl, 1 EDTA, 0.5 EGTA, 0.1% Na-Deoxycholate, 0.5% N-Lauroylsarcosine). For H3K27ac ChIP, 10 mM sodium butyrate (Fisher NC9851678) was added to all buffers.
Following sonication, chromatin was supplemented with 1% Triton and was incubated overnight with the following antibodies coupled to Protein A Dynabeads (Thermo Fisher, 10002D): H3K27ac (Abcam 4729, 0.2 μg for 2.5 million cells), 5–8 μg ARNT2 (in house protein A purified anti-serum; see below), 8 μg NPAS4 (custom made protein A purified anti-serum), NCoR2 (Millipore, 06891; 5 μg for 60–70 μg chromatin; Thermo PA1–843; 5 μg for 60–70 μg chromatin (40–50 million cells)); TBL1 (Abcam 24528; 5 μg for 60–70 μg chromatin (40–50 million cells)); HDAC3 (Santa Cruz sc11416X; 5 μg for 60–70 μg chromatin (40–50 million cells). ChIPs were performed at least twice with a given antibody from independent neuronal cultures. Libraries were generated using the Ovation Ultralow V2 kit (Nugen, 0344–32) according to the manufacturer’s instructions and PCR amplified for 13–16 cycles, depending on antibody. Library quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies). Seventy-five bp reads were generated on the Illumina Nextseq 500 and subsequently analyzed with our standardized ChIP-seq data analysis pipeline (below).
RNA-seq Library Preparation
Tissue from AAV-injected hippocampi was microdissected to include portions with maximal florescence while maintaining consistency in region between control and experimental groups. For in vivo Arnt2 knockdown RNA-sequencing experiments, 3 independent mice were injected with AAV2/1 expressing a non-targeting control shRNA (U6-control shRNA-CAG-Gfp) into one hippocampal hemisphere and an AAV2/1 expressing Arnt2 shRNA #1 (U6-Arnt2 shRNA #1-CAG-mcherry) into the contralateral side. Each hemisphere from a separate mouse constituted an independent replicate. For in vivo Ncor2 KO RNA-sequencing experiments, 3–5 independent mice were injected with AAV2/1 expressing truncated Cre (ΔCre-mCherry) into one hippocampal hemisphere and AAV2/1 expressing Cre-GFP into the contralateral side. Each hemisphere from a separate mouse constituted an independent replicate. Tissue was collected in Trizol reagent and RNA was extracted using the RNAeasy Kit (Qiagen) according to the manufacturer’s instructions. In the case of in vitro RNA-seq on NPAS4-overexpressing cells, separate neuronal cultures transduced on independent days constituted biological replicates. For RNA collection from cultured samples, cells were washed twice with PBS to remove dead cells and scraped immediately into Trizol. Total RNA (1 μg) was used to generate libraries following rRNA depletion (NEBNext, E6310X) according to the manufacturer’s instructions (NEBNext, E7420). Seventy-five bp reads were generated on the Illumina Nextseq 500 and subsequently analyzed with our standardized RNA-seq data analysis pipeline (below).
ATAC-seq Library Preparation
To assess regions of open chromatin, primary cultured neurons were washed twice with cold PBS and incubated in 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal-630 to release nuclei. Experiments were performed in duplicate from independent cultures of neurons on independent days. 50,000 nuclei per condition were resuspended in 20 μl of ice-cold water and transposed using the Nextera DNA Library Prep Kit (Illumina, FC-121–1030) as previously described (Buenrostro et al., 2013). Transposition was carried out for 30 min at 37ºC. Transposed DNA fragments from individual samples were purified, independently barcoded, and amplified for 11 cycles. ATAC-seq libraries were selected for fragments ranging 200–1000 bp by gel electrophoresis and sequenced on the Illumina Nextseq 500 with seventy-five bp single-end reads. Samples were aligned with our standard mapping pipeline (see below).
For in vivo Arnt2 knockdown ATAC-seq experiments, control mice were injected with AAV2/1 expressing a non-targeting control shRNA and separate mice were injected with AAV2/1 expressing Arnt2 shRNA #1. Cells from both hippocampi were pooled per mouse and independent replicates consisted of independent mice. For ATAC-seq libraries generated from CA1 pyramidal neurons in vivo, CamkIIa-expressing CA1 pyramidal neurons were isolated using the INTACT method (Mo et al., 2015). Briefly, the hippocampi were isolated from CamkIIaCre/-; Sun1-Gfpfl/- mice injected with AAVs expressing control shRNA or Arnt2 shRNA#1. Hippocampi were dounced 15X in buffer HB (0.25M sucrose, 25mM KCl, 5mM MgCl2, 20mM Tricine-KOH, pH 7.8, 1mM DTT, 0.15mM spermine, 0.5 mM spermidine) to release nuclei. Nuclei were purified by spinning through an iodixanol gradient at 10,000 rcf. Nuclei expressing SUN1-GFP on the nuclear membrane in a Cre-dependent manner were isolated by incubating the nuclear suspension with 5 μg of anti-GFP antibody (Invitrogen G10362) for 30 min at 4°C. Antibody-coated nuclei were subsequently captured by incubation with magnetic Protein G Dynabeads (Thermo Fisher). Following isolation and counting, approximately 20,000 nuclei per condition were lysed and transposed as described above.
Immunoprecipitation
To prepare antibody bead complexes, Protein A Dynabeads (Life Technologies) were incubated with antibodies [NPAS4 (20 μg/45–50 million cells), ARNT2 (20 μg/35–50 million cells), rabbit IgG (Santa Cruz H270 (sc-66931); 20 μg/45–50 million cells)] for 30 min at room temperature followed by washing in NE1 buffer (20 mM HEPES pH 7.9, 10 mM KCl, 1 mM MgCl2, 0.1% Triton X100, 1 mM DTT, 250 mM NaCl) to remove unbound antibody. Crude nuclear extracts from primary cortical neurons were prepared by lysing cells in NE1 buffer (20 mM HEPES pH 7.9, 10 mM KCl, 1 mM MgCl2, 0.1% Triton X100, 1 mM DTT) for 10 min. Nuclei were pelleted by gentle centrifugation (2000 rpm) and resuspended in 1 packed nuclear volume of NE1 buffer. To facilitate release of chromatin-associated proteins, cells were incubated for 30 min at 4 degrees with Benzonase endonuclease (Sigma E1014, >25KU (1 μL/60 million nuclei)) followed by addition of NaCl a final concentration of 300 mM. Following high speed centrifugation to remove insoluble material, lysates were diluted 1:2 and incubated for 2 hours with prepared antibody/Dynabead complexes at 4 degrees. Beads were washed 4 times with NE1 buffer containing 250 mM NaCl. To reduce the elution of antibody: target complexes that can obscure the detection of interacting proteins by mass spectrometry and/or western blot, immunoprecipitates were eluted by the addition of 0.1X LDS (Thermo, NP0007) diluted in PBS at room temperature for 10 min. In these elutions, bait protein (e.g. ARNT2 or NPAS4) and antibody complexes are largely retained on the beads while interacting factors are released into the supernatant. Following the 0.1X LDS elution, the proteins that remain bound to the beads were subsequently eluted by incubation of beads in LDS buffer containing 10% beta mercaptoethanol for 10 min at 95°C. Eluates from 0.1X LDS and boiled bead elutions were run on 10% Bis-Tris gels. Each sample was subdivided into 5–6 sections excised from the gel. For each sample, the peptides identified in each gel section were merged during subsequent analysis.
To perform IPs in tissue, 5–10 hippocampi were isolated from mice injected with 15–20 mg/kg kainic acid (Sigma-Aldrich, K0250) to induce strong NPAS4 expression. Hippocampi were dounced in NE1 buffer. IPs were performed as previously described for cell pellets post douncing. For isolation of NPAS4 interactors from Npas4-Flag-Ha (Npas4-FH) knockin mice (manuscript in progress), nuclear lysates from knockin mice were incubated overnight at 4°C with 60 μl of anti-M2-FLAG resin (Sigma-Aldrich, A2220). NPAS4 interacting proteins were competitively eluted off of M2 resin by incubation in 500 μg/ml 3X FLAG peptide (Sigma Aldrich, F4799) in NE1 buffer for 30 min at room temperature.
Immunoblotting
Whole-cell or nuclear extracts from primary neurons or brain tissue were resolved on 3–8% Tris-Acetate gels (Thermo Fisher, EA0375BOX) or 10% Tris-Glycine gels and transferred to nitrocelluose. Membranes were incubated overnight in the following primary antibodies: NPAS3 (Gift of Steven McKnight), NCoR2 (Thermo Fisher PA1–843 (Ab1); Millipore, 06891 (Ab2)), TBL1 (Abcam 24528 (Ab1); Santa Cruz (H-3): sc-137006 (Ab2)), HDAC3 (Santa Cruz sc11416X), β-TUBULIN3 (Covance), GAPDH (Sigma G9545), MYC (EMD Millipore 05–724, Developmental Studies Hybridoma Bank 9E10), FOS (Santa Cruz, sc7202X), Histone H3 (Abcam, ab1791). Following washing, membranes were incubated with secondary antibodies conjugated to IRdye 700 or 800 and imaged with LiCOR Odyssey.
Mass spectrometry
All samples were processed according to standard procedures of Taplin Mass Spectrometry Facility (Harvard University). Briefly, gel bands of 1 mm3 were washed and dehydrated with acetonitrile for 10 min before complete dehydration using a speed-vac. Rehydrated gel pieces were incubated with 50 mM ammonium bicarbonate solution containing 12.5 ng/μL modified sequencing-grade trypsin (Promega, Madison, WI) at 4ºC for 45 min. Following removal of excess trypsin solution, samples were placed at 37ºC in 50 mM ammonium bicarbonate solution. Digested peptides were recovered by removal of ammonium bicarbonate solution, followed by washing in 50% acetonitrile and 1% formic acid before dehydration.
For liquid chromatography, samples reconstituted in 5 – 10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid) were resolved using a nano-scale reverse-phase HPLC capillary column created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip. Samples were loaded via a Famos auto sampler (LC Packings, San Francisco CA), and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid). Eluted peptides were subjected to electrospray ionization followed by entry into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide.
Recombinant protein purification for ARNT2 antibody production
A fragment of ARNT2 encoding amino acids 312 to 617 (ARNT2312−617) was cloned into the pGEX bacterial expression system (GE Life Sciences) to create an N-terminal GST fusion protein. pGEXARNT2312−617 was introduced by standard transformation techniques into the Rossetta 2(DE3)pLysS bacterial strain. Bacteria were grown to an O.D600:~0.4–0.5 before recombinant protein being induced by introduction of 0.5 mM IPTG. Bacteria were grown for 3 hours at 37ºC prior to pelleting and snap-freezing in liquid nitrogen. Bacteria were lysed by sonication in lysis buffer (in mM: 50 Tris pH 8.0, 300 NaCl, 0.05% Triton X 100, 0.5% NP-40) and immobilized on Glutathione-Sepharose beads (GE Life Sciences). Non-specific proteins were washed on column with 3× 10–15 mL of wash buffer (in mM: 50 Tris pH 8.0, 150 NaCl). Purified recombinant protein was eluted from glutathione sepharose beads with elution buffer (in mM: 50 Tris pH 8.0, 150 NaCl, 20 Reduced Glutathione). Purified recombinant protein was dialyzed against 1x PBS and stored at 4C until usage. 5–10 mg of recombinant ARNT2312−617 was injected into immuncompromised rabbits and serum was collected serially. Final purification was achieved using a Protein A purification column.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
The statistical analysis for each experiment is detailed in the figure legends. For sequencing data, the comparison of distributions between distinct peak sets was calculated by the Wilcoxon rank sum test. For luciferase assays, statistical significance was calculated by paired, one-sided t-test with Benjamini-Hochberg multiple hypothesis correction. Statistical analyses for electrophysiological recordings were computed with paired two-tailed t-tests. No statistical methods were used to predetermine sample sizes, but replicate size and number follow guidelines of the ENCODE consortium (Consortium, 2012). No other sample randomization was performed. Experiments were not performed in a blinded fashion.
Electrophysiology data acquisition and analysis
Electrophysiology data were acquired using pClamp software and a Multiclamp 700B and digitized with a DigiData 1440 data acquisition board (Axon Instruments). Data were sampled at 10 kHz and filtered at 4 kHz. Off-line data analysis was performed using custom written scripts in Igor Pro or Axograph. Experiments were discarded if the holding current was greater than −500 pA or if the series resistance was greater than 25 mOhms. Recordings were discarded if the series resistance between pairs of neurons differed by >25%. All recordings were performed at room temperature (~20–22°C).
Luciferase data quantification
To calculate the relationship between transcription factor binding and the effect on luciferase reporter activity, the luciferase/renilla reading for each shRNA-treated well was divided by the luciferase/renilla reading for the non-targeting shRNA transfected within the same experiment. Binding strength was determined by normalized read counts overlapping with luciferase regions of interest using the HOMER software package (see below). Negative ratios indicate reduced luciferase activity in shRNA-treated samples relative to control shRNA-treated neurons. Linear regression between binding strength and the effect of shRNA treatment on luciferase activity was performed in R. The significance of the regression is reported with Pearson and Spearman correlation co-efficients. For individual sites, the luciferase/renilla ratios amongst a minimum of 3 biological replicates are plotted with the standard error of the mean (Figures S1F, S2L, S5F, S6E). Significance between control shRNA-transfected samples and samples transfected with Npas4, Arnt2, or Ncor2 shRNAs were calculated by a paired, one-sided t-test with Benjamini-Hochberg multiple hypothesis correction across all tested sites by shRNA for a given gene.
Peptide quantification mass spectrometry
To determine protein identity, peptide sequences were compared against protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific, Waltham, MA). Databases include a reversed version of all the sequences. Data was filtered to between a one and two percent peptide false discovery rate. For each sample (e.g. IgG versus ARNT2 IP), total peptides for a given protein identified in each gel section were merged to generate a list of total peptides associated with a given protein. Duplicate NPAS4 IP-MS and triplicate ARNT2 IP-MS experiments were performed from independently generated cortical neuron cultures. Peptide counts are provided in Table S2. The R package EdgeR (Robinson et al., 2010) was used to identify proteins significantly enriched in ARNT2 or NPAS4 IP samples relative to control samples (IgG) using the glmTREAT() function. For the NPAS4 IP, control samples included anti-IgG IP from stimulated neurons as well as anti-NPAS4 IP from unstimulated neurons where NPAS4 is not expressed. Proteins with an FDR of <0.1 are indicated as a colored dot on Volcano plots.
Sequencing, alignment, and genome browser track generation
All experiments were sequenced on the Nextseq 500 (Illumina). Seventy-five base pair single-end reads were obtained for all datasets. ATAC-seq experiments were sequenced to a minimum depth of 20 million reads; ChIP- and RNA-seq were sequenced to a depth of at least 30 million reads. All samples were aligned to the mm10 genome using default parameters for the Subread alignment software (subread-1.4.6-p3, (Liao et al., 2013)) after quality trimming with Trimmomatic v0.33 (Bolger et al., 2014) with the following command: java -jar trimmomatic-0.33.jar SE -threads 1 -phred33 [FASTQ_FILE] ILLUMINACLIP:[ADAPTER_FILE]:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:4:20 MINLEN:45. Truseq adapters were trimmed out in ChIP- and RNA-seq experiments; Nextera adapters were specified for ATAC-seq data.
To generate UCSC genome browser tracks for ChIP- an ATAC-seq data, all aligned bam files for each replicate of a given experiment were pooled and converted to BED format with bedtools bamtobed. The 75 base pair reads were extended in the 3’ direction to 200 basepairs (bp) (average fragment length for ChIP- and ATAC-seq experiments as measured by bioAnalyzer) with the bedtools slop command using the following parameters: -l 0 -r 125 -s. Published mm10 ChIP-seq blacklisted regions (Consortium, 2012) were filtered out using the following command: bedops –not-element-of 1 [BLACKLIST_BED].
The filtered BED files were converted to coverageBED format using the bedtools genomecov command with the following options: -scale [NORM_FACTOR to scale each library to 20M reads] –bg. Finally, bedGraphToBigWig (UCSC-tools) was used to generate the bigWIG files displayed on browser tracks throughout the manuscript.
RNA-seq quantification of gene expression
To quantify gene expression, we applied the featureCounts package (Liao et al., 2013) after aligning RNA-seq libraries to the genome as described above, using a custom filtered annotation file: gencode.v17.annotation.gtf filtered for feature_type=“gene”, gene_type=“protein_coding” and gene_status=“KNOWN”) to obtain gene counts for each sample. These counts tables were TMM-normalized using the EdgeR software analysis package (Robinson et al., 2010). Any genes that were not expressed in at least 3 samples with TMM-normalized CPM>1 were dropped from further analysis. Differential expression (DE) analyses were conducted using the voom/limma analysis software packages (requiring FDR-corrected q<0.05). In the case of in vivo RNA-sequencing data in kainate-treated animals, if the animal failed to induce activity-dependent genes in the control hemisphere, both control and shRNA/KO hemispheres were excluded from subsequent analysis. Results are plotted as volcano plots, with activity-dependent gene sets recalculated for each experimental condition.
Single-cell RNA-seq sample preparation, library preparation, sequencing, and processing
Libraries generated from 2 replicates each of approximately 3,000 cells obtained from 0, 1, and 6 hour KCl depolarized 7DIV hippocampal and cortical neuronal cultures were collected using the inDrops platform (Zilionis et al., 2017). Transcripts were quality trimmed, mapped, and assigned to cells using the published inDrops pipeline (https://github.com/indrops/indrops). Cells were clustered as described in (Hrvatin et al., 2018) using Approach 1 with the python implementation of bh-tsne (Maaten, 2014) and assigned cell types based on the expression of marker genes shown in Figure S1G.
ChIP-seq and ATAC-seq peak calling
Regions of ATAC-seq enrichment were determined using MACS2 (v 2.1.0) parameters -p 1e-5 --nolambda --keep-dup all --slocal 10000, as previously described (Buenrostro et al., 2013). Peaks from individual sample replicates were intersected to find only reproducible regions of enrichment. Blacklist regions were removed as previously described (Consortium, 2012). To identify sites with enriched ChIP-seq signal (peaks), we applied the IDR pipeline (Li, 2011) using the MACS2 peak calling algorithm (Zhang et al., 2008) with the following parameters: --nomodel --extsize 200 --keep-dup all. An IDR threshold of .01 was used for self-consistency, true replicate, and pooled-consistency analyses. The ‘optThresh’ cutoff was then used to obtain a final set of high-confidence, reproducible ChIP-seq peaks for each factor or chromatin modification at each stimulus condition tested.
Identification of the set of active elements
After calling ATAC-seq data and H3K27ac ChIP-Seq peaks, we constructed a set of all putative functional regulatory elements in the embryonic cultured neuronal system by taking the union of all sites that showed significant (IDR<0.01) ATAC or H3K27Ac ChIP-seq signal in any of the unstimulated, 2 hour KCl depolarized, or 6-hour KCl depolarized conditions ± any viral treatment, resulting in a total of 31,138 sites genome-wide which we defined as the set of active regulatory elements across our experimental paradigms (Figure S1C).
Calculations of significance for peak set overlap
We used a bootstrapping approach to determine the significance of overlap of NPAS4, ARNT2, and NCoR2 complex components as measured by ChIP-seq genome-wide (Figures 3F, S3D). For each factor, sample size-matched enhancer sets were randomly sampled without replacement from the list of all enhancers. These sets were then intersected and the number of three set intersections was measured across 10,000 such samples. The p-value reported represents the fraction of such samples that showed a greater degree of intersection than the observed value using the true NPAS4-, ARNT2-, and NCoR2-bound enhancer sets.
Identification of activity-dependent regulatory elements as measured by H3K27ac
The R package Edger (Robinson et al., 2010) was used to identify differentially acetylated regions after TMM normalization. P-values were calculated across the 2–0 KCl stimulus conditions using the glmTREAT() function with a fold change threshold of 1.5 and FDR < 0.05. Annotations that were significantly differentially acetylated using these thresholds constituted the set of inducible regulatory elements referenced in the paper.
Quantification of ChIP- and ATAC-seq signal for visualization
We used the HOMER software package to quantify the number of reads present across all active regulatory elements from all ChIP- and ATAC-seq experiments by constructing tag directories (makeTagDirectory –fragLength 200) and using the annotatePeaks.pl script (Heinz et al., 2010) with the following parameters to obtain raw counts tables by locus: -len 200 -size given -noadj -noann -nogene. Any annotations within 1 kb of an annotated TSS were considered ‘promoter’ regulatory elements; all other annotations were termed enhancers.
Quantile box-whisker plots were generated using the counts tables described above. Active regulatory elements with the desired features were ranked and partitioned according to NPAS4 2 hour KCl or ARNT2 0 hour KCl ChIP signal intensity. Negative regions were chosen as sites with the lowest NPAS4/ARNT2 ChIP-signal in our active regulatory element gene set. Plots display median and interquantile range, notches denote confidence interval of median.
Fixed line plots were generated with Homer’s annotatePeaks.pl script from the bioreplicate pooled bed files, using the following parameters: -len 200 -size 4000 –ghist -hist 25 -noann -nogene to generate a master table of normalized read density per bin at each of the 31,138 active elements. All samples were normalized to 10M reads per Homer’s default processing.
Aggregate plots were generated by averaging the count density tables output by the commands above across the peak set under consideration.
Motif discovery
Starting from the initial set of active regulatory elements, IDR-confirmed NPAS4 peaks (IDR <.01) from the 2 hour KCl depolarized condition were intersected with ARNT2 0 hour KCl peaks to identify NPAS4POS/ARNT2POS and NPAS4NEG/ARNT2POS peak sets. These were sorted by NPAS4 and ARNT2 ChIP-signal strength respectively, TSS-proximal elements were removed, and the top 5,000 sites were used as input for the KMACS motif-calling software (Guo et al., 2018) with the following arguments: --k_min 5 --k_max 13 --k_top 10 (only 4,632 NPAS4NEG/ARNT2POS active enhancers survived pre-filtering). For each peak set, the other was used as the negative control using the --neg_seq parameter to enrich for set-specific motifs. An output metric ranking motif results, the scaled output partial area under receiver operating characteristic (up to false positive rate of .1, auc), is displayed for each analysis.
DATA AND SOFTWARE AVAILABILITY
Raw and processed sequencing data are deposited in Gene Expression Omnibus accession number GSE121660. Analyzed single cell data from visual cortex (Hrvatin et al. 2018) is provided under GEO accession number GSE102827. Scripts for individual analyses are available upon request.
ADDITIONAL RESOURCES
N/A
Supplementary Material
TABLE S1. Related to Figures 1, 2, 5, 6. List of regions and associated primers cloned into pGL4.11 luciferase reporter construct.
TABLE S2. Related to Figures 3, 5. Full list of proteins and associated peptide counts in ARNT2 and NPAS4 IP-MS data from primary cortical neurons.
Highlights.
ARNT2:NCoR2 complexes suppress activity-dependent genes in the absence of activity
ARNT2 recruits NPAS4 or NCoR2 to induce activity-dependent transcription
ARNT2 controls the strength of somatic inhibition by this transcriptional regulation
Acknowledgements
We thank the Greenberg lab for feedback. We thank T. Vierbuchen, E. Ling, L. Boxer, E. Griffith, E. Greer, and D. Ginty for their critical reading of the manuscript. This work was supported by National Institutes of Health (NIH) grant R37 NS028829 and the ROADS Program of F. Hoffmann-La Roche Ltd. to M.E.G; NIH grant 5T32 NS007484-15 and Good Ventures Life Sciences Research Fellowship to E.A.P; NIH grant T32GM007753 to M.A.N; the Stuart H.Q. and Victoria Quan and the Department of Neurobiology Graduate Fellowships to E.L.Y.; and NIH grant 5T32AG000222-23 to S.H.
Footnotes
Declaration of interests:
M.E.G. is on the Board of Directors of and holds equity in Allergan, plc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adikesavan AK, Karmakar S, Pardo P, Wang L, Liu S, Li W, and Smith CL (2014). Activation of p53 transcriptional activity by SMRT: a histone deacetylase 3-independent function of a transcriptional corepressor. Mol Cell Biol 34, 1246–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghagen H, Ragnhildstveit E, Krogsrud K, Thuestad G, Apriletti J, and Saatcioglu F (2002). Corepressor SMRT functions as a coactivator for thyroid hormone receptor T3Ralpha from a negative hormone response element. J Biol Chem 277, 49517–49522. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, and Greenberg ME (2013). The activitydependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion AM, Link WA, Ledo F, Mellstrom B, and Naranjo JR (1999). DREAM is a Ca2+-regulated transcriptional repressor. Nature 398, 80–84. [DOI] [PubMed] [Google Scholar]
- Chawla S, Vanhoutte P, Arnold FJ, Huang CL, and Bading H (2003). Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem 85, 151–159. [DOI] [PubMed] [Google Scholar]
- Chen JD, and Evans RM (1995). A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377, 454–457. [DOI] [PubMed] [Google Scholar]
- Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, and Fan G (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JM, Kim TK, West AE, Nord AS, Markenscoff-Papadimitriou E, and Lomvardas S (2015). Genomic Views of Transcriptional Enhancers: Essential Determinants of Cellular Identity and Activity-Dependent Responses in the CNS. J Neurosci 35, 13819–13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, and Ziff EB (1984). Stimulation of 3T3 cells induces transcription of the c-fos protooncogene. Nature 311, 433–438. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB, and Greene LA (1986). Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science 234, 80–83. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, and Shiekhattar R (2000). A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev 14, 1048–1057. [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Consortium on the Genetics of, S., Group, P.S., et al. (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslin K, and Coutellier L (2017). Npas4 deficiency and prenatal stress interact to affect social recognition in mice. Genes Brain Behav. [DOI] [PubMed] [Google Scholar]
- Hill CS (2016). Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JY, Schaukowitch K, Farbiak L, Kilaru G, and Kim TK (2016). Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci 19, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, Gabel HW, Gilbert CS, Griffith EC, and Greenberg ME (2015). Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A 112, 6800–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenecke N, Johnston J, He Q, Meier S, and Zeitlinger J (2017). Drosophila poised enhancers are generated during tissue patterning with the help of repression. Genome Res 27, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JH, and Nedivi E (2011). Activity-regulated genes as mediators of neural circuit plasticity. Prog Neurobiol 94, 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, and Greenberg ME (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, et al. (2014). Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci 17, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly AR, Spiegel I, Patrizi A, Centofante E, Bazinet JE, Tzeng CP, Mandel-Brehm C, Harmin DA, Adesnik H, Fagiolini M, et al. (2016). Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature 531, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellstrom B, Sahun I, Ruiz-Nuno A, Murtra P, Gomez-Villafuertes R, Savignac M, Oliveros JC, Gonzalez P, Kastanauskaite A, Knafo S, et al. (2014). DREAM controls the on/off switch of specific activity-dependent transcription pathways. Mol Cell Biol 34, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, and Curran T (1986). Role of ion flux in the control of c-fos expression. Nature 322, 552555. [DOI] [PubMed] [Google Scholar]
- Mottis A, Mouchiroud L, and Auwerx J (2013). Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 27, 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ (2014). Epigenetic mechanisms of drug addiction. Neuropharmacology 76 Pt B, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, PlajzerFrick I, Shoukry M, Afzal V, et al. (2013). Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Pattabiraman K, Visel A, and Rubenstein JLR (2015). Genomic perspectives of transcriptional regulation in forebrain development. Neuron 85, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooe N, Saito K, and Kaneko H (2009). Characterization of functional heterodimer partners in brain for a bHLH-PAS factor NXF. Biochim Biophys Acta 1789, 192–197. [DOI] [PubMed] [Google Scholar]
- Ooe N, Saito K, Mikami N, Nakatuka I, and Kaneko H (2004). Identification of a novel basic helixloop-helix-PAS factor, NXF, reveals a Sim2 competitive, positive regulatory role in dendritic-cytoskeleton modulator drebrin gene expression. Mol Cell Biol 24, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, and Natoli G (2013). Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171. [DOI] [PubMed] [Google Scholar]
- Panteleeva I, Rouaux C, Larmet Y, Boutillier S, Loeffler JP, and Boutillier AL (2004). HDAC-3 participates in the repression of e2f-dependent gene transcription in primary differentiated neurons. Ann N Y Acad Sci 1030, 656–660. [DOI] [PubMed] [Google Scholar]
- Pattabiraman K, Golonzhka O, Lindtner S, Nord AS, Taher L, Hoch R, Silberberg SN, Zhang D, Chen B, Zeng H, et al. (2014). Transcriptional regulation of enhancers active in protodomains of the developing cerebral cortex. Neuron 82, 989–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, and Rosenfeld MG (2004). A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116, 511–526. [DOI] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, and Rosenfeld MG (2010). Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet 11, 109–123. [DOI] [PubMed] [Google Scholar]
- Peterson TJ, Karmakar S, Pace MC, Gao T, and Smith CL (2007). The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor alpha transcriptional activity. Mol Cell Biol 27, 5933–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons L, Cordier MP, Labalme A, Till M, Louvrier C, Schluth-Bolard C, Lesca G, Edery P, and Sanlaville D (2015). A new syndrome of intellectual disability with dysmorphism due to TBL1XR1 deletion. Am J Med Genet A 167A, 164–168. [DOI] [PubMed] [Google Scholar]
- Qiu Z, and Ghosh A (2008). A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 60, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthi K, Fropf R, Belfort GM, Fitzmaurice HL, McKinney RM, Neve RL, Otto T, and Lin Y (2011). Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science 334, 1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, et al. (2007). Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56, 517–529. [DOI] [PubMed] [Google Scholar]
- Sabatini PV, Speckmann T, Nian C, Glavas MM, Wong CK, Yoon JS, Kin T, Shapiro AMJ, Gibson WT, Verchere CB, et al. (2018). Neuronal PAS Domain Protein 4 Suppression of Oxygen Sensing Optimizes Metabolism during Excitation of Neuroendocrine Cells. Cell Rep 22, 163–174. [DOI] [PubMed] [Google Scholar]
- Sando R 3rd, Gounko N, Pieraut S, Liao L, Yates J 3rd, and Maximov A (2012). HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, and Greenberg ME (2014). Npas4 regulates excitatory-inhibitory balance within neural circuits through celltype-specific gene programs. Cell 157, 1216–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, et al. (2017). Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164 e1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci 20, 476483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabet AC, Leroy C, Dupont C, Serrano E, Hernandez K, Gallard J, Pouvreau N, Gadisseux JF, Benzacken B, and Verloes A (2014). De novo deletion of TBL1XR1 in a child with non-specific developmental delay supports its implication in intellectual disability. Am J Med Genet A 164A, 2335–2337. [DOI] [PubMed] [Google Scholar]
- Takata A, Ionita-Laza I, Gogos JA, Xu B, and Karayiorgou M (2016). De Novo Synonymous Mutations in Regulatory Elements Contribute to the Genetic Etiology of Autism and Schizophrenia. Neuron 89, 940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Carreira MB, Cooper YA, Bobadilla AC, Heinsbroek JA, Koike N, Larson EB, Balmuth EA, Hughes BW, Penrod RD, et al. (2017). HDAC5 and Its Target Gene, Npas4, Function in the Nucleus Accumbens to Regulate Cocaine-Conditioned Behaviors. Neuron 96, 130–144 e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh CH, Lam KK, Loh CC, Loo JM, Yan T, and Lim TM (2006). Neuronal PAS domain protein 1 is a transcriptional repressor and requires arylhydrocarbon nuclear translocator for its nuclear localization. J Biol Chem 281, 34617–34629. [DOI] [PubMed] [Google Scholar]
- Telese F, Ma Q, Perez PM, Notani D, Oh S, Li W, Comoletti D, Ohgi KA, Taylor H, and Rosenfeld MG (2015). LRP8-Reelin-Regulated Neuronal Enhancer Signature Underlying Learning and Memory Formation. Neuron 86, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho JH, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, et al. (2018). Different Neuronal Activity Patterns Induce Different Gene Expression Programs. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell 68, 1067–1082 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Su X, Potluri N, Kim Y, and Rastinejad F (2016). NPAS1-ARNT and NPAS3-ARNT crystal structures implicate the bHLH-PAS family as multi-ligand binding transcription factors. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, Guthrie AN, Oldenborg A, Heiney SA, Ohmae S, et al. (2016). Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kalkum M, Chait BT, and Roeder RG (2002). The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell 9, 611–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Related to Figures 1, 2, 5, 6. List of regions and associated primers cloned into pGL4.11 luciferase reporter construct.
TABLE S2. Related to Figures 3, 5. Full list of proteins and associated peptide counts in ARNT2 and NPAS4 IP-MS data from primary cortical neurons.
Data Availability Statement
Raw and processed sequencing data are deposited in Gene Expression Omnibus accession number GSE121660. Analyzed single cell data from visual cortex (Hrvatin et al. 2018) is provided under GEO accession number GSE102827. Scripts for individual analyses are available upon request.