

Abstract
GABAA receptors (GABAARs) are targets for important classes of clinical agents (e.g., anxiolytics, anticonvulsants, and general anesthetics) that act as positive allosteric modulators (PAMs). Previously, using photoreactive analogs of etomidate ([3H]azietomidate) and mephobarbital [[3H]1-methyl-5-allyl-5-(m-trifluoromethyl-diazirynylphenyl)barbituric acid ([3H]R-mTFD-MPAB)], we identified two homologous but pharmacologically distinct classes of general anesthetic binding sites in the α1β3γ2 GABAAR transmembrane domain at β+–α− (β+ sites) and α+–β−/γ+–β− (β− sites) subunit interfaces. We now use competition photolabeling with [3H]azietomidate and [3H]R-mTFD-MPAB to identify para-substituted propofol analogs and other drugs that bind selectively to intersubunit anesthetic sites. Propofol and 4-chloro-propofol bind with 5-fold selectivity to β+, while derivatives with bulkier lipophilic substitutions [4-(tert-butyl)-propofol and 4-(hydroxyl(phenyl)methyl)-propofol] bind with ∼10-fold higher affinity to β− sites. Similar to R-mTFD-MPAB and propofol, these drugs bind in the presence of GABA with similar affinity to the α+–β− and γ+–β− sites. However, we discovered four compounds that bind with different affinities to the two β− interface sites. Two of these bind with higher affinity to one of the β− sites than to the β+ sites. We deduce that 4-benzoyl-propofol binds with >100-fold higher affinity to the γ+–β− site than to the α+–β− or β+–α− sites, whereas loreclezole, an anticonvulsant, binds with 5- and 100-fold higher affinity to the α+–β− site than to the β+ and γ+–β− sites. These studies provide a first identification of PAMs that bind selectively to a single intersubunit site in the GABAAR transmembrane domain, a property that may facilitate the development of subtype selective GABAAR PAMs.
Introduction
GABAA receptors (GABAARs), the receptors primarily responsible for fast inhibitory transmission in the brain, are key targets for a structurally diverse class of drugs that act as general anesthetics, including fluorinated ethers, propofol, etomidate, barbiturates, and steroids, which all act as positive allosteric modulators (PAMs) (Franks, 2008; Zeller et al., 2008). Despite their general efficacy, general anesthetics are dangerous medicines that have therapeutic indices ranging from 2 to 4, and there is increasing concern about anesthetic toxicities and undesirable side effects that makes important the development of safer anesthetics (Forman, 2010; McKinstry-Wu et al., 2015). Other GABAAR PAMs act as anticonvulsants, with use limited by side effects (Rogawski, 2006; Greenfield, 2013). Members of the superfamily of pentameric ligand-gated ion channels, GABAARs are assembled from five identical or homologous subunits, and the 19 human GABAAR subunit genes (including α1–6, β1–3, and γ1–3) result in the expression of at least a dozen physiologic relevant subtypes that are widely distributed throughout the central nervous system (Chua and Chebib, 2017). With multiple GABAAR subtypes as targets in vivo, anesthetics and anticonvulsants may elicit desired actions through some subtypes and undesirable actions through others (Bonin and Orser, 2008; Greenfield, 2013). Therefore, development of GABAAR subtype selective PAMs may lead to therapeutic agents with fewer undesirable side effects.
Photolabeling studies with photoreactive anesthetics and mutational analyses, in conjunction with recently solved structures of GABAARs (Miller and Aricescu, 2014; Phulera et al., 2018; Zhu et al., 2018; Masiulis et al., 2019) and other pentameric ligand-gated ion channels, establish the presence of multiple binding sites for anesthetics and other PAMs within a single GABAAR (Sieghart, 2015; Chua and Chebib, 2017). In αβγ GABAARs, the most common subtype expressed at postsynaptic sites, GABA binding sites are located in the extracellular domain (ECD) at the two β+–α− subunit interfaces, and benzodiazepines bind to a homologous site at the α+–γ− subunit interface (Sigel and Ernst, 2018) (Fig. 1). In the transmembrane domain (TMD), M2 helices from each subunit come together to form the ion channel. Amino acids from the M1, M2, and M3 helices contribute to subunit interfaces, which contain binding sites for propofol, etomidate, and barbiturates in the extracellular third of the TMD (Chiara et al., 2013; Forman and Miller, 2016) and distinct binding sites for steroid anesthetics nearer the cytoplasmic surface (Hosie et al., 2006; Laverty et al., 2017; Miller et al., 2017). Photoaffinity labeling with analogs of etomidate ([3H]azietomidate) and mephobarbital [[3H]1-methyl-5-allyl-5-(m-trifluoromethyl-diazirynylphenyl)barbituric acid ([3H]R-mTFD-MPAB)] identified two homologous, pharmacologically distinct classes of intersubunit general anesthetic binding sites (Chiara et al., 2013) (Fig. 1). Azietomidate and etomidate bind with 100-fold selectivity to sites in the two β+–α− subunit interfaces, while R-mTFD-MPAB binds with 50-fold selectivity to the sites within the α+–β−/γ+–β− subunit interfaces. Competition photolabeling assays using these photoprobes established that other barbiturates (pentobarbital and thiopental) bind with less than 8-fold selectivity to the β− sites, while propofol binds with ∼2-fold selectivity to the β+ sites. Mutational analyses predict that the anticonvulsant PAMs loreclezole and tracazolate also bind to the β+ sites (Wingrove et al., 1994; Thompson et al., 2002). The α+–β−and γ+–β− sites are intrinsically nonequivalent due to differences in the amino acids contributed by αM3 and γM3 helices to the + side of β− interfaces, but in the presence of GABA, R-mTFD-MPAB binds with similar high affinity to both sites (Jayakar et al., 2015).
Fig. 1.

Locations in a α1β3γ2 GABAAR of binding sites in the ECD for GABA and benzodiazepines and in the TMD for general anesthetics. Subunits are arranged β–α–β–α–γ, counterclockwise, as viewed from the extracellular space. GABA binds to sites at the interface between the β and α subunits, referred to as the β+–α− subunit interface, and benzodiazepines bind to a homologous site at the α+–γ− interface. Indicated in the TMD are the four transmembrane helices (M1–M4) within each subunit and the high-affinity binding sites for azietomidate/etomidate at the β+–α− interfaces and for R-mTFD-MPAB at homologous sites at the α+–β− and γ+–β− subunit interfaces (Chiara et al., 2013; Forman and Miller, 2016). Propofol binds with similar affinity to the β+ and β− sites (Chiara et al., 2013; Forman and Miller, 2016), and steroid anesthetics bind to a distinct site at the β+–α− subunit interface (Hosie et al., 2006; Laverty et al., 2017; Miller et al., 2017).
To further our understanding of the structural determinants that contribute to selective binding of GABAAR PAMs to the intersubunit binding sites in the TMD, we used competition photolabeling assays with [3H]azietomidate and [3H]R-mTFD-MPAB to determine the affinities of a panel of propofol derivatives and structurally unrelated anticonvulsants for the β+ and β− sites and to identify drugs that bind nonequivalently to the β− intersubunit sites. Of the 11 agents studied, four had the highest affinity for the β+ sites, whereas seven had higher affinity for the β− sites. Four agents had different affinities for the α+–β− and γ+–β− sites, with two of these, 4-benzoyl-2,6-diisopropylphenol (4-benzoyl-propofol) and loreclezole, binding with the highest affinity to the γ+–β− and α+–β− sites, respectively.
Materials and Methods
Materials.
R-mTFD-MPAB (1-methyl-5-allyl-5-(m-trifluoromethyldiazirynylphenyl]barbituric acid), [3H]R-mTFD-MPAB (38 Ci/mmol, 26 μM in ethanol), and [3H]azietomidate (19 Ci/mmol, 53 μM in ethanol) were synthesized and tritiated previously (Savechenkov et al., 2012; Jayakar et al., 2014). 4-(Tert-butyl)-2,6-diisopropylphenol (4-(tert-butyl)-propofol) was from Chiron AS (Trondheim, Norway). 4-Acetyl-2,6-diisopropylphenol (4-acetyl-propofol), 4-chloro-2,6-diisopropylphenol (4-Cl-propofol), and 4-benozyl-propofol were synthesized as described previously (Trapani et al., 1998; de Lassauniere et al., 2005), with the identity and purity of the compounds confirmed by 1H NMR. 4-Benzyl-2,6-diisopropylphenol (4-benzyl-propofol), racemic 4-(hydroxyl(phenyl)methyl)-2,6-diisopropylphenol (4-[hydroxyl(phenyl)methyl]-propofol), and the S-enantiomer were synthesized as described previously (Jarava-Barrera et al., 2016). R-4-(hydroxyl(phenyl)methyl)-propofol was synthesized following the same procedure (Jarava-Barrera et al., 2016), and the purity and structure were verified by NMR spectroscopy and by measurement of specific rotation. (R)-Etomidate, propofol, GABA, stiripentol, topiramate, valnoctamide, felbamate, soybean asolectin, and the FLAG peptide (DYKDDDDK) were from Sigma-Aldrich. Loreclezole and tracazolate were from Tocris. Bicuculline methochloride was from Abcam. Ethyl 2-(4-bromophenyl)-1-(2,4-dichlorophenyl)-1H-4-imidazolecarboxylate (TG-41) was a gift from Dr. Douglas Raines (Department of Anesthesia and Critical Care, Massachusetts General Hospital). The detergents n-dodecyl β-d-maltopyranoside and 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (CHAPS) were from Anatrace-Affymetrix.
Expression and Purification of Human α1β3γ2 GABAARs.
α1β3γ2L GABAARs, with the α1 subunit containing a FLAG epitope at the N terminus of the mature subunit (MRK…SYGDYKDDDDKQPS…), were expressed in a tetracycline-inducible, stably transfected HEK293S cell line and purified on an anti-FLAG affinity resin as described previously (Chiara et al., 2013; Dostalova et al., 2014). In brief, membranes were solubilized for 2.5 hours in purification buffer (Chiara et al., 2013; Dostalova et al., 2014) supplemented with 30 mM n-dodecyl β-d-maltopyranoside. Column wash and elution buffers contained 5 mM CHAPS and 0.2 mM asolectin, with 1.5 mM FLAG peptide used for elution of GABAAR. The concentration of GABAAR at the different stages of purification was assayed by determination of [3H]muscimol binding by use of a filtration assay after precipitation with polyethylene glycol (Li et al., 2006; Chiara et al., 2013). The GABAAR binding site concentration was measured with 250 nM [3H]muscimol, with 1 mM GABA added to determine nonspecific binding. For each purification, etomidate modulation of 2 nM [3H]muscimol binding was determined as previously described (Chiara et al., 2013). For 16 independent purifications, each starting with membrane fractions containing ∼4 nmol of [3H]muscimol binding sites, final column eluates contained ∼1 nmol of binding sites. First and second elutions, each of 12 ml, contained site concentrations (mean ± S.D.) of 54 ± 8 and 31 ± 7 nM, respectively. For elutions 1 and 2, etomidate at 10 μM enhanced binding of 2 nM [3H]muscimol by 140% ± 20% and 134% ± 18%, respectively. Receptor aliquots (1 ml) were flash frozen in liquid nitrogen and stored at −80°C until use.
Photoaffinity Labeling of α1β3γ2 GABAARs.
To measure photoincorporation at the subunit level, 30–60 μl of α1β3γ2 GABAAR was photolabeled per condition with [3H]R-mTFD-MPAB (3 μCi) or [3H]azietomidate (2 μCi) at concentrations of 1–3 μM. After transferring appropriate volumes of [3H]azietomidate or [3H]R-mTFD-MPAB stock solutions to glass test tubes and evaporating solvent under an argon stream, the radioligand was resuspended with GABAAR (∼0.7 ml) for 30 minutes on ice with occasional vortexing. Aliquots of 0.35 ml were then equilibrated for 15 minutes with GABA (300 μM) or bicuculline methochloride (100 μM). During this time, appropriate volumes of nonradioactive drugs were added by use of a 1-μl syringe to 10-μl aliquots of GABAAR (without radioligand) in 350-μl sample vials (catalog number CERT5000-69LV; Thermo Fisher Scientific). GABAAR equilibrated with radioligand was then added to samples containing nonradioactive drugs and further incubated for 30 minutes on ice to allow all binding and conformational transitions to equilibrate before transfer to 96-well plastic plates (Corning catalog number 2797) for photolabeling. Samples were irradiated using a 365-nm lamp (Spectroline Model EN-16; Spectronics Corp, Westbury, NJ) for 30 minutes on ice at 0.5- to 1-cm distance from the lamp. Stock solutions of 1 M propofol or 60 mM nonradioactive R-mTFD-MPAB, loreclezole, tracazolate, ivermectin, and etomidate were prepared in ethanol. Stock solutions of the propofol analogs were prepared at 60 mM in methanol. TG-41 (25 mM) was prepared in dimethylsulfoxide. Bicuculline methochloride (6 mM) and GABA (100 mM) were prepared in water. Ethanol or methanol at 0.5% (v/v) (or 0.2% dimethylsulfoxide for TG-41 experiments) was present in all samples during photolabeling. Those solvent concentrations altered photolabeling by <5%. All additions of nonradioactive drugs were to GABAARs in elution buffer containing 0.2 mM asolectin and 5 mM CHAPS, a solution in which the drugs were soluble at the highest concentrations tested.
Photolabeled samples were solubilized in SDS sample buffer and incubated for 30–60 minutes at room temperature before fractionation by SDS-PAGE on Tris-glycine gels (6%) as previously described (Chiara et al., 2013). Gel bands were visualized with GelCode Blue Safe Protein Stain (Thermo Fisher Scientific), with the gels treated with prestain solution [50% (v/v) methanol and 10% (v/v) glacial acetic acid] for 3 hours prior to staining to reduce background staining. The three stained bands enriched in GABAAR α (56 kDa) and β (59 and 61 kDa) subunits were excised separately, and 3H incorporation into the excised gel bands was assayed by liquid scintillation counting as previously described (Chiara et al., 2013).
Allosteric Coupling and State Dependence of Drug Binding.
To quantify allosteric coupling between sites and the state dependence of drug binding, competition photolabeling experiments were carried out in parallel with GABAARs equilibrated with 300 μM GABA, which under our assay conditions was expected to stabilize receptors in the desensitized state, or with 100 μM bicuculline, an inverse agonist (Chang and Weiss, 1999; Thompson et al., 1999), which shifts receptors toward the resting state. For photolabeling at anesthetic concentrations (∼1–3 μM), we found that in 21 independent GABAAR purifications, the ratio of specific incorporation of [3H]azietomidate or [3H]R-mTFD-MPAB (i.e., inhibitable by 300 μM etomidate or 60 μM R-mTFD-MPAB) in the presence of bicuculline compared with GABA varied from 20% to 80%. The source of this variability is unknown, although the ratio appeared to increase as GABAARs were purified from membranes isolated from later passage cells. For the results reported here, 16 GABAAR purifications were used, each characterized by specific incorporation ratios of <45%. Each drug was characterized in at least three independent experiments utilizing at least two different receptor purifications. In four experiments using three of these GABAAR purifications, photolabeling was also determined in the absence of GABA or bicuculline (control). In these experiments, the ratio of labeling in the presence of bicuculline compared with control was 72% ± 14%, consistent with bicuculline acting as an inverse agonist, while labeling in the presence of GABA was 184% ± 21% of control.
In a representative experiment, control [3H]azietomidate α subunit photolabeling was 1333 ± 13 cpm (+GABA), 434 ± 27 cpm (+bicuculline), and 205 ± 23 cpm (nonspecific). For [3H]R-mTFD-MPAB, β subunit photolabeling was 4399 ± 359 cpm (+GABA), 2244 ± 310 cpm (+bicuculline), and 574 ± 72 cpm (nonspecific). Depending upon the receptor purification and the concentration of radioligand, control [3H]azietomidate and [3H]R-mTFD-MPAB photolabeling in the presence of GABA varied from 1100 to 3100 cpm and 2000–10,000 cpm, respectively, with labeling in the presence of bicuculline at 30%–40% of the GABA level. Nonspecific labeling was at 10%–20% of the GABA level for [3H]azietomidate and at 10%–15% for [3H]R-mTFD-MPAB.
Quantitation of Concentration-Dependent Inhibition or Enhancement of Photolabeling.
For GABAARs photolabeled with [3H]azietomidate, parameters for the concentration dependence of drug modulation were determined for 3H incorporation in the 56-kDa gel band, which reflects photolabeling of α1Met-236 in αM1 in the β+–α− interface sites. For [3H]R-mTFD-MPAB, parameters were determined for 3H incorporation in the 59- and 61-kDa gel bands, which reflects photolabeling of βM1 β3Met-227 in the homologous but nonequivalent sites at the α+–β−/γ+–β+ interfaces (Chiara et al., 2013). For photolabeling in the presence of GABA, the concentration dependence of inhibition was fit using nonlinear least squares to single- or two-site models using eqs. 1 and 2, respectively:
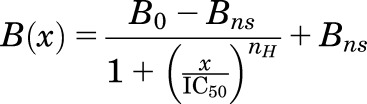 |
(1) |
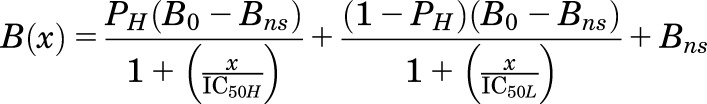 |
(2) |
where B(x) is the 3H in counts/min (cpm) incorporated into a subunit gel band at a total modulator concentration of x; and B0 is the 3H incorporation in the absence of inhibitor. Unless noted, the nonspecific incorporation (Bns) was equal to the incorporation observed in the presence of 300 μM etomidate for [3H]azietomidate or 60 μM R-mTFD-MPAB for [3H]R-mTFD-MPAB. IC50 is the total concentration of inhibitor reducing incorporated 3H by 50%, with H and L denoting high- and low-affinity binding sites; PH is the fraction of specific photolabeling associated with the high-affinity binding site; and nH is the Hill coefficient. To combine data from multiple independent experiments, for each experiment 3H incorporation at each inhibitor concentration was normalized to incorporation in the absence of inhibitor (as percentage), and the full data set was fit to eq. 1 or eq. 2 using GraphPad Prism 7.0. The data plotted in the figures are the mean (±S.D.) from N independent experiments. For all fits to models, the best fit values (±S.E. in Results, 95% asymmetric confidence interval in tables) of the variable parameters and the number of experiments are reported, with the plotted curves calculated from those parameters.
For drugs that inhibited photolabeling in the presence of bicuculline, data were also fit to eq. 1, with nH set to 1 because of the small signal size compared with that in the presence of GABA. When increasing drug concentrations first enhanced and then inhibited photolabeling in the presence of bicuculline, data were fit to a model that assumes enhancement of photolabeling occurs as a result of the tested drug binding with apparent affinity EC50 to a site different than the photolabeled site, where the drug binding inhibits photolabeling (eq. 3A):
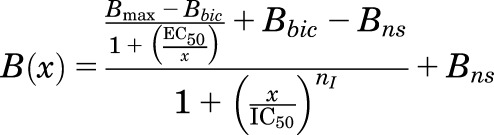 |
(3A) |
with Bmax either fixed at the control 3H cpm incorporated in the presence of GABA or, when noted, allowed to vary to determine whether a better fit can be obtained. Bbic is the control 3H cpm incorporated in the presence of bicuculline, and Bns is the nonspecific incorporation. EC50 and IC50 are the total drug concentrations producing half-maximal enhancement and inhibition of photolabeling, respectively. Data from multiple independent experiments were combined by normalizing (as percentage) to the control photolabeling in the presence of GABA alone. Unless noted, the Hill coefficient for inhibition (nI) was set to 1, and Bns was fixed at the value seen in the presence of 300 μM etomidate or 60 μM R-mTFD-MPAB.
For four drugs (4-benzoyl-propofol, loreclezole, tracazolate, and TG-41) that bind with different affinities to the two β− sites in the presence of GABA, enhancement and inhibition of [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline were fit to eq. 3A and to eq. 3B:
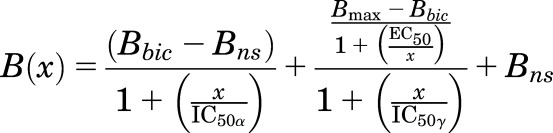 |
(3B) |
This model takes into account the nonequivalent binding of [3H]R-mTFD-MPAB to the β− sites in the presence of bicuculline (Jayakar et al., 2015). Under our photolabeling conditions at ∼1 μM [3H]R-mTFD-MPAB, it is the α+–β− site that is occupied and photolabeled (Bbic-Bns), and the enhanced photolabeling in the presence of GABA or other modulator primarily reflects increased binding to the γ+–β− site (Bmax-Bbic). IC50α and IC50γ are the apparent affinities for the α+–β− and γ+–β− sites, respectively, and EC50 is the concentration producing half-maximal enhancement of photolabeling. For a drug binding with high affinity at the α+–β− site that enhances photolabeling at the γ+–β− site, we expect that EC50 = ∼IC50α. For a drug binding with the highest affinity to the γ+–β− site, eq. 3B is not appropriate because the drug can only inhibit, not enhance, photolabeling at that site. A drug binding with the highest affinity to the γ+–β− site might produce a small enhancement of photolabeling at the α+–β− site, which is not fully occupied under our labeling conditions, in which case a fit to eq. 3A is appropriate, with EC50 a measure of the apparent affinity for the γ+–β− site and IC50 the affinity for the α+–β− site. If the enhancement of photolabeling is at the α+–β− site, the maximal enhancement from the fit (Bmax) should be ∼50%–60% of that seen in the presence of GABA, and values of EC50 and IC50 should be close to the values of IC50H and IC50L determined (eq. 2) by inhibition of [3H]R-mTFD-MPAB photolabeling (+GABA). If a drug binds nonequivalently to the β− sites with enhancement of photolabeling resulting from drug binding to the β+ sites, fit to eq. 3B is appropriate. In this case, EC50 should be close to the IC50 for the β+ sites, and the affinity for the α+–β− site (IC50α) may be less than or greater than the affinity for the γ+–β− site (IC50γ).
Data were fit to the models by nonlinear least squares using GraphPad Prism 7.0 or SigmaPlot 11.0 (Systat Software, San Jose, CA). For all fits, the best fit values of the variable parameters (±S.E. in Results, 95% asymmetric confidence interval in tables) and the number of independent experiments are reported. The extra sum of the squares principle (F test, α = 0.05) was used to compare nested models with nonequivalent degrees of freedom. Otherwise, the difference in corrected Akaike’s information criterion values (ΔAICc) was used (GraphPad Prism 7.0) with the value of ΔAICc and the predicted probability (%) reported. To compare inhibition of [3H]azietomidate and [3H]R-mTFD-MPAB photolabeling by a drug in the presence of GABA, an extra sum-of-squares F test (α = 0.05) was used to compare separate fits for each radioligand to eq. 1 (nH = 1) with the fit for the pooled data (null hypothesis). This F test was also used to compare the separate fits to fits of the pooled data (null hypothesis) to determine the significance for differences of inhibition of [3H]azietomidate or [3H]R-mTFD-MPAB photolabeling by a drug in the presence of GABA versus bicuculline. One-way ANOVA with Tukey’s multiple comparison test was used to determine the significance of differences (α = 0.05) of log IC50 values for pairs of propofol analogs as inhibitors of [3H]azietomidate or [3H]R-mTFD-MPAB photolabeling (+GABA).
Results
State Dependence of Photolabeling.
We first compared [3H]azietomidate and [3H]R-mTFD-MPAB photolabeling of α1β3γ2 GABAARs in the presence of GABA, which stabilizes the desensitized state, with that in the presence of bicuculline, an inverse agonist (Chang and Weiss, 1999; Thompson et al., 1999) that stabilizes the resting, closed-channel state (Masiulis et al., 2019) (Fig. 2, A and D). For [3H]azietomidate, 3H incorporation was quantified in the α subunit gel band, which reflects photolabeling of α1Met-236 in αM1 in the β+–α− interface sites. For [3H]R-mTFD-MPAB, 3H incorporation was quantified in the β subunit gel bands, which reflects photolabeling of β3Met-227 in βM1 in the homologous but nonequivalent sites at the α+–β−/γ+–β− interfaces (Chiara et al., 2013). For photolabeling at anesthetic concentrations (1–3 μM), specific photoincorporation of [3H]azietomidate or [3H]R-mTFD-MPAB (i.e., etomidate or R-mTFD-MPAB inhibitable) in the presence of bicuculline was ∼30% of that seen in the presence of GABA. The lower level of [3H]azietomidate photolabeling in the presence of bicuculline resulted from its decreased affinity for the β+–α− sites. In addition, etomidate was less potent as an inhibitor of [3H]azietomidate photolabeling in the presence of bicuculline (IC50 = 5.5 ± 1.4 μM) than in the presence of GABA (IC50 = 2.0 ± 0.1 μM). For [3H]R-mTFD-MPAB in the presence of GABA, the β subunit photolabeling resulted from labeling of the α+–β− and γ+–β− sites at similar efficiency (Chiara et al., 2013; Jayakar et al., 2015). R-mTFD-MPAB inhibited [3H]R-mTFD-MPAB photolabeling with IC50 = 0.66 ± 0.05 μM and a Hill coefficient close to 1 (nH = 1.2 ± 0.1), consistent with R-mTFD-MPAB binding with similar affinities to the two β− sites (Fig. 2D). In the presence of bicuculline, R-mTFD-MPAB inhibited photolabeling with IC50= 3.1 ± 1.0 μM.
Fig. 2.

Allosteric coupling between etomidate (β+) and R-mTFD-MPAB (β−) intersubunit anesthetic binding sites. α1β3γ2 GABAARs were photolabeled with [3H]azietomidate (A and C) or [3H]R-mTFD-MPAB (B and D) in the presence of etomidate (A and B) or nonradioactive R-mTFD-MPAB (C and D) and bicuculline (100 μM, ⬤) or GABA (300 μM, ○). The receptor subunits were resolved by SDS-PAGE, and covalent incorporation of 3H was determined by liquid scintillation counting of excised gel bands containing the GABAAR α ([3H]azietomidate) or β ([3H]R-mTFD-MPAB) subunits. Nonspecific photolabeling, indicated by the dashed lines, was determined in the presence of 300 μM etomidate or 60 μM R-mTFD-MPAB. For each independent experiment, data were normalized to the 3H cpm incorporated in the control condition (+GABA, no competitor), and the plotted data are the averages (±S.D.) from the independent experiments. As described under Materials and Methods, the pooled data from the independent experiments were fit to eq. 1 or, when enhancement of photolabeling was seen in the presence of bicuculline, to eq. 3A. Parameters for the fits and the number of independent experiments (N) are tabulated in Table 1. Based upon an F test comparison of fits of the data in the presence of GABA and bicuculline to eq. 1 to the same or separate IC50 values, separate fits were favored for etomidate inhibition of [3H]azietomidate [P = 0.0014, F(DFn,DFd) = 11.5(1,50)] and R-mTFD-MPAB inhibition of [3H]R-mTFD-MPAB photolabeling [P < 0.0001, F(DFn,DFd) = 246(1,68)].
Allosteric Coupling between Intersubunit Anesthetic Binding Sites.
In the presence of bicuculline, etomidate at concentrations from 1 to 100 μM increased [3H]R-mTFD-MPAB photolabeling to the level seen in the presence of GABA, with inhibition then seen at higher concentrations (Fig. 2B). R-mTFD-MPAB at concentrations up to 3 μM enhanced [3H]azietomidate photolabeling, inhibiting it at higher concentrations (Fig. 2C). The concentration-dependent enhancement of photolabeling by etomidate or R-mTFD-MPAB established that even in the presence of bicuculline, the binding of etomidate to the β+ sites is positively coupled energetically to the β− sites, and the binding of R-mTFD-MPAB to a β− site is positively coupled energetically to the β+ sites.
A Photolabeling Assay to Determine Selectivity of Drugs for Intersubunit Anesthetic Sites and to Detect Additional Sites.
In the presence of GABA, [3H]R-mTFD-MPAB photoincorporation in the β subunit resulted from labeling of the α+–β− and γ+–β− sites with similar efficiency (Chiara et al., 2013; Jayakar et al., 2015). The concentration dependence of inhibition of subunit photolabeling by drugs that bind nonequivalently to the two sites will be characterized by Hill coefficients less than 1. For such drugs, fits of the data to a two-site model will allow determination of the affinities (IC50 values) for the α+–β− and γ+–β− sites, and the IC50 for inhibition of [3H]azietomidate photolabeling defines the affinity for the β+ sites.
The enhancement of [3H]R-mTFD-MPAB or [3H]azietomidate photolabeling by drugs in the presence of bicuculline provides an assay to identify PAMs that act with a similar state dependence as GABA. Comparison of the concentration dependence of enhancement to the IC50 for inhibition of [3H]azietomidate or [3H]R-mTFD-MPAB photolabeling in the presence of GABA provides a test of whether the enhancement results from the drug binding to either the β+ or β− intersubunit sites. For example, if a drug enhanced photolabeling at the β+ (etomidate) sites at concentrations lower than those necessary to bind to the β− (R-mTFD-MPAB) sites, it would provide evidence that this PAM binds to sites other than the β+ and β− intersubunit anesthetic sites.
Propofol in the presence of GABA was more potent as an inhibitor of [3H]azietomidate (IC50 = 7.8 ± 0.8 μM) than of [3H]R-mTFD-MPAB labeling (IC50 = 44 ± 4 μM) (Fig. 3, A and B), consistent with previous results (Chiara et al., 2013). For both anesthetic photolabels, the concentration dependence of inhibition by propofol was fit by nH close to 1 (nH = 1.1 ± 0.1). In the presence of bicuculline, propofol from 3 to 30 μM increased [3H]R-mTFD-MPAB photolabeling, with inhibition occurring at higher concentrations (Fig. 3B). In contrast, propofol did not enhance [3H]azietomidate photolabeling but inhibited it at concentrations above 10 μM (Fig. 3A). Since propofol in the presence of GABA binds with 5-fold higher affinity to the β+ sites than to the β− sites, propofol enhancement of [3H]R-mTFD-MPAB photolabeling is consistent with positive allosteric coupling between the β+ and β− sites. The lack of enhancement of [3H]azietomidate photolabeling is qualitatively consistent with propofol’s lower affinity for the β− sites. In addition, this lack of enhancement provides evidence that there are no other sites that both bind propofol with higher affinity than the β+ sites and are coupled energetically to the β+ sites. For propofol and all other drugs tested in the presence of bicuculline, quantitative fits of enhancement/inhibition are presented later.
Fig. 3.

Propofol (A and B) and 4-Cl-propofol (4-Cl-PPF) (C and D) bind preferentially to the β+ ([3H]azietomidate) sites and equivalently to the α+/γ+–β− ([3H]R-mTFD-MPAB) GABAAR anesthetic sites. GABAARs were photolabeled with [3H]azietomidate (A and C) or [3H]R-mTFD-MPAB (B and D) in the presence of bicuculline (⬤) or GABA (○). As described in Fig. 2, the 3H cpm incorporation in the GABAAR subunits was determined by liquid scintillation counting, and the data were then normalized for individual experiments that were combined, with the dashed lines indicating the nonspecific subunit photolabeling. The plotted data are the averages (±S.D.) from the independent experiments. The full data sets for each condition were fit to eq. 1, or to eq. 3A for [3H]R-mTFD-MPAB (+bicuculline). Parameters for the fits and the number of independent experiments are tabulated in Table 1. Based upon an F test comparison of fits of the data in the presence of GABA and bicuculline to eq. 1 to the same or separate IC50 values, separate fits were favored for inhibition of [3H]azietomidate photolabeling by propofol [P = 0.003, F(DFn,DFd) = 9.2(1,69)] and 4-Cl-propofol [P = 0.016, F(DFn,DFd) = 6.2(1,62)].
Seeking para-Substituted Propofols that Bind Nonequivalently to α+–β− and γ+–β− Sites.
We characterized a panel of 4-substituted (or para-substituted) propofol analogs, since many, including 4-Cl- and 4-benzoyl-propofol, act as potent GABAAR PAMs and general anesthetics (Trapani et al., 1998; Krasowski et al., 2001a; Stewart et al., 2011). For five derivatives [4-Cl-propofol, 4-(tert-butyl)-propofol, 4-acetyl-propofol, 4-benzyl-propofol, and 4-[4-hydroxy(phenyl)methyl]-propofol], the concentration dependence for inhibition of [3H]azietomidate and [3H]R-mTFD-MPAB labeling in the presence of GABA was characterized by Hill coefficients close to 1 (Table 1). Similar to propofol, 4-Cl-propofol bound with 4-fold higher affinity to the β+ sites (IC50 = 5 ± 1 μM) than to the β− sites (Fig. 3, C and D). 4-Acetyl-propofol bound with little selectivity between the β+ and β− sites (IC50 values ∼40 μM; Fig. 4, A and B). 4-Benzyl-propofol (Table 1), 4-(tert-butyl)-propofol (Fig. 4, C and D), and 4-[4-hydroxy(phenyl)methyl]-propofol (Fig. 5, C and D) each bound with ∼10-fold higher affinity to the β− sites (IC50 = 5.7 ± 0.5, 17 ± 3, and 10 ± 1 μM, respectively). For each of the drugs tested, the difference in IC50 values for [3H]azietomidate and [3H]R-mTFD-MPAB was statistically significant. With the exception of 4-acetyl-propofol, there was no overlap of 95% confidence intervals (Table 1). Based upon F test comparisons of fits assuming common versus separate values of IC50, separate fits were preferred. For 4-acetyl-propofol, P < 0.002 [F(DFn,DFd) = 10.3(1,47)], and for the other drugs, P < 0.0001.
TABLE 1.
Inhibition and enhancement (+bicuculline) by para-substituted propofol analogs of α1β3γ2 GABAAR photolabeling by [3H]azietomidate and [3H]R-mTFD-MPAB
When drugs only inhibited photolabeling, data were fit to eq. 1 (Materials and Methods) to determine IC50, the total drug concentration producing 50% inhibition of GABAAR photolabeling, and Hill coefficients (nH) and, when indicated, to eq. 2, a two-site model. For inhibition in the presence of bicuculline, nH was set to 1. When enhancement was seen in the presence of bicuculline, data were fit to eqs. 3A or 3B, as noted in the figure legends, to determine EC50 and IC50 values for 50% maximal enhancement and inhibition. Parameters [best fit ± S.E. or 95% confidence interval (CI)] were determined from fits of data from N independent experiments from two or more GABAAR purifications. As bicuculline and GABA experiments were paired, their values of N are the same.
| Drug (NR-mTFD-MPAB, NAziET) | Ratio IC50(AziEt)/IC50(TFD-MPAB)a | [3H]R-mTFD-MPAB IC50 {95% CIs} (nH) |
[3H]R-Azietomidate IC50 {95% CIs} (nH) |
||
|---|---|---|---|---|---|
| +GABA |
+GABA |
+Bicuculline EC50/IC50 |
+GABA |
+Bicuculline EC50/IC50 |
|
| μM | μM | μM | μM | ||
| Etomidate (5, 3) | 0.01 ± 0.002 | 430 {330, 580} (1.5 ± 0.3) | 3.0 ± 0.7/440 ± 50 | 2.0 {1.8, 2.3} (1.00 ± 0.06) | 5.5 {3.4, 9} |
| R-mTFD-MPAB (5, 3) | 45 ± 12 | 0.7 {0.6, 0.8} (1.2 ± 0.1) | 3.1 {1.5, 5.0} | 38 {29, 50} (1.0 ± 0.1) | 1.1 ± 0.3/18 ± 3 |
| Propofol (6, 6)b | 0.2 ± 0.04 | 44 {36, 53} (1.1 ± 0.1) | 2.5 ± 0.8/57 ± 11 | 7.8 {6, 10} (1.1 ± 0.1) | 29 {12, 97} |
| 4-Cl-propofol (4, 4)b | 0.25 ± 0.08 | 20 {14, 28} (1.2 ± 0.2) | 1.5 ± 0.4/19 ± 3 | 4.9 {3.4, 7.0} (0.9 ± 0.2) | 20 {7, 90} |
| 4-acetyl-propofol (4, 3)c | 1.5 ± 0.4 | 32 {25, 41} (0.9 ± 0.1) | 250 {100, 790} | 48 {38, 61} (1.1 ± 0.3) | 17 ± 2/41 ± 4 |
| 4-(tert-butyl)-propofol (4, 3)c | 8 ± 3 | 17 {13, 23} (1.0 ± 0.1) | 120 {68, 220} | 134 {100, 226} (1.2 ± 0.2) | 5.1 ± 0.7/72 ± 8 |
| 4-benzyl-propofol (2, 3) | 5 ± 1 | 5.7 {4.7, 6.9} (1.3 ± 0.1) | ND | 30 {21, 53} (0.8 ± 0.1) | ND |
| 4-benzoyl-propofol (6, 5)d | 200 ± 140e | 21 {11, 38} (0.27 ± 0.04) eq. 2 IC50H = 0.5 ± 0.3 (45% ± 4%)/IC50L = 330 ± 120 | eq. 3A: 1.0 ± 0.9/156 ± 54 Bmax= 61% ± 5% eq. 3B: 1.0 ± 0.8/158 ± 54 Bmax= 87% ± 5% | 99 {82, 120} (2.0 ± 0.3) | 0.6 ± 0.2/89 ± 8 Bmax = 81% ± 3%f |
| 4-[hydroxyl(phenyl)methyl]-propofol (3,3) R-isomer (2) S-isomer (2)d | 16 ± 5 | 10 {8.5, 12} (1.1 ± 0.1) 12 ± 1; 7 ± 1 | 30 {15, 63} ND; ND | 157 ± 33 (1.1 ± 0.3) ND; ND | 8 ± 2/41 ± 8 ND; ND |
ND, not determined;
For the ratio of IC50 values (+GABA), uncertainties were determined by propagation of error from the individual parameter uncertainties.
Data from Fig. 3.
Data from Fig. 4.
Data from Fig. 5, with parameters for 4-benzoyl-propofol inhibition of [3H]R-mTFD-MPAB (+GABA) also fit to a two-site model, eq. 2.
The ratio calculated for the high-affinity component (IC50H) of [3H]R-mTFD-MPAB inhibition.
Parameters based upon fit of the data to eq. 3A with nI = 2, the Hill coefficient determined for inhibition in the presence of GABA.
Fig. 4.

4-Acetyl-propofol (A and B) binds nonselectively to β+ ([3H]azietomidate) and β− ([3H]R-mTFD-MPAB) intersubunit sites, but 4-(tert-butyl)-propofol (4-tert-butyl-PPF) (C and D) binds preferentially to the β− intersubunit anesthetic sites. GABAARs were photolabeled with [3H]azietomidate (A and C) or ([3H]R-mTFD-MPAB (B and D) in the presence of bicuculline (⬤) or GABA (○). As described in Fig. 2, the plotted data are the averages (±S.D.) from the independent experiments, with the dashed lines indicating the nonspecific subunit photolabeling. Data were fit to eq. 1 for [3H]R-mTFD-MPAB (both conditions) and to eqs. 1 and 3A for [3H]azietomidate in the presence of GABA and bicuculline, respectively. Parameters for the fits and the number of independent experiments are tabulated in Table 1. For fits of inhibition of [3H]R-mTFD-MPAB photolabeling in the presence of GABA vs. bicuculline by 4-acetyl-propofol and 4-tert-butyl-PPF, separate fits were favored [P < 0.0001, F(DFn,DFd) = 22(1,52) and 31(1,60), respectively].
Fig. 5.

4-Benzoyl-propofol (4-benzoyl-PPF) (A and B) binds nonequivalently to the β− ([3H]R-mTFD-MPAB) intersubunit anesthetic sites, but 4-[hydroxyl(phenyl)methyl]-propofol (C and D) binds equivalently. GABAARs were photolabeled with [3H]azietomidate (A and C) or ([3H]R-mTFD-MPAB (B and D) in the presence of bicuculline (⬤) or GABA (○). The plotted data are the averages (±S.D.) from independent experiments, with the dashed lines indicating nonspecific subunit photolabeling. For experiments in the presence of GABA, the pooled data from the independent experiments were fit to eq. 1 (Materials and Methods), and for 4-benzoyl-propofol ([3H]R-mTFD-MPAB) also to eq. 2, a two-site model (Table 1). Data for [3H]azietomidate (+bicuculline) were fit to eq. 3A, with 4-benzoyl-propofol data fit to Bmax = 100% [dotted line, EC50 = 2.1 ± 0.4 μM, IC50 = 63 ± 5 μM, nI = 2, (R2 = 0.87, F = 245)] or Bmax adjustable [solid line, Bmax = 81% ± 3%, EC50 = 0.6 ± 0.2 μM, IC50 = 89 ± 8 μM, nI = 2 (R2 = 0.93)], a fit that was favored [P < 0.0001, F(DFn,DFd) = 30.6 (1,37)]. Since 4-benzoyl-propofol bound nonequivalently to the β− sites in the presence of GABA, data for [3H]R-mTFD-MPAB photolabeling (+bicuculline) were fit to eqs. 3A and 3B, which fit the data similarly (ΔAICc = 0, (50%)). Equations 3A and 3B distinguish between preferential binding to the γ+–β− and α+–β− sites, respectively (see Results and Discussion).
4-Benzoyl-propofol was identified as a drug that distinguished strongly between the two β− sites. The concentration dependence of inhibition of [3H]R-mTFD-MPAB photolabeling was fit by a Hill coefficient (nH) of 0.27 ± 0.04 and IC50 = 21 ± 6 μM (Fig. 5B). In a two-site model (eq. 2, Materials and Methods), this inhibition was characterized by high-affinity (IC50H = 0.5 ± 0.3 μM; 45% ± 4% of specific incorporation) and low-affinity (IC50L = 320 ± 120 μM) components. 4-Benzoyl-propofol was also unusual in that it was the only propofol analog studied that inhibited [3H]azietomidate photolabeling (IC50 = 99 ± 8 μM) with a Hill coefficient significantly greater than 1 (nH = 2.0 ± 0.3, P < 0.0001 [F(DFn,DFd) = 25.6(1,38)]) (Fig. 5A).
For each propofol analog tested, the enhancement of [3H]azietomidate or [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline was qualitatively consistent with the ligand selectivity for the β+ or β− sites. 4-Cl-propofol, which bound preferentially to the β+ sites, strongly enhanced [3H]R-mTFD-MPAB photolabeling without enhancing [3H]azietomidate photolabeling (Fig. 3, C and D). 4-Acetyl-propofol and 4-(tert-butyl)-propofol clearly enhanced [3H]azietomidate photolabeling (Fig. 4, A and C) with little, if any, enhancement of [3H]R-mTFD-MPAB photolabeling (Fig. 4, B and D). 4-[4-Hydroxy(phenyl)methyl]-propofol (Fig. 5, C and D), which binds with 10-fold higher affinity to the β− sites, enhanced [3H]azietomidate photolabeling without enhancing [3H]R-mTFD-MPAB photolabeling. For drugs that only inhibited photolabeling in the presence of bicuculline, the differences in IC50 values in the presence of GABA versus bicuculline were highly significant. Based upon F test comparison of fits assuming common versus separate values of IC50, separate fits were preferred for [3H]azietomidate inhibition by propofol (P = 0.003) and 4-Cl-propofol (P = 0.016) and for [3H]R-mTFD-MPAB inhibition by 4-acetyl-propofol, 4-(tert-butyl)-propofol (each P < 0.0001), and 4-[4-hydroxy(phenyl)methyl]-propofol (P = 0.001).
4-Benzoyl-propofol, which binds with the highest affinity to one of the β− sites, strongly enhanced [3H]azietomidate photoincorporation, and at concentrations up to 10 μM, it also enhanced [3H]R-mTFD-MPAB photolabeling, with inhibition at higher concentrations as in the presence of GABA (Fig. 5, A and B). Interpretation of the effects of 4-benzoyl-propofol on [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline is complicated by the fact that the drug binds to one of the β− sites with the highest affinity, to the β+ sites with intermediate affinity, and to the other β− site with the lowest affinity. The assignment of the β− site associated with high-affinity binding will be considered later in conjunction with quantitative analyses of the enhancement/inhibition profiles for all drugs tested in the presence bicuculline.
Other Anesthetics and Sedative/Anticonvulsants Can Distinguish between β− Sites.
We also identified PAMs structurally unrelated to propofol that interact nonequivalently with the β− sites: loreclezole, a triazole sedative/anticonvulsant (Wauquier et al., 1990); tracazolate, a pyrazolopyridine anxiolytic and anticonvulsant with low sedative potency (Patel and Malick, 1982); and TG-41, an imidazolecarboxylate that is a potent general anesthetic (Mascia et al., 2005). Loreclezole and tracazolate inhibited [3H]azietomidate incorporation in the presence of GABA with IC50 values of 9.3 ± 0.9 μM (nH = 1.2 ± 0.1) and 8.0 ± 0.9 μM (nH = 1.0 ± 0.1), respectively (Fig. 6, A and C). They each interacted nonequivalently with the β− sites (Fig. 6, B and D). The concentration dependence of loreclezole inhibition of [3H]R-mTFD-MPAB photolabeling was characterized by a Hill coefficient (nH) of 0.43 ± 0.06 (IC50 = 116 ± 35 μM) (Fig. 6B). When fit to a two-site model, this inhibition was characterized by a high-affinity component (IC50H = 1.4 ± 1.2 μM) accounting for 29% ± 7% of specific incorporation and a low-affinity component (IC50L = 320 ± 120 μM). Tracazolate at concentrations of 100 and 300 μM inhibited [3H]R-mTFD-MPAB photolabeling maximally by ∼40%, with the observed inhibition characterized by IC50 = 18 ± 7 μM (Fig. 6D). In the presence of bicuculline, neither loreclezole nor tracazolate enhanced [3H]azietomidate labeling, while at concentrations from 1 to 30 μM, both enhanced [3H]R-mTFD-MPAB photolabeling, with inhibition at higher concentrations.
Fig. 6.

Loreclezole, tracazolate, and TG-41 bind with high affinity to the β+ ([3H]azietomidate) sites and to one β− ([3H]R-mTFD-MPAB) site. GABAARs were photolabeled with [3H]azietomidate (A, C, and E) or [3H]R-mTFD-MPAB (B, D, and F) in the presence of bicuculline (⬤) or GABA (○). As described in Fig. 2, the plotted data are the averages (±S.D.) from independent experiments, with the dashed lines indicating nonspecific subunit photolabeling. The data for [3H]azietomidate were fit to eq. 1. For [3H]R-mTFD-MPAB (+GABA), loreclezole data were fit to eqs. 1 and 2 (Table 2), while tracazolate and TG-41 data were fit to eq. 1 (nH = 1) with variable Bns. For tracazolate, Bns = 56% ± 5% and IC50 = 18 ± 7 μM (R2 = 0.68, F = 104); for TG-41, Bns = 67% ± 2% and IC50 = 0.1 ± 0.04 μM (R2 = 0.75, F = 89). For [3H]R-mTFD-MPAB (+bicuculline), fit of the loreclezole data to eq. 3B with α+–β− as the high-affinity site (EC50 = IC50α = 2.5 ± 0.8 μM, IC50γ = 160 ± 30 μM, R2 = 0.70, F = 68) was favored over fit to eq. 3A with γ+–β− as the high-affinity, enhancing site or to eq. 3B with γ+–β− as the high-affinity site and enhancement caused by binding to a site other than a β− site (see Results). For tracazolate and TG-41, which each bound with highest affinity to the β+ sites and only to one of the β− sites over the concentrations tested, data were fit to eq. 3B. For tracazolate, the fit with γ+–β− as the high-affinity site [Bmax = 100%, EC50 = 2.7 ± 0.6 μM, IC50γ = 24 ± 5 μM (R2 = 0.67, F = 96)] was strongly favored (ΔAICc = 12.7, (99.8%)) over the fit for α+–β− as the high-affinity site. For TG-41, the fit with α+–β− as the high-affinity site [Bmax = 100%, EC50 = 0.02 ± 0.007 μM, IC50α = 0.4 ± 0.2 μM (R2 = 0.73, F = 79)] was slightly favored (ΔAICc = 2.5, (78%)) over the best fit for a high-affinity γ+–β− site.
TG-41 bound with very high affinity to the β+ sites, inhibiting [3H]azietomidate photolabeling with IC50 values of 16 ± 3 nM (+GABA) and 220 ± 60 nM (+bicuculline) (Fig. 6E). TG-41 in the presence of GABA, similar to tracazolate, inhibited [3H]R-mTFD-MPAB maximally by only ∼35% at concentrations of 10–50 μM (Fig. 6F). The observed inhibition was characterized by an IC50 of 100 ± 40 nM. In the presence of bicuculline, TG-41 at concentrations above 30 nM increased [3H]R-mTFD-MPAB incorporation to the level seen at those concentrations in the presence GABA.
We also screened additional anticonvulsants that act as GABAAR PAMs but do not bind to the GABA or benzodiazepine binding sites (Bonin and Orser, 2008; Greenfield, 2013) (Fig. 7). Topiramate, felbamate, and valnoctamide did not inhibit photolabeling at concentrations where they act as GABAAR PAMs (Rho et al., 1997; Simeone et al., 2006; Spampanato and Dudek, 2014). Stiripentol, which acted as an α1β3γ2 GABAAR PAM at concentrations of 30–100 μM (Fisher, 2009), inhibited [3H]azietomidate and [3H]R-mTFD-MPAB photolabeling with IC50 values of 130 and 60 μM, respectively.
Fig. 7.

Inhibition of [3H]azietomidate and [3H]R-mTFD-MPAB photolabeling by sedatives/anticonvulsants. (A) GABAARs were photolabeled in the presence of GABA (300 μM) in the absence (control) or presence of 10 mM topiramate, 3 mM felbamate, 1 mM valnoctamide, or 300 μM stiripentol, and nonspecific photolabeling was determined in the presence of 300 etomidate or 60 μM R-mTFD-MPAB. Receptor subunits were resolved by SDS-PAGE, and covalent incorporation of 3H incorporation was determined by liquid scintillation counting of excised gel bands containing the GABAAR α ([3H]azietomidate) or β ([3H]R-mTFD-MPAB) subunits. Specific subunit photolabeling (3H cpm, Btot – Bns), normalized to the control value, is plotted for two independent experiments (means ± 1/2 range). (B and C) Stiripentol inhibition of [3H]azietomidate or [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline (100 μM, ⬤) or GABA (300 μM, ○). For each independent experiment, data were normalized to the 3H cpm incorporated in the control condition (+GABA, no competitor), and the plotted data are the averages (±S.D.) from the independent experiments. The pooled data were fit to eq. 1 or for [3H]azietomidate (+bicuculline) to eq. 3A (Table 2).
Allosteric Coupling and Selectivity of Drugs for the Two β− Intersubunit Sites, α+–β− or γ+–β−.
For propofol derivatives other than 4-benzoyl-propofol, which binds nonequivalently to the β− sites, the concentration dependence of enhancement and inhibition of [3H]R-mTFD-MPAB or [3H]azietomidate photolabeling in the presence of bicuculline was fit to a model (Materials and Methods, eq. 3A) that assumes one site associated with enhancement (EC50) and a second site associated with inhibition (IC50). Unless noted in figure legends, better fits were obtained with Bmax equal to 100%, the photolabeling level in the presence of GABA, than with variable Bmax. For the drugs that bound preferentially to the [3H]azietomidate (β+) sites (propofol, 4-Cl-propofol) or to the [3H]R-mTFD-MPAB (β−) sites [4-(tert-butyl)-propofol and 4-[hydroxyl(phenyl)methyl]-propofol], the IC50 values for inhibition of the high-affinity site were 2- to 3-fold greater than the EC50 values for enhancement at the low-affinity site (Fig. 8A; Table 1).
Fig. 8.

(A) Ratios of [3H]azietomidate IC50 (+GABA)/[3H]R-mTFD-MPAB EC50 (+bicuculline) and of [3H]R-mTFD-MPAB IC50 (+GABA)/[3H]azietomidate EC50 (+bicuculline) for drugs selective for β+ and β− subunit interfaces. Ratios and error bars are calculated from the parameter fits tabulated in Tables 1 and 2. (B) The relationship between a drug’s IC50 for inhibiting [3H]azietomidate (⬤) or [3H]R-mTFD-MPAB (○) photolabeling (+GABA) and the Connolly excluded volume of the propofol phenyl ring substituent. Substituent volume is calculated (Discovery Studio; Biovia, San Diego, CA) as the increase of the Connolly excluded volume over that of propofol. The lines are the linear least-squares fit of the data sets from Table 1, with the plotted value of IC50H for [3H]R-mTFD-MPAB/4-benozyl-propofol (4-benzoyl-PPF) excluded from the fit. One-way ANOVA with Tukey’s multiple comparison test was used to determine the significance of differences (α = 0.05) of log IC50 values for pairs of propofol analogs as inhibitors of [3H]azietomidate or [3H]R-mTFD-MPAB photolabeling (+GABA). For [3H]azietomidate, propofol/Cl- did not differ significantly (P = 0.16), but each differed from the other tested drugs (P < 0.0001). Other significant differences were as follows: acetyl- vs. t-butyl- (P = 0.0003), benzoyl- (P = 0.006), or HO-phenyl- (P < 0.0001); t-butyl- vs. benzyl- (P < 0.0001); benzyl- vs. benzoyl- or HO-phenyl- (P < 0.0001). For [3H]R-mTFD-MPAB (with omission of benzoyl-, which was fit to two-site model), propofol differed from each drug (P < 0.0001) except acetyl-. Other significant differences were: Cl- vs. benzyl- (P < 0.0001) or HO-phenyl (P = 0.008); acetyl- vs. t-butyl- (P = 0.0066), benzyl-, or HO-phenyl- (P < 0.0001); t-butyl- vs. benzyl- (P < 0.0001).
In the presence of GABA, 4-benzoyl-propofol distinguished strongly between the two β− sites, inhibiting [3H]R-mTFD-MPAB photolabeling with IC50H = 0.5 μM and IC50L = 330 μM, whereas at the β+ sites, it inhibited [3H]azietomidate photolabeling with IC50 = 99 μM (Fig. 5, A and B). In the presence of bicuculline, 4-benzoyl-propofol enhanced [3H]azietomidate photolabeling with EC50 = 0.6 ± 0.2 μM (fit to eq. 3A), which was consistent with this action being mediated by binding to the high-affinity β− site. In the presence of bicuculline, [3H]R-mTFD-MPAB photolabeling was enhanced by 4-benzoyl-propofol at concentrations sufficient to occupy the high-affinity β− site but 30-fold too low to occupy the β+ sites. To attempt to determine which of the two β− sites is the high-affinity site for 4-benzoyl-propofol, we fit our data to eqs. 3A and 3B, reasoning the following as described in Materials and Methods: 1) if the high-affinity site is the α+–β− site (eq. 3B), increasing 4-benzoyl-propofol concentrations would inhibit photolabeling at α+–β−, the site photolabeled in the presence of bicuculline (Jayakar et al., 2015), and enhance photolabeling at the γ+–β− site (α+–β−: IC50α = EC50 = 1.0 ± 0.8 μM, Bmax = 87% ± 5%), with inhibition at high concentrations (γ+–β−: IC50γ = 158 ± 54 μM); and 2) if the high-affinity site is the γ+–β− site (eq. 3A), 4-benzoyl-propofol cannot enhance photolabeling at γ+–β−, but it could produce small enhancement at the α+–β− site (γ+–β−: EC50 = 1.0 ± 0.8 μM, Bmax = 60% ± 5%), with inhibition at high concentrations (α+–β−: IC50 = 158 ± 44 μM). Based upon Akaike’s information criterion, eqs. 3A and 3B fit the data equally (ΔAICc = 0, (50%)), so our results do not allow an independent identification of the high-affinity β− site (but see Discussion).
Loreclezole, similar to 4-benzoyl-propofol, in the presence of GABA bound nonequivalently to the β− sites (IC50H = 1.4 μM, IC50L = 320 μM) but with the affinity for the β+ sites (IC50 = 9 ± 1 μM) closer to that for the high-affinity β− site (Fig. 7, A and B). In the presence of bicuculline, loreclezole enhanced [3H]R-mTFD-MPAB photolabeling at concentrations where it also bound to the β+ sites. In contrast to 4-benzoyl-propofol, loreclezole binding to its high-affinity β− site did not enhance [3H]azietomidate photolabeling. The fit of the [3H]R-mTFD-MPAB data to eq. 3B with Bmax = 100% (α+–β−, IC50α = EC50 = 2.5 ± 0.8 μM; γ+–β−, IC50γ= 159 ± 32 μM) was slightly favored (ΔAICC = 2.6, (79%)) over the fit to eq. 3A with variable Bmax (Bmax = 77% ± 7%, γ+–β−: EC50 = 2.2 ± 1.4 μM, α+–β−: IC50 = 172 ± 62 μM) and more strongly favored (ΔAICC = 4.4, (90%)) over a fit to eq. 3B with γ+–β− being the high-affinity site and enhancement not resulting from binding to a β− site (EC50 = 6 ± 2 μM, IC50α = 620 ± 480 μM, IC50γ= 26 ± 8 μM), a fit that resulted in a value for the high-affinity β− site not consistent with the value determined in the presence of GABA (IC50H = 1.4 μM). These results predict that α+–β− is the high-affinity site for loreclezole, although the limited selectivity of loreclezole for a β− site versus the β+ sites makes it likely that the enhancement of [3H]R-mTFD-MPAB photolabeling is caused primarily by loreclezole binding to the β+ sites. Consistent with this, loreclezole affinity for the β+ sites (IC50 = 9 μM) was ∼4-fold greater than the EC50 value of 2 μM for enhancement of [3H]R-mTFD-MPAB photolabeling from the preferred fit to eq. 3B, a ratio similar to that seen for propofol enhancement of [3H]R-mTFD-MPAB photolabeling.
Tracazolate and TG-41 in the presence of GABA both bound with the highest affinity to the β+ sites (Table 2), and consistent with this, they both enhanced [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline (Fig. 6, D and F). In the presence of GABA, high concentrations of either drug inhibited [3H]R-mTFD-MPAB photolabeling maximally by ∼40%. Control experiments established that this partial inhibition at high concentrations of either drug did not result from increased nonspecific photolabeling in the presence of 60 μM R-mTFD-MPAB. Thus, the incomplete inhibition at high concentrations provided evidence that each drug bound nonequivalently to the β− sites, and the data allowed determination of an IC50 only for the high-affinity site. Because of this partial inhibition, data for tracazolate and TG-41 inhibition of [3H]R-mTFD-MPAB in the presence of GABA were fit to eq. 1 with variable Bns rather than the value in the presence of 60 μM R-mTFD-MPAB and with nH = 1 because of the small amplitude of the signal. For tracazolate and TG-41, the IC50 values were 18 ± 7 and 0.1 ± 0.04 μM, respectively (Table 2).
TABLE 2.
Sedative/anticonvulsant inhibition and enhancement (+bicuculline) of α1β3γ2 GABAAR photolabeling by [3H]azietomidate and [3H]R-mTFD-MPAB
When drugs only inhibited photolabeling, data were fit to eq. 1 (Materials and Methods) to determine IC50, the total drug concentration producing 50% inhibition of GABAAR photolabeling, and Hill coefficients (nH) and, when indicated, to eq. 2, a two-site model. For inhibition in the presence of bicuculline, nH was set to 1. When enhancement was seen in the presence of bicuculline, data were fit to eq. 3B, as noted in the figure legends, to determine EC50 and IC50 values for 50% maximal enhancement and inhibition. Parameters (best fit ± S.E.) were determined from fits of data from N independent experiments, using two or more GABAAR purifications. As bicuculline and GABA experiments were paired, their values of N are the same.
| Drug (NR-mTFD-MPAB, Naziet) | Ratio IC50(AziEt)/IC50(R-mTFD-MPAB)a | [3H]R-mTFD-MPAB IC50 (nH) |
[3H]R-Azietomidate IC50 (nH) |
||
|---|---|---|---|---|---|
| +GABA |
+GABA |
+Bicuculline EC50/IC50 |
+GABA |
+Bicuculline EC50/IC50 |
|
| μM | μM | μM | μM | ||
| TG-41 (4, 3) | 0.2 ± 0.1 | 0.10 ± 0.04 (1) Bns = 67% ± 2% | eq. 3B 0.02 ± 0.007/0.4 ± 0.2 | 0.016 ± 0.003 (1) | 0.22 ± 0.06 |
| Loreclezole (4, 4)b | 6c | 116 ± 35 (0.43 ± 0.06) eq. 2 IC50H = 1.4 ± 1.2 (29% ± 7%)/IC50L = 320 ± 120 | eq. 3B 2.5 ± 0.8/160 ± 30 | 9.3 ± 0.9 (1.2 ± 0.1) | 44 ± 30 |
| Tracazolate (6, 3) | 0.4 ± 0.2 | 18 ± 7 (1) Bns = 56% ± 5% | eq. 3B 2.7 ± 0.6/24 ± 5 | 8.0 ± 0.9 (1.0 ± 0.1) | 18 ± 7 |
| Stiripentol (3, 3) | 2 ± 0.4 | 62 ± 7 (1.7 ± 0.3) | 73 ± 39 | 134 ± 14 (1.8 ± 0.3) | 70 ± 14/84 ± 14 |
For the ratio of IC50 values (+GABA), uncertainties were determined by propagation of error from the individual parameter uncertainties.
For loreclezole inhibition of [3H]R-mTFD-MPAB photolabeling (+GABA), parameters are also included for the fit of the data to a two-site model, eq. 2.
The ratio calculated for the high-affinity component (IC50H) for inhibition of [3H]R-mTFD-MPAB photolabeling.
Since tracazolate and TG-41 enhancement of [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline resulted from the drugs binding to the β+ sites, data were fit to eq. 3B to determine whether high-affinity binding to the α+–β− or γ+–β− site provided a better fit. For tracazolate, the fit with γ+–β− as the high-affinity site (Bmax = 100%, EC50 = 2.7 ± 0.6 μM, IC50γ = 24 ± 5 μM) was strongly favored (ΔAICc = 12.7, (99.8%)) over the best fit for the α+–β− site as high affinity. The value of IC50γ was close to the IC50 of 18 μM for [3H]R-mTFD-MPAB inhibition in the presence of GABA, and the value of EC50 was close to the IC50 of 8 μM for tracazolate inhibition of [3H]azietomidate photolabeling (+GABA). In contrast, for TG-41, fit to eq. 3B with α+–β− as the high-affinity site (Bmax = 100%, EC50 = 0.02 ± 0.007 μM, IC50α = 0.4 ± 0.2 μM) was slightly favored (ΔAICc = 2.5, (78%)) over the best fit for the γ+–β− site as high affinity (Bmax = 85% ± 3%, EC50 = 0.02 μM, IC50γ = 55 ± 27 μM). Consistent with identification of α+–β− as the high-affinity site, the value of IC50α was close to the IC50 of 0.1 μM for TG-41 inhibition of [3H]R-mTFD-MPAB photolabeling in the presence of GABA, whereas the best fit value of IC50γ was not.
Discussion
In this report, we used competition photolabeling assays to identify structural features of propofol analogs and other GABAAR PAMs that determine selectivity between the homologous intersubunit anesthetic binding sites in the α1β3γ2 GABAAR transmembrane domain. Within a panel of para-substituted propofol analogs, drugs were identified that bind with highest affinity to one (4-benzoyl-propofol) or two [β+: propofol, 4-Cl-propofol; β−: 4-(tert-butyl)-propofol, 4-[hydroxyl(phenyl)methyl]-propofol] sites per receptor, or with similar affinity to 4 (β+ and β−, 4-acetyl-propofol) sites. We also identified PAMs structurally unrelated to propofol that bind nonequivalently to the β− sites (loreclezole, tracazolate, and TG-41). Table 3 summarizes the relative affinities of the drugs for the β+ and β− intersubunit sites. Similar to GABA, the drugs studied bind preferentially to a desensitized state of the receptor compared with the resting state stabilized by bicuculline. However, electrophysiological studies are necessary to determine relative affinities of these drugs at each site for open-channel compared with resting states.
TABLE 3.
Relative affinities of GABAAR PAMs for intersubunit anesthetic sites in the presence of GABA
Data from Tables 1 and 2 are summarized by identifying the site with highest affinity (H), the sites binding with 2- to 10-fold lower affinity (I), or >15-fold lower affinity (L) than the high-affinity site. For the drugs that, in the presence of GABA, bind nonequivalently to the β− sites, prediction of the high-affinity β− site is based upon [3H]R-mTFD-MPAB photolabeling in the presence of bicuculline (see Results and Discussion).
| Ligands |
β+ Interfaces |
β− Interfaces |
|
|---|---|---|---|
|
β+–α− IC50 |
α+–β− IC50 |
γ+–β− IC50 |
|
| μM | μM | μM | |
| That distinguish the two β− sites | |||
| 4-benzoyl-propofol | L 100 | L 330 | H 0.5 |
| Loreclezole | I 9 | H 1 | L 300 |
| TG-41 | H 0.02 | I 0.1 | L > 100 |
| Tracazolate | H 8 | L > 300 | I 18 |
| That do not distinguish the two β− sites | |||
| Etomidate | H 2.0 | L 430 | L 430 |
| Propofol | H 7.8 | I 44 | I 44 |
| 4-Cl-propofol | H 4.9 | I 20 | I 20 |
| 4-acetyl-propofol | H 48 | H 32 | H 32 |
| 4-(tert-butyl)-propofol | I 130 | H 17 | H 17 |
| 4-benzyl-propofol | I 30 | H 6 | H 6 |
| Stiripentol | I 130 | H 60 | H 60 |
| 4-OHPheMe-propofol | L 160 | H 10 | H 10 |
| R-mTFD-MPAB | L 32 | H 0.7 | H 0.7 |
Structural Determinants for Selective Binding to β+ or β− Intersubunit Anesthetic Sites.
This work provides a first identification of small structural modifications of propofol that determine selectivity for the β+ versus β− intersubunit sites. Propofol and derivatives with para-substituents of small volume (H-, Cl-) bind with 5-fold selectivity to the β+ sites, while substituents with increasing volume bind with lower affinity to the β+ sites and with higher affinity to the β− sites (Table 1). The substituent volumes vary from 16 to 100 Å3, compared with propofol’s volume of 175 Å3, and over this range there is a reasonable linear correlation between substituent volume and log[IC50] for [3H]R-mTFD-MPAB (R2 = 0.75, P = 0.03) or [3H]azietomidate (R2 = 0.65, P = 0.05) inhibition (Fig. 8B). For para-substituted propofol analogs, including 4-Cl- and 4-benzoyl-propofol, as inhibitors of [35S]-tert-butylbicyclophosphorothionate binding to brain membranes, potency was enhanced by increased lipophilicity but reduced by substituent size (Trapani et al., 1998). Quantitative structure-activity analyses identified the hydrophobic pockets accommodating the 2- and 6-iso-propyl groups as key determinants of propofol’s anesthetic and GABAAR modulatory actions (Krasowski et al., 2002). While sec-butyl or tert-butyl replacements decreased affinity at the β+ and β− sites similarly (Chiara et al., 2013), studies with a broader range of substitutions can establish whether these positions determine selectivity for β+ versus β− sites.
Nonequivalent Binding to β− Intersubunit Sites.
This study’s novel finding is that some compounds bind selectively to the γ+–β− or α+–β− site. The preference of 4-benzoyl-propofol for a single β− site was unexpected in view of the nonselective binding of 4-benzyl-propofol (4-C6H5CH2-propofol) and 4-[hydroxyl(phenyl)methyl]-propofol. The most likely explanation for this difference is that, because of carbonyl-mediated electron resonance stabilization, 4-benzoyl-propofol’s structure is more rigid than the other structures. 4-Benzoyl propofol inhibited [35S]-tert-butylbicyclosphophorothionate binding to brain membranes at concentrations [IC50 = 0.9 μM (Trapani et al., 1998)] consistent with binding to the high-affinity β− site but 100-fold lower than those necessary to occupy the β+ sites. Thus, 4-benzoyl-propofol may serve as a lead compound to identify other propofol derivatives that bind nonequivalently to the γ+/α+–β− sites.
Amino acid differences on the plus side of the interface at homologous positions in αM3 (Ala, Tyr) and γM3 (Ser, Phe) contributing to the β− binding sites (Chiara et al., 2013) are likely to contribute to the nonequivalent binding of 4-benzoyl-propofol to the β− sites. Our results establish that 4-benzoyl-propofol binding to its high-affinity β− site is state-dependent and leads to enhancement of [3H]azietomidate photolabeling (+bicuculline). Unfortunately, the [3H]R-mTFD-MPAB/4-benzoyl-propofol photolabeling data were fit equally well by equations assuming high-affinity binding to the γ+–β− (eq. 3A) or α+–β− site (eq. 3B), which precludes identification of the high-affinity β− site. However, much published functional data suggest strongly that the γ+–β− site is the high-affinity site that enhances [3H]azietomidate photolabeling. Drug interactions with the γ+–β− interface have been identified as major determinants of αβγ GABAAR conformational transitions in other functional and photolabeling studies: 1) mutational analyses found the γ+–β− interface more important than the α+–β− interface for propofol enhancement (Maldifassi et al., 2016); 2) mutational analyses established that ivermectin binding to the γ+–β− interface produced persistent GABAAR activation in the absence of GABA, while binding to the α+–β− site enhanced GABA responses without persistent direct activation (Estrada-Mondragon and Lynch, 2015); and 3) 1-methyl-5-propyl-5-(m-trifluoromethyl-diazirynylphenyl)barbituric acid, a convulsant differing in structure from R-mTFD-MPAB by chirality and the presence of a 5-propyl rather than 5-allyl group, inhibited α1β3γ2 while enhancing α1β3 GABAAR responses (Desai et al., 2015; Jayakar et al., 2015), and α1β3γ2 GABAAR photolabeling established that 1-methyl-5-propyl-5-(m-trifluoromethyl-diazirynylphenyl)barbituric acid binds with highest affinity to the γ+–β− site in the presence of bicuculline, while R-mTFD-MPAB binds to that site preferentially in the presence of GABA and with little state dependence to the α+–β− site.
Loreclezole, tracazolate, and TG-41 also bind with high affinity to only one of the β− sites, with neither tracazolate nor TG-41 inhibiting photolabeling at the other β− site at the highest concentrations tested. Loreclezole bound to its high-affinity β− site with ∼5-fold higher affinity than to the β+ sites but did not enhance [3H]azietomidate photolabeling (+bicuculline). Fits of the [3H]R-mTFD-MPAB photolabeling data (+bicuculline) for loreclezole slightly favored the α+–β− site as its high-affinity β− site. That loreclezole did not enhance [3H]azietomidate photolabeling provides evidence that loreclezole, similar to R-mTFD-MPAB, binds with little state dependence to the α+–β− site. Tracazolate and TG-41 each bound with the highest affinity to the β+ sites and enhanced [3H]R-mTFD-MPAB photolabeling (+bicuculline). Fits of the [3H]R-mTFD-MPAB photolabeling data (+bicuculline) predicted strongly that γ+–β− is the high-affinity β− site for tracazolate (IC50γ ∼25 μM), while α+–β− is the high-affinity β− site for TG-41. For loreclezole, tracazolate, and TG-41, mutational analyses provided strong evidence that their enhancement of GABAAR responses partially results from binding to the β+ sites. Thus, substitutions at β2/3Asn-265, position 15′ in the βM2 helix, prevented enhancement by etomidate and propofol (Belelli et al., 1997; Krasowski et al., 2001b; Bali and Akabas, 2004) and reduced enhancement by loreclezole, tracazolate, and TG-41 (Wingrove et al., 1994; Thompson et al., 2002; Asproni et al., 2005; Groves et al., 2006). β3Asn-265 contributes directly to the [3H]azietomidate/etomidate binding site (Chiara et al., 2012; Chiara et al., 2013; Stewart et al., 2014). While loreclezole, tracazolate, and TG-41 bind nonequivalently to the β− sites, their high affinity for the β+ sites makes them less suited than 4-benzoyl-propofol as leads to developing drugs binding nonequivalently to the β− sites.
Allosteric Coupling between Intersubunit Anesthetic Binding Sites.
Our results provide direct evidence that even in the presence of bicuculline, PAM occupancy of β+ and β− sites in the TMD is coupled energetically. Cryo-electron microscopy analyses of αβγ GABAAR structures in the presence of bicuculline and etomidate or R-mTFD-MPAB will be necessary to determine whether structural changes in the TMD are coupled to those in the ECD. Positive energetic coupling was demonstrated by the close correlation between the IC50 values for PAM binding to its high-affinity β+ or β− site and the EC50 for enhancement of photolabeling at the other class of intersubunit sites (Fig. 8A; Table 1). For 4-benzoyl-propofol, the enhancement of [3H]azietomidate photolabeling was consistent with occupancy of the high-affinity β− site, most likely the γ+–β− site, but the enhancement of [3H]R-mTFD-MPAB photolabeling could not be accounted for by binding to the β+ sites. This enhancement indicated that binding to the γ+–β− site also enhanced binding to the α+–β− site.
Consistent with its preferential binding to the β+ sites, propofol enhanced [3H]R-mTFD-MPAB’s photolabeling but not [3H]azietomidate’s. However, other functionally important propofol binding sites have been predicted based upon mutational analyses identifying propofol sensitivity determinants in the α1 subunit cytoplasmic domain (Moraga-Cid et al., 2011), in the β subunit M4 helix (Richardson et al., 2007), and at the base of the ECD [βTyr-143 (Shin et al., 2018)] in proximity to the β− subunit interface and a residue photolabeled by a photoreactive propofol analog (Yip et al., 2013). That propofol did not enhance [3H]azietomidate photolabeling makes it unlikely that propofol binds to these sites in a state-dependent manner with higher affinity than the β+ sites.
Conclusions
Our study is the first to demonstrate that drugs can selectively target the β− interfaces in the GABAAR TMD that are flanked by the α1 or γ2 subunit. Together with previous work (Chiara et al., 2013; Jayakar et al., 2015), this makes it possible in principle to develop drugs selective for four of the five TMD subunit interfaces in synaptic GABAARs. The potential for targeting those interfaces is great because there are six α and three β subunits, and they appear in both α+–β− and β+–α− varieties. This principle has been recognized in the ECD, where agents are being developed that target sites homologous to the benzodiazepine site in the α+–γ− interfaces (Sieghart et al., 2012). However, the possibilities for allosteric action in the ECD are fewer because two of the interfaces contain the orthosteric agonist sites.
Abbreviations
- 4-acetyl-propofol
4-acetyl-2,6-diisopropylphenol
- ΔAICc
corrected Akaike’s information criterion
- 4-benzyl-propofol
4-benzyl-2,6-diisopropylphenol
- 4-benzoyl-propofol
4-benzoyl-2,6-diisopropylphenol
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- 4-Cl-propofol
4-chloro-2,6-diisopropylphenol
- ECD
extracellular domain
- GABAAR
GABA type A receptor
- 4-[hydroxyl(phenyl)methyl]-propofol
4-(hydroxyl(phenyl)methyl)-2,6-diisopropylphenol
- PAM
positive allosteric modulator
- R-mTFD-MPAB
1-methyl-5-allyl-5-(m-trifluoromethyl-diazirynylphenyl)barbituric acid
- 4-(tert-butyl)-propofol
4-(tert-butyl)-2,6-diisopropylphenol
- TG-41
ethyl 2-(4-bromophenyl)-1-(2,4-dichlorophenyl)-1H-4-imidazolecarboxylate
- TMD
transmembrane domain
Authorship Contributions
Participated in research design: Jayakar, Chiara, Cohen.
Conducted experiments: Jayakar, Chiara.
Contributed new reagents or analytic tools: Zhou, Jarava-Barrera, Savechenkov, Bruzik, Tortosa, Miller.
Performed data analysis: Jayakar, Cohen.
Wrote or contributed to writing of the manuscript: Jayakar, Chiara, Miller, Cohen.
Footnotes
This work was supported, in whole or in part, by the National Institutes of Health National Institute of General Medical Sciences [Grant GM-58448] (J.B.C., K.W.M.).
References
- Asproni B, Talani G, Busonero F, Pau A, Sanna S, Cerri R, Mascia MP, Sanna E, Biggio G. (2005) Synthesis, structure-activity relationships at the GABA(A) receptor in rat brain, and differential electrophysiological profile at the recombinant human GABA(A) receptor of a series of substituted 1,2-diphenylimidazoles. J Med Chem 48:2638–2645. [DOI] [PubMed] [Google Scholar]
- Bali M, Akabas MH. (2004) Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol 65:68–76. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. (1997) The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci USA 94:11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin RP, Orser BA. (2008) GABA(A) receptor subtypes underlying general anesthesia. Pharmacol Biochem Behav 90:105–112. [DOI] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. (1999) Allosteric activation mechanism of the α 1 β 2 γ 2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J 77:2542–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara DC, Dostalova Z, Jayakar SS, Zhou X, Miller KW, Cohen JB. (2012) Mapping general anesthetic binding site(s) in human α1β3 γ-aminobutyric acid type A receptors with [3H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry 51:836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB. (2013) Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem 288:19343–19357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua HC, Chebib M. (2017) GABAA receptors and the diversity in their structure and pharmacology. Adv Pharmacol 79:1–34. [DOI] [PubMed] [Google Scholar]
- de Lassauniere P-EC, Harnett J, Bigg D, Liberatore A, Pommier J, Lannoy J, Thurieau C, Dong ZX. (2005) inventors, Ipsen Pharma SAS, assignee. Derivatives of heterocycles with 5 members, their preparation and their use as medicaments. U.S. patent 20050038087A1. 2005 Feb 17.
- Desai R, Savechenkov PY, Zolkowska D, Ge RL, Rogawski MA, Bruzik KS, Forman SA, Raines DE, Miller KW. (2015) Contrasting actions of a convulsant barbiturate and its anticonvulsant enantiomer on the α1 β3 γ2L GABAA receptor account for their in vivo effects. J Physiol 593:4943–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostalova Z, Zhou X, Liu A, Zhang X, Zhang Y, Desai R, Forman SA, Miller KW. (2014) Human α1β3γ2L gamma-aminobutyric acid type A receptors: high-level production and purification in a functional state. Protein Sci 23:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada-Mondragon A, Lynch JW. (2015) Functional characterization of ivermectin binding sites in α1β2γ2L GABA(A) receptors. Front Mol Neurosci 8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JL. (2009) The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology 56:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA. (2010) Molecular approaches to improving general anesthetics. Anesthesiol Clin 28:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA, Miller KW. (2016) Mapping general anesthetic sites in heteromeric γ-aminobutyric acid type A receptors reveals a potential for targeting receptor subtypes. Anesth Analg 123:1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. (2008) General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 9:370–386. [DOI] [PubMed] [Google Scholar]
- Greenfield LJ., Jr (2013) Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure 22:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves JO, Guscott MR, Hallett DJ, Rosahl TW, Pike A, Davies A, Wafford KA, Reynolds DS. (2006) The role of GABAbeta2 subunit-containing receptors in mediating the anticonvulsant and sedative effects of loreclezole. Eur J Neurosci 24:167–174. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HMA, Smart TG. (2006) Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444:486–489. [DOI] [PubMed] [Google Scholar]
- Jarava-Barrera C, Parra A, López A, Cruz-Acosta F, Collado-Sanz D, Cárdenas DJ, Tortosa M. (2016) Copper-catalyzed borylative aromatization of p-quinone methides: enantioselective synthesis of dibenzylic boronates. ACS Catal 6:442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB. (2014) Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J Biol Chem 289:27456–27468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakar SS, Zhou X, Savechenkov PY, Chiara DC, Desai R, Bruzik KS, Miller KW, Cohen JB. (2015) Positive and negative allosteric modulation of an α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor by binding to a site in the transmembrane domain at the γ+-β- interface. J Biol Chem 290:23432–23446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Hong X, Hopfinger AJ, Harrison NL. (2002) 4D-QSAR analysis of a set of propofol analogues: mapping binding sites for an anesthetic phenol on the GABA(A) receptor. J Med Chem 45:3210–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. (2001a) General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther 297:338–351. [PubMed] [Google Scholar]
- Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. (2001b) Methionine 286 in transmembrane domain 3 of the GABAA receptor β subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology 41:952–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty D, Thomas P, Field M, Andersen OJ, Gold MG, Biggin PC, Gielen M, Smart TG. (2017) Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat Struct Mol Biol 24:977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-D, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. (2006) Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci 26:11599–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldifassi MC, Baur R, Sigel E. (2016) Functional sites involved in modulation of the GABAA receptor channel by the intravenous anesthetics propofol, etomidate and pentobarbital. Neuropharmacology 105:207–214. [DOI] [PubMed] [Google Scholar]
- Mascia MP, Asproni B, Busonero F, Talani G, Maciocco E, Pau A, Cerri R, Sanna E, Biggio G. (2005) Ethyl 2-(4-bromophenyl)-1-(2,4-dichlorophenyl)-1H-4-imidazolecarboxylate is a novel positive modulator of GABAA receptors. Eur J Pharmacol 516:204–211. [DOI] [PubMed] [Google Scholar]
- Masiulis S, Desai R, Uchański T, Serna Martin I, Laverty D, Karia D, Malinauskas T, Zivanov J, Pardon E, Kotecha A, et al. (2019) GABAA receptor signalling mechanisms revealed by structural pharmacology [published correction appears in Nature (2019) 566:E8]. Nature 565:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry-Wu AR, Bu W, Rai G, Lea WA, Weiser BP, Liang DF, Simeonov A, Jadhav A, Maloney DJ, Eckenhoff RG. (2015) Discovery of a novel general anesthetic chemotype using high-throughput screening. Anesthesiology 122:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR. (2014) Crystal structure of a human GABAA receptor. Nature 512:270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Scott S, Masiulis S, De Colibus L, Pardon E, Steyaert J, Aricescu AR. (2017) Structural basis for GABAA receptor potentiation by neurosteroids. Nat Struct Mol Biol 24:986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraga-Cid G, Yevenes GE, Schmalzing G, Peoples RW, Aguayo LG. (2011) A Single phenylalanine residue in the main intracellular loop of α1 γ-aminobutyric acid type A and glycine receptors influences their sensitivity to propofol. Anesthesiology 115:464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JB, Malick JB. (1982) Pharmacological properties of tracazolate: a new non-benzodiazepine anxiolytic agent. Eur J Pharmacol 78:323–333. [DOI] [PubMed] [Google Scholar]
- Phulera S, Zhu H, Yu J, Claxton DP, Yoder N, Yoshioka C, Gouaux E. (2018) Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho JM, Donevan SD, Rogawski MA. (1997) Barbiturate-like actions of the propanediol dicarbamates felbamate and meprobamate. J Pharmacol Exp Ther 280:1383–1391. [PubMed] [Google Scholar]
- Richardson JE, Garcia PS, O’Toole KK, Derry JMC, Bell SV, Jenkins A. (2007) A conserved tyrosine in the β2 subunit M4 segment is a determinant of γ-aminobutyric acid type A receptor sensitivity to propofol. Anesthesiology 107:412–418. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. (2006) Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res 69:273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savechenkov PY, Zhang X, Chiara DC, Stewart DS, Ge R, Zhou X, Raines DE, Cohen JB, Forman SA, Miller KW, et al. (2012) Allyl m-trifluoromethyldiazirine mephobarbital: an unusually potent enantioselective and photoreactive barbiturate general anesthetic. J Med Chem 55:6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DJ, Germann AL, Johnson AD, Forman SA, Steinbach JH, Akk G. (2018) Propofol is an allosteric agonist with multiple binding sites on concatemeric ternary GABAA receptors. Mol Pharmacol 93:178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W. (2015) Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv Pharmacol 72:53–96. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Ramerstorfer J, Sarto-Jackson I, Varagic Z, Ernst M. (2012) A novel GABA(A) receptor pharmacology: drugs interacting with the α(+) β(-) interface. Br J Pharmacol 166:476–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Ernst M. (2018) The benzodiazepine binding sites of GABAA receptors. Trends Pharmacol Sci 39:659–671. [DOI] [PubMed] [Google Scholar]
- Simeone TA, Wilcox KS, White HS. (2006) Subunit selectivity of topiramate modulation of heteromeric GABA(A) receptors. Neuropharmacology 50:845–857. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Dudek FE. (2014) Valnoctamide enhances phasic inhibition: a potential target mechanism for the treatment of benzodiazepine-refractory status epilepticus. Epilepsia 55:e94–e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DS, Pierce DW, Hotta M, Stern AT, Forman SA. (2014) Mutations at beta N265 in γ-aminobutyric acid type A receptors alter both binding affinity and efficacy of potent anesthetics. PLoS One 9:e111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DS, Savechenkov PY, Dostalova Z, Chiara DC, Ge R, Raines DE, Cohen JB, Forman SA, Bruzik KS, Miller KW. (2011) p-(4-Azipentyl)propofol: a potent photoreactive general anesthetic derivative of propofol. J Med Chem 54:8124–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Smith MZ, Wingrove PB, Whiting PJ, Wafford KA. (1999) Mutation at the putative GABA(A) ion-channel gate reveals changes in allosteric modulation. Br J Pharmacol 127:1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Wingrove PB, Connelly L, Whiting PJ, Wafford KA. (2002) Tracazolate reveals a novel type of allosteric interaction with recombinant γ-aminobutyric acid(A) receptors. Mol Pharmacol 61:861–869. [DOI] [PubMed] [Google Scholar]
- Trapani G, Latrofa A, Franco M, Altomare C, Sanna E, Usala M, Biggio G, Liso G. (1998) Propofol analogues. Synthesis, relationships between structure and affinity at GABAA receptor in rat brain, and differential electrophysiological profile at recombinant human GABAA receptors. J Med Chem 41:1846–1854. [DOI] [PubMed] [Google Scholar]
- Wauquier A, Fransen J, Melis W, Ashton D, Gillardin JM, Lewi PJ, Van Clemen G, Vaught J, Janssen PAJ. (1990) Loreclezole (R 72 063): an anticonvulsant chemically unrelated to prototype antiepileptic drugs. Drug Dev Res 19:375–392. [Google Scholar]
- Wingrove PB, Wafford KA, Bain C, Whiting PJ. (1994) The modulatory action of loreclezole at the γ-aminobutyric acid type A receptor is determined by a single amino acid in the β 2 and β 3 subunit. Proc Natl Acad Sci USA 91:4569–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip GMS, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, Townsend RR, Fuchs K, Sieghart W, Evers AS, et al. (2013) A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat Chem Biol 9:715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller A, Jurd R, Lambert S, Arras M, Drexler B, Grashoff C, Antkowiak B, Rudolph U. (2008) Inhibitory ligand-gated ion channels as substrates for general anesthetic actions. Handb Exp Pharmacol 182:31–51. [DOI] [PubMed] [Google Scholar]
- Zhu S, Noviello CM, Teng J, Walsh RM, Jr, Kim JJ, Hibbs RE. (2018) Structure of a human synaptic GABAA receptor. Nature 559:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]