

Abstract
Bacterial pathogens evolve during the course of infection as they adapt to the selective pressures that confront them inside the host. Identification of adaptive mutations and their contributions to pathogen fitness remains a central challenge. Although mutations can either target intergenic or coding regions in the pathogen genome, studies of host adaptation have focused predominantly on molecular evolution within coding regions, whereas the role of intergenic mutations remains unclear. Here, we address this issue and investigate the extent to which intergenic mutations contribute to the evolutionary response of a clinically important bacterial pathogen, Pseudomonas aeruginosa, to the host environment, and whether intergenic mutations have distinct roles in host adaptation. We characterize intergenic evolution in 44 clonal lineages of P. aeruginosa and identify 77 intergenic regions in which parallel evolution occurs. At the genetic level, we find that mutations in regions under selection are located primarily within regulatory elements upstream of transcriptional start sites. At the functional level, we show that some of these mutations both increase or decrease transcription of genes and are directly responsible for evolution of important pathogenic phenotypes including antibiotic sensitivity. Importantly, we find that intergenic mutations facilitate essential genes to become targets of evolution. In summary, our results highlight the evolutionary significance of intergenic mutations in creating host-adapted strains, and that intergenic and coding regions have different qualitative contributions to this process.
Keywords: intergenic evolution, cis-regulatory elements, bacterial adaptation, gene expression, antibiotic resistance
Introduction
Bacterial pathogens evolve during infection as they adapt to the environment inside the host (Didelot et al. 2016). Because the bacterial phenotypes selected in vivo may have profound impact on disease severity and progression (Hoffman et al. 2009; Das et al. 2016) as well as response to antibiotic therapy (Honsa et al. 2017), identification and analysis of the full range of genetic changes that underlie host adaptation is of importance.
Adaptive mutations may potentially change the sequences of either coding regions or nontranslated intergenic regions and thus affect protein function or gene expression, respectively. Nevertheless, studies of pathogen adaptation during infection of host tissues have focused predominantly on molecular evolution within coding regions (Marvig et al. 2015; Klemm et al. 2016), whereas the role of adaptive mutations in intergenic regions has received comparably less attention. The shortage of systematic, genome-wide analyses of intergenic evolution in bacterial pathogens is surprising, given the fact that these regions are home to a large number of functional elements required for expression of virulence and resistance determinants in vivo and that intergenic regions are maintained by purifying selection in many bacterial species (Molina and van Nimwegen 2008; Kim et al. 2012; Thorpe et al. 2017). Moreover, cis-regulatory mutations are known to play an important role in phenotypic evolution in eukaryotic organisms (Wray 2007). Overall, it remains unclear to what extent intergenic mutations contribute to the evolutionary response of pathogens to the host environment and whether intergenic mutations have a qualitatively distinct role in host adaptation.
There are clear, albeit few, examples of intergenic regions that evolve under selection within the host. For example, evolution of a novel regulatory interaction between the virulence regulator SsrB and the promoter of the sfrN gene was shown to result in enhanced within-host fitness in Salmonellatyphimurium (Osborne et al. 2009). In Pseudomonas aeruginosa, evolution of the intergenic region of the phuR-phuSTUVW genes during host colonization was shown to increase transcription of the Phu heme uptake system and improve the ability of the pathogen to acquire iron from hemoglobin (Marvig et al. 2014). Also, mutations in the 5′ untranslated region of the fatty acid biosynthesis gene cluster accBC in P. aeruginosa were shown to reduce translation efficiency of accBC, contributing to evolution of small-colony variant phenotypes (Blanka et al. 2015). In Mycobacterium tuberculosis, evolution of ethambutol resistance has been linked to the empAB promoter region acquiring mutations that enhance expression of genes encoding enzymes essential for the synthesis of cell wall arabinogalactan (Cui et al. 2014). In addition to these examples from pathogens evolving within hosts, several cases of adaptive intergenic mutations have also been documented in experimental laboratory evolution studies in which microbial populations are evolving under controlled in vitro conditions (Stoebel et al. 2009; Blank et al. 2014; Lamrabet et al. 2019).
Overall, these and other examples point towards an evolutionary significant role of mutations in intergenic regions in connection to bacterial pathogenesis and justify a broader analysis of this type of mutations.
One reason for the paucity in genome-wide analysis of intergenic evolution is related to the inherent difficulties in inferring function directly from the sequence within intergenic regions and, consequently, differentiating adaptive mutations with functional effects from neutral mutations that have been fixed by chance. Also, intergenic regions cannot readily be analyzed for selection by the same metrics as for coding regions (e.g., measuring the rate of nonsynonymous substitutions relative to the rate of synonymous substitutions [dN/dS]). An analogous approach for determining the selection in intergenic regions has been recently described and involves measuring the ratio of intergenic mutations to synonymous substitutions (dI/dS) (Thorpe et al. 2017). Where data sets with appropriate metadata and phylogenetic information are available, an alternative method for studying intergenic evolution is to identify signatures of parallel evolution in intergenic regions, which is the approach utilized in the present study.
Here, we harnessed the combination of data mining and functional genomics to identify intergenic regions under selection in the genome of the opportunistic pathogen P. aeruginosa during the process of host adaptation in multiple cystic fibrosis (CF) patients. Our study reveals that adaptive intergenic mutations represent an underappreciated aspect of host adaptation in P. aeruginosa, and that intergenic and coding region mutations may contribute differently to this process.
Materials and Methods
Bacterial Strains and Growth Conditions
Luria-Bertani (LB) and ABT minimal medium supplemented with 1% glucose and 1% casamino acids (ABTGC) (Yang et al. 2008) were routinely used for growth of Escherichiacoli and P. aeruginosa strains. Escherichiacoli CC118 λpir was used for maintenance of recombinant plasmids supplemented with 8 μg/ml of tetracycline. Pseudomonasaeruginosa PAO1 strain was used for phenotypic investigation of intergenic mutations. For final marker selection of P. aeruginosa, 50 μg/ml of tetracycline was used.
Assembly of the Data Set Used for Identification of Adaptive Intergenic Regions
We imported previously called variants in the intergenic regions of CF-adapted P. aeruginosa isolates from six longitudinal studies (Smith et al. 2006; Chung et al. 2012; Marvig et al. 2013; Jeukens et al. 2014; Marvig et al. 2015). In order to generate a systematic analysis of the shared intergenic regions, we only considered those regions where both flanking genes were present in P. aeruginosa PAO1 (Stover et al. 2000) genome and omitted all others. In addition, Marvig et al. (Marvig et al. 2013) reported draft genome sequences of four P. aeruginosa B3 strains isolated from a chronically infected Danish CF patient, who underwent antibiotic chemotherapy over a period of 4 years. Here, we called the variants in the genomes of these isolates and identified a total of 315 mutations (237 SNPs and 78 indels) when mapping the reads to the reference PAO1 genome. In total, we identified 3,489 intergenic mutations across 44 different clonal lineages. Detailed description of the data set can be found in supplementary tables S1 and S2, Supplementary Material online. To establish existing genetic variation between all 44 recognized lineages of P. aeruginosa used in this study and to avoid parallel observation of identical lineages, we performed MLST analysis on the genome of each lineage using the P. aeruginosa MLST website (Jolley et al. 2010). Sequence reads from Chung P5, Chung P6, and Chung P7 lineages were unavailable and the determined ST are reported by the publication itself (Chung et al. 2012). Overview of MLST results can be found in supplementary table S3, Supplementary Material online.
Identification of Adaptive Intergenic Regions
Adaptive intergenic regions in bacteria are characterized as regions important for adaptation to the host environment (i.e., likely to be under positive selection). They are therefore expected to be targeted by multiple mutations acquired in parallel by isolates from different lineages. In order to distinguish such mutations from random mutations introduced by genetic drift, we defined an intergenic region as adaptive when it is targeted by mutations found in isolates of three or more clonal lineages within a window of <30 bp apart from each other. This is because most regulatory elements in bacterial genomes are <30 bp in length (Stewart et al. 2012). As an additional criterion, the window has to be <200 bp away from at least one of the flanking genes for an intergenic region to be considered adaptive. To rule out possible sequencing artefacts among intergenic mutations, we excluded 13 regions where identical indel mutations occurred within isolates of the same data set (Marvig et al. 2015). As P. aeruginosa PAO1 genome has 4,682 intergenic regions constituting a total of 631,498 bp, we expect 0.0055 clone type mutation/bp rate (3,489 mutations in total) for intergenic regions. However, observing three or more mutations within a 30-bp intergenic region window (0.1 mutation/bp) is 18-fold higher than what would be expected by chance and represents a significant increase in mutation density [P(X ≥ 3) ∼ pois(X; 0.17) =6.70e‒4, where P(X ≥ 3) is the probability of observing ≥3 mutations given a Poisson distribution with a mean of 0.17 mutations (0.0055 mutation/bp ×30 bp)]. Applying these criteria, we identified 47 intergenic regions. In addition, we also included 16 regions in which two or more consecutive 30 bp intergenic windows each carrying 2 clone type mutations was observed. Although the occurrence of two clone type mutations within a single 30 bp intergenic window itself represents a significant increase in mutation density (Poisson, P = 0.01), we imposed the extra criterion of the presence of at least 2 such 30 bp windows with mutations to increase specificity of our selection.
Finally, for the identification of adaptive regions selected within each clonal lineage, we applied the same criteria as described above but only included cases of the presence of at least 3 distinct isolate type mutations within a narrow window of <30 bp. While this led to identification of 41 intergenic regions selected within clonal lineages, we manually inspected the mutations within each of these regions to exclude cases where linkage between isolates of the same lineage might have falsely introduced three or more mutations. To exclude such cases, we imposed an extra criterion in which each of the three or more mutations should not be identically present in all isolates of the same lineage and the mutations should be present in isolates from more than one patient. For cases where the phylogeny of isolates within a clonal lineage is known, we counted homoplastic mutations occurring in isolates of different patients as independent mutations. This manual search led to the exclusion of 13 regions selected within lineages.
Identification of Putative Intergenic Elements
The position of putative intergenic elements including core promoters, transcription factor binding sites, transcriptional terminators, invert repeats, small RNAs (sRNAs) and Shine-Dalgarno sequences were mapped within adaptive regions selected in parallel across different lineages (n = 63). We used BPROM (Solovyev and Salamov 2011), CollecTF (Kiliç et al. 2014), PRODORIC (Münch et al. 2003), RegTransBase (Kazakov et al. 2007), and the Pseudomonas Genome Database (PGD) (Winsor et al. 2016) to map putative promoters, transcription factor binding sites, Shine-Dalgarno sequences, and invert repeats. To increase the number of annotated promoters in P. aeruginosa, we utilized the findings of a recent study that validated putative binding sites of sigma factors in P. aeruginosa genome with RNA-seq and/or ChIP-seq (Schulz et al. 2015). A detailed description of present promoters and whether they have been targeted by intergenic mutations are available in supplementary table S6, Supplementary Material online. We also used ARNold and PGD (Gautheret and Lambert 2001; Macke et al. 2001; Winsor et al. 2016) for the identification of putative transcriptional terminators. Presence of sRNAs within adaptive intergenic regions was confirmed by a recent study reporting over 500 novel sRNAs within intergenic regions in P. aeruginosa genome (Gómez-Lozano et al. 2012). The presence of 232 sRNAs in P. aeruginosa expressed in response to different stress conditions (Gómez-Lozano et al. 2014) was also inspected within adaptive regions. We mapped the position of mutations to the identified putative elements (supplementary table S5, Supplementary Material online).
Construction of Reporter Fusions
We randomly selected 25 potentially adaptive intergenic regions upstream of 32 genes from DK2 isolates. We also included regions upstream of ampC and ampR from DK1-P43-M2-2002 (Andersen et al. 2015). Mutated intergenic regions upstream of 32 genes were amplified from genomic DNA of corresponding isolates (supplementary table S7, Supplementary Material online) using Phusion polymerase and primers described in supplementary table S9, Supplementary Material online. The PCR fragments and the pHK-CTX2-lux (Marvig et al. 2014) plasmid were doubled digested with restriction enzymes XhoI and PstI and ligated together with T4 DNA ligase (ThermoScientific). Similarly, wild-type regions upstream of all 32 genes were also amplified from DK2-CF30-1979 and cloned upstream of lux in pHK-CTX2-lux. The presence of mutations and the intergenic regions in resulting plasmids was verified using Sanger sequencing at LGC Genomics. The fusions were integrated at the neutral attB site in the chromosome of P. aeruginosa PAO1 by transformation as previously described (Choi and Schweizer 2006).
Measurements of Growth and Luminescence in Reporter Fusion Strains
Overnight cultures of reporter fusion strains were diluted 200 times in fresh LB medium and aliquots of 100 μl were transferred to black clear-bottom 96-well microtiter plate (Greiner). Three biological replicates were prepared for each fusion on the same day and measurements of growth (OD600) and luminescence were recorded by Cytation 5 multimode reader (BioTek) every 6 min for 8 h at 200 rpm and 37 °C. For cases with marginally altered expression, the measurements were repeated five times. The luminescence values were normalized by cell density (at OD600 =0.15) and compared for all fusions. Background luminescence from a PAO1 strain containing the promoterless lux cassette was measured in the same way and it was used for correcting luminescence expression of all strains. Data were analyzed using a custom-made script in R, version 3.1.3 (R Core Team 2013).
This script (supplementary file, Supplementary Material online) imports data for luminescence and absorbance (OD600) every 6 min for 8 h of 96 wells in a microtiter plate and calculates luminescence/OD600 at OD600 =0.15 for each well of a microtiter plate. These values are further analyzed using Microsoft Excel, where two-tailed Student’s t test is performed to examine the statistical difference between the means of three biological replicates from the mutant and wild-type allele fusions. The fold change of expression of mutant alleles was calculated relative to the expression of the wild-type fusions. Bonferroni correction of the raw P-value from Student’s t test has been performed when necessary.
Allelic Replacement of Intergenic Region Upstream of ampC and ampR in PAO1
A 1,361-bp fragment containing the intergenic region upstream of ampC and ampR was amplified from genomic DNA of DK1-P43-M2-2002 and DK2-CF173-1995 using Phusion polymerase and primers ampRi-F-XbaI and ampCi-R-SacI (supplementary table S10, Supplementary Material online). The PCR fragments and vector pNJ1 (Yang et al. 2012) were doubled digested with XbaI and SacI and ligated together using T4 DNA ligase. As the sequence of the ampC gene from the laboratory strain PAO1 differed from that of DK2 and DK1 isolates, we amplified the 1,361-bp fragment from DK2-CF30-1979 to obtain a pNJ1 plasmid with wild-type copy of the ampR//ampC intergenic region. Moreover, an additional mutation (G7A) was found at the start of ampC in DK1-P43-M2-2002. To account for possible epistatic effects, we created the ampC mutation (G7A) in the pNJ1 plasmid containing wild-type region using QuickChange Lightning Multi site-directed mutagenesis kit (Agilent Technologies). All ligation mixes were electroporated into E. coli CC118 λpir (Herrero et al. 1990) and transferred into the PAO1 strain (Holloway et al. 1979) by triparental mating using helper strain E. coli HB101/pRK600 (Kessler et al. 1992). After incubation overnight, merodiploid mutants were selected by plating the conjugation mixture on LB agar plate with 50 μg/ml tetracycline. Colonies were streaked on 6% (w/v) sucrose-LB plates without NaCl for several times until they became sensitive to tetracycline. Sucrose-resistant/tetracycline-sensitive colonies were finally streaked on sucrose-LB plates and allelic replacement mutants were verified by Sanger sequencing at LGC Genomics.
Minimum Inhibitory Concentrations
Minimum inhibitory concentration (MIC) was determined in two ways. For MICs of imipenem and ampicillin standard broth microdilution was used. Overnight cultures of PAO1 strains with and without intergenic mutations upstream of ampR and ampC were diluted in Mueller‒Hinton (MH) broth to an OD600 =0.02. Serial dilutions were performed in clear 96-well microtiter plates (Greiner) to obtain gradient concentrations of imipenem and ampicillin in MH broth. Aliquots of 100 μl were inoculated in each well containing 100 μl of MH broth with different concentrations of imipenem and ampicillin. We inoculated two technical replicates of each strain on each microtiter plate. Microtiter plates were incubated overnight at 37 °C with 200 rpm. MIC was defined as the lowest concentration of antibiotic where visible growth was observed. We repeated the experiment five times to obtain five biological replicates. For ceftazidime, MIC was determined using the E-test based on the manufacturer’s protocol (BioMerieux). Briefly, cultures of strains grown overnight in MH broth were diluted to OD600 =0.5, 100 μl was spread on MH agar plates and a sterile strip of ceftazidime E-test was placed on the plate. The values were measured after 22-h incubation of the plates at 37 °C and the E-test was performed in triplicate.
Results
Data Set Compilation for Identification of Parallel Intergenic Evolution
To investigate the contribution of intergenic mutations to bacterial adaptation to the host environment, we first compiled data from seven longitudinal studies (Smith et al. 2006; Chung et al. 2012; Marvig et al. 2015), in which multiple clonal P. aeruginosa isolates had been sampled and sequenced during the course of infection in different CF patients. These seven studies include 534 isolates from 44 different P. aeruginosa clonal lineages (each a distinct MLST), for which 22,491 mutations (SNPs and indels) were recorded as they accumulated in the lineages during adaptation to their individual hosts (n = 68). In total, 3,866 of the 22,491 mutations were located in intergenic regions. Here, we focused only on intergenic regions that were present in the laboratory strain PAO1. While this led to the exclusion of lineage-specific intergenic regions, it also enabled streamlined comparison between the 44 clonal lineages. Overall, this approach reduced the number of intergenic mutations to 3,489 (2,024 SNPs and 1,465 indels), distributed in a total of 1,610 regions.
In CF infections, the host environments in individual subjects represent parallel selective conditions by which evolution is directed, and the identification of parallel genetic evolution in the same loci in bacteria from independent infections is strongly suggestive of positive selection at these loci (Marvig et al. 2013). Analysis of such parallel genetic changes across multiple clonal lineages has previously led to the identification of several adaptive mutations within genes (Marvig et al. 2015). Here, we adopt a similar approach to identify likely intergenic targets of selection and only focus on intergenic mutations found in parallel in different lineages or in parallel among strains within the same lineage (fig. 1a). In addition, we hypothesized that adaptive intergenic mutations would predominantly target regulatory elements. Because the majority of regulatory elements in the bacterial genome range between 5 and 30 bp in length (Stewart et al. 2012), we hypothesized that the position of adaptive intergenic mutations would be constrained by a 30-bp boundary.
Fig. 1.

—Approach for identifying adaptive intergenic loci across 44 clonal lineages of P. aeruginosa. (a) An illustration of adaptive mutations occurring either across or within clonal lineages. The different lineages are represented by phylogenetic trees in green, blue and red and each genome within a lineage is displayed as a circle. Orange and yellow stripes within circles represent adaptive mutations. (b) The criteria for identifying adaptive mutations in our data set. As shown in the upper panel, three distinct clonal lineage mutations need to cluster within a narrow 30 bp window of an intergenic region in order to be considered adaptive. An additional criterion, as displayed in the lower panel, is that two distinct clonal lineage mutations need to occur within each of two narrow 30 bp windows within an intergenic region. In both cases, the window of mutations is less than 200 bp from at least one of the flanking genes.
We chose to look at cases of parallel intergenic evolution where three or more mutations occurred. Considering the total number of intergenic mutations found within the data set and the total length of intergenic regions in the genome, we calculated that three or more such mutations in a 30-bp intergenic window were 18-fold less likely to occur by chance (see Materials and Methods). In connection to analysis of parallel mutations across different lineages, we also included cases in which at least two 30-bp windows within the same intergenic region contained two mutations (fig. 1b) (see Materials and Methods). The final criterion that we imposed was that all clusters of mutations should be positioned <200 bp from at least one of the flanking genes.
Parallel Intergenic Evolution between Clonal Lineages
Applying these criteria, we found 63 intergenic regions with parallel genetic evolution between different clonal lineages (black and striped squares in fig. 2). The most frequently mutated region was between genes PA4786 and PA4787 (labeled PA4786//PA4787 in fig. 2). Isolates from 12 different clonal lineages had acquired mutations within this region during infection. Interestingly, the PA4786//PA4787 region has previously been shown to contain a small RNA gene of unknown function, which is differentially expressed in response to antibiotic exposure (Gómez-Lozano et al. 2014) (fig. 3a). Also, among the 63 regions was the phuR//phuSTUVW region in which mutations have previously been shown to increase expression of the operon and enhance the ability of the cell to acquire iron (Marvig et al. 2014) (fig. 3b). Further examples include mutations in the intergenic region of the genes encoding the antibiotic resistance regulator AmpR and the AmpC β-lactamase (ampR//ampC) (fig. 3c), as well as the intergenic region between motY and pyrC (fig. 3d). MotY is required for flagella-based motility, whereas PyrC is a dihydroorotase essential for survival and proliferation in CF sputum (Turner et al. 2015).
Fig. 2.

—A matrix showing 77 intergenic regions targeted by adaptive mutations across or within 44 clonal lineages. The black squares in the matrix show the intergenic regions where adaptive mutations were present in a single isolate of a respective clonal lineage and in isolates from other clonal lineages. The red squares in the matrix show the intergenic regions with adaptive mutations present only within isolates of a single respective clonal lineage. Squares with striped red color indicate intergenic regions in which adaptive mutations occurred both within isolates of a respective clonal lineage and in isolates from other clonal lineages.
Fig. 3.

—Examples of intergenic regions targeted by parallel evolution. The five intergenic regions have acquired parallel mutations across multiple clonal lineages (a‒d) or within isolates from a single clonal lineage (e). The orientation and the annotation of the flanking genes are shown for each of the five regions. The number of clonal lineages in which mutations are found is shown above each region. Occurrences of mutations in regulatory elements are indicated below the intergenic regions. A blue arrow illustrates the small regulatory RNA asRNA206.
Parallel Intergenic Evolution within Clonal Lineages
Because certain P. aeruginosa clonal lineages are transmissible and can form clinic-specific outbreaks among patients (Marvig et al. 2013), we also analyzed if distinct intergenic mutations had accumulated in parallel among clonal isolates within each of the 44 lineages. To exclude mutation signatures introduced by linkage within all isolates of the same lineage, we imposed the extra criterion that each of the three or more mutations should not be represented by all isolates of the same lineage and be present across different isolates from more than one patient (see Materials and Methods). We identified 28 intergenic regions in which 3 or more distinct mutations (<30 bp apart) had accumulated in isolates of the same clonal lineage (labeled with red or striped squares in fig. 2). Interestingly, 14 of these regions are also represented among the 63 regions identified in our analysis of parallel mutations between lineages, providing further support for the importance of these mutations in adaptation of P. aeruginosa to the CF environment (striped squares in fig. 2). Among the 14 adaptive regions that are only found within specific lineages, we identified mutations in the PA4838//zrmA region which were recently shown to increase expression of the zrmABCD operon (Hermansen et al. 2018) (fig. 3e). The zrmABCD operon (also called cntOLMI) encodes a metallophore system involved in zinc acquisition (Lhospice et al. 2017).
In total, we identified 77 intergenic regions that had likely evolved under the pressure of natural selection within the hosts. These adaptive intergenic regions and their flanking genes display potential importance for pathogen adaptation and thus provide a prioritized list for experimental testing of hypothesis regarding the selective forces that operate on the pathogen. We note, however, that there are different sources of error inherent between the 63 regions selected by mutations across different lineages and the 14 regions selected only within each lineage, which could potentially lead to incorrect conclusions when they are grouped together. For this reason, we treat the 2 groups separately and only consider the 63 regions selected across different lineages for further systematic molecular analyses of intergenic regions.
Intergenic Mutations Frequently Target Promoter Sequences
We next analyzed the genomic distribution of the identified intergenic mutations. Intergenic regions are distributed across the genome in three possible orientations: 1) upstream of two genes, 2) upstream of one gene and downstream of another gene, and 3) downstream of two genes, where the second orientation may include short regions within an operon (fig. 4a). We found an over-representation of mutations upstream of two genes among the adaptive regions (Fisher’s exact test, P = 0.01, n = 63; fig. 4b). This bias toward selection of intergenic mutations upstream of genes suggests that the majority of adaptive intergenic mutations target potential cis-regulatory elements such as the core promoter, transcription factor binding sites, ribo-regulators, or translational elements, and consequently influence gene expression levels by affecting transcriptional or post-transcriptional processes.
Fig. 4.

—Orientation of intergenic regions and presence of regulatory elements. (a) Overview of the three different orientations of intergenic regions and the possible locations of potential regulatory elements within each type. (b) Distribution of different orientations of intergenic regions (I‒III) within PAO1 genome and the adaptive intergenic regions. Two-tailed Fisher’s exact test is performed to analyze over-representation or under-representation of certain orientations within adaptive intergenic regions (n = 63). (c) Pie chart demonstrating the distribution of putative intergenic elements targeted by adaptive mutations in our data set (n = 24).
To further explore this hypothesis, we analyzed the complete set of 63 adaptive regions for the presence of predicted regulatory elements (see Materials and Methods) and mapped the overlap between these putative regulatory sites and the identified adaptive mutations. While bacterial intergenic regions are home to a wide range of regulatory elements, many of which are not well characterized, we nevertheless observed 24 regions (38%), in which the cluster of adaptive mutations was positioned within one or more putative regulatory elements. The majority of mutations within these 24 regions target the putative core promoter alone or in combination with other elements (fig. 4c), suggesting that intergenic mutations frequently target sequences important for transcriptional processes. In support of this, we observed that intergenic mutations were more frequently located upstream of known transcriptional start sites (TSS) (29 cases) than downstream (9 cases) (supplementary table S6, Supplementary Material online).
Adaptive Intergenic Mutations Change Transcriptional Activity of Genes Involved in Host Interaction, Metabolism and Antibiotic Susceptibility
To further explore the potential relationship between intergenic mutations and transcription, we quantified the effects of a subset of intergenic mutations on transcription of downstream genes. To this end, we constructed transcriptional fusions of both wild-type and mutant intergenic alleles with the luciferase reporter (luxCDABE) genes and integrated single copies of the fusions at the neutral attB site (Marvig et al. 2014) in the chromosome of P. aeruginosa PAO1 (Stover et al. 2000). We measured the transcriptional activity of 25 different intergenic regions in which adaptive mutations were located upstream of either one or 2 genes. These 25 mutated intergenic alleles were randomly chosen from the DK2 lineage and thus represent an analysis of the majority (25 out of 41) of adaptive intergenic regions (both from across and within-lineage data sets) found in this particular lineage. In addition, for one of the intergenic regions (ampR//ampC), we tested an additional allele from another clonal lineage (supplementary table S7 and fig. S1, Supplementary Material online). In total, this selection resulted in 34 transcriptional fusions.
Measurements of lux expression during exponential growth in LB medium and ABTGC minimal medium revealed statistically significantly altered expression in 16 of the 34 tested fusions in at least 1 of the 2 conditions (Student t test, P < 0.05; fig. 5). Expression changes were observed in both directions (seven mutant alleles resulted in increased expression and nine mutant alleles resulted in decreased expression) (fig. 5), suggesting that adaptive intergenic mutations may equally well either enhance or weaken gene expression. Altered expression was in most cases moderate (<5-fold change) and the fold change ranged from −5.0 to 23.4 for the mutant alleles compared with that of wild-type (fig. 5). Interestingly, 10 of these 16 fusions exhibited altered expression only in either LB or ABTGC minimal medium, but not in both conditions, which suggests that many adaptive intergenic mutations alter transcriptional levels without interfering with conditional gene expression control mechanisms.
Fig. 5.

—Intergenic mutations with functional effects on transcription. Luminescence from transcriptional lux reporter fusions with mutated and wild-type alleles was measured at OD600=0.15 and normalized by cell density. Transcriptional fusions were examined in Luria-Bertani (LB) and ABTGC minimal media. Normalized mean luminescence was calculated for three biological replicates, followed by calculating the relative fold change of mutant versus wild-type allele. Statistical analysis of the difference between two means was performed by a two-tailed Student’s t test and the asterisk denotes P < 0.05.
Several of the 16 fusions with altered expression relate to genes that encode proteins with known functions in bacteria‒host interactions, cellular metabolism and antibiotic resistance. For example, cerN encodes a ceramidase involved in utilization of host-produced sphingolipids (LaBauve and Wargo 2014), exsC encodes a protein involved in positive regulation of the type III secretion system (Dasgupta et al. 2004), and zrmA (PA4837) is the first gene in an operon (PA4837-34) involved in expression of a metalophore system essential for survival in airway mucus secretions (Gi et al. 2015; Mastropasqua et al. 2017). In a recent parallel study, we have further explored the effects of mutations in the zrmA promoter and shown that additional mutant alleles of the PA4836//zrmA region also result in increased expression of zrmA (PA4837) (Hermansen et al. 2018). Other genes are known to play a role in pyrimidine and aromatic amino acid metabolism (pyrC and hmgA, respectively). Finally, two genes are linked to antibiotic resistance: rluC (Toh and Mankin 2008) and ampR (Kumari et al. 2014). Seven genes encode proteins of unknown functions and their role in relation to host adaptation is thus not clear.
These results show that a substantial fraction of the intergenic mutations are associated with functional (transcriptional) effects despite the fact that we recorded these effects in the non-native PAO1 genetic background (i.e., with removal of potential epistatic effects from the additional variants found in the clinical isolates) and in a narrow range of conditions, which most likely means that we are not capturing the full spectrum of functional effects connected to the intergenic mutations.
Mutations Upstream of ampR and ampC Enhance Resistance to Several Antibiotics
Next, we explored the direct effects of intergenic mutations on the physiology of the pathogen. As resistance towards antibiotics is a common phenotype that emerges during CF infections, we selected the mutations found in the two alleles of the ampR//ampC intergenic region for further study. Mutations in this intergenic region resulted in enhanced expression of ampR, which encodes a global antibiotic resistance regulator, but had no direct effect on expression of a β-lactamase-encoding gene ampC (fig. 5). We introduced these mutations in the genome of P. aeruginosa PAO1 through allelic replacement (see Materials and Methods). Because an SNP mutation (G7A) was present at the start of the ampC gene in one of the alleles, we also made an allelic replacement of this mutation alone in the PAO1 genome to separate the effects caused by the intergenic mutations (supplementary fig. S1, Supplementary Material online). For each strain and their isogenic wild-type, we measured the MIC of various β-lactam antibiotics such as imipenem, ceftazidime, and ampicillin from carbapenem, cephalosporin, and penicillin classes of β-lactams, respectively. For both intergenic alleles, we observed a small but significant increase in the MIC of imipenem (2.4-fold) and ampicillin (2.2-fold) (Student’s t test, P < 0.01; fig. 6), but not ceftazidime. AmpR is a global transcriptional factor that regulates β-lactam resistance both through direct regulation of ampC expression as well as via an AmpC-independent manner (Kong et al. 2005; Kumari et al. 2014). Irrespective of the mechanism, our results show that acquisition of intergenic mutations between ampR and ampC is directly linked to a host-relevant phenotypic alteration (i.e., reduced β-lactam susceptibility).
Fig. 6.

—Mutations in the intergenic region between ampC and ampR cause an increased tolerance towards imipenem and ampicillin. The values for minimal inhibitory concentration (MIC) are shown on the y axis and the constructed mutations in each strain of PAO1 are displayed under the y axis. Mutation G-98A upstream of ampC derives from isolate DK2-CF173-1995. Three mutations G-38A, C-66T and G-78A upstream of ampC originate from isolate DK1-P43-M2-2002. An SNP mutation at the start of ampC (G7A) in DK1-P43-M2-2002 was also constructed in laboratory strain PAO1 to isolate the effect of this mutation and the effect of intergenic mutations from DK1-P43-M2-2002. Error bars indicate standard deviation from three different biological replicates. Double asterisk indicates statistically significant difference between mean MIC of the strains (two-tailed Student’s t test, P < 0.01).
Intergenic Evolution Targets Essential Genes
Finally, we explored potential differences in the contribution of coding and intergenic mutations to pathogen adaptation. We focused on a subset of isolates (n = 474) included in this study, in which 52 coding regions had been previously described to be under positive selection during host adaptation in another study (Marvig et al. 2015). In these isolates, we identified 35 adaptive intergenic regions (see Materials and Methods; supplementary table S8, Supplementary Material online).
We analyzed qualitative differences between coding and intergenic mutations by determining the presence of essential genes among the adaptive coding and intergenic regions. By cross-referencing the 35 adaptive intergenic regions to the list of 445 genes previously shown to be essential for survival of P. aeruginosa PAO1 in the CF sputum environment (Turner et al. 2015), we found that 7 of the 38 genes (18%) located immediately downstream of the 35 adaptive intergenic regions are essential (supplementary table S8, Supplementary Material online). Two of these genes (pyrC and PA5492) showed altered expression as a consequence of adaptive mutations in their intergenic region, demonstrating that such mutations can indeed modulate expression of essential genes (fig. 5). Importantly, the association between adaptive intergenic regions and essential genes at a level of 18% represents a significant overrepresentation from the normal prevalence of CF sputum essential genes (8%) in the P. aeruginosa PAO1 genome (fig. 7; Fisher’s exact test, P = 0.029). In contrast, there were no CF sputum essential genes within 52 adaptive coding regions, demonstrating a significant underrepresentation of these genes within adaptive coding regions (fig. 7; Fisher’s exact test, P = 0.033).
Fig. 7.
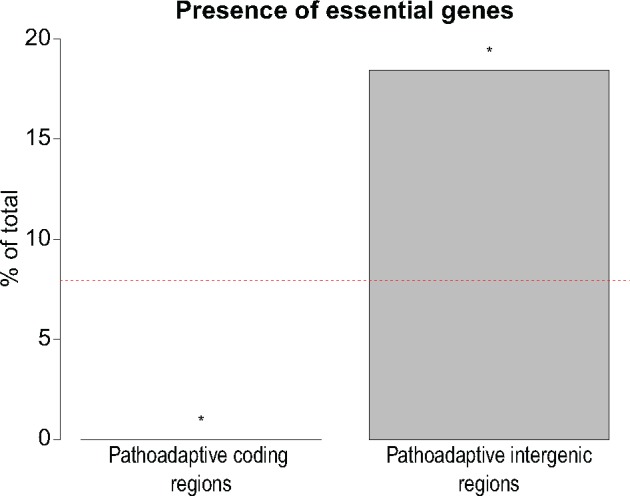
—Qualitative function of adaptive coding and intergenic mutations. A total of 52 coding regions were previously shown to be under positive selection in a selected subset of our data set comprised of 474 long-term CF-adapted isolates of P. aeruginosa (Marvig et al. 2015). In the same isolates, we identify 35 intergenic regions under positive selection for adaptive mutations (supplementary table S8, Supplementary Material online). 445 genes are essential for survival of P. aeruginosa in CF sputum environment (Turner et al. 2015). The percentage of these essential genes within 52 adaptive genes and 38 genes downstream of 35 adaptive intergenic regions is demonstrated. Asterisk denotes P < 0.05 from two-tailed Fisher’s exact test. Red dashed line indicates the percentage of CF essential genes in PAO1.
Discussion
In this study, we show that intergenic mutations constitute an important part of the genetic basis for host adaptation in P. aeruginosa. Generally, the contribution of intergenic regions to evolution of host-adapted variants has received little attention. However, because development of predictive models of pathogen evolution and discovery of new therapeutic targets rely on understanding the evolutionary response of pathogens to the host environment (Smith and Romesberg 2007; Yen and Papin 2017), identification of the full range of adaptive mutations in both coding and noncoding regions is important. Here, our genome-wide identification of intergenic regions under selection within the host was made possible by combining analysis of parallel evolution across a large number of infected individuals with functional genomics. This approach may be useful for analysis of intergenic regions in connection to host adaptation in other pathogens or niche adaptation in general.
It should be noted that our analysis relies upon the different sequencing and variant-calling methods of the previously published studies. Thus, it is expected that there will be a certain degree of false negative and false positive adaptive mutations in the data set. To try to mitigate this issue and to minimize potential errors, we used a set of strict criteria that took into account prior knowledge of the evolutionary processes in CF infections as well as positional information of intergenic mutations. This possible limitation can be additionally addressed in further studies by utilizing data sets that have been produced using the same sequencing technology, which would then enable us to systematically call variants with the same algorithms and parameters.
In addition, our data set was comprised of intergenic regions that were only present in the PAO1 genome. While this can lead to exclusion of some intergenic regions not found in PAO1, but present in the studied isolates, this approach has allowed us to perform a systematic analysis of intergenic mutations in shared regions and has also facilitated testing the effects of specific intergenic mutations in the PAO1 background.
Our list of potentially adaptive intergenic regions in P. aeruginosa provides insight into the cellular functions targeted by intergenic mutations (fig. 2) and thus points to the selective pressures that confront the pathogen within its CF host. For example, adaptive mutations were found to alter expression of genes such as cerN (involved in sphingolipid utilization) (LaBauve and Wargo 2014), phuR-phuSTUVW (involved in iron acquisition) (Marvig et al. 2014), and PA4837-34 (involved in zinc acquisition) (Mastropasqua et al. 2017; Hermansen et al. 2018), which strongly indicates that metabolic adaptation for a better exploitation of available nutrients in the host is an important evolutionary driver. Similarly, we observed that mechanisms of the development of tolerance to antibiotics and other inhibitors in the host are also frequent targets of intergenic molecular evolution. Similar functional categories have been found in studies focusing on adaptive mutations within coding regions (Marvig et al. 2013, 2015; Jeukens et al. 2014), suggesting that key selective pressures such as nutrient availability and antibiotic stress can be mitigated both by intergenic and coding region evolution in P. aeruginosa.
At the functional level, we show that intergenic evolution predominantly targets transcriptional processes to alter the downstream gene expression. Interestingly, we also found two transcriptional terminators targeted by mutations. We additionally saw indication of parallel evolution in three intergenic small RNAs, as well as several cases of mutations located downstream of transcriptional start sites (supplementary tables S5 and S6, Supplementary Material online), which suggests that adaptive mutations may indeed also target elements that control protein synthesis at the post-transcriptional level. Importantly, we have shown here and in a previous study that intergenic mutations can be directly responsible for the evolution of important pathogenic traits such as reduced sensitivity to antibiotics (fig. 6) and increased iron uptake (Marvig et al. 2014). Further studies, in particular of adaptive regions upstream of genes with unknown functions, will most likely uncover new mechanisms central to CF host colonization and pathogenesis, and assist in identifying the full complement of stressors present in the host, most of which are currently unknown.
Our study also reveals important qualitative differences between the intergenic and coding region mutations. A generally accepted model is that intergenic mutations would typically confer local and subtle regulatory effects primarily on the immediate downstream genes, whereas mutations in coding regions—with their potential to inactivate entire pathways—would be more likely to cause systemic changes of the physiology of the cell (Wray 2007; Coombes 2013). One prediction from this model is that intergenic mutations are associated with less antagonistic effects relative to coding region mutations. While this prediction is difficult to test, our observation of enrichment of essential genes for which intergenic evolution occurred is a clear illustration of this point (fig. 7). The finding that intergenic mutations can bypass the deleterious effects of coding region mutations, thus helping essential genes to become targets for evolutionary changes, appears to reveal an important aspect of the role of intergenic mutations as well as one of the key functional differences between intergenic and coding region mutations.
We hypothesize that the relative contribution of coding and intergenic mutations is variable and depends on a set of identifiable factors of either environmental nature (e.g., niche complexity) or intrinsic to the bacterial pathogen (e.g., genome size, and the number of transcriptional regulatory systems and essential genes encoded in the genome). Although the precise factors that influence the relative contribution of the two types of mutations may be difficult to disentangle, we speculate that in the case of P. aeruginosa CF infections, a major contributing factor is the composition of the adaptive environment in the host. The CF host niche is characterized by a complex combination of multiple stressors that must be mitigated for successful bacterial colonization (Folkesson et al. 2012). In such environments, mutations in intergenic regions that tune expression levels while maintaining responsiveness to environmental and host-derived cues may result in less pleiotropic effects than mutations that change protein structure or function (Coombes 2013). Further studies of P. aeruginosa adaptation in other infections and host environments such as chronic wounds and ulcerative keratitis (Winstanley et al. 2005) are required to identify factors that may influence the relative contribution of intergenic and coding region evolution.
Our documentation of the evolutionary significance of intergenic mutations was obtained in the particular genetic and ecological context of P. aeruginosa adaptation to the CF airway niche. Nevertheless, our study provides insight into the contribution and functionality of intergenic versus intragenic mutations, which is of broader relevance in connection to bacterial evolution in natural environments. This is supported by recent observations indicating that other contexts may also promote intergenic evolution. For example, our study resonates well with results showing that adaptive intergenic mutations contribute to innovation of novel metabolic functions in laboratory-evolving E.coli (Blank et al. 2014), evidence of a signal of positive selection in M. tuberculosis intergenic regions (Thorpe et al. 2017), and the suggestion that intergenic evolution may mitigate detrimental fitness effects associated with acquisition of novel genetic material (McNally et al. 2016). We suggest that adaptive mutations in intergenic regions represent an important but underappreciated aspect of bacterial evolution not only in connection to host colonization but also niche adaptation in other natural environments.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Lea M. Sommer and Anders Norman for technical advices in bioinformatics approaches, Esben V. Nisted for help in design of primers, Nicoline Uglebjerg and Caroline A. S. Lauridsen for assistance in identification of putative intergenic elements. We also thank Grith Hermansen, Geoff Winsor, Ed Feil, Dominique Schneider, and Thomas Hindré for valuable discussions. This work was supported by the Danish Council for Independent Research (6108-00300A) and the Villum Foundation (VKR023113).
Authors’ Contributions
S.M.H.K. and L.J. conceived study and designed research. S.M.H.K. performed research. S.M.H.K., P.S., and L.J. analyzed data and wrote the manuscript.
Literature Cited
- Andersen SB, Marvig RL, Molin S, Krogh Johansen H, Griffin AS.. 2015. Long-term social dynamics drive loss of function in pathogenic bacteria. Proc Natl Acad Sci USA. 112(34):10756–10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank D, Wolf L, Ackermann M, Silander OK.. 2014. The predictability of molecular evolution during functional innovation. Proc Natl Acad Sci USA. 111(8):3044–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanka A, et al. 2015. Constitutive production of c-di-GMP is associated with mutations in a variant of Pseudomonas aeruginosa with altered membrane composition. Sci Signal. 8(372):ra36.. [DOI] [PubMed] [Google Scholar]
- Choi KH, Schweizer HP.. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc. 1(1):153–161. [DOI] [PubMed] [Google Scholar]
- Chung JCS, et al. 2012. Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J Bacteriol. 194(18):4857–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes BK. 2013. Regulatory evolution at the host–pathogen interface. Can J Microbiol. 59(6):365–367. [DOI] [PubMed] [Google Scholar]
- Cramer N, et al. 2011. Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol. 13(7):1690–1704. [DOI] [PubMed] [Google Scholar]
- Cui Z, et al. 2014. Mutations in the embC-embA intergenic region contribute to Mycobacterium tuberculosis resistance to ethambutol. Antimicrob Agents Chemother. 58(11):6837–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, et al. 2016. Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proc Natl Acad Sci USA. 113(22):E3101–E3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta N, Lykken GL, Wolfgang MC, Yahr TL.. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol. 53(1):297–308. [DOI] [PubMed] [Google Scholar]
- Didelot X, Walker AS, Peto TE, Crook DW, Wilson DJ.. 2016. Within-host evolution of bacterial pathogens. Nat Rev Microbiol. 14(3):150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson A, et al. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. 10(12):841–851. [DOI] [PubMed] [Google Scholar]
- Gautheret D, Lambert A.. 2001. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol. 313(5):1003–1011. [DOI] [PubMed] [Google Scholar]
- Gi M, et al. 2015. A novel siderophore system is essential for the growth of Pseudomonas aeruginosa in airway mucus. Sci Rep. 5(October):14644.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Lozano M, Marvig RL, Molin S, Long KS.. 2012. Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ Microbiol. 14(8):2006–2016. [DOI] [PubMed] [Google Scholar]
- Gómez-Lozano M, Marvig RL, Tulstrup MV, Molin S.. 2014. Expression of antisense small RNAs in response to stress in Pseudomonas aeruginosa. BMC Genomics 15(1):783.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermansen GMM, Hansen ML, Khademi SMH, Jelsbak L.. 2018. Intergenic evolution during host adaptation increases expression of the metallophore pseudopaline in Pseudomonas aeruginosa. Microbiology 164(8):1038–1047. [DOI] [PubMed] [Google Scholar]
- Herrero M, de Lorenzo V, Timmis KN.. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J Bacteriol. 172(11):6557–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, et al. 1994. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 125(2):167–188. [Google Scholar]
- Hoffman LR, et al. 2009. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J Cyst Fibros. 8(1):66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway BW, Krishnapillai V, Morgan AF.. 1979. Chromosomal genetics of Pseudomonas. Microbiol Rev. 43(1):73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsa ES, et al. 2017. RelA mutant enterococcus faecium with multiantibiotic tolerance arising in an immunocompromised host. MBio 8(1):e02124–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeukens J, et al. 2014. Comparative genomics of isolates of a Pseudomonas aeruginosa epidemic strain associated with chronic lung infections of cystic fibrosis patients. PLoS One 9(2):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, et al. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11(1):595.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazakov AE, et al. 2007. RegTransBase—a database of regulatory sequences and interactions in a wide range of prokaryotic genomes. Nucleic Acids Res. 35(Database issue):D407–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler B, de Lorenzo V, Timmis KN.. 1992. A general system to integrate lacZ fusions into the chromosomes of gram-negative eubacteria: regulation of the Pm promoter of the TOL plasmid studied with all controlling elements in monocopy. Mol Gen Genet. 233(1–2):293–301. [DOI] [PubMed] [Google Scholar]
- Kiliç S, White ER, Sagitova DM, Cornish JP, Erill I.. 2014. CollecTF: a database of experimentally validated transcription factor-binding sites in Bacteria. Nucleic Acids Res. 42(D1):D156–D160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2012. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 8(8):e1002867.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm EJ, et al. 2016. Emergence of host-adapted Salmonella Enteritidis through rapid evolution in an immunocompromised host. Nat Microbiol. 1(3):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K-F, et al. 2005. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother. 49(11):4567–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H, Balasubramanian D, Zincke D, Mathee K.. 2014. Role of Pseudomonas aeruginosa AmpR on β-lactam and non-β-lactam transient cross-resistance upon pre-exposure to subinhibitory concentrations of antibiotics. J Med Microbiol. 63(Pt 4):544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBauve AE, Wargo MJ.. 2014. Detection of host-derived sphingosine by Pseudomonas aeruginosa is important for survival in the murine lung. PLoS Pathog. 10(1):e1003889.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamrabet O, et al. 2019. Plasticity of promoter-core sequences allows bacteria to compensate for the loss of a key global regulatory gene. Mol Biol Evol., doi:10.1093/molbev/msz042 [DOI] [PubMed] [Google Scholar]
- Lesnik EA, et al. 2001. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 29(17):3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhospice S, et al. 2017. Pseudomonas aeruginosa zinc uptake in chelating environment is primarily mediated by the metallophore pseudopaline. Sci Rep. 7(1):17132.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macke TJ, et al. 2001. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 29(22):4724–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig RL, et al. 2013. Draft genome sequences of Pseudomonas aeruginosa B3 strains isolated from a cystic fibrosis patient undergoing antibiotic chemotherapy. Genome Accounc. 1(5):e00804–e00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig RL, Johansen HK, Molin S, Jelsbak L.. 2013. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 9(9):e1003741.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig RL, Sommer LM, Molin S, Johansen HK.. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet. 47(1):57–64. [DOI] [PubMed] [Google Scholar]
- Marvig RL, et al. 2014. Within-host evolution of Pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. MBio 5(3):e00966–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastropasqua MC, et al. 2017. Growth of Pseudomonas aeruginosa in zinc poor environments is promoted by a nicotianamine-related metallophore. Mol Microbiol. 106(4):543–561. [DOI] [PubMed] [Google Scholar]
- McNally A, et al. 2016. Combined analysis of variation in core, accessory and regulatory genome regions provides a super-resolution view into the evolution of bacterial populations. PLOS Genet. 12(9):e1006280.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina N, van Nimwegen E.. 2008. Universal patterns of purifying selection at noncoding positions in bacteria. Genome Res. 18(1):148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch R, et al. 2003. PRODORIC: prokaryotic database of gene regulation. Nucleic Acids Res. 31(1):266–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne SE, et al. 2009. Pathogenic adaptation of intracellular bacteria by rewiring a cis-regulatory input function. Proc Natl Acad Sci USA. 106(10):3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2013. R: a language and environment for statistical computing. Vienna (Austria: ): R Found Stat Comput; Available from: http://www.r-project.org/, last accessed April 18, 2019. [Google Scholar]
- Schulz S, et al. 2015. Elucidation of sigma factor-associated networks in Pseudomonas aeruginosa reveals a modular architecture with limited and function-specific crosstalk. PLoS Pathog. 11(3):e1004744.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA, Romesberg FE.. 2007. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat Chem Biol. 3(9):549–556. [DOI] [PubMed] [Google Scholar]
- Smith EE, et al. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci USA. 103(22):8487–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V, Salamov A.. 2011. Automatic annotation of microbial genomes and metagenomic sequences. In: Li RW, editor. Metagenomics and its applications in agriculture, biomedicine and environmental studies. Hauppauge (NY): Nova Science Pub Inc., p. 61–78.
- Stewart AJ, Hannenhalli S, Plotkin JB.. 2012. Why transcription factor binding sites are ten nucleotides long. Genetics 192(3):973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoebel DM, Hokamp K, Last MS, Dorman CJ.. 2009. Compensatory evolution of gene regulation in response to stress by Escherichia coli lacking RpoS. PLoS Genet. 5(10):e1000671.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, et al. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406(6799):959–964. [DOI] [PubMed] [Google Scholar]
- Thorpe HA, Bayliss SC, Hurst LD, Feil EJ.. 2017. Comparative analyses of selection operating on nontranslated intergenic regions of diverse bacterial species. Genetics 206(1):363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh S-M, Mankin AS.. 2008. An indigenous posttranscriptional modification in the ribosomal peptidyl transferase center confers resistance to an array of protein synthesis inhibitors. J Mol Biol. 380(4):593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M.. 2015. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci USA. 112(13):4110–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor GL, et al. 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44: 646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley C, et al. 2005. Genotypic and phenotypic characteristics of Pseudomonas aeruginosa isolates associated with ulcerative keratitis. J Med Microbiol. 54(6):519–526. [DOI] [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 8(3):206–216. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. 2008. In situ growth rates and biofilm development of Pseudomonas aeruginosa populations in chronic lung infections. J Bacteriol. 190(8):2767–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, et al. 2012. Polysaccharides serve as scaffold of biofilms formed by mucoid Pseudomonas aeruginosa. FEMS Immunol Med Microbiol. 65(2):366–376. [DOI] [PubMed] [Google Scholar]
- Yen P, Papin JA.. 2017. History of antibiotic adaptation influences microbial evolutionary dynamics during subsequent treatment. PLoS Biol. 15(8):e2001586.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.