

Summary
Background
NLRP3 inflammasome-directed pyroptotic cell death drives ineffective haemopoiesis in myelodysplastic syndromes. During inflammasome assembly, the apoptosis-associated speck-like protein containing a CARD (PYCARD, commonly known as ASC) adaptor protein polymerises into large, filamentous clusters termed ASC specks that are released upon cytolysis. Specks are resistant to proteolytic degradation because of their prion-like structure, and therefore might serve as a biomarker for pyroptotic cell death in myelodysplastic syndromes.
Methods
This observational cohort study was done at the H Lee Moffitt Cancer Center (Tampa, FL, USA). Patients with myelodysplastic syndromes, healthy controls, and patients with non-myelodysplastic syndrome haematological cancers or type 2 diabetes were recruited. We used confocal and electron microscopy to visualise, and flow cytometry to quantify, ASC specks in peripheral blood and bone marrow plasma samples. Speck percentages were compared by t test or ANOVA, correlations were assessed by Spearman’s rank correlation coefficient, and biomarker efficiency was assessed by receiver operating characteristics and area under the curve (AUC) analysis.
Findings
Between Jan 1, 2005, and Jan 12, 2017, we obtained samples from 177 patients with myelodysplastic syndromes and 29 healthy controls for the discovery cohort, and 113 patients with myelodysplastic syndromes and 31 healthy controls for the validation cohort. We also obtained samples from 22 patients with del(5q) myelodysplastic syndromes, 230 patients with non-myelodysplastic syndrome haematological cancers and 23 patients with type 2 diabetes. After adjustment for glucose concentration, the log10-transformed mean percentage of peripheral blood plasma-derived ASC specks was significantly higher in the 177 patients with myelodysplastic syndromes versus the 29 age-matched, healthy donors (−0.41 [SD 0.49] vs −0.67 [0.59], p=0.034). The percentages of ASC specks in samples from patients with myelodysplastic syndromes were significantly greater than those in samples from individuals with every other haematological cancer studied (all p<0.05) except myelofibrosis (p=0.19). The findings were confirmed in the independent validation cohort (p<0.0001). Peripheral blood plasma danger-associated molecular pattern protein S100-A8 and protein S100-A9 concentrations from 144 patients with myelodysplastic syndromes from the discovery cohort directly correlated with ASC speck percentage (r=0.4, p<0.0001 for S100-A8 and r=0.2, p=0.017 for S100-A9). Patients with at least two somatic gene mutations had a significantly greater mean percentage of peripheral blood plasma ASC specks than patients with one or no mutation (−0.22 [SD 0.63] vs −0.53 [0.44], p=0.008). The percentage of plasma ASC specks was a robust marker for pyroptosis in myelodysplastic syndromes (AUC=0.888), in which a cutoff of 0.80 maximised sensitivity at 0.84 (95% CI 0.65–0.91) and specificity at 0.87 (0.58–0.97).
Interpretation
Our results underscore the pathobiological relevance of ASC specks and suggest that ASC specks are a sensitive and specific candidate plasma biomarker that provides an index of medullary pyroptotic cell death and ineffective haemopoiesis in patients with myelodysplastic syndromes.
Introduction
Pyroptosis is mediated by the formation of NLRP3 inflammasome complexes, which function as redox-sensitive, cytosolic sensors of danger signals.1 Uponstimulation, NLRP3 recruits the adaptor protein apoptosis-associated speck-like protein containing a CARD (PYCARD, commonly known as ASC), triggering ASC polymerisation to create large, cytoplasmic filaments.2 ASC filaments stack to form a cytoplasmic cluster, referred to as specks, which are generally described as about 1–3 μm in diameter and are released into the extracellular space after pyroptotic cytolysis.2,3 Because oftheir prion-like structure, specks are resistant to proteolytic degradation.3 Extracellular ASC specks retain catalytic activity with abundant caspase-1 activation sites, fostering inflammasome signal strengthening that augments the inflammatory response.2–4 Pyroptosis occurs after assembly of the inflammasome complex comprised of NLRP3 that is bound to caspase-1 via the adaptor molecule ASC. Caspase-1 then undergoes autocatalytic cleavage that activates the proenzyme that cleaves and generates mature pro-interleukin iβ. Inflammasome activation induces cell swelling, pore formation, and caspase-1-dependent pyroptotic cytolysis with expulsion of intracellular contents.1–3
The diagnosis of myelodysplastic syndromes can be challenging because, in addition to the substantial disease heterogeneity, diagnosis relies on subjective morphological assessment of cytological dysplasia and bone marrow aspirates of variable technical quality.5–7 Furthermore, many haematological conditions can display dysplastic features, including overlap syndromes such as chronic myelomonocytic leukaemia and various benign conditions.8 Myelodysplastic syndromes are characterised by ineffective haemopoiesis arising in part from caspase-1-dependent pyroptotic death of stem and progenitor cells, as well as erythroid and myeloid bone marrow cell compartments.9 We previously published a study describing the role of pyroptosis in the underlying mechanism ofineffective haemopoiesis in patients with myelodysplastic syndromes. To our knowledge, that was the first study to implicate this novel form of cell death in the pathobiology ofmyelodysplastic syndromes.9 Myelodysplastic syndrome-related somatic gene mutations can trigger NADPH oxidase-dependent generation of reactive oxygen species10,11 that leads to NLRP3 inflammasome formation, resulting in caspase-1 and β-catenin activation, and pyroptosis. This process occurs in the context of common myelodysplastic syndrome somatic gene mutations of varied functional classes, including U2 small nuclear RNA auxiliary factor 1 (U2AF1), splicing factor 3b subunit 1 (SF3B1), serine and arginine rich splicing factor 2 (SRSF2), additional sex combs like 1, transcriptional regulator (ASXL1), and Tet methylcytosine dioxygenase 2 (TET2), recognised as splicing, methylation, and transcription factor genes.9 On the basis of these investigations showing that NLRP3 inflammasome activation directs pyroptotic cell death in myelodysplastic syndrome bone marrow progenitors,9 we rationalised that this would generate higher percentages of bone marrow and peripheral blood specks in patients with myelodysplastic syndromes than in healthy donors and patients with other haematological cancers. ASC specks might be a surrogate marker for the magnitude of intramedullary pyroptosis in bone marrow samples from patients with myelodysplastic syndromes.9 Furthermore, given their resistance to protease degradation, we hypothesised that ASC specks from peripheral blood plasma might also be a biologically rational biomarker for the diagnosis and monitoring of ineffective haemopoiesis in patients with myelodysplastic syndromes. The main objective of this study was to establish whether ASC specks, a measure of pyroptosis, are significantly increased in peripheral blood plasma from patients with myelodysplastic syndromes as compared with healthy controls and patients with other haematological cancers.
Methods
Study design and participants
This observational cohort study was done at the H Lee Moffitt Cancer Center (Tampa, FL, USA). Patients with myelodysplastic syndromes, non-myelodysplastic syndrome haematological cancers, and type 2 diabetes, and healthy controls were recruited. Myelodysplastic syndrome diagnoses were done by morphological assessment according to the 2016 revision of the WHO Classification of Myelodysplastic Syndromes.12 Patients with myelodysplastic syndromes who had a chromosome 5 deletion (del[5q]) were analysed separately from non-del[5q] patients because of differences in disease biology. Inclusion criteria for healthy controls were age of at least 60 years and no history of type 2 diabetes or cancer. Inclusion criteria for all disorders only required physician diagnosis with no exclusion based on treatment status; patients with both diabetes and haematological cancer diagnoses were eligible. Patients and healthy donors gave written informed consent for participation in the study. The protocol was approved by the Institutional Review Board or equivalent regulation board for each institution that provided specimens. All patients except for those for the validation cohort were acquired by the H Lee Moffitt Cancer Center.
Procedures
Peripheral blood plasma samples from patients with non-del(5q) myelodysplastic syndromes were age matched to samples from healthy controls, patients with del (5q) myelodysplastic syndromes, patients with other haematological cancers, or individuals with type 2 diabetes. Bone marrow plasma samples were taken from patients with non-del(5q) myelodysplastic syndromes. These patients were stratified according to the International Prognostic Scoring System. We defined lower-risk patients as those with low or intermediate-1 risk and higher-risk patients as those with intermediate-2 or high risk.
Microscopy
We compared percentage of ASC specks in bone marrow and peripheral blood samples from patients with non-del(5q) myelodysplastic syndromes with those from healthy donors and patients with other haematological cancers. We used confocal immune-fluorescence and electron microscopy to visualise intracellular ASC specks, which are distinguished as solitary punctate cytoplasmic ASC aggregates (figure 1; appendix). Slides were imaged using a Leica SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany). Speck puri-fication was performed as previously described13 and imaged using a haemocytometer and the EVOS Autoimager (Thermo Fisher Scientific, Waltham, MA, USA) or, after being allowed to dry on a formvar-coated grid, using a Jeol JEM1400 transmission electron microscope (Jeol USA Inc, Peabody, MA, USA; appendix).
Figure 1: Visualisation of ASC specks in patients with myelodysplastic syndromes.
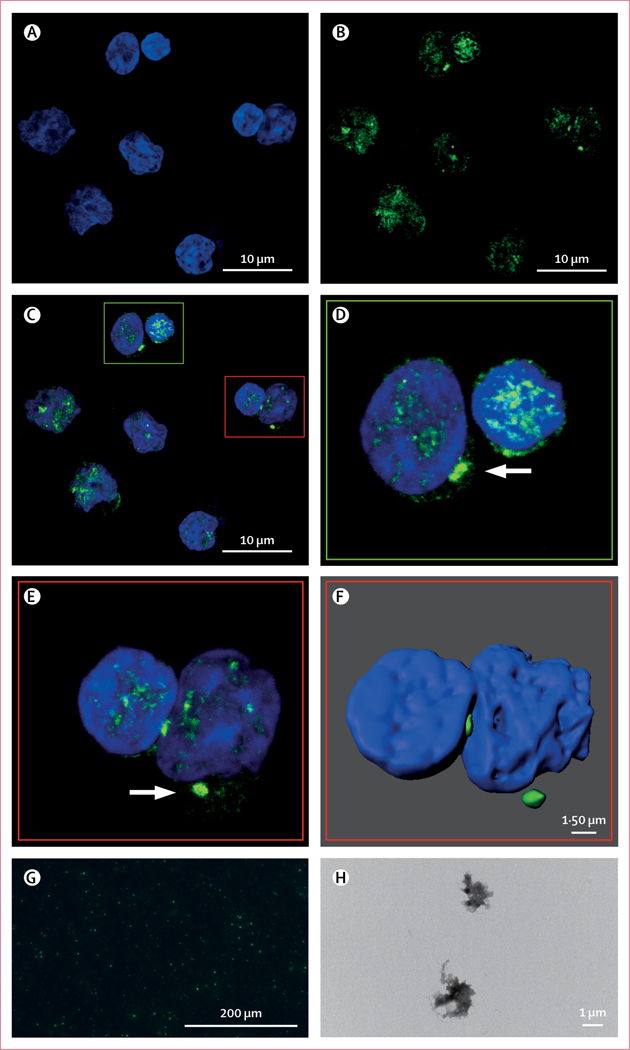
Representative confocal microscopy micrographs of ASC specks in bone marrow mononuclear cells from three patients with myelodysplastic syndromes from the discovery cohort. (A) DAPI nuclear staining. (B) Anti-ASC antibody staining. (C) Merged DAPI and anti-ASC image (A–C × 2100 magnification). (D and E) Digitally enlarged images of parts of C to highlight specks (white arrows). (F) Surface rendering of E. (G) Purified GFP-ASC specks from lipopolysaccharide-stimulated or nigericin-stimulated THP-1-GFP-ASC cell lines. (H) Transmission electron microscopy image of GFP-ASC specks isolated from stimulated THP-1 cells (× 15 000 magnification).
ASC=apoptosis-associated speck-like protein containing a CARD. GFP=green fluorescent protein
Flow cytometry
To assess the relationship between reported genetic drivers ofmyelodysplastic syndromes and inflammasome-derived ASC specks, we analysed the glucose-adjusted log10-transformed percentage of ASC specks in peripheral blood plasma in all patients and healthy controls. Flow cytometry was performed with modification to previously described methods (appendix).3,14 We did not implement a size threshold to identify specks that would favour only the largest aggregates. The percentage of ASC specks was quantified using a secondary antibody-only tube as the negative control. Data were analysed using FloJo software and reported as percentage of positive events or percentage of specks. These data were then adjusted for total glucose concentrations and transformed by log10 for analysis. We compared matched samples of peripheral blood and bone marrow plasma from the patients with myelodysplastic syndromes; and peripheral blood samples from patients with non-del(5q) myelodysplastic syndromes versus samples from healthy controls, patients with different haematological cancers, and patients with type 2 diabetes (appendix).
ELISA
The danger-associated molecular pattern (DAMP) protein S100-A9 initiates NLRP3 inflammasome activation anxptotic death in haemopoietic stem cells and progenitor cells in patients with lower-risk myelodysplastic syndromes.9 To confirm the specificity of NLRP3 inflammasome-derived ASC specks as a measure of ineffective haemopoiesis, we measured protein S100-A8 and protein S100-A9 concentrations by ELISA with CircuLex ELISA kits (MBL International Corporation, Woburn, MA, USA) in the peripheral blood samples from the patients with non-del(5q) myelodysplastic syndromes only. Peripheral blood plasma glucose concentrations were measured using a glucose colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA).
Sequencing
Gene sequencing was performed in the patients with non-del(5q) myelodysplastic syndromes on bone marrow mononuclear cells as previously described15 or using a next-generation sequencing-targeted gene panel, which included up to 54 genes with a variant allele frequency threshold of 5% and a 500×minimum depth of coverage.
Statistical analysis
The discovery cohort included all patients with non-del(5q) myelodysplastic syndromes for whom peripheral blood plasma samples were available in our laboratory. To optimise the assay for disease-specific pyroptosis, ASC speck percentage was normalised to plasma glucose concentration to adjust for possible confounding effects of hyperglycaemia. Hyperglycaemia is a known activator of the NLRP3 inflammasome, and in myelodysplastic syndromes, bone marrow plasma glucose concentrations are greatly increased as a result of inflammasome-driven insulin resistance.16–18 Therefore, we adjusted for glucose concentration to distinguish disease-specific inflamma-some activation.
Paired samples (peripheral blood and bone marrow) were compared using paired t tests. The two-sided t test of log10-transformed peripheral blood plasma specks normalised to glucose concentration was used to compare patients with myelodysplastic syndromes with healthy controls. Wilcoxon signed-rank test was used to compare diseases with fewer than ten patients. Correlations between log10-normalised percentage of ASC specks and log10-normalised protein S100-A8 and protein S100-A9 concentrations were analysed using Spearman’s rank correlation coefficient. Biomarker efficiency was analysed using receiver operating characteristics (ROC) and areas under the curve (AUC) as effective measures of accuracy for myelodysplastic syndromes versus controls, or by k-fold cross-validation when comparing patients with myelodysplastic syndromes to those with non-myelodysplastic syndrome haematological cancers. To power the validation cohort, a sample of 39 cases from the positive group and 39 from the negative group achieved 81% power to detect a difference of 0 2 between the AUC under the null hypothesis of 0.65 and an AUC under the alternative hypothesis of 0.8 using a one-sided z test at a significance level of 0.05. The ratio of the standard deviation of the responses in the negative group to the standard deviation of the responses in the positive group was 1.00. Mutation analyses were performed by one-way ANOVA or paired group t test. Statistical analyses were done using R, version 3.4.2.
Role of the funding source
The funders of the study had no role in study design, data collection, analysis, or interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Specimens collected between Jan 1, 2005, and Jan 12, 2017, were used for this study. Specimens were from 290 patients with non-del(5q) myelodysplastic syndromes, 60 healthy controls, 22 patients with del(5q) myelodysplastic syndromes, 230 patients with non-myelodysplastic syndrome haematological cancers, and 23 patients with type 2 diabetes (table 1; appendix). The discovery cohort consisted of 177 patients with myelodysplastic syndromes (mean age 70 years [SD 8.4]; 162 [92%] with lower-risk and 15 [8%] with higher-risk disease) and 29 healthy controls with no previous or current cancer or diabetes diagnosis (mean age 65 years [5.0]). The validation cohort consisted of 113 patients with myelodysplastic syndromes (mean age 72 years [7.2]; 66 [58%] with lower-risk and 47 [42%] with higher-risk disease) and 31 healthy controls with no previous or current cancer or diabetes diagnosis (mean age 74 years [6.0]). Distribution of morphological subtypes according to the 2016 revision of the WHO Classification of Myelodysplastic Syndromes12 included patients with myelodysplastic syndromes with single-lineage dysplasia (SLD), with SLD and ringed sideroblasts (SLD-RS), with multilineage dysplasia (MLD), with MLD-RS, with excess blasts, patients with unclassified myelodysplastic syndromes, and patients with myelodysplastic syndromes not otherwise specified.
Table 1:
Baseline cohort characteristics
| Discovery cohort |
Validation cohort |
|||
|---|---|---|---|---|
| Patients with myelodysplastic syndromes (n=177) |
Controls (n=29) |
Patients with myelodysplastic syndromes (n=113) |
Controls (n=31) |
|
| Mean age (SD), years | 70 (8.4) | 65 (5–0) | 72 (7–2) | 74 (6.0) |
| Sex | ||||
| Men | 100 (56%) | 0 | 61 (54%) | 4 (13%) |
| Women | 43 (24%) | 29 (100%) | 23 (20%) | 27 (87%) |
| Unknown | 34 (19%) | 0 | 29 (26%) | 0 |
| Risk category | ||||
| Higher-risk | 15 (8%) | NA | 47 (42%) | NA |
| Lower-risk | 162 (92%) | NA | 66 (58%) | NA |
| WHO myelodysplastic syndromes subtypes | ||||
| Single-lineage dysplasia | 9 (5%) | NA | 4 (4%) | NA |
| Single-lineage dysplasia with ringed sideroblasts | 30 (17%) | NA | 36 (32%) | NA |
| Multilineage dysplasia | 36 (20%) | NA | 12 (11%) | NA |
| Multilineage dysplasia with ringed sideroblasts | 14 (8%) | NA | 10 (9%) | NA |
| Excess blasts | 23 (13%) | NA | 19 (17%) | NA |
| Not otherwise specified | 59 (33%) | NA | 30 (27%) | NA |
| Unclassified | 6 (3%) | NA | 2 (2%) | NA |
NA=not applicable.
Extracellular ASC specks were quantified using flow cytometry in matched peripheral blood and bone marrow plasma samples from 38 patients with myelodysplastic syndromes. Peripheral blood plasma had a significantly higher mean percentage of specks than bone marrow plasma (24.9% [SD 12.4] vs 15.3% [9.7]; p<0.0001; figure 2A). Given the accessibility of peripheral blood for diagnostic screening, further investigations should focus solely on peripheral blood plasma-derived ASC specks rather than bone marrow plasma samples.
Figure 2: Quantification of ASC specks in bone marrow and peripheral blood from patients with myelodysplastic syndromes relative to patients with other haematological cancers, type 2 diabetes, or healthy donors.
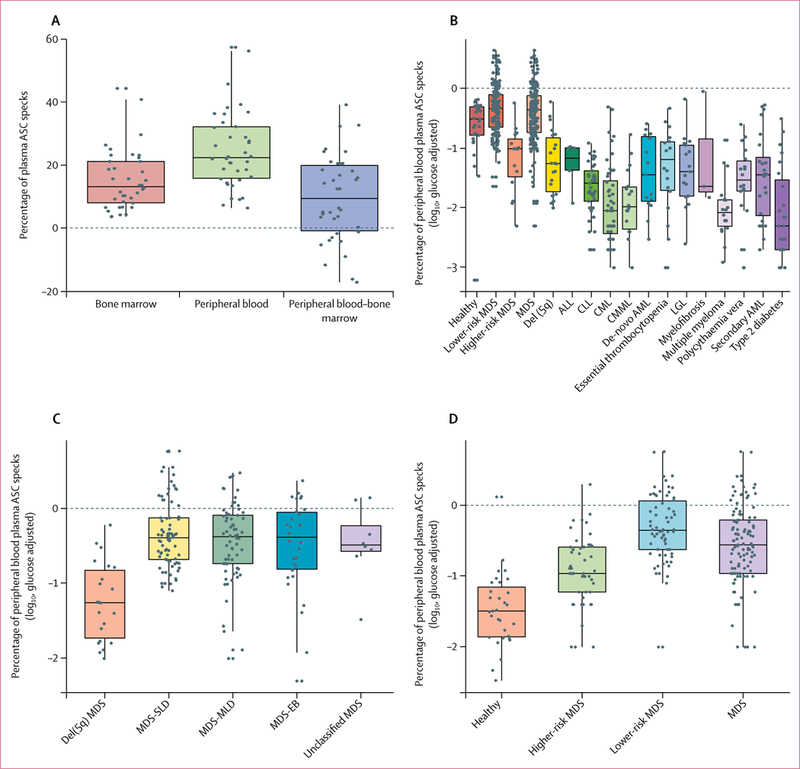
ASC=apoptosis-associated speck-like protein containing a CARD. MDS=myelodysplastic syndromes. del(5q)=chromosome 5 deletion. ALL=acute lymphocytic leukaemia. CLL=chronic lymphocytic leukaemia. CML=chronic myelogenous leukaemia. CMML=chronic myelomonocytic leukaemia. AML=acute myeloid leukaemia. LGL=large granular lymphocytic leukaemia. SLD=single-lineage dysplasia. MLD=multilineage dysplasia. EB=excess blasts. Boxes show median (centre line) and IQR (outer lines) and whiskers show SD. (A) Glucose-adjusted percentage of ASC specks in bone marrow and peripheral blood plasma, and difference between peripheral blood and bone marrow in samples from patients with myelodysplastic syndromes in the discovery cohort, quantified by flow cytometry. (B) Glucose-adjusted log10-transformed percentage of ASC specks in peripheral blood plasma from healthy controls and patients with myelodysplastic syndromes in the discovery cohort, as well as patients with non-myelodysplastic syndrome haematological cancers and type 2 diabetes, quantified by flow cytometry. (C) Glucose-adjusted log10-transformed percentage of ASC specks quantified by flow cytometry in patients with different subtypes of myelodysplastic syndromes from the discovery cohort and patients with del(5q) mutation, as categorised by 2016 WHO guidelines. (D) Glucose-adjusted log10-transformed percentage of ASC specks quantified by flow cytometry in peripheral blood plasma samples from patients with myelodysplastic syndromes versus healthy controls in the validation cohort.
The log10-transformed and glucose-adjusted mean percentage of peripheral blood ASC specks was significantly greater in peripheral blood samples from patients with non-del(5q) myelodysplastic syndromes (−0.36 [SD 0.41], p=0.034), and in those with lower-risk disease (−0.33 [0.34], p=0.0075) versus the healthy donors (−0.51 [0.67]; figure 2B). Consistent with our previous findings of a higher pyroptotic cell fraction in patients with lower-risk myelodysplastic syndromes,9 a higher mean percentage of ASC specks were present in lower-risk than in higher-risk myelodysplastic syndrome plasma samples (lower-risk −0.33 [SD 0–33] vs higher-risk −1.01 [0.55], p<0.0001; figure 2B). We found that the percentage of ASC specks was significantly lower in patients with myelodysplastic syndromes with isolated del(5q) than in the non-del(5q) patients (with single-lineage dysplasia, multilineage dysplasia, excess blasts, or unclassified subtypes), with no discernible differences among any of the non-del(5q) subtypes (figure 2C). The mean percentage of ASC specks was −1.243 (SD 0.52) in patients with del(5q) myelodysplastic syndromes, −0.506 (0.63) for those with myelodysplastic syndromes with excess blasts, −0.437 (0.52) for those with MLD, −0.348 (0.45) for those with SLD, and −0.466 (0.51) for unclassified myelodysplastic syndromes (figure 2C).
In the analysis of peripheral blood ASC specks as a diagnostic tool, the glucose-adjusted log10 percentage of ASC specks in samples from patients with non-del(5q) myelodysplastic syndromes was significantly greater than the percentage of specks from those with del(5q) myelodysplastic syndromes and all other haematological cancers we studied (all p<0.05), with the exception of myelofibrosis, which was present in only three participants in our study (p=0.19; table 2, figure 2B). In peripheral blood samples from patients with type 2 diabetes, ASC specks were significantly fewer than those from healthy control samples (p<0.0001) or samples from patients with non-del(5q) myelodysplastic syndrome (p<0.0001).
Table 2:
Log10-normalised percentage of peripheral blood plasma ASC specks in patients versus healthy participants
| n | Mean (SD) | Median (IQR) | p value vs normal |
p value vs myelodysplastic syndromes |
p value vs lower-risk |
|
|---|---|---|---|---|---|---|
| Healthy | 29 | −0·67 (0·59) | −0·51 (−0·78 to −0·31) | ·· | 0·034 | 0·0075 |
| Lower-risk myelodysplastic syndromes | 162 | −0·33 (0·42) | −0·33 (−0·65 to −0·10) | 0·0075 | ·· | ·· |
| Higher-risk myelodysplastic syndromes | 15 | −1·18 (0·55) | −1·01 (−1·48 to −0·85) | 0·0073 | ·· | <0·0001 |
| Myelodysplastic syndromes | 177 | −0·41 (0·49) | −0·35 (−0·74 to −0·12) | 0·034 | ·· | ·· |
| Del(5q) | 22 | −1·24 (0·52) | −1·26 (−1·73 to −0·82) | 0·00057 | <0·0001 | <0·0001 |
| Acute lymphocytic leukaemia* | 6 | −1·27 (0·37) | −1·17 (−1·37 to −0·10) | 0·0010 | 0·00030 | <0·0001 |
| Chronic lymphocytic leukaemia | 45 | −1·66 (0·43) | −1·59 (−1·89 to −1·38) | <0·0001 | <0·0001 | <0·0001 |
| Chronic myelogenous leukaemia | 48 | −1·94 (0·65) | −2·05 (−2·43 to −1·54) | <0·0001 | <0·0001 | <0·0001 |
| Chronic myelomonocytic leukaemia | 18 | −1·91 (0·56) | −1·98 (−2·35 to −1·65) | <0·0001 | <0·0001 | <0·0001 |
| De-novo acute myeloid leukaemia | 15 | −1·43 (0·62) | −1·44 (−1·89 to −0·81) | 0·00054 | <0·0001 | <0·0001 |
| Essential thrombocytopenia | 18 | −1·37 (0·72) | −1·19 (−1·71 to −0·90) | 0·0016 | <0·0001 | <0·0001 |
| Large granular lymphocytic leukaemia | 17 | −1·38 (0·57) | −1·39 (−1·80 to −0·95) | 0·00030 | <0·0001 | <0·0001 |
| Myelofibrosis* | 3 | −1·17 (0·98) | −1·64 (−1·73 to −0·84) | 0·46 | 0·19 | 0·15 |
| Multiple myeloma | 18 | −2·02 (0·50) | −2·08 (−2·31 to −1·86) | <0·0001 | <0·0001 | <0·0001 |
| Polycythaemia vera | 18 | −1·56 (0·61) | −1·53 (−1·72 to −1·21) | <0·0001 | <0·0001 | <0·0001 |
| Secondary acute myeloid leukaemia | 24 | −1·48 (0·72) | −1·44 (−2·13 to −1·15) | <0·0001 | <0·0001 | <0·0001 |
| Type 2 diabetes | 23 | −2·09 (0·78) | −2·3 (−2·70 to −1·53) | <0·0001 | <0·0001 | <0·0001 |
| Normal discovery† | 29 | −0·67 (0·59) | −0·51 (−0·78 to −0·31) | ·· | ·· | ·· |
| Normal validation† | 31 | −1·47 (0·52) | −1·49 (−1·86 to −1·15) | ·· | ·· | ·· |
| Myelodysplastic syndromes discovery‡ | 177 | −0·41 (0·49) | −0·35 (−0·74 to −0·12) | ·· | ·· | ·· |
| Myelodysplastic syndromes validation‡ | 113 | −0·56 (0·57) | −0·55 (−0·96 to −0·20) | ·· | ·· | ·· |
ASC=apoptosis-associated speck-like protein containing a CARD. del(5q)=chromosome 5 deletion.
p values represent Wilcoxon significance values because of low number of patients.
p<0·0001 for patients with myelodysplastic syndromes in the discovery versus validation cohorts.
p=0·02 for healthy controls in the discovery versus validation cohorts.
To validate these findings, we studied samples from the validation cohort of patients with non-del(5q) myelodysplastic syndromes and age-matched healthy controls. We confirmed that ASC specks were significantly increased in the plasma of patients with myelodysplastic syndromes compared with healthy controls (p<0.0001; figure 2D). A ROC-AUC analysis was done in the validation and discovery cohorts to assess the specificity and sensitivity of ASC specks as a possible pyroptosis-specific biomarker in myelodysplastic syndromes. ROC-AUC analysis illustrates the diagnostic accuracy of a two-class prediction of a validation cohort using the logistic regression model on the discovery cohort. By ROC-AUC analyses, we showed that ASC specks could be considered as a robust biomarker of pyroptosis linked to myelodysplastic syndromes (AUC=0.888; figure 3), and was highly sensitive for lower-risk myelodysplastic syndromes (AUC=0.951). Our calculations allowed us to establish that a log10-transformed, glucose-adjusted percentage of speck threshold of 0.80 maximised sensitivity (0.84 [95% CI 0.65–0.91]) and specificity (0.87 [0.58–0.97]) for all myelodysplastic syndromes, whereas a threshold of 0.761 maximised sensitivity (0.92 [0.17–0.98]) and specificity (0.94 [0.77–1.0]) for lower-risk myelodysplastic syndromes. The threshold output is the point at which both the sensitivity and specificity have the greatest values. Adjustment of this threshold can increase either the sensitivity or specificity but not both, and therefore, the provided threshold depicts where both are maximised. Additionally, in the absence of an independent cohort of non-haematological disorders, we did a five-fold cross-validation (k=5), which was repeated 30 times to compare patients with myelodysplastic syndromes in the discovery cohort with the control patients who had other haematological cancers, yielding a mean AUC of 0.934 (data not shown), showing the specificity of this marker with respect to other haematological disorders.
Figure 3: Sensitivity and specificity of ASC specks in peripheral blood plasma from patients with myelodysplastic syndromes.
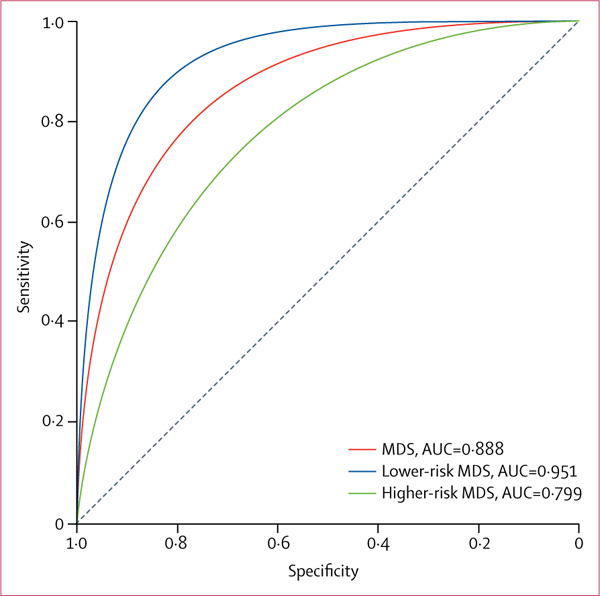
Receiver operating characteristic AUC analysis of glucose-adjusted log10-normalised ASC speck percentage in patients with lower-risk, higher-risk, or any myelodysplastic syndromes from the discovery and validation cohorts.
ASC=apoptosis-associated speck-like protein containing a CARD.
MDS=myelodysplastic syndromes. AUC=area under the curve.
Protein S100-A8 and protein S100-A9 concentrations were measured in 144 peripheral blood plasma samples from patients with myelodysplastic syndromes. Protein S100-A8 (r=0.4, p<0.0001) and protein S100-A9 (r=0.2, p=0.017) concentrations directly correlated with glucose-adjusted ASC speck percentages. The concentrations of the two S100 proteins were also closely correlated (r=0.5, p<0.0001), consistent with the propensity of these proteins to heterodimerise.
In the analysis of the relationship between genetic drivers ofmyelodysplastic syndromes (table 3), comparing patients with myelodysplastic syndromes from the validation and discovery cohorts with a specific mutation to all those without any mutation (n=4), ASC specks in peripheral blood plasma were significantly increased in patients who have somatic mutations involving pre-mRNA splicing genes (p=0·019), methylation genes (p=0·011), transcription factor genes (p=0·019), and signalling genes (p=0·029). Further-more, when we compared the 78 patients with RNA splicing mutations with the 18 patients without this type of mutation, patients with splicing gene mutations had a significantly higher mean percentage of plasma ASC specks than participants without (−0·25 [SD 0·61] vs −0·54 [0·44], p=0·026). Similarly, when we compared the 52 patients with any methylation mutation with the 44 patients without, the mean percentage of ASC specks was significantly higher in patients with methylation gene mutations (−0·20 [SD 0·69] with these mutations vs −0·44 [0·43] without, p=0·039; table 3). The non-founder gene mutation classes (defined as non-founder because they are not driver mutations), such as signalling and transcriptional genes, did not have significantly different percentages of ASC specks from the other classes (table 3). Furthermore, irrespective of mutation class, patients with at least two mutations had a significantly greater percentage of peripheral blood plasma ASC specks than patients with one or no mutation (−0·22 [SD 0·63] vs −0·53 [0·44], p=0.008; table 3). However, in patients with more than two total mutations, additional somatic gene mutations did not have an additive effect (p=0.207 for total number of mutations vs percentage of ASC specks; figure 4).
Table 3:
Log10-normalised percentage of peripheral blood plasma ASC specks by classes of somatic gene mutations in patients with myelodysplastic syndromes
| n | Mean (SD) | Median (IQR) | p value | |
|---|---|---|---|---|
| Splicing gene mutations* | ||||
| Yes | 78 | −0·25 (0·61) | −0·35 (−0·62 to 0·07) |
0·026 |
| No | 18 | −0·54 (0·44) | −0·52 (−0·80 to −0·24) |
·· |
| Methylation gene mutations† | ||||
| Yes | 52 | −0·20 (0·69) | −0·32 (−0·61 to 0·11) |
0·039 |
| No | 44 | −0·44 (0·43) | −0·42 (−0·75 to −0·13) |
·· |
| Transcriptional gene mutations‡ | ||||
| Yes | 37 | −0·29 (0·66) | −0·34 (−0·62 to 0·08) |
0·821 |
| No | 59 | −0·32 (0·56) | −0·40 (−0·70 to −0·04) |
·· |
| Signalling gene mutations§ | ||||
| Yes | 11 | −0·26 (0·66) | −0·44 (−0·79 to −0·11) |
0·787 |
| No | 85 | −0·31 (0·59) | −0·36 (−0·67 to 0·03) |
·· |
| ≥1 mutations | ||||
| Yes | 92 | −0·28 (0·59) | −0·35 (−0·63 to 0·03) |
0·024 |
| No | 4 | −0·90 (0·32) | −0·86 (−1·08 to −0·67) |
·· |
| ≥2 mutations | ||||
| Yes | 68 | −0·22 (0·63) | −0·32 (−0·60 to −0·11) |
0·008 |
| No | 28 | −0·53 (0·44) | −0·50 (−0·85 to −0·31) |
·· |
| Total number of mutations | ||||
| 0 | 4 | −0·90 (0·32) | −0·86 (−1·08 to −0·67) |
0·207 |
| 1 | 26 | −0·45 (0·43) | −0·44 (−0·76 to −0·12) |
·· |
| 2 | 39 | −0·23 (0·54) | −0·34 (−0·50 to 0·07) |
·· |
| 3 | 13 | −0·15 (0·60) | −0·08 (−0·62 to 0·20) |
·· |
| 4 | 11 | −0·25 (1·02) | −0·43 (−0·73 to −0·03) |
·· |
| 5 | 3 | −0·17 (0·30) | −0·24 (−0·34 to −0·04) |
·· |
ASC=apoptosis-associated speck-like protein containing a CARD.
SF3B1, SRSF2, U2AF, or ZRSR2 mutations.
Tet2, IDH, DNMT3A, ASXL1, or EZH2 mutations.
SetBP1, TP53, PHF6, RUNX1, ETV6, or NPM1 mutations.
CBL, NRAS, KIT, JAK2, or MPL mutations.
Figure 4: Percentage of peripheral blood plasma ASC specks from patients with myelodysplastic syndromes according to number of somatic gene mutations.
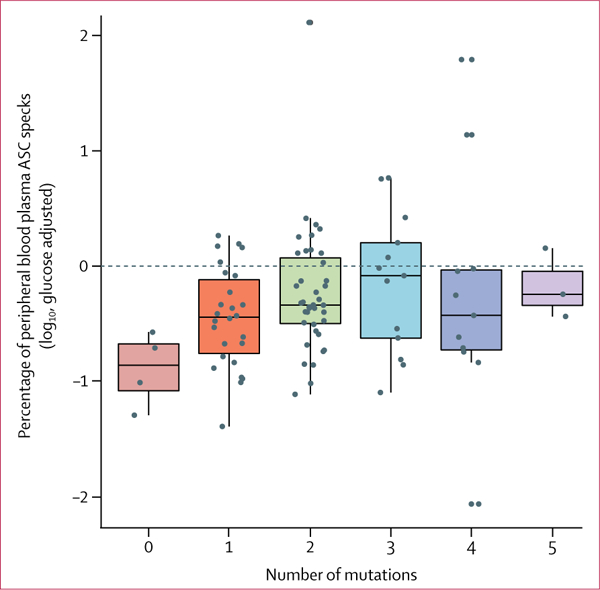
Boxes show median (centre line) and IQR (outer lines) and whiskers show SD.
Data are from the discovery and validation cohorts (n=96).
ASC=apoptosis-associated speck-like protein containing a CARD.
Discussion
Collectively, our results indicate that NLRP3 inflammasome-derived ASC specks are highly specific for pyroptosis associated with myelodysplastic syndromes. We also highlight here the relationship between the presence of myelodysplastic syndrome-related somatic gene mutations and peripheral blood plasma ASC speck percentages, which lends further support to the relevance of this candidate biomarker as a plausible index of pyroptosis in myelodysplastic syndromes.
Because of their prion-like structure, ASC specks are resistant to proteolytic degradation, and therefore persist in the environment long after cell death.3,4 Indeed, experiments in reporter mice—including a heterozygous mouse model bearing insertion of a green fluorescent protein transgene into lysosomes and in wild-type mice3—have shown that specks can remain in tissues for up to 96 h after ear injection of fluorescent ASC specks, indicating that inflammasome-initiated pyroptosis might have long-lasting effects on adjacent stromal cells.3 This might impair stromal support for normal haemopoiesis in patients with myelodysplastic syndromes, which is being investigated.3,4 Extracellular ASC specks can be engulfed by resident macrophages and visualised in phago-lysosomal compartments.3 This observation might explain the lower percentages of ASC specks in bone marrow versus peripheral blood plasma noted in our study, although a direct analysis of ASC speck phagocytosis by myelodysplastic syndrome macrophages remains to be done.
Because ASC specks are only released during pyroptotic cytolysis,3 they can function as a surrogate marker of the magnitude of pyroptosis in bone marrow haemopoietic precursors. Using an optimised flow cytometry assay, we showed that the percentage of peripheral blood plasma ASC specks was significantly increased in patients with myelodysplastic syndromes compared with an age-matched, healthy control population, in both our discovery and validation cohorts. We hypothesised that ASC specks might be more common in peripheral blood than bone marrow plasma because the specks can be engulfed by resident macrophages in the bone marrow, and we did find a higher percentage of specks in peripheral blood samples relative to the bone marrow samples. We observed a greater percentage of specks in patients with lower-risk myelodysplastic syndromes than in those with higher-risk myelodysplastic syndromes, consistent with a higher pyroptotic cell fraction in patients with lower-risk disease. Decreased pyroptosis in patients with higher-risk myelodysplastic syndromes might be attributed to decreased S100-A9 concentrations and bone marrow myeloid-derived suppressor cells, as well as increased survival signals in people with higher-risk myelodysplastic syndromes, but this remains to be shown. However, we did not observe differences in ASC speck percentages between patients with different WHO subtype classifications except del(5q) myelodysplastic syndromes, which might be attributable to the low number of participants with each subtype. As expected, we observed decreased ASC speck percentages in peripheral blood from patients with type 2 diabetes as compared with individuals with myelodysplastic syndromes, because of the low levels of pyroptotic execution despite inflammasome activation. Notably, patients with type 2 diabetes also had decreased speck percentages as compared with the healthy control cohort, possibly because some of the participants with type 2 diabetes might have received active glycaemia control with hypoglycaemic drugs or insulin. The results from the 5-fold crossvalidation indicated that the percentage of specks had potential use as a pyroptosis marker in myelodysplastic syndromes versus other haematological cancers, showing the specificity of ASC specks in samples from patients with myelodysplastic syndrome as compared with other haematological cancers. Furthermore, as concentrations of the DAMPs S100-A8 and S100-A9 have been shown to correlate directly with the extent of pyroptotic death,9 we also showed a correlation between percentage of ASC specks and S100-A8 and S100-A9 concentrations in plasma. Our results underscore the biological relevance of peripheral blood plasma ASC specks as a surrogate marker for the index of pyroptosis induced by S100-A8 and S100-A9 in peripheral blood plasma. This analysis, as well as thorough analysis of common myelodysplastic syndrome somatic gene mutations linked to pyroptosis, further support the possibility of using measurement of ASC specks as a biomarker of pyroptosis in myelodysplastic syndromes. Our results showed that the percentage of ASC specks in patients with somatic gene mutations was significantly increased compared with patients who had no gene mutations. Percentage of peripheral blood plasma ASC specks was significantly greater in patients who had at least two somatic gene mutations than in patients with one or no mutation, although percentages did not increase significantly beyond three mutations, which might reflect the smaller sample size of these groups or the class of passenger gene mutations. Because transcriptional mutations and signalling mutations are not driver mutations, and are therefore referred to as non-founder gene mutations, it seems logical that they were not associated with a significant difference in plasma ASC speck percentages, perhaps also reflecting the pro-survival effect of these classes of somatic gene mutations. However, studies with a larger sample size are needed to assess the relationship between specific gene mutations or mutation classes and the extent of pyroptosis.
An assay for cell pyroptosis in myelodysplastic syndromes that is independent of cell morphology could be invaluable for diagnosis. Peripheral blood plasma-derived ASC specks might serve as an index of pyroptosis and could be the first biomarker for pyroptosis associated with myelodysplastic syndromes that offers a high degree of specificity and sensitivity. Newly developed assays such as an ASC ELISA might be better at quantifying ASC specks than the flow cytometry assay used in our study, and might offer greater clinical utility. Immunofluorescence detection of cytoplasmic specks in bone marrow precursors could be an alternative approach for validation of the diagnosis of pyroptosis associated with myelodysplastic syndromes in patients presenting with subtle features of cytological dysplasia. The results from our study suggest that the detection of ASC specks might be a biologically rational peripheral blood biomarker for pyroptosis associated with myelo-dysplastic syndromes, and could also serve as an index of intramedullary bone marrow pyroptosis. Inflammasome inhibition is an attractive strategy for the treatment of patients with myelodysplastic syndromes, which has been shown in vitro to restore haemopoiesis,9 and the diagnostic technique we have identified might help assess responses to therapy. Limitations of this study include the overlap in ASC speck percentage values we noted among different cancers, which warrants further investigation. Furthermore, it must be noted that the use of other biomarkers in combination with the detection of ASC specks is likely to improve the specificity of detection of pyroptosis, and this merits further attention. Alternative assays to flow cytometry are likely to improve quantitative and accurate assessment of ASC specks. Despite these limitations, this study emphasises the potential use of detection of ASC specks as a candidate biomarker ofcell death and a complementary assessment tool that might facilitate diagnosis of myelodysplastic syndromes.
Supplementary Material
Research in context.
Evidence before this study
On June 15, 2018, we searched PubMed for publications on ASC specks in myelodysplastic syndromes, using the search terms “ASC”, “inflammasome”, and “MDS”, with no language or publication date restrictions. The only relevant study was our previous study, which identified NLRP3 inflammasome-directed pyroptotic cell death as a driver of ineffective haemopoiesis in patients with myelodysplastic syndromes, and described pyroptosis in the context of non-myelodysplastic syndrome haematological cancers. During inflammasome assembly the apoptosis-associated speck-like protein containing a CARD (ASC) adaptor protein polymerises into large, filamentous clusters termed ASC specks that are released during cytolysis and are resistant to proteolytic degradation. Because specks are readily quantified by flow cytometry and measured independently of cell morphology, we hypothesised that ASC specks might serve as a candidate biomarker associated with pyroptotic cell death in myelodysplastic syndromes, which might provide an index of ineffective haemopoiesis and so be used in the diagnosis of these cancers.
Added value of this study
By showing the use of ASC specks as a surrogate marker of pyroptosis with specificity for myelodysplastic syndromes, our findings suggest that the detection of ASC specks in peripheral blood from patients with myelodysplastic syndromes is a candidate biomarker for pyroptosis. We also hypothesise that ASC specks could be used as an index of intramedullary bone marrow pyroptosis, which might be used in monitoring responses to therapy. This hypothesis merits further investigation.
Implications of all the available evidence
These data identify a potential diagnostic companion that distinguishes patients with myelodysplastic syndromes from those with other bone marrow cancers. ASC speck quantification has potential as a surrogate for pyroptosis that might be applied to assess therapeutic responses.
Acknowledgments
AAB was supported by T32 Training Grant (NIH/NCI 5T32 CA115308–08). This work is supported in part by the Edward P Evans Foundation, The Taub Foundation Grants Program, and the Flow Cytometry, Analytic Microscopy, and Tissue Core Facilities at the H Lee Moffitt Cancer Center and Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292). We thank Benjamin Meyer and Amanda Garces from the Lisa Muma Weitz Advanced Microscopy and Cell Imaging Core Laboratory at Morsani College of Medicine USF Health for assistance with electron microscopy.
Funding T32 Training Grant (NIH/NCI 5T32 CA115308–08), Edward P Evans Foundation, The Taub Foundation Grants Program, the Flow Cytometry, Analytic Microscopy, and Tissue Core Facilities at the H Lee Moffitt Cancer Center and Research Institute, a National Cancer Institute-designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Declaration of interests
We declare no competing interests.
References
- 1.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16: 407–20. [DOI] [PubMed] [Google Scholar]
- 2.Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014; 156: 1193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franklin BS, Bossaller L, De Nardo D, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 2014; 15: 727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 2016; 7: 11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Font P, Loscertales J, Soto C, et al. Interobserver variance in myelodysplastic syndromes with less than 5% bone marrow blasts: unilineage vs. multilineage dysplasia and reproducibility of the threshold of 2% blasts. Ann Hematol 2015; 94: 565–73. [DOI] [PubMed] [Google Scholar]
- 6.Naqvi K, Garcia-Manero G, Sardesai S, et al. Association of comorbidities with overall survival in myelodysplastic syndrome: development of a prognostic model. J Clin Oncol 2011; 29: 2240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bejar R Myelodysplastic syndromes diagnosis: what is molecular testing? Curr Hematol Malig Rep 2015; 10: 282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Font P, Loscertales J, Benavente C, et al. Inter-observer variance with the diagnosis of myelodysplastic syndromes (MDS) following the 2008 WHO classification. Ann Hematol 2013; 92: 19–24. [DOI] [PubMed] [Google Scholar]
- 9.Basiorka AA, McGraw KL, Eksioglu EA, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016; 128: 2960–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rassool FV, Gaymes TJ, Omidvar N, et al. Reactive oxygen species, DNA damage, and error-prone repair: a model for genomic instability with progression in myeloid leukemia? Cancer Res 2007; 67: 8762–71. [DOI] [PubMed] [Google Scholar]
- 11.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett 2008; 270: 1–9. [DOI] [PubMed] [Google Scholar]
- 12.Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med 2017; 5: 139–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Sanchez F, Gomez A, Pelegrin P. Isolation of particles of recombinant ASC and NLRP3. Bio Protoc 2015; 5: e1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoss F, Rolfes V, Davanso MR, Braga TT, Franklin BS. Detection of ASC speck formation by flow cytometry and chemical cross-linking. Methods Mol Biol 2018; 1714: 149–65. [DOI] [PubMed] [Google Scholar]
- 15.Chesnais V, Renneville A, Toma A, et al. Effect of lenalidomide treatment on clonal architecture of myelodysplastic syndromes without 5q deletion. Blood 2016; 127: 749–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei S, Eksioglu EA, Chen X, et al. Inflammaging-associated wmetabolic alterations foster development of the MDS genotype. Blood 2015; 126: 144.25990863 [Google Scholar]
- 17.Tseng HH, Vong CT, Kwan YW, Lee SM, Hoi MP. TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci Rep 2016; 6: 35016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013; 62: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.