

Abstract
Bovine respiratory disease complex (BRDC) is one of the most significant diseases of cattle. Bacterial pathogens involved in BRDC include Mannheimia haemolytica, Mycoplasma bovis, Histophilus somni, and Pasteurella multocida. We developed and evaluated a multiplexed real-time hydrolysis probe (rtPCR) assay using block-based Peltier and rotary-based thermocycling on lung tissue, nasal swabs, and deep nasopharyngeal swabs. The rtPCR results were compared to culture or a gel-based M. bovis PCR using statistical analysis to determine optimum quantification cycle (Cq) cutoffs to maximize agreement. The limits of detection were 1.2–12 CFU/reaction for each pathogen. M. haemolytica was the most prevalent organism detected by rtPCR, and was most frequently found with P. multocida. The rtPCR assay enabled enhanced levels of detection over culture for all pathogens on both thermocycling platforms. The rotary-based thermocycler had significantly lower Cq cutoffs (35.2 vs. 39.7), which maximized agreement with gold standard culture or gel-based PCR results following receiver operating characteristic analysis to maximize sensitivity (Se) and specificity (Sp). However, overall assay Se and Sp were similar on both platforms (80.5% Se, 88.8% Sp vs. 80.1% Se, 88.3% Sp). Implementation of these tests could enhance the detection of these pathogens, and with high-throughput workflows could reduce assay time and provide more rapid results. The assays may be especially valuable in identifying coinfections, given that many more antemortem samples tested in our study were positive for 2 or more pathogens by rtPCR (n = 125) than were detected using culture alone (n = 25).
Keywords: Bacterial pathogens, bovine respiratory disease, clinical specimens, real-time PCR, receiver operating characteristic analysis
Introduction
Bovine respiratory disease complex (BRDC) is the most significant disease in beef cattle production, which in 2011 was worth an estimated retail value of $79 billion in the United States. BRDC is estimated to cost producers more than $13 per animal, making the economic impact to cattle production tremendous.24 The total overall cost to the United States is estimated to be >$1 billion as a result of production losses, death, labor, and drug costs.7,9 Although effective antimicrobials and vaccines for BRDC have been licensed and are available, the incidence of BRDC in feedlot cattle remains high, with 62–72% of animals having lung lesions at slaughter indicative of prior BRDC, and with 61–68% of these never having been treated.23,25 This tremendous cost and high incidence of undiagnosed disease underscores the need for new rapid and accurate tools to mitigate the impact of BRDC on beef cattle production and enable targeted prevention and treatment strategies.
Multiple pathogens, including both viruses and bacteria, contribute to BRDC. The complex is frequently initiated by infection with a virus that is subsequently exacerbated by stress from transport and commingling. Such stress disrupts local defenses and causes immune suppression, which enables bacteria that are normal nasopharyngeal commensals to colonize deeper respiratory tissues, replicate, and secrete toxins in lower airways causing tissue destruction, inflammation, and pneumonia.18 Bacterial pathogens that are isolated frequently in association with BRDC include: Mannheimia haemolytica, Pasteurella multocida, Histophilus somni, and Mycoplasma bovis. A 2014 study investigating a large number of BRDC mortalities in North American bovine feedlots showed M. haemolytica to be the most frequently isolated (91%), followed by M. bovis (63%), H. somni (57%), and P. multocida (13%).12 This supports other studies showing frequent isolation or detection of these agents in North American feedlots.2,12 Identification of the pathogens responsible for BRDC is critical to development of effective prevention and treatment strategies, because some therapies, such as beta-lactam antimicrobials, are ineffective against M. bovis, and several of the pathogens have shown in vitro resistance to available antimicrobial therapies.16,21
Classically, bacterial pathogens of BRDC are identified by isolation of that pathogen from lungs that have gross or histologic lesions consistent with BRDC. However, tissues or clinical specimens from diagnostic submissions are frequently from animals that have been treated with antimicrobials prior to submission and/or are chronic lesions. Many specimens have undergone autolysis and have bacterial overgrowth or environmental contamination from field postmortem collection before receipt at a diagnostic laboratory. Some specimens have undergone temperature extremes during shipping that may impede the ability to isolate pathogens in culture. A 2014 study has demonstrated that gel-based PCR methodology was able to enhance the detection of bacterial pathogens in pneumonic lung tissues by ~20%, indicating that the potential for a large number of false-negative tests exists when relying on culture alone.1 Frequently, only the predominant organism is found given the overgrowth of a single agent; therefore PCR has been explored as a tool to identify potential coinfections in lung tissues.1
Real-time hydrolysis probe PCR (rtPCR) is a rapid tool that enables the relative abundance of these organisms to be calculated using quantification cycle (Cq) values, and thus provide additional data to assist the clinician in determining clinical relevance. Antemortem clinical samples, such as nasal swabs (NS) and deep nasopharyngeal swabs (DNPS) are frequently submitted to diagnostic laboratories for culture testing. The use of guarded swabs to sample deep nasopharyngeal passages has been shown to readily obtain M. bovis and M. haemolytica, although swab isolates may not always reflect those populations found in deeper tissues such as lung.3,8
We determined the utility of a multiplex rtPCR assay to detect bacterial pathogens of BRDC in clinical specimens (NS, DNPS, and lung tissue) commonly submitted to diagnostic laboratories for testing. We performed receiver operating characteristic (ROC) analyses to determine the quantification cycle (Cq) value as the optimal cutoff of a multiplex rtPCR assay by maximizing its test accuracy (both sensitivity [Se] and specificity [Sp] as the primary accuracy indices) compared to gold standards. Additionally, we compared 2 rtPCR platforms, one that utilizes air heating and cooling on a rotary-based design with high-power light-emitting diodes (RGQ; Rotor-Gene Q, Qiagen, Hilden, Germany) with a traditional block-based Peltier heating and cooling system (ABI; ABI 7500 Fast, Thermo Fisher Scientific, Waltham, MA) to determine if the type of rtPCR instrumentation has an impact on results.
Materials and methods
Reference strains
Strains used for limit of detection analyses and preliminary validation of the assays included: P. multocida (ATCC BAA-1113), M. haemolytica (ATCC BAA-410), H. somni (129Pt), and M. bovis (ATCC 25223; American Type Culture Collection, Manassas, VA). A collection of field isolates and off-target controls were also employed to ensure assay Sp (Table 1) and included Mannheimia granulomatis, Mannheimia varigena, Mannheimia sp. Angen Group V, Bibersteinia trehalosi, Gallibacterium anatis, and Gallibacterium sp. All isolates, if not ATCC-sourced strains, were identified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS; Biotyper, Bruker Daltonics, Billerica, MA) to the species level with scores of 2.0 or higher and/or subjected to whole genome sequencing (Mannheimia sp. Angen Group V, Mannheimia varigena). The M. haemolytica isolates represented all major genotypes and subtypes of M. haemolytica as described previously, and were identified to the species level using both MALDI-TOF MS and whole genome sequencing.4,15
Table 1.
Target and non-target isolates used to validate assay specificity for Mycoplasma bovis, Mannheimia haemolytica, Histophilus somni, and Pasteurella multocida. Template cell lysate was prepared using a 1 McFarland standard of pure culture growth. Quantification cycle (Cq) values for each target are reported using the Rotor-Gene Q instrument using threshold values of 0.1, except for red, which was set to 0.25, and green, which was set to 0.15. Dynamic tube normalization was used for all analyses.
| Species/strain | Cq value | |||
|---|---|---|---|---|
| M. bovis | M. haemolytica | H. somni | P. multocida | |
| M. bovis | ||||
| UNL1 | 18.86 | – | – | – |
| M. haemolytica | ||||
| USMARC 28284 | – | 20.02 | – | – |
| USMARC 28454 | – | 19.1 | – | – |
| USMARC 28229 | – | 20.06 | – | – |
| USMARC 34142 | – | 19.30 | – | – |
| USMARC 33749 | – | 19.14 | – | – |
| USMARC 23580 | – | 19.19 | – | – |
| USMARC 23533 | – | 18.73 | – | – |
| USMARC 23570 | – | 19.16 | – | – |
| USMARC 23217 | – | 18.53 | – | – |
| USMARC 23473 | – | 23.22 | – | – |
| USMARC 22604 | – | 16.81 | – | – |
| USMARC 39434 | – | 17.26 | – | – |
| USMARC 33204 | – | 19.2 | – | – |
| USMARC 23299 | – | 23.06 | – | – |
| USMARC 23311 | – | 20.61 | – | – |
| USMARC 23348 | – | 20.56 | – | – |
| USMARC 28226 | – | 25.99 | – | – |
| USMARC 23309 | – | 19.73 | – | – |
| USMARC 33982 | – | 23.54 | – | – |
| USMARC 34180 | – | 18.40 | – | – |
| USMARC 1590 | – | 17.92 | – | – |
| USMARC 1621 | – | 20.08 | – | – |
| USMARC 1562 | – | 22.24 | – | – |
| USMARC 32563 | – | 24.86 | – | – |
| USMARC 32635 | – | 21.14 | – | – |
| USMARC 33170 | – | 22.44 | – | – |
| USMARC 23456 | – | 19.63 | – | – |
| USMARC 32864 | – | 20.69 | – | – |
| USMARC 1629 | – | 20.55 | – | – |
| USMARC 22549 | – | 20.23 | – | – |
| H. somni | ||||
| ATCC43625 | – | – | 16.79 | |
| ATCC 700002 | – | – | 17.97 | |
| UNL22 | – | – | 13.41 | – |
| UNL21 | – | – | 13.13 | – |
| UNL20 | – | – | 22.86 | – |
| UNL19 | – | – | 22.98 | – |
| UNL18 | – | – | 26.61 | – |
| UNL17 | – | – | 14.34 | – |
| UNL16 | – | – | 21.75 | – |
| P. multocida | ||||
| UNL15 | – | – | – | 10.07 |
| UNL14 | – | – | – | 13.73 |
| UNL13 | – | – | – | 13.37 |
| UNL12 | – | – | – | 12.65 |
| UNL11 | – | – | – | 15.02 |
| UNL10 | 10.90 | |||
| UNL9 | – | – | – | 11.95 |
| UNL8 | 17.40 | |||
| UNL7 | – | – | – | 15.44 |
| UNL6 | – | – | – | 16.62 |
| Mannheimia sp. Angen V | ||||
| USMARC 28377 | – | – | – | – |
| USMARC 23448 | – | – | – | – |
| Mannheimia varigena | ||||
| USMARC 33577 | – | – | – | – |
| USMARC 23064 | – | – | – | – |
| Mannheimia granulomatis | ||||
| ATCC 49244 | – | – | – | – |
| Actinobacillus pleuropneumoniae | ||||
| ATCC 27050 | – | – | – | – |
| Bibersteinia trehalosi | ||||
| ATCC 29703 | – | – | – | – |
| UNL5 | – | – | – | – |
| UNL4 | – | – | – | – |
| Gallibacterium anatis | ||||
| UNL3 | – | – | – | – |
| Gallibacterium sp. | ||||
| UNL2 | – | – | – | – |
Dash (–) = no amplification detected (Cq of 0). Instrument source: Rotor-Gene Q, Qiagen, Hilden, Germany.
Isolation and identification of bacteria from clinical specimens
Diagnostic specimens (NS, DNPS, and lung tissue) consisted of submissions to the University of Nebraska–Lincoln Veterinary Diagnostic Center for bovine respiratory disease testing. Samples included NS (n = 43), lung tissues (n = 47), and DNPS (n = 95). Although detailed clinical information was not available for many submissions, most of these samples were most likely collected from non-healthy clinically ill animals. Specimens were accessioned, processed, and tested by trained personnel following validated and approved standard operating procedures in an American Association of Veterinary Laboratory Diagnosticians (AAVLD)-accredited diagnostic laboratory. For bacterial culture, fresh lung tissues were trimmed, dipped in alcohol, the exterior was flame sterilized, bisected, and the cut surface inoculated onto culture media. Swabs were directly inoculated onto plates if submitted in a liquid medium, or suspended in a small volume of phosphate-buffered saline (PBS). Culture media, including tryptic soy agar with 5% sheep blood, chocolate agar, and MacConkey agar (Remel, Lenexa, KS), were inoculated and streaked for isolation. Samples were incubated in 5% CO2 and examined at 24 and 48 h after inoculation. Colonies with morphology consistent with M. haemolytica, P. multocida, and/or H. somni were subjected to biochemical and/or other phenotypic or MALDI-TOF MS testing validated for definitive identification of these organisms.
Nucleic acid extraction
For preliminary validations, nucleic acids were extracted from reference strains in pure subculture growth by picking several well-isolated colonies with a sterile stick and resuspending in 100 µL of nuclease-free water to a 1–2 McFarland standard turbidity. Cell suspensions were boiled at 100°C for 10 min to lyse bacterial cells. Cell debris was clarified by centrifugation at 15,700 RCF for 2 min. Swab samples were resuspended in 200 µL of nuclease-free water and vortexed. One hundred µL of this eluent was transferred to a sterile 2-mL microcentrifuge tube. Lung tissue was placed in a filter Whirl-Pak (Nasco, Fort Atkinson, WI) with 1–5 mL of sterile PBS and placed in a stomacher for 30–60 s. Once stomached, as much fluid as possible was taken from the Whirl-Pak bag and placed in a 2-mL microcentrifuge tube. The tube was then spun at full speed (~13,500 × g) for 2 min. After centrifugation, 100 µL of the eluent was transferred to a sterile 2-mL microcentrifuge tube. These samples were then subjected to DNA extraction (QiaCube, DNeasy blood and tissue kit, Qiagen).
M. bovis gel-based PCR
The presence or absence of M. bovis was categorized using the previously validated PCR assay used currently in our laboratory (Lauerman L, et al. Nucleic acid amplification assays for diagnosis of animal diseases. Am Assoc Vet Lab Diagnosticians Workshop; 1998; Minneapolis MN). The assay included 25 µL of master mix, which contained 16.9 µL of nuclease-free water, 2.5 µL of 10× PCR Rxn buffer, 2 µL of 50 mM MgCl2, 0.5 µL each of F primer (100 mM) and R primer (100 mM), 0.5 µL of dNTPs (100 mM; Invitrogen, Carlsbad, CA), 0.1 µL of Taq DNA polymerase (Invitrogen), and 2 µL of template. The mix was then subjected to the following thermocycling conditions: 95°C for 3 min, followed by 45 cycles at 94°C for 40 s, 55°C for 30 s, and 72°C for 1.5 min, ending at 72°C for 5 min. PCR products were analyzed using capillary gel electrophoresis (QIAxcel, Qiagen) and compared to a reference isolate used as a positive control.
Real-time PCR assay design
Primers and probes for the multiplex PCR assay were designed using a previously described assay for M. bovis.22 Targets for M. haemolytica,6 P. multocida,14 and H. somni26 were selected from previously described gene targets found to discriminate these organisms with additional reference sequences found in GenBank (Supplementary Table 1). Gene sequences from these targets were identified using BLASTn (https://blast.ncbi.nlm.nih.gov/), aligned, and utilized for primer design using PrimerQuest software with settings for a probe-based assay design (Integrated DNA Technologies [IDT], Iowa City, IA; Table 2). Primer sequences were evaluated in silico with reference sequences where available. The sequences included 21 fully assembled genomes of P. multocida, 4 of which were from cattle (Supplementary Fig. 1), 9 fully assembled genomes of H. somni (Supplementary Fig. 2), 14 fully assembled genomes of M. haemolytica (Supplementary Fig. 3), and 9 fully assembled genomes of M. bovis (Supplementary Fig. 4). Additionally, the lktD locus of 1,133 M. haemolytica isolates sequenced in a previous study was used for screening of potential polymorphisms at the primer-binding sites.4
Table 2.
Oligonucleotide sequences for primers and probes used in the assay. For Histophilus somni probe, JOE-labeled probe was used in the ABI assay, and the Cy5.5-labeled probe was used for the RGQ assay.
| Target/primer or probe | Sequence (5’–3) | Size | Reference |
|---|---|---|---|
| Mycoplasma bovis (oppD) | |||
| PMB996-F | TCAAGGAACCCCACCAGAT | 71 | 22 |
| PMB1066-R | AGGCAAAGTCATTTCTAGGTGCAA | ||
| Mbovis1016 (FAM/TAMRA) | TGGCAAACTTACCTATCGGTGACCCT | ||
| Mannheimia haemolytica (LktD) | |||
| F | CTGCAACAAAGCCGATATCTTT | 95 | Current study |
| R | TACGACTGCTGAAACCTTGAT | ||
| P (CY5/BHQ) | ACACATCGTCTTCCGGCACAATGA | ||
| Histophilus somni (31kD) | |||
| F | GCAATGATGTACCWGCCAAAG | 111 | Current study |
| R | CCTTCAGCTCACCATTACCATA | ||
| P (JOE/BHQ) | TTGCTTACGTCCAAACCGTCGTGT | ||
| P2 (Cy5.5/BHQ) | TTGCTTACGTCCAAACCGTCGTGT | ||
| Pasteurella multocida (Pm1231) | |||
| F | ATCCCTGCGTTACAGAGTTTAG | 110 | Current study |
| R | GACGYGGGYAGTACCATAAA | ||
| P (Texas Red/BHQ) | TTGATGCCTTCTTTGCGGGTTTCG | ||
| Internal control | |||
| IC-1F | GACCACTACCAGCAGAACAC | 177 | 11 |
| IC-2R | CTTGTACAGCTCGTCCATGC | ||
| PNED/MGBNFQ | AGCACCCAGTCCGCCCTGAGCA | ||
| M. bovis | |||
| M. bovis-F | TGATAGCAATATCATAGCGGC | * | |
| M. bovis-R | GTAGCATCATTTCCTATGCTAC | 415 | |
ABI = ABI 7500 Fast, Thermo Fisher Scientific, Waltham, MA; RGQ = Rotor-Gene Q, Qiagen, Hilden, Germany.
Lauerman L, et al. Nucleic acid amplification assays for diagnosis of animal diseases. Am Assoc Vet Lab Diagnosticians Workshop; 1998; Minneapolis MN.
Primers and probe sequences were selected that were unique and did not possess significant identity to other sequences in GenBank, with the exception of M. haemolytica (Table 2). M. haemolytica had some sequence identity with closely related species; however, some polymorphisms were present in primer and probe-binding sites for non-target species (Supplementary Fig. 5). All of the primers and probes were synthesized and purchased from IDT, except minor groove binder probes used for the internal control (Molecular Probes, Thermo Fisher Scientific). These primers and probes were combined with a heterologous internal control target system11 (intype IC DNA, Qiagen) and run on a rotary real-time PCR instrument (hereafter RGQ; Rotor-Gene Q, Qiagen) in a single 5-plex reaction, or run on a block-based Peltier real-time thermocycling system in a 4-plex reaction without an internal control (hereafter ABI; ABI 7500 Fast, Thermo Fisher Scientific).
The number of detection channels, requirements for passive reference dyes, light source, and temperature kinetics of the 2 instruments varied; therefore, separate protocols were used. Protocols were independently optimized for primer and probe concentrations and annealing temperatures, and subsequently evaluated utilizing different instruments. For ABI, the 25-µL PCR master mix contained 6.1 μL of nuclease-free H2O, 12.5 μL of 2× Quantifast multiplex PCR master mix (Qiagen), 0.4 μL of 50× ROX solution, 1 µL of each primer–probe mix (4 total µL) containing F (10 μM), R (10 μM), and P (10 μM), and 2.0 µL of template DNA. For RGQ, the 25-µL PCR master mix contained 10 μL of 2× Quantifast multiplex PCR master mix (Qiagen), 1.0 μL of each primer–probe mix (5 μL total) containing F (10 μM), R (10 μM), and P (10 μM), and 5.0 µL of template DNA. The PCR reactions were subjected to thermocycling under the following conditions: 95°C for 5 min, then 45 cycles of 95°C for 15 s, 60°C for 45 s. Cq was established by subtracting 10% from the peak fluorescence intensity of the positive control for ABI. This was set manually by adjusting the baseline up to the peak fluorescence of the positive control curve and then taking 10% of that value, resulting in a Cq of 0.01–0.1. The Cq threshold for the RGQ was set at a fixed value of 0.1 for all detection channels, except crimson, which was set at 0.05 in clinical samples. For undiluted DNA extracted from pure culture, thresholds were set at threshold values of 0.1, except for red, which was set to 0.25, and green, which was set to 0.15. Dynamic tube normalization was used for all analyses on RGQ.
Preliminary assay validation and analytical Se
The multiplex PCR assays were initially validated using ATCC control organisms and reference bacterial isolates. Target and off-target validation controls were run in undiluted clarified cell lysate suspension in single reaction on the RGQ platform (Table 1). For analytical Se, ATCC reference isolates were used to generate standards and run on both instruments. Briefly, sterile brain–heart infusion (BHI) broth (P. multocida and M. haemolytica; Oxoid, Hants, UK), BHI supplemented with 0.5% yeast extract and 0.05% thiamine monophosphate (H. somni), and mycoplasma broth (M. bovis; Udder Health Systems, Meridian, ID) was inoculated with several isolated colonies of fresh growth and shaken at 200 rpm for ~4 h at 37°C at a 1:10 flask-to-media ratio under atmospheric conditions (P. multocida and M. haemolytica) or incubated in 5% CO2 without shaking (H. somni and M. bovis). Cultures of M. bovis were incubated for ~24 h. Optical density from cultures was measured and adjusted to ~0.26 absorbance (OD600) using a spectrophotometer (UV GENESYS 140, Thermo Fisher Scientific). Cultures were serially diluted in 10-fold dilutions in sterile PBS and plated onto trypticase soy agar with 5% sheep blood (P. multocida and M. haemolytica), chocolate agar (H. somni; Remel), or mycoplasma agar (M. bovis; Udder Health Systems) incubated at 37°C in 5% CO2. Colonies enumerated and colony-forming units (CFU)/mL were calculated. Remaining dilutions were immediately placed on ice, and nucleic acid was extracted as described above.
Statistical analysis
Statistical analyses were conducted to 1) determine the optimal thresholds for the detection of H. somni, M. haemolytica, and P. multocida, for RGQ and ABI rtPCR assays compared to culture or gel-based PCR (M. bovis) as gold standards, respectively, and 2) to evaluate the concordance between RGQ and ABI assays. Analyses were performed using either all data or data stratified by pathogens and sample matrices. All statistical analyses were performed in R v.3.2.3 (https://www.r-project.org/).
Threshold determination
ROC curves were plotted to determine pathogen-detection thresholds, by using the pROC package in R.20 ROC curve is a plot of the true-positive rate (Se) against the false-positive rate (1 − Sp) for a series of possible cut-points of a test by comparison to the gold standards, which facilitated setting the optimal thresholds for the tests by balancing the tradeoff of test Se and Sp. For M. haemolytica, P. multocida, and H. somni, culture isolation and identification were used as the gold standard. For M. bovis, the gel-based PCR approach was used in lieu of culture as a gold standard, because this was the validated standard approach to M. bovis detection used in our laboratory.
The Cq value of “0” for RGQ and ABI rtPCR assays was replaced by “50” in the whole dataset, which was larger than the largest observed Cq value in the dataset. A rtPCR result of “0” indicated the absence of DNA products, which should be referred to as a negative result. However, the calculations in the roc() function assumed a Cq value equal to or smaller than a cut-point referred to a positive result. With this assumption, a PCR result of “0” would be referred to as a positive detection. Changing zeros to a value greater than the largest Cq value in the whole dataset guaranteed a consistent analysis that accurately reflected the PCR results.
Specificity and Se values of the optimal thresholds were computed and presented as percentages. The 95% confidence intervals (CI) of the Se and Sp values were computed with 2,000 stratified bootstrap replicates using the ci.thresholds() function in the {pROC} package. ROC curves were plotted with the indication of the Se (%) and Sp (%) of computed optimal thresholds.
Concordance analysis
Performance of RGQ and ABI rtPCR assays were evaluated by comparing the difference in the mean Cq values of both assays by ANOVA and Cohen kappa statistic.5 A p ⩽ 0.05 was considered statistically significant.
Linear regression models were developed using the “lm()” function to compare RGQ and ABI real-time Cq values using ANOVA. The equations included “organism” and/or “source” variables as potential modifiers on the association between Cq values obtained by RGQ and ABI assays, which were all categorical data with more than 2 mutually exclusive categories. For example, the factor “organism”, contained 4 categories of “H. somni”, “M. haemolytica”, “M. bovis”, and “P. multocida”. A dummy coding approach was used to develop the linear regression models to estimate the impact of effect modifiers on the association of interest. To dummy code the k categories, where k denotes the number of levels of a specific factor (for example regarding the “organism”, k = 4), k – 1 dummy variables needed to be constructed (Di, i = 1, …, k – 1, where Di denoted dummy variables for a specific factor), with Di set to 1 if a case was in category i, otherwise to 0.10 Regarding the factor “organism” for example, dummy variables of “M. haemolytica”, “M. bovis”, and “P. multocida” were constructed, which means “H. somni” was set as the reference category. Hence, pathogen dummy variables (“M. haemolytica”, “M. bovis”, and “P. multocida”) are all set to “0” for cases in the category of “H. somni”. In the case of M. haemolytica–associated test results, the pathogen dummy variable setting was “M. haemolytica” = 1, “M. bovis” = 0 and “P. multocida” = 0. The same approach was applied to variable “source”. The comparison was done by “Anova” function in the “{car}” package. For example, ABI results were the response values in the linear regression model for the dataset containing all of the information for the sample source “lung tissue”, where RGQ results and organism type were the explanatory variables. ANOVA calculated a p value determining the significance of the coefficient for the RGQ test results, where p ⩽ 0.05 indicated a significant association between the Cq values of RGQ and ABI. For ANOVA, Cq values of “0” were kept as they were, and the equations showing the significant relationship between ABI and RGQ values for each dataset were included in the results.
Cohen kappa statistic was used to explore the concordance between RGQ and ABI results, as well as the comparisons of RGQ and ABI (at their optimal thresholds) with the gold standard results, respectively. Cq values of “50” were used to replace “0” for the same aforementioned reason. The following categories were assigned for the agreement associated with kappa statistic: <0 indicating no agreement, 0–0.20 slight, 0.21–0.40 fair, 0.41–0.60 moderate, 0.61–0.80 substantial, and 0.81–1 almost perfect agreement.13 The “kappa2” function in the {“irr”} package was used for the calculations.10
Results
Analysis performed in silico demonstrated that all primer sequences used for the assay had 100% identity to all reference sequences isolated from the target species (bovine). Preliminary validation of test Sp demonstrated no amplification of non-target species (Cq of 0) for each non-target reference culture isolate tested (Table 1). However, in silico analysis indicated that closely related species, such as Mannheimia glucosida and Mannheimia ruminalis, have high sequence identity with target sequences, where in some cases only a single polymorphism was present in the primer–probe sequences (Supplementary Fig. 5).
The assay limits of detection were calculated using reference bacterial isolates (Table 3). The assays were characterized as having limits of detection and corresponding Cq values for M. haemolytica as 24 CFU/reaction (ABI Cq 33.9) or 2.4 CFU/reaction (RGQ Cq 37.6); for P. multocida 12 CFU/reaction (ABI Cq 36.3) or 1.2 CFU/reaction (RGQ Cq 31.7); for H. somni 12 CFU/reaction (ABI Cq 34.7) or 1.2 CFU/reaction (RGQ Cq 37.6); and for M. bovis 12 CFU/reaction (ABI Cq 37.4) or 1.2 CFU/reaction (RGQ Cq 36.3).
Table 3.
Sensitivity and limit of detection in colony-forming units per reaction (CFU/rxn) for both assays using characterized reference strains of each isolate. Limit of detection utilized reference strains Pasteurella multocida (ATCC BAA-1113), Mannheimia haemolytica (ATCC BAA-410), Histophilus somni (129Pt), and Mycoplasma bovis (ATCC 25223).
| Organism | CFU/rxn | Cq |
RBQ |
ABI |
|||
|---|---|---|---|---|---|---|---|
| ABI | RGQ | R 2 | RE | R 2 | RE | ||
| M. haemolytica | 2,400 | 28.2 | 27.4 | ||||
| 240 | 31.69 | 31.38 | |||||
| 24 | 33.96 | 34.43 | |||||
| 2.4 | – | 37.67 | 0.998 | 1.01 | 0.998 | 0.996 | |
| P. multocida | 1,200 | 30.46 | 26.08 | ||||
| 120 | 34.21 | 29.05 | |||||
| 12 | 36.38 | 31.72 | |||||
| 1.2 | – | – | 0.999 | 1.00 | 0.993 | 0.97 | |
| H. somni | 1,200 | 30.60 | 28.02 | ||||
| 120 | 33.06 | 30.49 | |||||
| 12 | 36.26 | 35.05 | |||||
| 1.2 | – | 37.6 | 0.999 | 1.02 | 0.993 | 1.06 | |
| M. bovis | 1,200 | 30.12 | 25.98 | ||||
| 120 | 33.67 | 29.97 | |||||
| 12 | 37.4 | 33.31 | |||||
| 1.2 | – | 36.34 | 0.998 | 0.95 | 0.997 | 0.91 | |
ABI = ABI 7500 Fast, Thermo Fisher Scientific; Cq = quantification cycle; dash (–) = no amplification detected; RE = reaction efficiency (10^(–1/m) – 1); RGQ = Rotor-Gene Q, Qiagen.
The most prevalent finding in diagnostic specimens using the culture or gel-based PCR approach was P. multocida (n = 88), followed by M. haemolytica (n = 63), M. bovis (n = 23), and H. somni (n = 14; Table 4). When classified according to rtPCR results only, levels of detection increased, with M. haemolytica the most prevalent (n = 136), followed by P. multocida (n = 119), M. bovis (n = 54), and H. somni (n = 48; Table 4). Co-detections were frequently found (Table 4), with the most frequent combination being M. haemolytica and P. multocida (n = 98). Thus, compared to rtPCR, gold standard testing detected 29%, 43%, 46%, and 74% of H. somni, M. bovis, M. haemolytica, and P. multocida positive samples, respectively.
Table 4.
Frequency of coinfections across all sample types. PCR results were categorized as positive if pathogen was detected in ⩽40 quantification cycles on either instrument.
|
M. haemolytica
|
P. multocida
|
H. somni
|
M. bovis
|
|||||
|---|---|---|---|---|---|---|---|---|
| PCR | Culture | PCR | Culture | PCR | Culture | PCR | Conv PCR | |
| Mannheimia haemolytica | (136) | (63) | 98 | 27 | 38 | 1 | 27 | 7 |
| Pasteurella multocida | x | x | (119) | (88) | 36 | 6 | 24 | 7 |
| Histophilus somni | x | x | x | x | (48) | (14) | 20 | 6 |
| Mycoplasma bovis | x | x | x | x | x | x | (54) | (23) |
Conv PCR = conventional PCR; x = 185 total samples. Numbers in parentheses are the total number of positives detected.
When examining co-detections based on sample type, the number of lung tissues that were positive for 2 or more pathogens using the least stringent criterion (RGQ platform with a Cq value of ⩽ 40) was 26. This was an additional 8 co-detections over the 18 using culture and gel-based PCR. For antemortem samples, there were 125 specimens with 2 or more pathogens detected, which was much more than the 25 co-detections that were found with culture and gel-based PCR. Much of this enhanced detection may have been the result of detection of non-viable organisms that could not be isolated using the culture approach. However, one exception was H. somni, in which case 4 lung samples were positive by culture, but negative by rtPCR on both platforms (Supplementary Table 3). These isolates were subsequently re-grown in pure culture; the lysates were tested using both PCR platforms, and were positive with comparable Cq values to other H. somni run in pure culture.
Other discrepant results with negative rtPCR and positive culture results were one M. haemolytica isolate from a NS and a DNPS, a P. multocida isolated from a lung, and 3 from DNPS that were cultured but not detected by either PCR assay (Supplementary Table 3). These isolates were lost to follow-up, so they could not be further evaluated by testing isolates. However, discrepant results may be the result of misclassification of the organism from the initial isolate identification, primer–probe mismatch with the isolate, or extraction errors.
Cq cutoff values were calculated to maximize agreement between culture results (gold standard test) and rtPCR results using ROC analysis. Values ranged from a minimum of 29.8 for M. haemolytica in lung matrices using the RGQ assay to a maximum of 44.9 for P. multocida in NS matrices run on the ABI (Supplementary Table 2). Pathogen-specific cutoff Cq values across matrices ranged from a low of 33.4 for M. haemolytica on the ABI to 44.9 for P. multocida on the ABI (Supplementary Table 2). Overall Cq values for all assays were significantly lower (p ⩽ 0.05) on the RGQ (35.2) than the ABI (39.7), indicating a significantly lower detection limit threshold of the RGQ, in general. As the ROC-based Cq values were calculated by maximizing agreement of rtPCR tests and the gold standard culture tests, the values are not indicative of the analytical cutoffs of CFU numbers beyond which quantitative results from rtPCR are valid. Instead, the Cq values refer to cutoffs that would need to be applied to enable the rtPCR methods to produce a result similar to the gold standard from both Se and Sp perspectives, and were calculated for comparative purposes.
ROC analysis of the 4 pathogens across all 3 sample types demonstrated very similar levels of Se and Sp between the 2 instruments compared to the gold standard (Fig. 1; represented by overall equations of ABI = 0.27 + 0.46 × RGQ + 2.49 × M. bovis + 5.37 × M. haemolytica + 7.17 × P. multocida – 3.33 × lung − 0.83 × nasal), where ABI and RGQ represented the Cq values measured by these 2 types of instruments; M. bovis, M. haemolytica, and P. multocida were the dummy variables for pathogen types; and lung and nasal were the dummy variables for sample matrices. This equation could be used to quantify the association between Cq values measured by ABI and RGQ. For example, to examine the association between M. haemolytica in lung tissues, the pathogen dummy variables were set as “M. haemolytica” = 1, “P. multocida” = 0, and “M. bovis” = 0, and matrices dummy variables were set as “Lung” = 1 and “Nasal” = 0. This resulted in ABI = 2.31 + 0.46 × RGQ.
Figure 1.
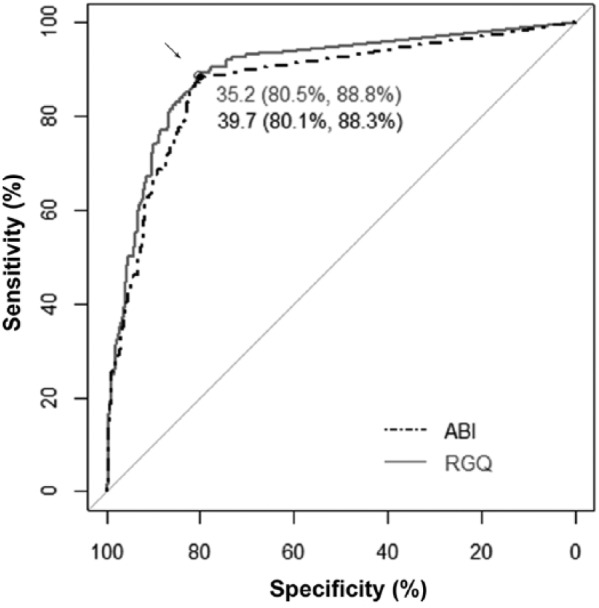
Receiver operating characteristic curves depicting the concordance between the ABI and RGQ real-time PCR (rtPCR) assays based on a crude comparison regardless of pathogens and sample types. The rtPCR assays evaluated were the block-based Peltier real-time thermocycling system (ABI 7500 Fast, Thermo Fisher Scientific) and the rotary real-time PCR instrument (Rotor-Gene Q, Qiagen). The gray diagonal line represents agreement between assays no better than chance alone. The gray solid and black dashed lines are the observed ROC curves for RGQ and ABI assays, respectively. The gray circle and black solid circle (arrow) have the greatest perpendicular distances from the diagonal line, suggesting the optimal quantification cycle values with the sensitivities and specificities of the RGQ and ABI rtPCR assays for pathogen detection.
The agreement between instruments on each assay was also very high, with substantial to almost perfect agreement for all pathogens and matrices by kappa statistic for inter-instrument agreement (⩾0.784), except for H. somni (Supplementary Table 2). For H. somni in lung and DNPS matrices, agreement was only moderate (0.619 and 0.54, respectively). Additionally, for all pathogens except H. somni, agreement with lung tissue gold standard testing was highest, and had substantial or nearly perfect agreement with gold standard testing. DNPS also provided mostly moderate levels of agreement (0.525–0.664) with gold standard testing for pathogens, except for H. somni, which had substantial levels of agreement with gold standard testing for both assays (0.694 and 0.789). Nasal swab matrices had the lowest overall agreement with gold standard testing, with fair-to-moderate levels of agreement (0.353–0.525).
Discussion
In our study, the culture-based result for M. haemolytica, H. somni, and P. multocida, or gel-based PCR result for M. bovis, was considered the gold standard, and both gave a dichotomous outcome of positive or negative for the target pathogen. The rtPCR assay produced a continuous outcome (i.e., the Cq value), which allowed flexibility to design the assay for multiple purposes by varying the Cq cutoff to transfer the continuous outcome into an answer of positive or negative. For example, the optimal cutoff value of Cq of the multiplex rtPCR approach in our study was determined by trading off Se and Sp using ROC to maximize the similarity of results produced by the multiplex rtPCR assay and the gold standard. Because the rtPCR is much more sensitive than the gold standard for detecting these bacterial species, the Se and Sp for rtPCR look low. Therefore, it may not always be reasonable to compare the superiority of a PCR assay over the gold standard based on the Se and Sp values determined by our approach.
If the purpose was to design a “rule-out” test with a high Se to detect a pathogen, a higher Cq cutoff could be used. When a cutoff of 40 was used for M. haemolytica in DNPS, the multiplex rtPCR assay had fewer false-negatives compared to the gold standard. As a result, a very high Se of 97.4% was found for the new test (vs. 74% based on the optimal cutoff), which indicates that a true infection can be detected at a probability of 97.4%, although it is compromised by a lower Sp of 41% (vs. 78% based on the optimal cutoff). This is the ideal property of a test to rule out the presence of the infection by a particular pathogen when a negative result is observed. Conversely, lower cutoffs would be more likely to identify infections with a particular pathogen of interest; in other words, higher Sp giving a better “rule-in” test with a compromise of lower Se. Given that BRDC is comprised of multiple opportunistic pathogens that may exist in normal animals at various levels, especially in antemortem clinical samples, interpretation of testing results is complex, and therefore multiple strategies were used to assess the rtPCR test and to compare results to traditional detection techniques. The Cq cutoff applied should reflect the goals of the test, and could be applied to either a “rule-in” or a “rule-out” diagnostic approach. Further clinical trials could also be conducted to relate shedding of BRD pathogens in antemortem samples to find Cq cutoffs that maximize agreement with clinical signs and/or the presence of postmortem lesions.
In general, lung tissues had higher agreement than other samples with the culture or gel-based PCR, followed by DNPS and NS. However, some discrepancies, such as those found with H. somni, were attributed to sampling differences between the tissue utilized for culture versus extracted for PCR. H. somni infections may be more prone to this type of sampling error, because the lung lesions can be more variable in distribution and type. Bovine respiratory disease caused by H. somni is typically a bilateral lobar pneumonia in the cranioventral lung; however, chronic infections may induce necrotic foci that are distributed unevenly. Additionally, given that H. somni can cause septicemia, pneumonia may also be interstitial, which may have significantly different pathogen distribution within tissues.17
Less agreement in DNPS and NS was likely the result of contaminating environmental organisms that are typically present in the upper respiratory tract, which can rapidly outgrow pathogens on culture plates and lead to false-negative culture results. Survival and subsequent recovery of these organisms in swabs and in transport media can also be lower when compared to tissues, leading to false-negative culture results. Additionally, some of the pathogens, such as P. multocida, can grow rapidly and be very mucoid, making culture assessment of mixed pathogen cultures challenging. Therefore, although the agreement was lowest in swab samples, the PCR methodology may be most useful for these given the potential for high false-negative culture rates in these specimen types. However, even though detection is enhanced using a PCR-based approach, phenotypic testing of susceptibility would not be possible without an isolate, therefore culture is still required to guide antimicrobial therapy, and as stated above, mere detection of these pathogens may not indicate clinical infection.
The RGQ yielded significantly lower Cq cutoffs that maximized agreement with gold standards over the ABI instrument, in agreement with previous work.19 This is potentially because the more precise temperature control and rapid temperature adjustment enhanced overall PCR efficiency. Some differences may be the result of how the Cq threshold values were applied; however, the threshold levels used for the ABI were uniformly lower, and thus would likely have lowered overall Cq compared to the RGQ. However, these differences in corresponding Cq values did not affect the overall Se and Sp of the assays, whereby the ABI and RGQ were nearly identical when the Cq cutoff values that maximized these values were applied.
Detection of pathogens in samples with rtPCR, especially in the absence of postmortem lesions, may not indicate the presence of disease. For antemortem samples, further research is needed to determine if a specific Cq cutoff used to assess nasal shedding levels can be used to maximally predict a clinical infection, given that many of these agents colonize the upper respiratory tract in normal, clinically healthy animals; stressed and sick cattle may have higher levels of shedding, and thus lower Cq values. Currently, pathogen burdens and shedding can be estimated based on the Cq value and copy number, but how to best apply this information to clinical treatment is an area for further study, because relevant clinical data that may allow for classification of these specimens is unavailable. However, the samples tested were from submissions from presumably non-healthy cattle, and therefore may not be representative of normal healthy animal populations. A multiplex hydrolysis probe rtPCR assay when utilized in either an RGQ or ABI instrument heavily increases the number of bovine respiratory pathogens that can be detected in NS, DNPS, and lung tissues over culture and gel-based PCR assays. The utilization of high-throughput extraction methods in conjunction with this assay would provide results in a matter of hours, rather than days, over culture methodology. This would strongly enhance the ability to detect pathogens in highly mixed specimens such as NS and DNPS, enabling veterinarians and clinicians to diagnose disease and administer treatment or implement prevention strategies more rapidly.
Supplementary Material
Acknowledgments
We thank students Joshua Payne and Kara Robbins for technical assistance, and veterinary diagnostic center staff Marijana Bradaric, Jamie Bauman, Matt Quinn, and Debra Royal.
Footnotes
Declaration of conflicting interests: Dr. Loy has served as a consultant for, and thus has disclosed a significant financial interest in, Harrisvaccines (Ames, IA). In accordance with its Conflict of Interest policy, the University of Nebraska–Lincoln’s Conflict of Interest in Research Committee has determined that this must be disclosed.
Funding: This project is based on research that was partially supported by the Nebraska Experiment Station with funding from the Hatch Act (accession 1007070) and the Animal Health and Disease Research (section 1433) capacity funding program (accession 1002196) through the USDA National Institute of Food and Agriculture. Qiagen Inc. provided some of the reagents for evaluation at no cost.
ORCID iD: John D. Loy  https://orcid.org/0000-0002-7282-096X
https://orcid.org/0000-0002-7282-096X
References
- 1. Bell CJ, et al. Investigation of polymerase chain reaction assays to improve detection of bacterial involvement in bovine respiratory disease. J Vet Diagn Invest 2014;26:631–634. [DOI] [PubMed] [Google Scholar]
- 2. Booker CW, et al. Microbiological and histopathological findings in cases of fatal bovine respiratory disease of feedlot cattle in western Canada. Can Vet J 2008;49:473–481. [PMC free article] [PubMed] [Google Scholar]
- 3. Capik SF, et al. Characterization of Mannheimia haemolytica in beef calves via nasopharyngeal culture and pulsed-field gel electrophoresis. J Vet Diagn Invest 2015;27:568–575. [DOI] [PubMed] [Google Scholar]
- 4. Clawson ML, et al. Genomic signatures of Mannheimia haemolytica that associate with the lungs of cattle with respiratory disease, an integrative conjugative element, and antibiotic resistance genes. BMC Genomics 2016;17:982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Meas 1960;20:37–46. [Google Scholar]
- 6. Davies RL, et al. Mosaic structure and molecular evolution of the leukotoxin operon (lktCABD) in Mannheimia (Pasteurella) haemolytica, Mannheimia glucosida, and Pasteurella trehalosi. J Bacteriol 2002;184:266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fulton RW, et al. Evaluation of health status of calves and the impact on feedlot performance: assessment of a retained ownership program for postweaning calves. Can J Vet Res 2002;66:173–180. [PMC free article] [PubMed] [Google Scholar]
- 8. Godinho KS, et al. Use of deep nasopharyngeal swabs as a predictive diagnostic method for natural respiratory infections in calves. Vet Rec 2007;160:22–25. [DOI] [PubMed] [Google Scholar]
- 9. Griffin D, et al. , Bacterial pathogens of the bovine respiratory disease complex. Vet Clin North Am Food Anim Pract 2010;26:381–394. [DOI] [PubMed] [Google Scholar]
- 10. Hayes AF, Preacher KJ. Statistical mediation analysis with a multicategorical independent variable. Br J Math Stat Psychol 2014;67:451–470. [DOI] [PubMed] [Google Scholar]
- 11. Hoffmann B, et al. A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J Virol Methods 2006;136:200–209. [DOI] [PubMed] [Google Scholar]
- 12. Klima CL, et al. Pathogens of bovine respiratory disease in North American feedlots conferring multidrug resistance via integrative conjugative elements. J Clin Microbiol 2014;52:438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics 1977;33:159–174. [PubMed] [Google Scholar]
- 14. Liu D, et al. Specific PCR identification of Pasteurella multocida based on putative transcriptional regulator genes. J Microbiol Methods 2004;58:263–267. [DOI] [PubMed] [Google Scholar]
- 15. Loy JD, Clawson ML. Rapid typing of Mannheimia haemolytica major genotypes 1 and 2 using MALDI-TOF mass spectrometry. J Microbiol Methods 2017;136:30–33. [DOI] [PubMed] [Google Scholar]
- 16. Lubbers BV, Hanzlicek GA. Antimicrobial multidrug resistance and coresistance patterns of Mannheimia haemolytica isolated from bovine respiratory disease cases—a three-year (2009–2011) retrospective analysis. J Vet Diagn Invest 2013;25:413–417. [DOI] [PubMed] [Google Scholar]
- 17. O’Toole D, et al. Histophilosis as a natural disease. In: Inzana TJ, ed. Histophilus somni: Biology, Molecular Basis of Pathogenesis, and Host Immunity. Heidelberg, Germany: Springer, 2016:15–48. [Google Scholar]
- 18. Panciera RJ, Confer AW. Pathogenesis and pathology of bovine pneumonia. Vet Clin North Am Food Anim Pract 2010;26:191–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reynisson E, et al. , Evaluation of probe chemistries and platforms to improve the detection limit of real-time PCR. J Microbiol Methods 2006;66:206–216. [DOI] [PubMed] [Google Scholar]
- 20. Robin X, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenbusch RF, et al. In vitro antimicrobial inhibition profiles of Mycoplasma bovis isolates recovered from various regions of the United States from 2002 to 2003. J Vet Diagn Invest 2005;17:436–441. [DOI] [PubMed] [Google Scholar]
- 22. Sachse K, et al. Use of a novel real-time PCR technique to monitor and quantitate Mycoplasma bovis infection in cattle herds with mastitis and respiratory disease. Vet J 2010;186:299–303. [DOI] [PubMed] [Google Scholar]
- 23. Schneider MJ, et al. An evaluation of bovine respiratory disease complex in feedlot cattle: impact on performance and carcass traits using treatment records and lung lesion scores. J Anim Sci 2009;87:1821–1827. [DOI] [PubMed] [Google Scholar]
- 24. Snowder GD, et al. Bovine respiratory disease in feedlot cattle: environmental, genetic, and economic factors. J Anim Sci 2006;84:1999–2008. [DOI] [PubMed] [Google Scholar]
- 25. Wittum TE, et al. Relationships among treatment for respiratory tract disease, pulmonary lesions evident at slaughter, and rate of weight gain in feedlot cattle. J Am Vet Med Assoc 1996;209:814–818. [PubMed] [Google Scholar]
- 26. Won J, Griffith RW. Cloning and sequencing of the gene encoding a 31-kilodalton antigen of Haemophilus somnus. Infect Immun 1993;61:2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.