

Abstract
Nanoparticles can be used to accomplish antigen-specific immune tolerance in allergic and autoimmune disease. The available options for custom-designing tolerogenic nanoparticles (NPs) include the use of nanocarriers that introduce antigens into natural tolerogenic environments, such as the liver, where antigen presentation promotes tolerance to self or foreign antigens. Here, we demonstrate the engineering of a biodegradable polymeric poly (lactic-co-glycolic acid) (PLGA) nanocarrier for the selective delivery of the murine allergen, ovalbumin (OVA), to the liver. This was accomplished by developing a series of NPs in the 200–300 nm size range as well as decorating particle surfaces with ligands that targets scavenger and mannose receptors on specialized tolerogenic liver sinusoidal endothelial cells (LSECs). LSECs represent a major antigen presenting cell (APC) type in the liver capable of generating regulatory T-cells (Tregs). In vitro exposure of LSECs to NPOVA induced abundant TGF-β, IL-4 and IL-10 production, which was further increased by surface ligands. Further animal experiments showed that in the chosen size range, NPOVA was almost exclusively delivered to the liver, where the co-localization of fluorescent-labeled particles with LSECs could be seen to increase by surface ligand decoration. Moreover, prophylactic treatment with NPOVA in OVA-sensitized and challenged animals (aerosolized inhalation), could be seen to significantly suppress anti-OVA IgE responses, airway eosinophilia and TH2 cytokine production in the bronchoalveolar lavage fluid. The suppression of allergic airway inflammation was further enhanced by attachment of surface ligands, particularly for particles decorated with the ApoB peptide, which induced high levels of TGF-β production in the lung along with the appearance of Foxp3+ Tregs. The ApoB peptide coated NPs could also interfere in allergic airway inflammation when delivered post-sensitization. The significance of these findings are that liver and LSEC targeting PLGA NPs could be used for therapy of allergic airway disease, in addition to using their tolerogenic effects for other disease applications.
Keywords: LSECs, immune tolerance, nanoparticles, immunosuppression, immunotherapy
Graphical Abstract

There is an unmet need for developing new treatment approaches for autoimmune and allergic disorders that goes beyond current therapeutic efforts of utilizing anti-inflammatory, immunosuppressive, targeted monoclonal antibody or immunomodulatory approaches. While most of these therapies provide symptomatic relief and a temporary abatement of disease activity, they do not provide long-term suppression of chronic disease activity or the prospect of a cure. However, there is growing awareness of the power of regulatory T-cell (Treg) biology to provide antigen-specific immune tolerance for autoimmune disease (e.g., rheumatoid arthritis) and allergic disorders (e.g., food allergy, anaphylaxis, asthma).1, 2 One approach for inducing antigen-specific tolerance is to use biodegradable nanoparticles to initiate and sustain immunomodulatory responses, based on the ability of these carriers to encapsulate disease-related antigens that are delivered to antigen presenting cells (APC).3–7 The tolerogenic properties of the liver is well known for this organ’s role in preventing immune responses to exogenous food antigens coming from the gastrointestinal tract and portal venous system, as well as promoting the persistence of tumor metastases to this organ.8 Moreover, the liver also enjoys immune privilege during organ transplantation, requiring less immunosuppressive therapy than kidney or heart transplants.9 It has also been demonstrated that concurrent transplant of a kidney or a heart with a liver is less prone to undergo immunological rejection, compared to an isolated organ transplant.10, 11 The hepatic expression and ability of liver APCs to present myelin basic protein (MBP) to the immune system has likewise been demonstrated to control experimental allergic encephalomyelitis (an autoimmune disorder that simulates multiple sclerosis) in mice.12
The immunosuppressive effects of the liver can, in part, be ascribed to its unique system of antigen-presenting cells (APCs), including natural tolerogenic APCs such as Kupffer cells (KC), dendritic cells (DC) and liver sinusoidal endothelial cells (LSECs).13, 14 These tolerogenic APCs constitute an integral component of the liver’s reticuloendothelial system (RES), which has the key function of clearing foreign materials, degradation products and toxins from sinusoidal blood by phagocytic uptake as well as endocytic processing.14 Moreover, while professional phagocytes (KC and DC) preferentially eliminate circulating micronscale particulate materials through phagocytosis, LSECs are more proficient in eliminating soluble macromolecules and particulates in the 200 nm size range by clathrin-mediated endocytosis.15, 16 From an immunoregulatory perspective, LSECs play a key role in inducing immune suppression of CD8+ and CD4+ populations through the generation of antigen-specific regulatory T-cells (Tregs), TGF-β production and upregulation of the ligand (PD-L1) for the programmed cell death protein 1 (PD-1) receptor.17–19 It is also of further interest that LSECs obtained from Foxp3gfp/KI transgenic mice were shown to be more capable of generating antigen-specific CD4+/Foxp3+ regulatory T-cells (Tregs) compared to KC or liver DC from the same animals.18, 20 Thus, the ability of LSECs to control the function of antigen-specific Tregs should be considered for the treatment of autoimmune and allergic disease manifestations.
The use of nanoparticles to induce immune tolerance is an active area of investigation and includes approaches such decorating particle surfaces with peptide/major histocompatibility (MHC) complexes, serving as a surrogate antigen presentation platform for immune tolerization in the absence of costimulation.21–25 Other approaches include the incorporation of food allergens or autoimmune proteins/peptides (e.g., type II collagen) in orally administered nanoparticles;26–28 harnessing apoptotic cell death (e.g., apoptotic cell-peptide conjugates or liposomes containing phosphatidylserine);29–33 targeting B-cell-specific tolerance via the CD22 receptor;34, 35 or encapsulating pharmacological agents (e.g., rapamycin) that induce tolerogenic states in APC by impacting antigen presentation, maturation and/or the expression of costimulatory molecules.36–38 While these nano-enabled immunotherapy approaches are yielding promising results, our preferred approach is to use the natural tolerogenic effects of the liver, which can be exploited by a versatile nanoparticle platform constructed from a biodegradable FDA-approved polymer, poly(lactic-co-glycolic acid) (PLGA). One approach could be to target mannose or scavenging receptors (SR) that are involved in endocytosis of circulating antigens, extracellular macromolecules, protein degradation products, and lipoproteins by LSECs.15 While the stabilin-1 and stabilin-2 SRs are exclusively expressed on LSECs, the mannose receptor also appears in lesser quantities on the KC surface.15 These receptors can be targeted by placing an apolipoprotein B (Apo B) peptide sequence or mannan, respectively, on the PLGA particle surface.
In this communication, we demonstrate the design and synthesis of PLGA nanoparticles for delivering ovalbumin (OVA) in a murine model of OVA-induced airway allergic inflammation to investigate whether surface coating with mannan or an ApoB peptide could change disease outcome by inducing a Treg-mediated tolerogenic effect. We demonstrate that ligand-coated NPs can improve OVA delivery to LSECs in vitro and in vivo, with the ability to induce TGF-β production and antigen-specific immune tolerance. While tolerization could be obtained by OVA delivery to the liver in non-decorated 200–300 nm NPs, the tolerogenic effects could be dramatically enhanced by the attachment of surface ligands. These results demonstrate the feasibility of developing a tolerogenic PLGA nanoparticle platform for allergen delivery to LSECs by controlling particle size and decorating the particle surface with ligands that target the liver.
Results
Synthesis of the PLGA NP platform for OVA delivery
The PLGA polymer was selected for NP synthesis because it is biodegradable, biocompatible, and FDA approved for drug delivery.39–41 A double-emulsion method, involving solvent evaporation, was used to fabricate PLGA NPs entrapping 50 μg/mg OVA. Since most NPs in the size range above 500 nm are phagocytosed by KC in the liver or macrophages in the liver and spleen,15, 21 our aim was to develop smaller, easy-to-synthesize particles using a water-in-oil-in-water (w/o/w) emulsion method (Fig. 1A). We settled on the synthesis of a primary particle size of 230 nm (Fig. 1B, Table 1), which yielded a size range of 246–297 nm following the attachment of surface ligands that could target mannose and stabilin receptors on liver cells.42, 43 Mannan (man) was selected to target the mannose receptor, while an ApoB peptide (ApoBP), RLYRKRGLK, was used as a ligand for stabilin receptors.15, 44–46 While the stabilin receptors are exclusively expressed on LSECs, the mannose receptor is also present in low abundance on the surface of KCs. Mannan attachment to PLGA particle surfaces was achieved either by physical adsorption or using a one-step covalent conjugation method to yield NPOVA/mannc and NPOVA/manc NP, respectively. For the attachment of ApoBP peptide, lower and higher molar ratios were used for conjugation to the polymer backbone by a two-step procedure, making use of a NAEM spacer; these particles were designated as NPOVA/ApoBPlo and NPOVA/ApoBPhi, respectively (Fig. 1A).
Figure 1.
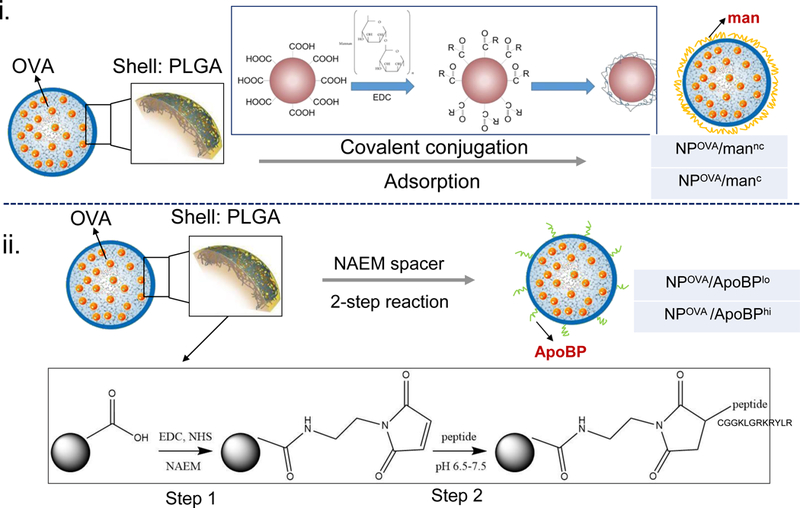


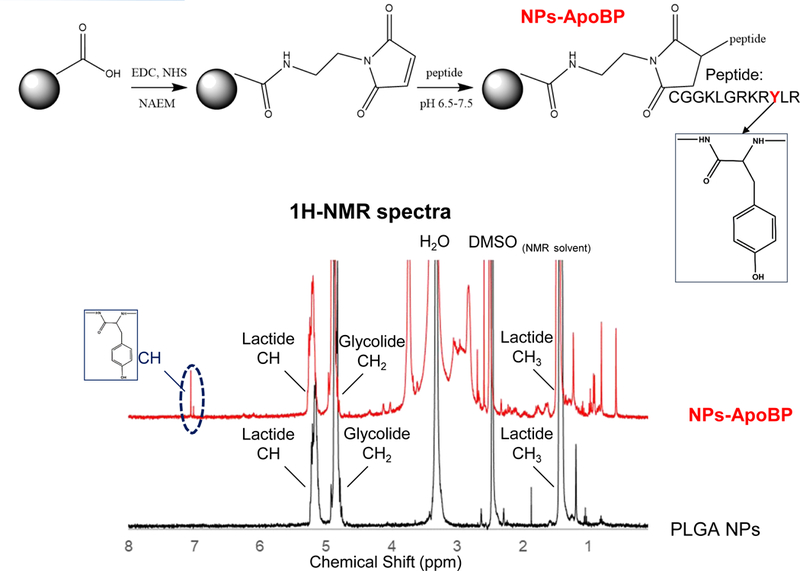
Synthesis and characterization of the LSEC-targeting PLGA NP platform for OVA delivery. (A) Schematic showing |particle surface decoration with mannan and ApoBP: (i) mannan (man) was either physically adsorbed to the particle surface or its hydroxyl-terminus used for covalent conjugation to the PGLA COOH-terminal groups; (ii) ApoBP was linked to the NP surface by a NAEM spacer, using a two-step conjugation process between the ApoBP cysteine tag and the NAEM maleimide group. (B) SEM pictures to show NP morphology, in the presence of attached ligands. (C) FTIR spectra of the NAEM-conjugated NPs. (D) 1H-NMR spectra of the synthesized particles with and without the ApoBP attachment, showing the appearance of the newly conjugated peptide at 7 ppm.
Table 1.
Comparison of different NP formulations for hydrodynamic size, zeta potential, OVA content, ligand content and molar ratios.
| Nanoparticle | Hydrodynamic size (nm) | Polydispersity index (PDI) | Zeta potential (mV) | OVA content (μg/mg NPs) | Ligand content (μg/mg NPs) | mol% of PLGA |
|---|---|---|---|---|---|---|
| NP only | 231.2 ± 2.17 | 0.096 | −42.55 ± 2.99 | NA | NA | NA |
| NPOVA | 246.5 ± 3.01 | 0.105 | −44.37 ± 2.81 | 51.61 ± 2.32 | NA | NA |
| NPOVA/mannc | 279.5 ± 2.74 | 0.121 | −51.63 ± 4.05 | 50.31 ± 3.52 | 139 ± 21 | NA |
| NPOVA/manc | 297.2 ± 2.98 | 0.113 | −54.82 ± 5.18 | 50.98 ± 3.88 | 346 ± 52 | NA |
| NPOVA/ApoBPlo | 268.8 ± 4.96 | 0.109 | −8.63 ± 1.38 | 50.77 ± 3.08 | 6.86 ± 0.89 | 2.8 |
| NPOVA/ApoBPhi | 270.8 ± 4.96 | 0.113 | −4.56 ± 2.25 | 50.12 ± 2.18 | 12.96 ± 0.77 | 5.3 |
The NP formulations were characterized for hydrodynamic size, zeta potential, OVA content, and ligand density as outlined in Table 1. The physicochemical characterization demonstrated a mean hydrodynamic diameter of 230–290 nm, the larger size being due to the attachment of surface ligands. NP without OVA (designated NP-only) or encapsulating OVA only (NPOVA) exhibited a negative surface charge in water, with zeta potentials of −42.55 mV and −44.37 mV, respectively. Mannan attachment increased the negative charge, while ApoBP conjugation brought the zeta potential to −8.63 mV and −4.56 mV for NPOVA/ApoBPlo and NPOVA/ApoBPhi, respectively.
The relative abundance of mannan incorporation was assessed by calculating the difference between the total amount offered for conjugation vs. the amount recovered in the supernatant. Mannan quantity was determined by a colorimetric method (using phenol-sulfuric acid45) to demonstrate a man content of 346±52 μg per mg of NPs after covalent attachment, while physical adsorption amounted to 139±21 μg/mg. The ApoBP conjugation to the NAEM spacer was determined by FTIR, which demonstrated demonstrating the presence of two amide bonds (stretching peaks at~1600 cm−1) as well as a maleimide ring (vibration peak at ~1100 cm−1) (Fig. 1C). We also assessed the proton NMR spectra of the pristine particles, showing the presence of the lactide (-CH and –CH3) and glycolide (-CH2) peaks in the PLGA backbone, as well as the appearance of an ApoBP tyrosine peak (~7 ppm) in conjugated particles (Fig. 1D). The abundance of peptide conjugation was assessed by a microBCA assay and a nanodrop method. Both methods showed a peptide quantity of ~2.8 and 5.3 mol% in the completed PLGA construct.
SEM was performed to show the morphology of the spherical NPs, which were demonstrated to be of uniform size (Fig. 1B). The biocompatibility of these materials in LSECs and Kupffer cells were assessed by using an ATP assay, following particle addition at concentrations ranging from 25 to 300 μg/mL. No evidence of cytotoxicity was observed (Fig. S1).
NP-mediated OVA uptake in LSECs and Kupffer (KUP5) cells
The encapsulation of fluorescein isothiocyanate (FITC)-labeled OVA in the NPs allowed us to use flow cytometry to follow NPOVA uptake in LSEC and KUP5 cells. This demonstrated a time-depended increase in OVA fluorescence intensity in LSECs, with the ApoBP decorated particle showing significantly higher uptake than either non-decorated (p<0.001) or mannan-decorated particles (p<0.01) (Fig. 2). The maximum particle uptake was seen for NPOVA/ApoBPhi, which showed a ~2-fold increase in the percentage of OVA-positive cells compared to NPOVA without ligands. No significant uptake was seen for non-encapsulated FITC-OVA. In contrast to LSECs, the uptake of encapsulated OVA in KUP5 cells was poor, with only 5–10 % of the cells showing a fluorescence signal, mostly for particles decorated with the ApoBP ligand. All considered, these data demonstrate that ApoBP and (to lesser extent) mannan are capable of augmenting OVA uptake in LSECs but not Kupffer cells.
Figure 2.
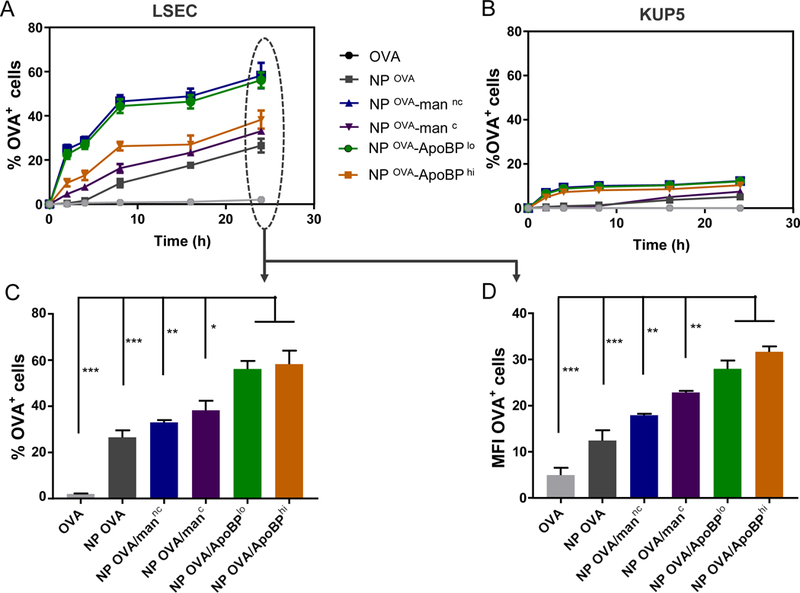
Quantitative analysis of particle-associated antigen uptake in LSECs and KUP5 cells, using flow cytometric analysis. Following OVA FITC-labeling, the antigen was encapsulated in NPs as described in Material and Methods. LSECs and KUP5 cells were incubated with FITC-OVA or NPFITC-OVA for the indicated time periods. After washing in PBS, the % FITC-OVA+ cells and the cellular mean fluorescence intensity (MFI) were analyzed in a BD LSRII (IMED) analytic flow cytometer by flowjo software. (A) and (B): Kinetics of NP-mediated antigen uptake in LSECs and KUP5 cells, respectively. (C) and (D): Histograms depicting the % and MFI of antigen-positive LSECs. Data are expressed as the mean ± SEM (n = 6). *p < 0.05; **p < 0.01; ***p < 0.00 (one-way ANOVA followed by a Tukey’s test).
Induction of a tolerogenic cytokine profile in LSECs and Kupffer cells by ligand-decorated NPs
APCs in the liver are capable of promoting immune tolerance by a variety of mechanisms, including TGF-β production.18, 19, 46 LSECs, in particular, are capable of converting CD4+/Foxp3- T-cells into CD4+/Foxp3+ Tregs through tethered TGF-β on their cell surfaces.15, 18, 20 Moreover, following antigen capture, LSECs are capable of producing anti-inflammatory cytokines to further assist generation of antigen-specific immune tolerance.47 In order to determine if PLGA NPs impact the production of tolerogenic/anti-inflammatory (TGF-β, IL-10, IL-4) or pro-inflammatory (e.g., IL-1 beta, IL-6, IL-12p70, and TNF-α) cytokines, LSECs and KUP5 cells were exposed for 24 h, and cellular supernatants collected for the performance of ELISA (Fig. 3). All NP formulations induced a significant increase (p<0.001) in TGF-β production compared to OVA alone in LSECs (Fig. 3A). Moreover, the cytokine response was significantly higher for NPOVA/ApoBPlo than NPOVA (p<0.01) or NPOVA/man (p<0.05). A similar trend was seen for IL-4 and IL-10 production in LSECs, with NPOVA/ApoBPlo inducing more robust responses than NPOVA/ApoBPhi (p<0.05) or any other particles in the panel (p<0.05). In contrast, the quantities of TGF-β and IL-4 released from KUP5 cells were much lower, with NPOVA/manc inducing more IL-4 and IL-10 than other NPs (Fig. 3B). This is consistent with the ability of covalently-attached mannan to improve OVA uptake in KC (Fig. 2). In contrast to the effect on anti-inflammatory cytokines, all NPs (irrespective of ligation status) trended towards decreasing the production of IL-1β, IL-6, IL-12p70, and TNF-α in LSECs and KC (Fig. S2). All considered, these data demonstrate that ligand-decorated nanoparticles induce significantly more tolerogenic cytokines and TGF-β in LSECs than KC, suggesting that in vivo targeting of sinusoidal endothelial cells could provide an effective means of inducing immune tolerization.
Figure 3.
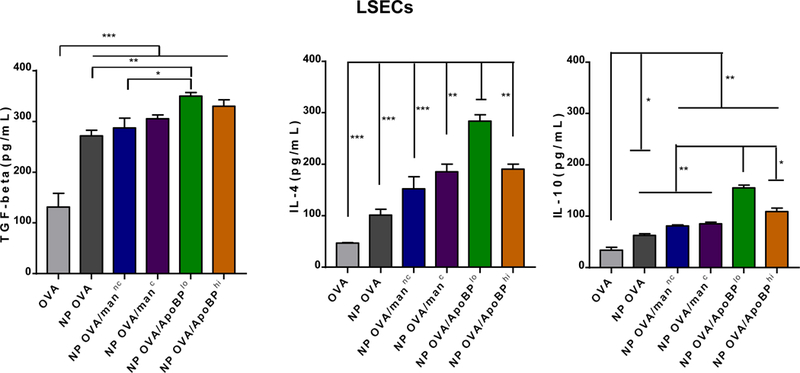
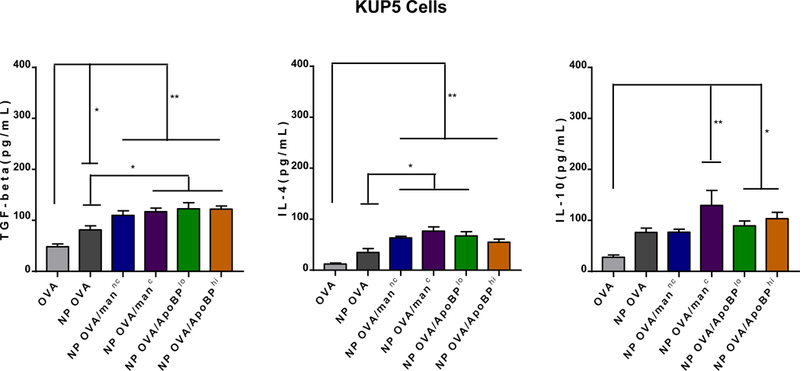
Assessment the effect of NPs on tolerogenic cytokine production, including TGF-β, IL-4, IL-6 and IL-10, in LSECs (A) and KUP5 cells (B). These cells were treated with NPs for 24 h, before removal of supernatants and assessment of cytokine content, using ELISA kits according to the manufacturer’s instructions. Data are expressed as the mean ± SEM (n = 6). *p < 0.05; **p < 0.01; ***p < 0.00 (one-way ANOVA followed by a Tukey’s test).
Hepatic biodistribution of NPs decorated with surface ligands
In order to image the in vivo biodistribution of decorated or non-decorated NPs, DyLight 680-labeled OVA was incorporated into the particles. These particles were intravenously (IV) injected in mice to deliver 25 µg OVA in 500 µg NPs per animal, before sacrifice after 24 h. Major organs such as the liver, spleen, heart, lung and kidney were harvested and mounted in a Petri dish for IVIS imaging. Compared to the distribution of non-encapsulated fluorescent OVA to the kidney, NP-encapsulated OVA mainly accumulated in the liver, with lower levels in the lung and the spleen; little or no distribution occurred to the heart and kidney (Fig. 4A, left panel). Quantitative expression of fluorescence intensity demonstrated that ApoBP-decorated particles show the highest liver accumulation, compared to particles decorated with mannan or non-decorated NPOVA (Fig. 4A, right panel).
Figure 4.
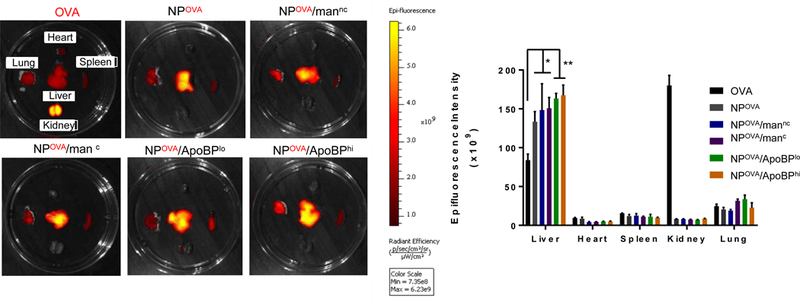
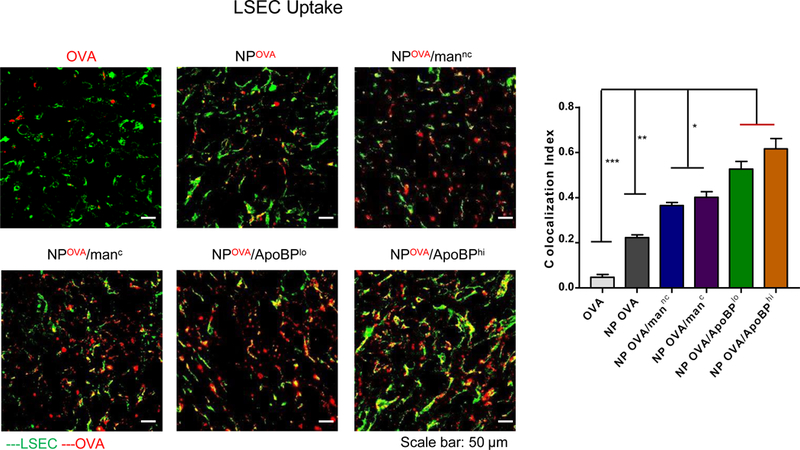
(A) Representative ex vivo IVIS images of the explanted hearts, livers, spleens, lungs, and kidneys collected from animals 24 hr after injecting each animal with 500 μg NPs, containing 25 μg Dylight680-labeled OVA (n=6). The histogram in the panel on the right hand side shows the fluorescence intensity of the particles in the main organs. (B) Confocal microscopy to show the intrahepatic distribution of free or encapsulated labeled OVA. Encapsulation of the labeled protein did not change the NP properties. The red and green fluorescence colors represent Dylight680-labeled OVA and isolectin B4 stained LSECs, respectively. Scale bars correspond to 50 μm. Data are expressed as the mean ± SEM (n = 6). *p < 0.05; **p < 0.01; ***p < 0.00 (one-way ANOVA followed by a Tukey’s test).
In order to visualize the intrahepatic distribution of the labeled NPs, isolectin B4 immunostaining was used to locate sinusoidal endothelial cells in liver tissue sections (Fig. 4B and Fig. S4). Confocal microscopy showed that, compared to fluorescent labeled OVA only or DyLight-labeled NPOVA, the co-localization of mannan or ApoBP-coated particles with isolectin-stained endothelial cells could be seen to increase (Fig. 4B and Fig. S3). Calculation of the co-localization frequency (Pearson correlation coefficient), using Image-Pro Plus 6.0 software, demonstrated a significantly higher co-localization index for the ApoBP-decorated particles than particles decorated with mannan (p<0.5) or delivering OVA only (p<0.01). These results are in agreement with the expression of stabilin-1 and stabilin-2 scavenger as well as mannose receptors on LSECs. Similar analysis was performed to evaluate the co-localization of the Dyligh-labeled NPs with KC, which were immunostained with F4/80 (Fig. S4). While confocal microscopy demonstrated that NPOVA are taken up by KC, the frequency of co-localization was increased for NPOVA/mannc and NPOVA/manc (Fig. S4). In contrast, there was no change in co-localization of ApoBP-decorated NPs with KC, which lack stabilin receptors.
Pretreatment with NPs exhibit tolerogenic effects on the humoral immune response and generation of allergic inflammation in the lungs of OVA-sensitized mice
Based on the observation that mannan and ApoBP-decorated NPs are capable of LSEC targeting and induction of tolerogenic cytokines, we asked whether these particles could be used for tolerance induction in a mouse model of OVA-induced allergic inflammation in the lung. This was accomplished by pretreating Balb/c mice on days 0 and 7 with OVA-encapsulated NPs, with and without surface ligand decoration (Fig. 5A). The controls included animals receiving no particles or PLGA particles that do not contain OVA. The mice were subsequently sensitized to OVA by intraperitoneal (IP) administration of the antigen on days 14 and 21, followed by inhalation challenge with aerosolized OVA on days 35–37. The animals were sacrificed on day 40 for performance of bronchoalveolar lavage (BAL) and organ harvesting (Fig. 5A). The allergic response to OVA is accompanied by TH2-driven IgE and IgG1 antibody production, which could be measured in the serum (Fig. 5B). This demonstrated that the IgE response to OVA could be significantly decreased by pretreatment with OVA-containing nanoparticles, with the strongest (15-fold) response observed in animals pretreated with NPOVA/ApoBPhi (p<0.001). No decline was seen in the group receiving NPs w/o OVA. A similar trend was seen for the IgG1 response to OVA, with antibody titer decreasing ~10-fold in animals pretreated with NPOVA/ApoBPhi (Fig. 5B). In contrast, the TH1-mediated IgG2a antibody response to OVA was not affected by pretreatment (not shown).
Figure 5.

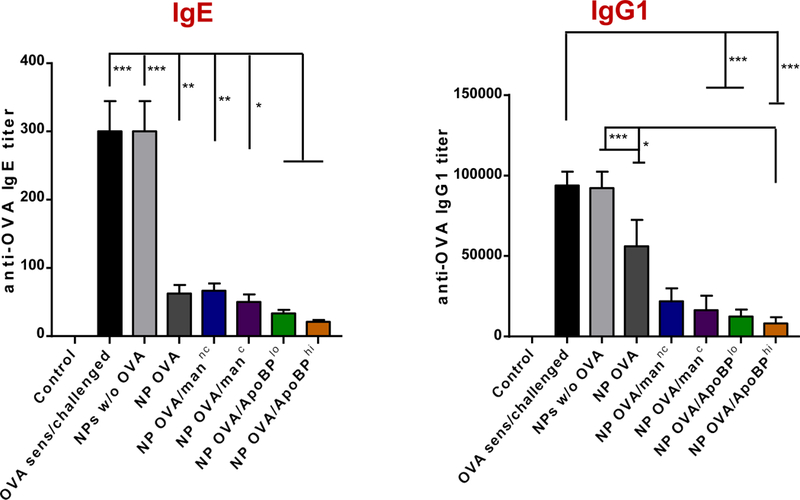
NP pretreatment interferes in OVA-induced antibody responses in an OVA sensitization model. (A) Outline of the experimental animal protocol. Six to 8 week old C57/BL6 mice received IV injection of NPOVA to deliver 25 μg OVA in 500 μg particles per mouse on days 0 and 7. The animals were subsequently sensitized two IP doses of OVA (10 μg/mouse) on days 14 and 21, prior to being exposed to aerosolized OVA inhalation (10 mg/mL) for 20 min on days 35–37. Animals were sacrificed for tissue harvesting and BALF on day 40. The treatment groups (n=6) in the experiment included: (1) a control group without NP pretreatment, sensitization or challenge; (2) no pretreatment before sensitization and challenge; (3) pretreatment with NPs w/o OVA before sensitization and challenge; or pretreatment with (4) NPOVA, (5) NPOVA/mannc, (6) NPOVA/manc, (7) NPOVA/ApoBPlo, (8) NPOVA/ApoBPhi before sensitization and challenge. (B) Serum anti-OVA IgE and IgG1 antibody titers were determined by ELISA. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.00 (one-way ANOVA followed by a Tukey’s test).
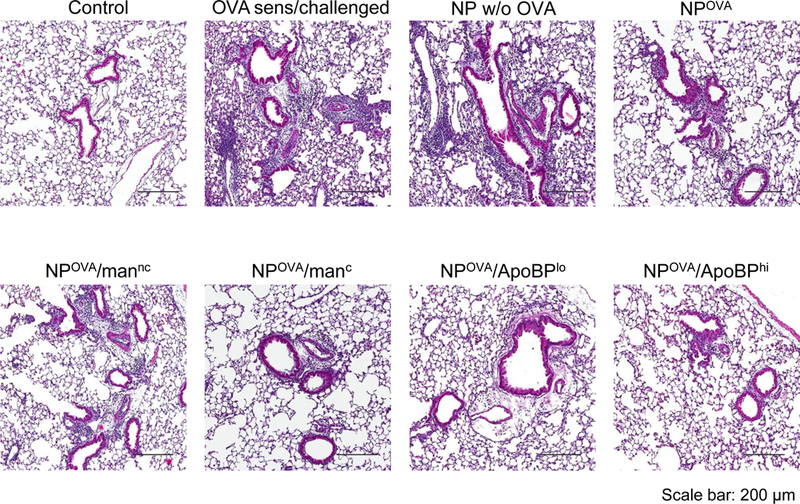
The assessment of BAL cell counts in the same experiment (Fig. 5A) demonstrated that the rise in eosinophil cell number was significantly suppressed by pretreatment with OVA-containing NPs, leading to almost total disappearance of the cell type from the BAL fluid (BALF) of animals prior treated with ApoBP-decorated NPs (Fig. 6A). Similar effects were seen for neutrophil influx, which was less abundant in the BALF. There was also a significant reduction in macrophage cell counts in animals treated with NPOVA/ApoBPhi, NPOVA/ApoBPlo and NPOVA/manc (Fig. 6A). The performance of lung histology and H&E staining demonstrated that, compared to the lack of inflammation in non-sensitized animals, the lungs of OVA-sensitized and challenged mice showed extensive mononuclear and eosinophilia inflammation with perivascular and peribronchial cuffing (Fig. 6B). While animals pretreated with NPs w/o OVA showed equally severe inflammation, mice receiving NPOVA, NPOVA/manC, and NPOVA/manNC showed a dramatic reduction in the eosinophilic pulmonary infiltrates. Strikingly, pretreatment with NPOVA/ApoBPlo and NPOVA/ApoBPhi could reduce the tissue inflammation to near-background levels.
Figure 6.
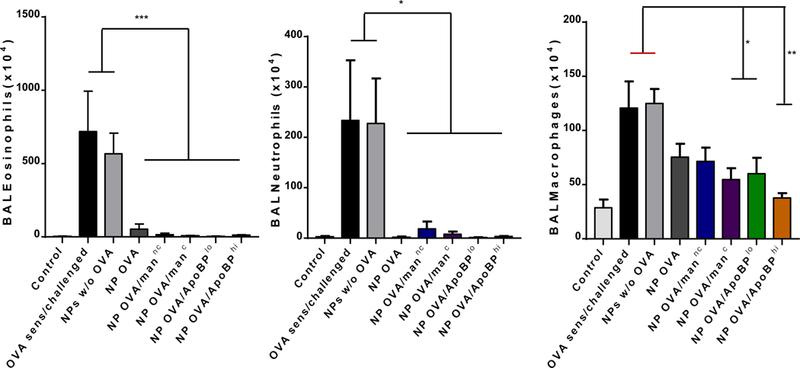
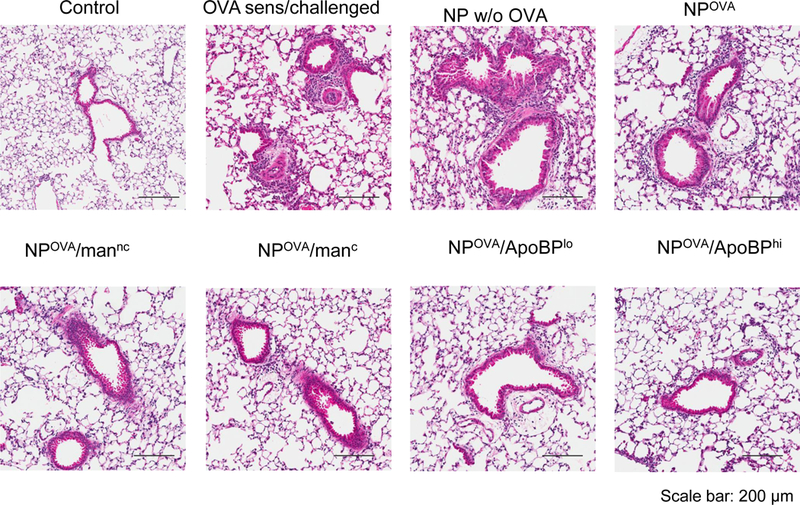
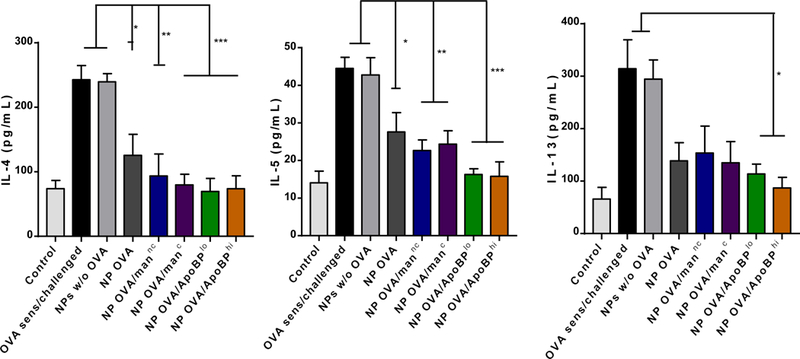
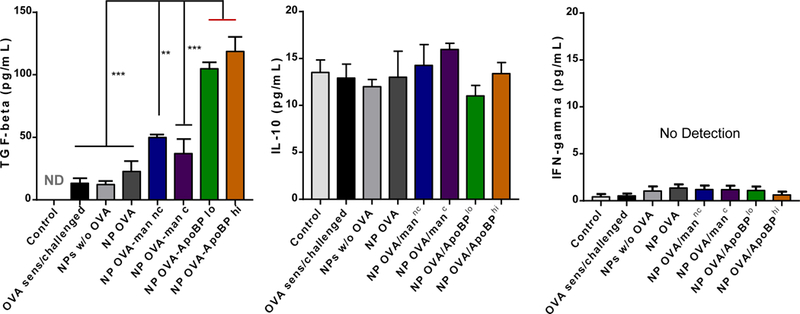
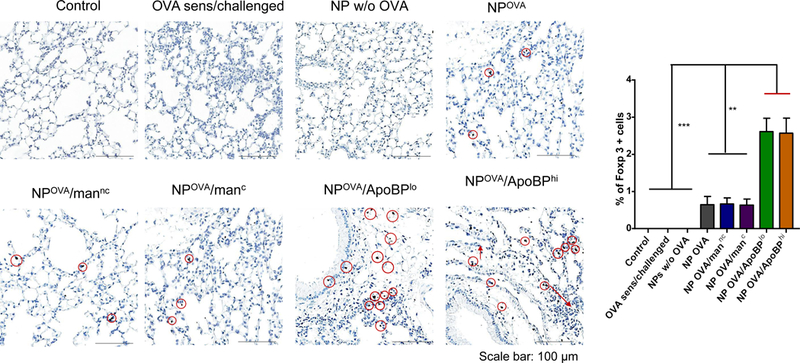
NP pretreatment alleviates allergic airway inflammation. Lung tissue and BALF from the experiment in Fig. 5A were used for the following analysis: (A) Differential eosinophil, neutrophil and macrophage cell counts on BALF. BAL was performed using 1 mL of PBS buffer for each animal, following which the BALF was cytospun onto slides for staining and counting. (B). Representative histological sections of lung tissue used for formalin fixing and H&E staining. The scale bars represent 200 μm. (C) TH2 cytokine (IL-4, IL-5, and IL-13) levels in the BALF, determined by ELISA; (D) TGF-β, IL-10 and INF-γ levels in the BALF, determined by ELISA. (E) Foxp3+ T-cell recruitment to the lung in response to pretreatment with tolerogenic NP. The lung tissues from the experiment in Fig. 5A were used for the IHC staining protocol to detect Foxp3+ T-cells. The scale bar represents 100 µm. Image-Pro Plus 6.0 software was used to detect cell nuclei and calculation of the percent-positive cells, under 10x magnification. A total of 12 independent fields were counted for each experimental group. The histogram on the right shows the % Foxp3+ T-cells in each group. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.00 (one-way ANOVA followed by a Tukey’s test).
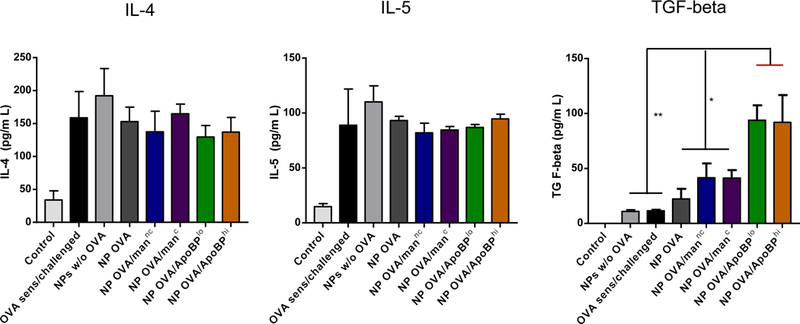
OVA challenge in prior sensitized animals (Fig. 5A), could also be seen to induce TH2 cytokine responses, as evidenced by increased IL-4, IL-5, and IL-13 levels in the BALF (Fig. 6C). Compared to the animals receiving no pretreatment, IL-4, IL-5, and IL-13 levels were significantly reduced by pretreatment with OVA-containing NPs. For NPOVA/ApoBPhi pretreated animals, this is achieved significant levels of p<0.001 for IL-4 and IL-5, while the corresponding level for IL-13 was p<0.05. No effect was seen in the animal group treated with NPs w/o OVA. In addition to TH2 cytokines, BALF was also used to assess TGF-β and IL-10 levels. This demonstrated a significant increase in TGF-β levels in animals pretreated with NPOVA/ApoBPlo or NPOVA/ApoBPhi compared to treatment with mannan-decorated particles or NPOVA (Fig. 6D). In contrast, there was no significant effect on IL-10 levels or the TH1 cytokine, IFN-γ (Fig. 6D).
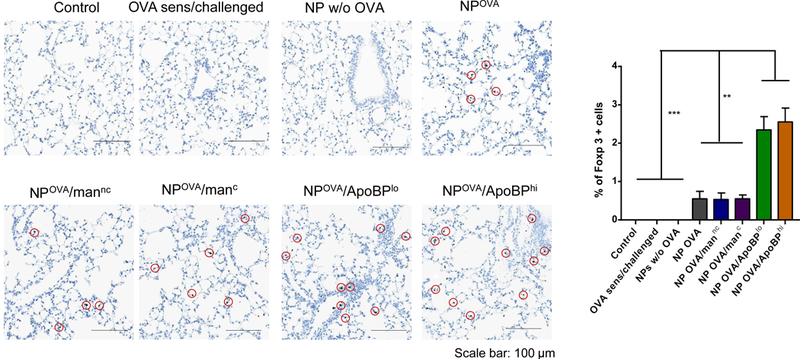
Based on the decline in allergic inflammation and an increase in TGF-β production in the lung, IHC staining was performed to assess Foxp3+ cell expression (Fig. 6E and Fig. S6A). While relatively sparse compared to the density of eosinophilic airway infiltrates, the presence of Foxp3+ cells in the lung could clearly be confirmed in animals exposed to OVA-containing NPs of all varieties (Fig. 6E and Fig. S6A). The increase was especially prominent in animals prior treated with ApoBP-coated NPs. Quantitative assessment of the number of positively stained cells, expressed as the % Foxp3+ T-cells for the total number of cells observed under 10X magnification, showed a statistically significant (p<0.01) increase in NPOVA/ApoBP pretreated animals compared to those receiving mannan-coated particles (Fig. 6E and Fig. S6A). These data are compatible with Treg generation in the liver, leading to suppression of allergic inflammation in the lung.
Post-sensitization NP treatment has a tolerogenic effect in allergic inflammation
To determine whether NPOVA could induce tolerogenic effects in already-sensitized animals, the particles were IV administered on days 14 and 21 to animals prior sensitized by OVA injection IP on days 0 and 7 (Fig. S5A). Inhalation OVA challenge was performed as before. While there was a trend towards decreased anti-OVA IgE and IgG1 levels in animals treated with ApoBP-coated NPs, the decline was not statistically significant (Fig. S5B). We did, however, observe decreased eosinophil numbers in the BALF in all animals exposed to NPOVA, without a differential effect of decorated vs. non-decorated particles (Fig. 7A). No significant effects were seen on neutrophil or macrophage cell numbers. Nonetheless, histological analysis revealed a noticeable reduction in airway inflammation and perivascular cuffing in all animal lungs receiving post-sensitization NPOVA administration (Fig. 7B). This was particularly obvious for animals treated with ApoBP-decorated NP and less so for mannan-coated particles (Fig. 7C). It could also be demonstrated that the reduction in the severity of airway inflammation showed excellent correlation with TGF-β levels in the BALF for animals treated with ApoBP-decorated NP, but less so for mannan-coated NPs (Fig. 7C). Interestingly, the reduction in airway inflammation was not accompanied by decreases in IL-4 and IL-5 production. Finally, performance of IHC staining to discern the appearance of Foxp3+ cells, demonstrated the appearance of Tregs for decorated NPs, particularly ApoBP-coated NPs (Fig. 7D and Fig. S6B).
Figure 7.

NP treatment post-sensitization alleviates allergic airway inflammation. The scheme is outlined in Fig. S5A. Briefly, 6–8-week-old C57/BL6 mice were IP sensitized with OVA (10 μg/mouse) on days 0 and 7. Subsequently, the animals received IV injection of NPOVA (with or without ligands) on two occasions days 14 and 21. The administered OVA and NP doses were similar as in Fig. 5. The post-treatment groups included: (1) a control group without any pretreatment or any sensitization or challenge; (2) no pretreatment before sensitization and challenge; (3) NPs w/o OVA; (4) NPOVA; (5) NPOVA/mannc; 6) NPOVA/manc; 7) NPOVA/ApoBPlo; 8) NPOVA/ApoBPhi. Finally, the animals received aerosolized OVA inhalation on days 35–37, as described in Fig. 5. Subsequent to animal sacrifice, BALF and lung tissue were harvested on day 40 to study the following endpoints: (A) Differential cell counts in the BALF. (B) Representative lung histology; scale bars correspond to 200 μm. (C) TH2 cytokine (IL-4 and IL-5) and TGF-β levels in BALF by ELISA. (D) IHC for Foxp3+ T-cell recruitment to the lung. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.00 (one-way ANOVA followed by a Tukey’s test).
A possible explanation for the difference of the tolerizing effect of the NPs on humoral vs. cell-mediated immunity is that IgE class switching in B-lymphocytes is more difficult to down regulate once induced by TH2 stimuli than the already-triggered allergic inflammatory T-cell responses with eosinophilic infiltrates in the lung (Figures 7A and B). The same finding has also been demonstrated by Smarr et al.48 While the exact explanation for this finding is unknown, it is well-known that it is easier to tolerize a primary immune response than an already induced immune response in the setting of autoimmune disease.20, 36 One possibility is that it is more difficult to turn off a clonally diverse immune responses that involve by epitope spreading compared to an epitope-restricted primary immune response.49
Discussion
In this study, we took advantage of the liver’s natural ability to generate systemic immune tolerance to alleviate allergic airway inflammation by LSEC-targeting nanoparticles. These carriers were produced by using biodegradable PLGA polymers to encapsulate OVA in 230–290 nm nanoparticles, which were decorated with surface ligands targeting mannose and scavenger receptors. We confirmed particle distribution to the liver, where the presence of mannan and ApoBP contributed to increased uptake by LSECs. APC processing of the encapsulated OVA induced the production of anti-inflammatory cytokines as well as of the tolerogenic growth factor, TGF-β. These effects were dramatically enhanced in vitro and in vivo by targeting ligands, allowing NPOVA to dramatically suppress allergen-induced allergic airway inflammation by tissue infiltrating Tregs. These findings demonstrate the potential utility of LSEC-targeting NPs for tolerogenic immunotherapy.
The major finding in the study is the use liver-targeting NPs, capable of inducing tolerogenic effects that suppress TH2-induced allergic airway disease in the lung. Although it is well-known that liver can induce immune tolerance, this knowledge has not been effectively translated into immunotherapy for allergic disorders in the clinic, including for respiratory allergies. While there is cumulative evidence indicating that oral administration of peanut allergen can induce immune tolerance to prevent a potentially fatal food allergic disorder in children,50 it still needs to be confirmed that the liver is involved. There are limited reports using a liver targeting approach to treat experimental allergic encephalomyelitis (EAE) in an animal model, where IV injection of small (~10 nm) non-targeted superparamagnetic iron oxide NPs (coated with a polymer) could be used to deliver myelin basic protein (MBP) peptides to the liver.20 Although the mechanism of biodistribution was uncertain, confocal microscopy confirmed decorated iron oxide NP co-localization with LSECs.20 Nonetheless, the iron oxide NPs were effective in preventing EAE when administered early after MBP exposure, also providing therapeutic benefit in animals with established disease.20 Treatment efficacy correlated with the induction of Tregs in the spleens of the NPMBP exposed mice.20 In contrast to the EAE study, we used larger particles comprised of an FDA-approved polymer, PLGA. These particles mainly distributed to the liver, where KC and LSEC uptake was enhanced by surface coating with mannan, while the attachment of a stabilin receptor ligand selectively promoted LSEC uptake (Fig. 4). We further demonstrated that OVA encapsulation and targeted delivery to LSECs was effective for reducing the allergic inflammation in the lung, accompanied by increased TGF-β production and the recruitment of Foxp3+ T-cells (Fig. 7). While a decrease of allergic inflammation in the lung in OVA-sensitized mice was previously demonstrated through the use of ~700 nm OVA-encapsulating PLGA NPs, there was no demonstration of particle biodistribution to the liver, use of a targeting ligand or the induction of Tregs.48 We propose that the combination of particle composition, smaller size and decorating the particle surface with ligands played a key role in targeting and endocytic uptake in LSECs, which sets our study apart from prior publications.
While there are a variety of cell types that can serve as APCs in the liver, LSECs are well-known for their capacity to induce immune tolerance.13, 51 Not only are LSECs the most abundant cell type in the liver, other than hepatocytes, but their flattened shape provides a large surface area, allowing them to engage in more frequent interactions with antigens and particulate matter than KC.46, 52 LSECs do not display a basal lamina and exhibits numerous fenestrae (~150 nm) that act as a selective ultrafiltration barrier, allowing the transition of molecules from the sinusoidal lumen to adjacent hepatocytes.15 Moreover, different from KCs, LSECs rarely phagocytose particles > 500 nm in size and are extremely active in clathrin-mediated uptake, and endocytic mechanism that is frequently used for soluble macromolecules, waste products, viruses and smaller particles (~200 nm).15, 53–55 This allows LSECs to play an assist role in the reticuloendothelial system, which differs from the effects of professional phagocytes such as KC, macrophages, and DC. One of the distinctive functions of LSECs is antigen uptake by scavenger receptors, which is further assisted by low-level expression of MHC-II and costimulatory molecules (e.g., CD40, CD80 and CD86).19 However, different from DC, LSECs do not participate in antigen presentation to CD4+ T-cells that control TH1 or TH17 immunity. Instead, LSECs present antigens to CD4+/Foxp3- non-Tregs, which are induced to transform into CD4+/Foxp3+ Tregs.14, 47 In the process, LSECs rely on TGF-β tethering to their surface membrane.18, 56 Moreover, LSECs also cross-present exogenous antigens to CD8+ T-cells for induction of CD8+ mediated tolerance.17 LSECs are also capable of producing anti-inflammatory cytokines (e.g., IL-10) that participate in tolerance induction.
In addition to constructing 200–300 nm PLGA nanoparticles for antigen delivery to LSECs, we show that surface coating with mannan and ApoBP is important for achieving their full potential of exerting tolerogenic effects in the liver. While the mannose receptor is expressed in several mammalian cell types such as LSECs, lymph node sinusoidal endothelial cells, or immature DC subpopulations, this receptor is not expressed in human KC and only present in low abundance on murine KC.15 This is compatible with the demonstration of increased OVA co-localization with KC in mannan-coated particles (Fig. S4). It is also worth commenting that KC contributes to tolerogenic effects in the liver, in addition to providing an exogenous source of TGF-β that can be used by LSECs.13, 57 Noteworthy, the ability to induce TGF-β production and Foxp3+ infiltration in the lung was significantly augmented by covalent attachment of ApoBP (Figs. 6D and 6E). ApoBP interacts with stabilin-1 and stabilin-2 receptors, which plays a role in clathrin-mediated uptake of oxidized LDL in the liver. 15, 58 Recent studies have shown that besides LSECs and KCs, other immunosuppressive cell types such as myeloid-derived suppressor cells (MDSCs) may be involved in suppressing allergic inflammation in asthma.59, 60 However, MDSCs have mostly been shown to be involved in inflammatory conditions of the liver or in liver fibrosis,61–63, a setting that likely differs from the animals with normal liver function used in this study.
Our study introduces a approach for employing tolerogenic nanoparticles to treat allergic disease, and the platform could also apply to the immunotherapy of autoimmune disease or transplant rejection. Another approach that holds great promise is the use of nanoparticles encapsulating pharmaceutical agents with an antigen, thereby locking APCs into a functional or metabolic state that favors antigen presentation to Tregs.21 For instance, the encapsulation of rapamycin by PLGA nanoparticles has shown to induce tolerization to porcine uricase (a highly immunogenic enzyme), leading to successful treatment of patients with disfiguring, tophaceous gout.64 These particles are currently being tested in phase 2 clinical trials. It is also possible to use nanoparticles for delivery of antigens to lymphocytes by targeting tolerogenic receptors or exhibiting surface coating with peptide-MHC class I complexes for antigen presentation to T-cells in the absence of costimulatory molecules.34, 35, 65 Attempts have also been made to induce oral tolerance through the use of PLGA particles that contain type II collagen for treatment of collagen-induced arthritis.26
Conclusion
In summary, we show that LSEC-targeting PLGA nanoparticles provide a safe and efficient approach for inducing antigen-specific immune tolerance in allergic airway disease. Proof-of-concept cellular studies demonstrated that antigen delivery to these cells induce OVA-specific tolerogenic effects in vitro. Moreover, animal studies confirmed that the particles are preferentially taken up by the liver, where surface coating with mannan and ApoB peptide enhances LSEC targeting, and are capable of inducing Tregs that suppress allergic inflammation in the lung. Not only could prophylactic administration of the OVA encapsulating particles prevent the generation of allergic airway inflammation, but it was also possible, through stabilin-targeting, to substantially suppress allergic information in already-sensitized animals. All considered, tolerogenic LSEC-targeting NPs could have far-reaching implications for treatment of allergic disease, food allergies, the drug reactions leading to anaphylaxis, and treatment of autoimmune disorders.
Materials and Methods
Reagents
The PLGA formulation obtained from Sigma (St. Louis, MO) has a lactide:glycolid molar ratio of 50:50, a viscosity of 0.45–0.60 dL/g, and includes a premixed content of ~5kDa PEG. Ovalbumin (OVA), dichloromethane, sodium cholate (used as a stabilizer in the outer water phase), mannan (mw 35–60 kDa), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), fluorescein isothiocyanate isomer I (FITC), N-hydroxysuccinimide (NHS), and N-(2-Aminoethyl) maleimide (NAEM) were purchased from Sigma. The Immortalized Mouse Hepatic Sinusoidal Endothelial Cells - SV40 (LSEC), Prigrow medium, and flasks for growing LSECs were purchased from Applied Biological Materials (Vancouver, BC, Canada). The mouse Kupffer cell line, KUP5, was purchased from RIKEN Cell Bank (Japan). The ATPlite luminescence assay kit was purchased from PerkinElmer (Santa Clara, CA). Hoechst 33342, DyLight 680 NHS-Ester, and the isolectin GS-IB4 were purchased from Thermo Fisher Scientific (Waltham, MA). The ApoB peptide (ApoBP), RLYRKRGLK, containing a GGC tag was synthesized by Biomatik (Cambridge, Ontario, Canada). ELISA kits for the measurement of murine TGF-β, IL-4, IL-10, IL-5, IL-13, TNF-α, IL-6, IFN-γ, and IL-1β were purchased from R&D (Minneapolis, MN). The horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody for detection of IgG2a (A-10685) and IgE (PA1–84764) were purchased from Invitrogen (Waltham, MA). The secondary antibody for detection of IgG1 (ab97240) was from Abcam (Cambridge, MA). The 3,3’,5,5’-tetramethylbenzidine (TMB) substrate kit was purchased from BD Biosciences (San Jose, CA).
Fabrication and characterization of the LSEC-targeting NPs
Pristine PLGA NPs were fabricated using a double-emulsion, w/o/w method, combined with solvent evaporation. 200 mg PLGA was dissolved in 10 mL dichloromethane. 30 mg of an OVA solution (1 mL) was added to the PLGA solution to form the primary emulsion (w/o), which was sonicated for 40 s, using a probe sonicator that delivers a power output of 60W and a 4/4 s on/off working pulse. The primary emulsion was poured into 60 mL 1% sodium cholate solution, and the mixture was sonicated for 2 min and added into 90 mL 0.5% sodium cholate solution. The double emulsion was stirred overnight to allow the evaporation of dichloromethane. The mixture was centrifuged and washed using DI water 5 times at 10000 g for 10 min to remove the non-encapsulated OVA, before suspension in DI water or PBS, as indicated.
Mannan attachment to the particle surface was achieved either through physical adsorption or covalent attachment. For physical adsorption, freeze-dried NPs (10 mg) were mixed with mannan (20 mg in 2 mL PBS, pH 5.0) and stirred overnight at room temperature. The NPs were collected and washed to remove excess mannan by centrifugation at 35000 g for 15 min. Conjugation chemistry was performed using the COOH-terminus of PLGA for covalent attachment of mannan. Briefly, mannan, EDC and sulfo-NHS were added to freeze-dried NPs [n(man): n(COOH): n(EDC) =0.4:1:10] and dispersed in PBS (2 mL, pH 5.0). The mixture was stirred overnight at room temperature. The NPs were collected and the excess mannan removed by centrifugation (35000 g, 15 min). The acquired NPs were designated as NPOVA/manC and NP OVA/mannc.
For peptide conjugation, ApoBP was conjugated to NPOVA by a two-step reaction, using a N-(2-aminoethyl) maleimide (NAEM) spacer. The COOH-terminal PLGA groups in the NPs were attached to the succinimidyl ester, using EDC (n(COOH): n(EDC) = 1:10) and NHS. NAEM was subsequently added (n(COOH): n(NAEM) =3:5), and the mixture stirred for 2h to decorate the particle surfaces with NAEM (NPs-NAEM). To remove the excess NAEM, NPOVA/NAEM was purified by spinning at 35,000 g for 15 min. The ApoBP solution, containing a cysteine tag at the N-terminal end, was added to the suspension and stirred for an additional 2 h. This allowed the maleimide group on NAEM to react with the cysteine sulfhydryl group to form a stable thioether bond. The final product was washed to remove excess reactants. Two doses of ApoBP were used (10 or 20 mg/mL) to generate NPs with different ligand density, which were designated as NPOVA/ApoBPlo and NPOVA/ApoBPhi.
The purified NPs were fully characterized using dynamic light scattering (DLS) to determine particle size and surface charge, while scanning electron microscopy (SEM) was performed to visualize particle morphology. The microBCA assay was used to detect OVA loading capacity and the conjugation efficiency of the peptide ligand, while the phenol-sulfuric acid method was used to determine mannan concentration. The endotoxin level was verified by a chromogenic LAL assay.
Cell culture
LSECs were grown in Prigrow medium, supplemented with 10% fetal bovine serum (FBS, Gemini, Sacramento, CA) and 100 U/mL-100 μg/mL of penicillin-streptomycin (Gibco, Waltham, MA). KUP5 cells were grown in high-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 250 μM 1-thioglycerol, 10 μg/mL bovine insulin, and 100 U/mL-100 μg/mL of penicillin-streptomycin.
Determination of NP cytotoxicity
Cytotoxicity assays were performed in LSECs and KUP5 cells, using the ATP assay. Following the exposure of the cells to NPs at different concentrations for 24 hr in a 96-well plate, the cell culture medium was replaced with an ATP solution. After centrifugation in a microplate centrifuge, 100 μL of each supernatant was removed to determine the luminescence intensity in a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
Quantification of NP associated OVA uptake by LSEC and Kupffer cells
To quantitatively evaluate antigen uptake into cells, OVA was labeled with FITC as previously described. Following the incorporation of FITC-OVA into NPs, LSECs and KUP5 cells were incubated with FITC-OVA or the labeled particles for 24 h. Cells were collected, washed with PBS and analyzed in a BD LSRII (IMED) analytic flow cytometer. Flowjo software (Ashland, OR) was used to quantify the percentage and mean fluorescence intensity of the FITC-OVA+ labeled cells.
Determination of NP-induced cytokine production by LSEC and Kupffer cells
LSEC and KUP5 cells were treated with NPs for 24 h. The culture medium supernatants were removed, and cytokines quantified by ELISA kits for the detection of mouse TGF-β, IL-4, IL-10, TNF-α, IL-6, and IL-1β, according to the manufacturer’s instructions.
Labeled PLGA nanoparticles biodistribution to the liver after IV injection
The near-infrared fluorescent dye, Dylight680 (Ex: 692, Em: 712), was used to label OVA, which was subsequently encapsulated in the NPs. Animals were IV injected with 25 mg/kg free OVA or 500 µg ligand-decorated or non-decorated NPOVA, containing the same amount of antigen (n=6). The mice were sacrificed after 24 h, followed by ex vivo IVIS imaging of organs (liver, lung, spleen, heart, and kidney) explanted in a Petri dish. The data were analyzed using Living Image software (PerkinElmer, version 4.5), and were expressed as the fluorescence intensity in the region of interest (ROI). Liver slices were soaked in OCT in stainless-steel molds, frozen and sent to UCLA Jonsson Comprehensive Cancer Center Translational Pathology Core Laboratory. Liver sections of 4 μm thickness were mounted on glass lives, which were fixed in pre-cooled acetone for 10 min. The slides were brought to room temperature, rinsed with PBS and incubated with a diluted solution of Hoechst 33342 and AlexaFluor 488 conjugated isolectin B4 in PBST (phosphate buffered saline containing 0.05% Tween-20) for 1 h. For KC staining, rat anti-mouse F4/80 antibody and AlexaFluor 594 conjugated goat anti-rat secondary antibodies were used for overnight or 2 h incubation, respectively. The slides were washed in tap water and PBS, dried and mounted in a Prolong gold antifade solution (Invitrogen, Waltham, MA), before the addition of coverslips. A Leica SP8-MD confocal microscope was used to visualize the slides. High magnification images were obtained under the 40 × objective lens.
Use of ligand-decorated or non-decorated NPOVA to induce tolerance in a murine allergic airway disease model
We used 6–8-week-old C57/BL6 mice to establish an OVA sensitization and challenge model that results in TH2 skewing of the immune response and generation of allergic airway disease. The basic sensitization protocol for the pretreatment approach with NPOVA involved intra-peritoneal (IP) administration of 0.5 mg/kg OVA on days 14 and 21, followed by aerosolized OVA inhalation (10 mg/mL) for 20 min on days 35 to 37. OVA nebulization was performed with a Schuco 2000 (Allied Health Care Products, St. Louis, MO), delivering OVA with a flow rate of 6 L/min at the nebulizer cup.66, 67 Animals were sacrificed on day 40, followed by collection of BALF (1 mM EDTA in PBS) and lung tissues for histology and immunohistochemistry. In order to assess the tolerogenic effects of NPOVA in the pretreatment protocol, mice received IV injection of NPOVA (OVA: 1.25 mg/kg, NPs: 25 mg/kg) on days 0 and 7. In order to determine the tolerogenic effects of the particles in a post-sensitization protocol, animals were sensitized by IP OVA on days 0 and 7, followed by IV particle administration on days 14 and 21. The animals were subsequently challenged by OVA inhalation as described above. Animal care was conducted according to the “Principles of Laboratory Animal Care” of the National Society for Medical Research (USA). The experimental protocol was approved by Division of Laboratory Animals Medicine (University of California, Los Angeles).
BAL cell counts, lung histology and quantification of cytokines
Total BALF cell counts were performed on cytospun samples, followed by fixing and staining with Hema3 solutions I and II (Fisher Healthcare, Waltham, MA). Differential cell counts were performed under a Fisherbrand microscope (Waltham, MA). Lungs were collected, fixed in formalin, embedded in paraffin, and stained with H&E. Slides were scanned by Aperio AT Turbo Digital Pathology Scanner (Leica Biosystems) at 10×magnification. Cell-free BALF was assayed for the release of IL-4, IL-5, IL-13, TGF-β, IL-10, and IFN-γ by ELISA (R&D), as previously described.
Quantification of OVA-specific antibody titers
Blood was collected from sacrificed animals and the serum fraction used to quantify OVA-specific antibodies by enzyme-linked immunosorbent assays (ELISA). Briefly, 96-well microplates were coated overnight with 2 μg of OVA per well in coating buffer (0.05 M CBS, pH 9.6) at 4°C. Plates were washed with PBST (0.01 M PBS containing 0.05 % [m/v] Tween 20, pH 7.4) and blocked by incubating with 1% (m/v) BSA (Gemini, West, Sacramento, CA) in PBST for 60 min at 37°C. After washing with PBST, 100 μL of serum diluted in PBST containing 0.1% [m/v] BSA were added to each well, and incubated for 30 min at 37°C. Plates were washed and incubated with 100 μL HRP-conjugated goat antibodies against either mouse IgE, IgG1, IgG2a (IgE diluted 1:500; IgG1 diluted 10000; IgG2a diluted 1:2000) for 30 min at 37°C. The plates were washed with PBST, and 100 μL TMB substrate was added to each well and incubated for 20 min at room temperature. The reaction was stopped by the addition of 50 μL of 2 M H2SO4 to each well, and the optical density (OD, 450 nm) read in a SpectraMax M5 microplate reader. Antibody titers were expressed as the highest sample dilution resulting in a duplication of the OD values over serum obtained from non-treated animals.
Hematoxylin-eosin (H&E) staining and immunohistochemistry (IHC) analysis
In order to visualize the presence of allergic airway inflammation, lungs were collected from sacrificed animals and fixed in 10% formalin followed by paraffin embedding. Sections of 4 μm thickness were mounted on glass slides by the UCLA Jonsson Comprehensive Cancer Center Translational Pathology Core Laboratory for H&E staining. We also performed IHC analysis to determine the appearance of Foxp3+ cells in the lung, using a standardized protocol.68 Briefly, the slides were deparaffinized, incubated in 3% methanol-hydrogen peroxide, followed by 10 mM EDTA (pH = 8) or 1 mM sodium citrate (pH = 6) at 95 °C using the Decloaking NxGen Chamber (Biocare Medical, DC2012). The slides were brought to room temperature, rinsed in PBST (containing 0.05% Tween-20) and then incubated with a ½00 dilution of the primary anti-Foxp3 antibody from eBioscience (Thermo Fisher, Waltham, MA) for 1 h. The slides were rinsed with PBST and incubated with the appropriate HRP-conjugated secondary antibody from Dako (Dako, K4003) for 30 min. After rinsing with PBST, the slides were incubated with DAB (3,3-diaminobenzidine) (Biocare Medical, FR805) for visualization. After washing in tap water, the slides were counterstained with Harris’ Hematoxylin, dehydrated in ethanol, and mounted in media, before being scanned by an Aperio AT Turbo Digital Pathology Scanner (Leica Biosystems) for interpretation by an experienced veterinary pathologist.
Statistical analysis
Statistical analysis was performed on GraphPad Prism 7 software (GraphPad Software, La Jolla, CA) using one-way ANOVA or the Student t test for determination of significance. The results were expressed as mean ± SEM of at least three independent experiments. Statistical significance thresholds were set at *p < 0.05; **p < 0.01; ***p < 0.001.
Supplementary Material
Acknowledgment
Research reported in this publication was supported, in part, by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number (U01ES027237). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank the CNSI Advanced Light Microscopy/Spectroscopy and Electron Imaging Center for NanoMachines Core Facilities, the Flow Cytometry Core Facility of Jonsson Comprehensive Cancer Center, and the Translational Pathology Core Laboratory (TPCL) Research Facility at UCLA.
Footnotes
Competing financial interests: Andre E. Nel and Huan Meng are co-founders and equity holders in Westwood Biosciences Inc and Nammi Therapeutics. The remaining authors declare no conflict of interest.
Supporting Information Available: Additional figures, table, and results as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wing K; Sakaguchi S, Regulatory T Cells Exert Checks and Balances on Self Tolerance and Autoimmunity. Nat. Immunol 2010, 11, 7–13. [DOI] [PubMed] [Google Scholar]
- 2.Sharabi A; Tsokos MG; Ding Y; Malek TR; Klatzmann D; Tsokos GC, Regulatory T Cells in the Treatment of Disease. Nat. Rev. Drug Discovery 2018, 17, 823–844. [DOI] [PubMed] [Google Scholar]
- 3.Smith DM; Simon JK; Baker JR Jr, Applications of Nanotechnology for Immunology. Nat. Rev. Immunol 2013, 13, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Getts DR; Shea LD; Miller SD; King NJC, Harnessing Nanoparticles for Immune Modulation. Trends Immunol 2015, 36, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes AC; Mohsen M; Bachmann MF, Harnessing Nanoparticles for Immunomodulation and Vaccines. Vaccines 2017, 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irvine DJ; Hanson MC; Rakhra K; Tokatlian T, Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev 2015, 115, 11109–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irvine DJ; Swartz MA; Szeto GL, Engineering Synthetic Vaccines Using Cues from Natural Immunity. Nat. Mater 2013, 12, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Racanelli V; Rehermann B, The Liver as an Immunological Organ. Hepatology 2006, 43, S54–S62. [DOI] [PubMed] [Google Scholar]
- 9.Madariaga MLL; Kreisel D; Madsen JC, Organ-Specific Differences in Achieving Tolerance. Curr. Opin. Organ Transplant 2015, 20, 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beal EW; Mumtaz K; Hayes D; Whitson BA; Black SM, Combined Heart–Liver Transplantation: Indications, Outcomes and Current Experience. Transplant. Rev 2016, 30, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puri V; Eason J, Simultaneous Liver—Kidney Transplantation. Current Transplantation Reports 2015, 2, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lüth S; Huber S; Schramm C; Buch T; Zander S; Stadelmann C; Brück W; Wraith DC; Herkel J; Lohse AW, Ectopic Expression of Neural Autoantigen in Mouse Liver Suppresses Experimental Autoimmune Neuroinflammation by Inducing Antigen-Specific Tregs. J. Clin. Invest 2008, 118, 3403–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomson AW; Knolle PA, Antigen-Presenting Cell Function in the Tolerogenic Liver Environment. Nat. Rev. Immunol 2010, 10, 753–766. [DOI] [PubMed] [Google Scholar]
- 14.Crispe IN, Liver Antigen-Presenting Cells. J. Hepatol 2011, 54, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sørensen KK; McCourt P; Berg T; Crossley C; Couteur DL; Wake K; Smedsrød B, The Scavenger Endothelial Cell: A New Player in Homeostasis and Immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol 2012, 303, R1217–R1230. [DOI] [PubMed] [Google Scholar]
- 16.Desjardins M; Griffiths G, Phagocytosis: Latex Leads the Way. Curr. Opin. Cell Biol 2003, 15, 498–503. [DOI] [PubMed] [Google Scholar]
- 17.Limmer A; Ohl J; Kurts C; Ljunggren H-G; Reiss Y; Groettrup M; Momburg F; Arnold B; Knolle PA, Efficient Presentation of Exogenous Antigen by Liver Endothelial Cells to Cd8+ T Cells Results in Antigen-Specific T-Cell Tolerance. Nat. Med 2000, 6, 1348–1354. [DOI] [PubMed] [Google Scholar]
- 18.Carambia A; Freund B; Schwinge D; Heine M; Laschtowitz A; Huber S; Wraith DC; Korn T; Schramm C; Lohse AW; Heeren J; Herkel J, Tgf-Β-Dependent Induction of Cd4+Cd25+Foxp3+ Tregs by Liver Sinusoidal Endothelial Cells. J. Hepatol 2014, 61, 594–599. [DOI] [PubMed] [Google Scholar]
- 19.Tiegs G; Lohse AW, Immune Tolerance: What Is Unique About the Liver. J. Autoimmun 2010, 34, 1–6. [DOI] [PubMed] [Google Scholar]
- 20.Carambia A; Freund B; Schwinge D; Bruns OT; Salmen SC; Ittrich H; Reimer R; Heine M; Huber S; Waurisch C; Eychmüller A; Wraith DC; Korn T; Nielsen P; Weller H; Schramm C; Lüth S; Lohse AW; Heeren J; Herkel J, Nanoparticle-Based Autoantigen Delivery to Treg-Inducing Liver Sinusoidal Endothelial Cells Enables Control of Autoimmunity in Mice. J. Hepatol 2015, 62, 1349–1356. [DOI] [PubMed] [Google Scholar]
- 21.Kishimoto TK; Maldonado RA, Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front. Immunol 2018, 9, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams AB; Ford ML; Larsen CP, Costimulation Blockade in Autoimmunity and Transplantation: The CD28 Pathway. J. Immunol 2016, 197, 2045–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esensten Jonathan H.; Helou Ynes A.; Chopra G; Weiss A; Bluestone Jeffrey A., CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clemente-Casares X; Blanco J; Ambalavanan P; Yamanouchi J; Singha S; Fandos C; Tsai S; Wang J; Garabatos N; Izquierdo C; Agrawal S; Keough MB; Yong VW; James E; Moore A; Yang Y; Stratmann T; Serra P; Santamaria P, Expanding Antigen-Specific Regulatory Networks to Treat Autoimmunity. Nature 2016, 530, 434. [DOI] [PubMed] [Google Scholar]
- 25.Tsai S; Shameli A; Yamanouchi J; Clemente-Casares X; Wang J; Serra P; Yang Y; Medarova Z; Moore A; Santamaria P, Reversal of Autoimmunity by Boosting Memory-Like Autoregulatory T Cells. Immunity 2010, 32, 568–580. [DOI] [PubMed] [Google Scholar]
- 26.Kim W-U; Lee W-K; Ryoo J-W; Kim S-H; Kim J; Youn J; Min S-Y; Bae E-Y; Hwang S-Y; Park S-H; Cho C-S; Park J-S; Kim H-Y, Suppression of Collagen-Induced Arthritis by Single Administration of Poly(Lactic-Co-Glycolic Acid) Nanoparticles Entrapping Type II Collagen: A Novel Treatment Strategy for Induction of Oral Tolerance. Arthritis Rheum 2002, 46, 1109–1120. [DOI] [PubMed] [Google Scholar]
- 27.Dhadwar SS; Kiernan J; Wen J; Hortelano G, Repeated Oral Administration of Chitosan/DNA Nanoparticles Delivers Functional FvIII with the Absence of Antibodies in Hemophilia a Mice. J. Thromb. Haemost 2010, 8, 2743–2750. [DOI] [PubMed] [Google Scholar]
- 28.Goldmann K; Ensminger SM; Spriewald BM, Oral Gene Application Using Chitosan-DNA Nanoparticles Induces Transferable Tolerance. Clin. Vaccine Immunol 2012, 19, 1758–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramani K; Miclea RD; Purohit VS; Mager DE; Straubinger RM; Balu-Iyer SV, Phosphatidylserine Containing Liposomes Reduce Immunogenicity of Recombinant Human Factor Viii (Rfviii) in a Murine Model of Hemophilia a**Karthik Ramani and Razvan D. Miclea Contributed Equally to the Manuscript. J. Pharm. Sci 2008, 97, 1386–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birge RB; Boeltz S; Kumar S; Carlson J; Wanderley J; Calianese D; Barcinski M; Brekken RA; Huang X; Hutchins JT; Freimark B; Empig C; Mercer J; Schroit AJ; Schett G; Herrmann M, Phosphatidylserine Is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Differ 2016, 23, 962–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearson RM; Casey LM; Hughes KR; Miller SD; Shea LD, In Vivo Reprogramming of Immune Cells: Technologies for Induction of Antigen-Specific Tolerance. Adv. Drug Delivery Rev 2017, 114, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearson RM; Casey LM; Hughes KR; Wang LZ; North MG; Getts DR; Miller SD; Shea LD, Controlled Delivery of Single or Multiple Antigens in Tolerogenic Nanoparticles Using Peptide-Polymer Bioconjugates. Mol. Ther 2017, 25, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kontos S; Kourtis IC; Dane KY; Hubbell JA, Engineering Antigens for in Situ Erythrocyte Binding Induces T-Cell Deletion. Proc. Natl. Acad. Sci. U. S. A 2013, 110, E60–E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duong BH; Tian H; Ota T; Completo G; Han S; Vela JL; Ota M; Kubitz M; Bovin N; Paulson JC; Nemazee D, Decoration of T-Independent Antigen with Ligands for Cd22 and Siglec-G Can Suppress Immunity and Induce B Cell Tolerance in Vivo. J. Exp. Med 2010, 207, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macauley MS; Pfrengle F; Rademacher C; Nycholat CM; Gale AJ; von Drygalski A; Paulson JC, Antigenic Liposomes Displaying CD22 Ligands Induce Antigen-Specific B Cell Apoptosis. J. Clin. Invest 2013, 123, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maldonado RA; LaMothe RA; Ferrari JD; Zhang A-H; Rossi RJ; Kolte PN; Griset AP; O’Neil C; Altreuter DH; Browning E; Johnston L; Farokhzad OC; Langer R; Scott DW; von Andrian UH; Kishimoto TK, Polymeric Synthetic Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E156–E165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tostanoski Lisa H.; Chiu Y-C; Gammon Joshua M.; Simon T; Andorko James I.; Bromberg Jonathan S.; Jewell Christopher M. , Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance That Is Systemic and Antigen Specific. Cell Rep 2016, 16, 2940–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaMothe RA; Kolte PN; Vo T; Ferrari JD; Gelsinger TC; Wong J; Chan VT; Ahmed S; Srinivasan A; Deitemeyer P; Maldonado RA; Kishimoto TK, Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Front. Immunol 2018, 9, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson JM; Shive MS, Biodegradation and Biocompatibility of PLA and PLGA Microspheres. Adv. Drug Delivery Rev 1997, 28, 5–24. [DOI] [PubMed] [Google Scholar]
- 40.Kumari A; Yadav SK; Yadav SC, Biodegradable Polymeric Nanoparticles Based Drug Delivery Systems. Colloids Surf., B 2010, 75, 1–18. [DOI] [PubMed] [Google Scholar]
- 41.Soppimath KS; Aminabhavi TM; Kulkarni AR; Rudzinski WE, Biodegradable Polymeric Nanoparticles as Drug Delivery Devices. J. Controlled Release 2001, 70, 1–20. [DOI] [PubMed] [Google Scholar]
- 42.Li R; Oteiza A; Sørensen KK; McCourt P; Olsen R; Smedsrød B; Svistounov D, Role of Liver Sinusoidal Endothelial Cells and Stabilins in Elimination of Oxidized Low-Density Lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G71–G81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Berkel TJ; De Rijke YB; Kruijt JK, Different Fate in Vivo of Oxidatively Modified Low Density Lipoprotein and Acetylated Low Density Lipoprotein in Rats. Recognition by Various Scavenger Receptors on Kupffer and Endothelial Liver Cells. J. Biol. Chem 1991, 266, 2282–2289. [PubMed] [Google Scholar]
- 44.Hirose M; Nishikawa M; Qian W; Haque A; Mashimo M; Inoue M, Mannose-Conjugated Alendronate Selectively Depletes Kupffer Cells and Inhibits Endotoxemic Shock in the Mice. Hepatol. Res 2006, 36, 3–10. [DOI] [PubMed] [Google Scholar]
- 45.Yamane S; Iwasaki N; Majima T; Funakoshi T; Masuko T; Harada K; Minami A; Monde K; Nishimura S.-i., Feasibility of Chitosan-Based Hyaluronic Acid Hybrid Biomaterial for a Novel Scaffold in Cartilage Tissue Engineering. Biomaterials 2005, 26, 611–619. [DOI] [PubMed] [Google Scholar]
- 46.Crispe IN; Giannandrea M; Klein I; John B; Sampson B; Wuensch S, Cellular and Molecular Mechanisms of Liver Tolerance. Immunol. Rev 2006, 213, 101–118. [DOI] [PubMed] [Google Scholar]
- 47.Crispe IN, Hepatic T Cells and Liver Tolerance. Nat. Rev. Immunol 2003, 3, 51–62. [DOI] [PubMed] [Google Scholar]
- 48.Smarr CB; Yap WT; Neef TP; Pearson RM; Hunter ZN; Ifergan I; Getts DR; Bryce PJ; Shea LD; Miller SD, Biodegradable Antigen-Associated PLG Nanoparticles Tolerize Th2-Mediated Allergic Airway Inflammation Pre- and Postsensitization. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanderlugt CL; Miller SD, Epitope Spreading in Immune-Mediated Diseases: Implications for Immunotherapy. Nat. Rev. Immunol 2002, 2, 85–95. [DOI] [PubMed] [Google Scholar]
- 50.Hofmann AM; Scurlock AM; Jones SM; Palmer KP; Lokhnygina Y; Steele PH; Kamilaris J; Burks AW, Safety of a Peanut Oral Immunotherapy Protocol in Children with Peanut Allergy. J. Allergy Clin. Immunol 2009, 124, 286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klugewitz K; Blumenthal-Barby F; Schrage A; Knolle PA; Hamann A; Crispe IN, Immunomodulatory Effects of the Liver: Deletion of Activated Cd4+Effector Cells and Suppression of Ifn-Γ-Producing Cells after Intravenous Protein Immunization. J. Immunol 2002, 169, 2407–2413. [DOI] [PubMed] [Google Scholar]
- 52.Elvevold K; Smedsrød B; Martinez I, The Liver Sinusoidal Endothelial Cell: A Cell Type of Controversial and Confusing Identity. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 294, G391–G400. [DOI] [PubMed] [Google Scholar]
- 53.Falkowska-Hansen B; Falkowski M; Metharom P; Krunic D; Goerdt S, Clathrin-Coated Vesicles Form a Unique Net-Like Structure in Liver Sinusoidal Endothelial Cells by Assembling Along Undisrupted Microtubules. Exp. Cell Res 2007, 313, 1745–1757. [DOI] [PubMed] [Google Scholar]
- 54.Kjeken R; Mousavi SA; Brech A; Gjøen T; Berg T, Fluid Phase Endocytosis of [125I]Iodixanol in Rat Liver Parenchymal, Endothelial and Kupffer Cells. Cell Tissue Res 2001, 304, 221–230. [DOI] [PubMed] [Google Scholar]
- 55.Hansen B; Longati P; Elvevold K; Nedredal G-I; Schledzewski K; Olsen R; Falkowski M; Kzhyshkowska J; Carlsson F; Johansson S; Smedsrød B; Goerdt S; Johansson S; McCourt P, Stabilin-1 and Stabilin-2 Are Both Directed into the Early Endocytic Pathway in Hepatic Sinusoidal Endothelium Via Interactions with Clathrin/Ap-2, Independent of Ligand Binding. Exp. Cell Res 2005, 303, 160–173. [DOI] [PubMed] [Google Scholar]
- 56.Andersson J; Tran DQ; Pesu M; Davidson TS; Ramsey H; O’Shea JJ; Shevach EM, CD4+Foxp3+Regulatory T Cells Confer Infectious Tolerance in a TGF-β–Dependent Manner. J. Exp. Med 2008, 205, 1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bissell DM; Wang SS; Jarnagin WR; Roll FJ, Cell-Specific Expression of Transforming Growth Factor-Beta in Rat Liver. Evidence for Autocrine Regulation of Hepatocyte Proliferation. J. Clin. Invest 1995, 96, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akhter A; Hayashi Y; Sakurai Y; Ohga N; Hida K; Harashima H, Ligand Density at the Surface of a Nanoparticle and Different Uptake Mechanism: Two Important Factors for Successful Sirna Delivery to Liver Endothelial Cells. Int. J. Pharm 2014, 475, 227–237. [DOI] [PubMed] [Google Scholar]
- 59.Shi M; Shi G; Tang J; Kong D; Bao Y; Xiao B; Zuo C; Wang T; Wang Q; Shen Y; Wang H; Funk CD; Zhou J; Yu Y, Myeloid-Derived Suppressor Cell Function Is Diminished in Aspirin-Triggered Allergic Airway Hyperresponsiveness In Mice. J. Allergy Clin. Immunol 2014, 134, 1163–1174. [DOI] [PubMed] [Google Scholar]
- 60.Hirose K; Iwata A; Tamachi T; Nakajima H, Allergic Airway Inflammation: Key Players Beyond the Th2 Cell Pathway. Immunol. Rev 2017, 278, 145–161. [DOI] [PubMed] [Google Scholar]
- 61.Cripps JG; Gorham JD, Mdsc in Autoimmunity. Int. Immunopharmacol 2011, 11, 789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hammerich L; Tacke F, Emerging Roles of Myeloid Derived Suppressor Cells in Hepatic Inflammation and Fibrosis. World J. Gastrointest. Pathophysiol 2015, 6, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Höchst B; Mikulec J; Baccega T; Metzger C; Welz M; Peusquens J; Tacke F; Knolle P; Kurts C; Diehl L; Ludwig-Portugall I, Differential Induction of Ly6G and Ly6C Positive Myeloid Derived Suppressor Cells in Chronic Kidney and Liver Inflammation and Fibrosis. PLoS One 2015, 10, e0119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kishimoto TK; Ferrari JD; LaMothe RA; Kolte PN; Griset AP; O’Neil C; Chan V; Browning E; Chalishazar A; Kuhlman W; Fu F.-n.; Viseux N; Altreuter DH; Johnston L; Maldonado RA, Improving the Efficacy and Safety of Biologic Drugs with Tolerogenic Nanoparticles. Nat. Nanotechnol 2016, 11, 890–899. [DOI] [PubMed] [Google Scholar]
- 65.Shen C; He Y; Cheng K; Zhang D; Miao S; Zhang A; Meng F; Miao F; Zhang J, Killer Artificial Antigen-Presenting Cells Deplete Alloantigen-Specific T Cells in a Murine Model of Alloskin Transplantation. Immunol. Lett 2011, 138, 144–155. [DOI] [PubMed] [Google Scholar]
- 66.Whitekus MJ; Li N; Zhang M; Wang M; Horwitz MA; Nelson SK; Horwitz LD; Brechun N; Diaz-Sanchez D; Nel AE, Thiol Antioxidants Inhibit the Adjuvant Effects of Aerosolized Diesel Exhaust Particles in a Murine Model for Ovalbumin Sensitization. J. Immunol 2002, 168, 2560–2567. [DOI] [PubMed] [Google Scholar]
- 67.Hao M; Comier S; Wang M; Lee JJ; Nel A, Diesel Exhaust Particles Exert Acute Effects on Airway Inflammation and Function in Murine Allergen Provocation Models. J. Allergy Clin. Immunol 2003, 112, 905–914. [DOI] [PubMed] [Google Scholar]
- 68.Lu J; Liu X; Liao Y-P; Salazar F; Sun B; Jiang W; Chang CH; Jiang J; Wang X; Wu AM; Meng H; Nel AE, Nano-Enabled Pancreas Cancer Immunotherapy Using Immunogenic Cell Death and Reversing Immunosuppression. Nat. Commun 2017, 8, 1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.