

Snail is a target of USP1 that links platinum response to metastasis in ovarian cancer.
Abstract
Resistance to platinum-based chemotherapy is a common event in patients with cancer, generally associated with tumor dissemination and metastasis. Whether platinum treatment per se activates molecular pathways linked to tumor spreading is not known. Here, we report that the ubiquitin-specific protease 1 (USP1) mediates ovarian cancer cell resistance to platinum, by regulating the stability of Snail, which, in turn, promotes tumor dissemination. At the molecular level, we observed that upon platinum treatment, USP1 is phosphorylated by ATM and ATR and binds to Snail. Then, USP1 de-ubiquitinates and stabilizes Snail expression, conferring resistance to platinum, increased stem cell–like features, and metastatic ability. Consistently, knockout or pharmacological inhibition of USP1 increased platinum sensitivity and decreased metastatic dissemination in a Snail-dependent manner. Our findings identify Snail as a USP1 target and open the way to a novel strategy to overcome platinum resistance and more successfully treat patients with ovarian cancer.
INTRODUCTION
Platinum compounds, including cisplatin (CDDP), carboplatin (CBDCA), and oxaliplatin, are frontline anticancer therapies and constitute part of the treatment regimen for several oncological patients with different types of solid tumors (1). Although most of the patients initially respond very well to these treatments, development of platinum-resistant tumor recurrences unfortunately represents a very frequent event (1, 2). Epithelial ovarian cancer (OC), the fourth leading cause of cancer death in women, is the prototype of cancer in which platinum resistance is associated with very poor prognosis (3). In OC, platinum-resistant recurrences grow as metastatic disease, mainly invading the peritoneum, the omentum, and the organs located in the peritoneal cavity (4).
Both chemoresistance and tumor spreading have been linked to the epithelial-to-mesenchymal transition (EMT), a complex molecular and biological program by which cancer cells acquire aggressive features, including cell plasticity, invasiveness, and resistance to apoptosis (5). Whether and how this phenotype is linked to an alteration of the DNA damage response (DDR) activated by chemotherapy are still unclear, and possible molecular mediators connecting EMT and DDR are still unknown.
Among the EMT inducers and regulators of cell plasticity, the zinc finger transcription factor Snail can contribute to platinum resistance in OC by sustaining cell survival and the acquisition of stem cell–like characteristics (6, 7). Snail is a short-living protein that is rapidly polyubiquitinated and degraded by the proteasome through the activity of different E3 ubiquitin ligases (8, 9).
The ubiquitin-specific protease 1 (USP1) is a de-ubiquitinase (DUB) known to regulate the mono-ubiquitination of FANCD2/FANCI and PCNA in response to DNA damage (10, 11), and its activity is critical for the establishment of a proficient DDR (12, 13). Moreover, it has been proposed that USP1 regulates the EMT process and the stem cell maintenance by controlling the ubiquitination levels of the inhibitor of DNA binding (ID) proteins (14). Here, we demonstrate that, following platinum treatment of OC cells, USP1 is able to de-ubiquitinate and stabilize Snail, eventually resulting in platinum resistance and metastatic dissemination of OC cells.
Our work highlights a novel function of USP1, acting as a key factor that links platinum response to tumor spreading through the stabilization of Snail protein. These results uncover the intriguing possibility of combining platinum treatment with the inhibition of USP1 to overcome platinum resistance and to prevent metastasis formation in many patients with cancer.
RESULTS
USP1 is a key mediator of platinum response and stem cell–like features in OC cells
To identify novel mediators of platinum response in OC cells, we used a short hairpin RNA (shRNA)–based loss-of-function screening to target 680 genes belonging to the apoptosis and DDR pathways (fig. S1A). Using two different OC cell lines transduced with the shRNA library and treated or not with suboptimal doses of CBDCA (~20% reduction in cell viability), we identified 50 candidate genes whose loss increased CBDCA activity in at least one cell line. A validation screening was then carried out in four different OC cell lines, using five specific shRNAs per gene. By this approach, we confirmed five genes and selected USP1 for further studies, since it optimally satisfied prespecified conditions, i.e., to increase CBDCA activity in at least three cell lines when silenced with at least three different shRNAs (fig. S1B). Further validation experiments demonstrated that USP1 silencing decreased CBDCA half-maximal inhibitory concentration (IC50) in MDAH-2774 cells by about threefold (i.e., 90 μg/ml in control and 41 and 37 μg/ml in shUSP1#2 and #3, respectively). Consistent results were obtained using CDDP (Fig. 1, A and B), which is more active than CBDCA in in vitro experiments.
Fig. 1. USP1 is a key mediator of platinum sensitivity in OC cells.
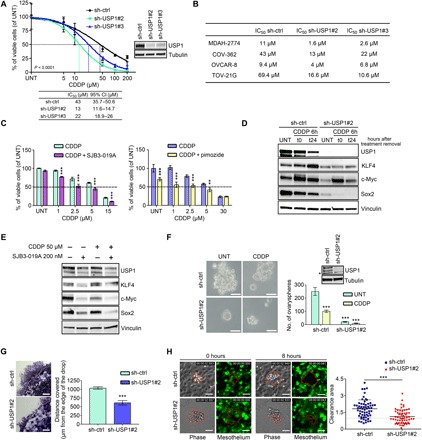
(A) Nonlinear regression analyses of cell viability assay in COV-362 cells transduced with control shRNA (sh-ctrl) or USP1 shRNAs and treated with increasing doses of CDDP for 16 hours. Data are expressed as percentage of viable cells with respect to the untreated (UNT) cells and represent the mean (±SD) of three biological replicates. The table shows the IC50 and the confidence interval (CI) of each condition. Inset: Western blot (WB) analysis reporting USP1 expression in the used cells. (B) Table showing the CDDP IC50 of the indicated OC cells calculated by transducing and treating the cells as described in (A). (C) Graph reporting MDAH-2774 cell viability after treatment with USP1 inhibitors SJB3-019A (left graph) and pimozide (right graph) in combination or not with increasing doses of CDDP as indicated. (A and C) Data report the percentage of viable cells with respect to the untreated cells and represent the mean (±SD) of three independent experiments. (D) Western blot analysis of the indicated proteins in COV-362 stably silenced for USP1 and treated or not with CDDP for 6 hours (t0) and allowed to repair for 24 hours (t24), as indicated. (E) Western blot analysis of the indicated proteins in MDAH-2774 cells treated with SJB3-019A in combination or not with CDDP for 6 hours. Lysates were harvested 24 hours after treatment. (F) Representative phase-contrast images of ovaryspheres formed by COV-362 cells described in (A) and treated or not with CDDP for 3 hours. Scale bars, 50 μm. On the right, the graph reports the quantification of ovarysphere formation and represents the mean (±SD) of three independent experiments. Inset: Western blot analysis reporting USP1 expression in the used cells. The asterisk indicates a nonspecific band. (G) Matrigel evasion assay of COV-362 cells described in (A). The graph on the right reports the distance covered by the individual cells from the edge of the drop (mean ± SD of three independent experiments). Scale bars, 500 μm. (H) Typical images of mesothelial cell clearance assay. The graph on the right shows the cleared area of mesothelial cell monolayers after coculture for 8 hours with COV-362 spheroids, described in (A) (mean ± SD of three independent experiments). In the figure, vinculin or tubulin was used as a loading control. In (A), Fisher’s exact test was used to calculate the global P value reported in the graph. Otherwise, statistical significance was determined by a two-tailed, unpaired Student’s t test (**P < 0.01, ***P < 0.001).
USP1 was expressed at a similar level in a panel of OC cell lines and only slightly less in normal epithelial OC cells (fig. S1C). We silenced USP1 expression using two different shRNAs in four different OC cell lines, chosen to encompass the three most common OC histotypes (serous, OVCAR-8; endometrioid, MDAH-2774 and COV-362; clear cell, TOV-21G). Upon CDDP treatment, we confirmed that USP1 silencing significantly reduced CDDP IC50 in all tested cell lines (Fig. 1, A and B). Accordingly, treatment with USP1 inhibitors SJB3-019A and pimozide enhanced the sensitivity of OC cells to CDDP (Fig. 1C and fig. S1, D and E). These data were consistent with the known role of USP1 in the regulation of the DDR pathway via regulation of FANCD2 mono-ubiquitination (11). However, in our model, USP1 silencing reduced viability but did not increase FANCD2 (or FANCD2-L) mono-ubiquitination in TOV-21G cells, which are reported to be FANCD2-L defective (15) and had only minor effects on FANCD2 mono-ubiquitination in OVCAR-8 cells (fig. S1F), therefore suggesting that USP1 could also have FANCD2-independent function(s) in regulating cell survival following CDDP treatment.
In addition to its implication in the DDR pathway, recent reports suggest a role for USP1 in EMT and stem cell state regulation (14, 16, 17). Accordingly, we observed that USP1 genetic and pharmacological inactivation decreased basal and CDDP-induced expression of genes associated with cancer stem cell–like phenotype, including KLF4, Sox2, and c-Myc (Fig. 1, D and E, and fig. S1G). Furthermore, spheroid formation assay, commonly used to evaluate cancer stem cell–like features, showed that USP1-silenced cells formed less spheroids than control cells, under both untreated and CDDP-treated conditions, supporting a role for USP1 in the acquisition of a stem cell–like phenotype (Fig. 1F). It has been proposed that the metastatic dissemination of OC in the peritoneal cavity is driven by the formation of tumor cell spheroids that detach from the primary tumor, survive in suspension, and then adhere and grow on the mesothelial cell layer of the peritoneum (18). Thus, we tested control and USP1-silenced cells in a three-dimensional (3D) Matrigel evasion assay (19) and in an in vitro mesothelial cell clearance assay (20) to model the initial steps of the OC metastatic process and evaluate USP1 role in this context. USP1-silenced cells displayed decreased ability to evade from the 3D matrix and also to attach, digest, and invade the mesothelial cell monolayer (Fig. 1, G and H). Together, these data support a role for USP1 in the regulation of CDDP sensitivity and in the control of stem cell–like and invasive features of OC cells.
USP1 is necessary for Snail protein up-regulation in CDDP-treated cells
Colonization of the mesothelium by OC cells is promoted by EMT transcription factors Zeb1, Snail, and Twist-1 (21). Several evidences indicate that these transcription factors are also activated by platinum treatment (22) and could control chemoresistance (23). Accordingly, we observed a substantial up-regulation of Snail, Slug, and Twist-1 (but not Zeb1) expression in CDDP-treated OC cells, but only Snail was consistently up-regulated by CDDP in all tested models (Fig. 2A and fig. S2A). USP1 silencing reduced both basal and CDDP-induced Snail expression while having no effect on Twist-1, Slug, and Zeb1 (Fig. 2B and fig. S2B). Similar effects were observed using USP1 inhibitors SJB3-019A and pimozide, either alone or after CDDP treatment, confirming that the reduction of Snail expression was a direct consequence of reducing USP1 activity (Fig. 2C and fig. S2C). Time-course analyses of cells exposed to CDDP for 6 hours and then allowed to repair DNA damages for 16 hours demonstrated that USP1 and Snail expression paralleled the activation of DDR, as evidenced by the phosphorylation of CHK1 and γH2AX, used as DDR markers (Fig. 2D and fig. S2D). In CDDP-treated cells, USP1 silencing not only prevented Snail protein up-regulation but also decreased CHK1 phosphorylation and increased γH2AX expression, suggesting that USP1 could act at the crossroads of DDR and EMT regulation (Fig. 2E and fig. S2E).
Fig. 2. Snail is a USP1 target protein.
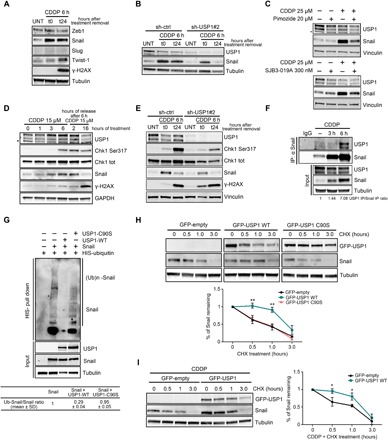
(A) Western blot analysis of the indicated transcription factors and the DNA damage marker γH2AX in COV-362 cells treated with CDDP for 6 hours (t0) and then allowed to repair for 24 hours (t24), as indicated. (B) Western blot analysis of Snail and USP1 expression in COV-362 cells silenced for USP1 and then treated or not with CDDP for 6 hours (t0) and allowed to repair for 24 hours (t24). (C) Western blot analysis of Snail and USP1 expression in COV-362 cells treated with pimozide (top) or SJB3-019A (bottom) in the presence or in the absence of CDDP for 6 hours. Lysates were harvested 24 hours after treatment. (D) Time-course analysis of USP1 and Snail protein expression in MDAH-2774 cells treated with CDDP as indicated. Phosphorylated Chk1 (S317) and γH2AX were used as a marker of DDR. (E) Western blot analysis of Snail, USP1, phosphorylated Chk1 (S317), and γH2AX expression in OVCAR-8 cells stably silenced for USP1 and treated with CDDP for 6 hours (t0) and allowed to repair for 24 hours (t24). (F) Co-IP analyses of endogenous USP1 and Snail proteins in OVCAR-8 cells treated or not with CDDP for 3 and 6 hours. Densitometric analysis of normalized USP1 IP/Snail IP expression is reported. (G) Ubiquitination assay of MYC-Snail in 293T/17 cells cotransfected with His-tagged ubiquitin, MYC-Snail, USP1WT, or USP1C90S and treated with MG132 for 6 hours. Densitometric analysis of Ub-Snail/Snail ratio of three independent experiments is reported in the table. In (E) and (F), input indicates the expression of Snail and USP1 in corresponding cell lysates. Immunoglobulin G (IgG) represents the control IP using an unrelated antibody. (H and I) Western blot analysis of USP1 and Snail expression in OVCAR-8 stably transfected with the indicated plasmids and exposed to a time-course treatment with CHX (H) or treated with CDDP and then with CHX for the indicated times (I). The densitometric analysis of Snail expression (normalized with respect to tubulin expression) is reported in the bottom graphs and represents the mean (±SD) of three independent experiments. Statistical significance was determined by a two-tailed, unpaired Student’s t test (*P < 0.05, **P < 0.01). In the figure panels, an asterisk indicates nonspecific bands, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), tubulin, or vinculin was used as a loading control.
USP1 de-ubiquitinates and stabilizes Snail protein
Following USP1 silencing, Snail mRNA levels did not change (fig. S2F), suggesting that protein down-regulation was controlled at the posttranscriptional level. By treating cells with cycloheximide (CHX), we observed that Snail protein half-life was decreased in USP1-silenced cells (fig. S2G). Moreover, when treated with the proteasome inhibitor MG132, USP1-silenced cells displayed accumulation of Snail, suggesting that Snail could be regulated by proteasomal degradation (fig. S2H), as already reported in other contexts (9). We thus hypothesized that USP1 may regulate Snail expression by controlling its ubiquitination levels and observed that USP1 readily coprecipitated with Snail, both at exogenous and endogenous levels, and their interaction was enhanced by CDDP treatment (Fig. 2F and fig. S2, I to K). Consistent with its DUB role, USP1 wild type (USP1WT), but not the de-ubiquitinase inactive mutant USP1C90S, markedly reduced the amount of ubiquitinated Snail in 293T/17 cells transfected with His-ubiquitin (Fig. 2G). Furthermore, Snail half-life was prolonged in USP1WT but not in USP1C90S-overexpressing OC cells, both under basal conditions and after CDDP treatment (Fig. 2, H and I), demonstrating that USP1 de-ubiquitinase activity was necessary to increase Snail expression after CDDP treatment. Together, these experiments demonstrated that Snail is a new USP1 target.
Snail mediates the protective effects of USP1 from CDDP-induced cell death
In addition to its well-known role of EMT inducer, Snail has been shown to mediate platinum resistance in several cancer models (6, 23, 24). To evaluate whether Snail could mediate, at least in part, the effects of USP1 in cell survival after CDDP treatment in our model, we silenced Snail expression in control and USP1-silenced cells. Snail silencing increased the sensitivity to CDDP (Fig. 3A) and resulted in reduced CHK1 phosphorylation and increased γH2AX expression, likely linked to a decreased expression of USP1 (Fig. 3B). The combined depletion of both USP1 and Snail did not result in enhanced cell death compared to the silencing of USP1 or Snail alone (fig. S3A). On the other hand, overexpression of Snail in USP1-silenced cells increased the IC50 for CDDP in USP1-silenced cells by about fivefold (Fig. 3C). Together, these results suggest that USP1 and Snail reside along the same axis and Snail mediates most of the effects of USP1 with respect to CDDP sensitivity in OC cells.
Fig. 3. Snail mediates the USP1 effects on CDDP sensitivity and cell invasion.
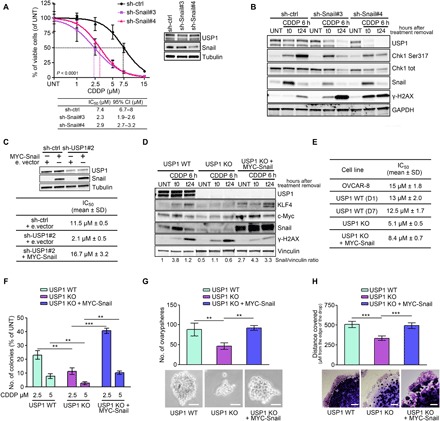
(A) Nonlinear regression analyses of cell viability assay in OVCAR-8 cells transduced with control or Snail shRNAs, as indicated, and treated with increasing doses of CDDP for 16 hours. Data are expressed as percentage of viable cells with respect to the untreated cells and represent the mean (±SD) of three biological replicates. The table shows the IC50 and the CI of each condition. Inset: Western blot analysis reporting USP1 and Snail expression in the used cells. (B) Western blot analysis of Snail, USP1, phosphorylated Chk1 (S317), and γH2AX expression in OVCAR-8 cells transduced as in (A). (C) Table showing the CDDP IC50 of MDAH-2774 cells transfected with an empty vector or with MYC-Snail and transduced with control or USP1 shRNAs, as indicated. The Western blot analyses of USP1 and Snail expression in the used cells are shown on top. (D) Western blot analyses evaluating the expression of USP1, KLF4, c-Myc, Snail, and γH2AX in CRISPR-Cas9 OVCAR-8 USP1 WT and USP1 KO clones and USP1 KO stably transfected with MYC-Snail. Cells were treated or not with CDDP for 6 hours (t0) and allowed to repair for 24 hours (t24). Densitometric analysis of Snail expression (normalized to vinculin) is reported under the blot and represents the mean of three independent experiments. (E) Table showing the CDDP IC50 of the indicated OVCAR-8 parental cells or CRISPR-Cas9 clones calculated by treating cells with increasing doses of CDDP for 16 hours. (F) Graph showing the number of colonies formed USP1 WT and USP1 KO and USP1 KO re-expressing MYC-Snail clones treated with the indicated CDDP doses. Data are expressed as percentage of colony formed in treated with respect to untreated cells. (G) Graph (top) and representative phase-contrast images (bottom) showing the ovarysphere-forming ability of USP1 WT and USP1 KO and USP1 KO re-expressing MYC-Snail clones. Scale bars, 50 μm. (H) Matrigel evasion assay of USP1 WT, USP1 KO, and USP1 KO re-expressing MYC-Snail. Graph (top) reports the distance covered by the individual cells from the edge of the drop. Representative phase-contrast images are reported in the bottom panels. Scale bar, 500 μm. In (A) to (D), GAPDH, tubulin, or vinculin was used as loading control. In (A), Fisher’s exact test was used to calculate the global P value reported in the graph. Otherwise, data represent the mean (±SD) of three independent experiments, and statistical significance was determined by a two-tailed, unpaired Student’s t test. Error bars denote SD (**P < 0.01, ***P < 0.001).
USP1 knockout OC cells are highly sensitive to CDDP and fail to up-regulate Snail in response to CDDP
To confirm our data, we exploited the CRISPR-Cas9 technology in the OVCAR-8 cell line to generate USP1 knockout (KO) cells. Different cell clones, either USP1 KO or WT, were isolated and compared with parental cells to verify that clonal selection per se did not induce substantial changes in Snail expression and/or in the biological behavior of these cells (fig. S3B). We observed that USP1 KO cells expressed lower Snail basal levels and failed to up-regulate Snail, KLF4, and c-Myc after CDDP treatment (Fig. 3D). When compared to USP1 WT, USP1 KO cells were more sensitive to CDDP treatment, both in drug-response curves and in colony assays (Fig. 3, E and F, and fig. S3, C and D). Furthermore, they formed less spheroids (ovaryspheres), both under basal conditions and when treated with CDDP (Fig. 3G and fig. S3, E and F), and were less invasive when included in 3D Matrigel (Fig. 3H). These USP1 KO cell phenotypes were all rescued by overexpressing Snail, confirming that Snail mediated the effects of USP1 after CDDP treatment (Fig. 3, D to H, and fig. S3, D to F). Overall, these experiments confirmed that USP1 expression in OC cells is necessary to acquire stem cell–like features.
ATM/ATR phosphorylate USP1 and favor USP1/Snail interaction in response to CDDP treatment
Snail protein is highly phosphorylated by kinases that promote either its degradation or stabilization, thereby modulating its half-life (25). To evaluate whether Snail phosphorylation mediated its interaction with USP1, we extracted proteins from Snail/USP1-transfected cells treated or not with CDDP and treated lysates with λ-phosphatase before performing co-immunoprecipitation (co-IP) for Snail and USP1. Phosphatase treatment only slightly decreased basal Snail/USP1 interaction but almost completely prevented their association following CDDP treatment, suggesting that phosphorylation of Snail, USP1, or both was needed (Fig. 4A). It is well known that, soon after CDDP treatment, ATM/ATR apical kinases are activated and phosphorylate downstream proteins of the DDR pathway on S/TQ motifs (2, 26). Both USP1 and Snail protein sequences contain phosphorylable serines within S/TQ motifs, respectively two (S42 and S331) and one (S100) [not shown and (27)]. We used an anti-pS/TQ antibody to evaluate whether these residues became phosphorylated after CDDP treatment. No S/TQ phosphorylation on Snail was detectable, while USP1 was considerably phosphorylated on S/TQ motif(s), particularly after CDDP treatment, both in exogenously overexpressed and in endogenous protein (fig. S4, A and B).
Fig. 4. ATR/ATM-mediated USP1 phosphorylation is required for USP1/Snail interaction.
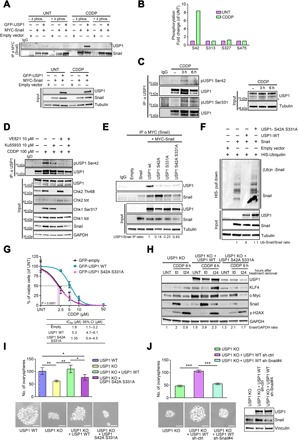
(A) Co-immunoprecipitation (Co-IP) analyses of MYC-Snail and GFP-USP1 in cells treated or not with CDDP. Cell lysates were collected and treated with or without λ phosphatase before the IP, as indicated. (B) Graph reporting the fold changes of the phosphorylation of the indicated USP1 serines, as evaluated by mass spectometry in MDAH-2774 cells untreated or treated with CDDP for 3 hours, as indicated. (C) Western blot analysis of phosphorylated USP1 (S42 and S331) in MDAH-2774 cells treated or not with CDDP for 3 or 6 hours. Cell lysates were immunoprecipitated with an anti-USP1 antibody and probed with anti-pS42, anti-pS331USP1, or total USP1 protein (right). (D) Western blot of pS42-USP1 expression in MDAH-2774 cells treated or not with CDDP for 3 hours in the presence of Ku55933 (ATM inhibitor), VE-821 (ATR inhibitor), or both, as indicated. Cell lysates were immunoprecipitated with an anti-USP1 antibody and probed with anti-pSer42 or total USP1 protein (top). The correspondent cell lysates (Input) were tested for the expression of USP1 and Snail, and the expression and phosphorylation of CHK1 and CHK2 were used as readouts of ATM and ATR activation and inhibition. (E) Co-IP analyses of USP1 and Snail in cells transfected with MYC-Snail and GFP-tagged USP1WT and USP1 point mutants, as indicated (top). Densitometric analysis of normalized USP1 IP/Snail IP expression is reported. (F) Ubiquitination assay of MYC-Snail in 293T/17 cells cotransfected with His-tagged ubiquitin, MYC-Snail, USP1WT, or USP1S42A/S331A and treated with MG132 for 6 hours. Polyubiquitinated Snail protein was visualized by blotting with an anti-Snail–specific antibody. Densitometric analysis of the Ub-Snail/Snail ratio is reported. (G) Nonlinear regression analyses of cell viability assay in OVCAR-8 cells transfected as indicated and treated with increasing doses of CDDP for 16 hours. Data are expressed as percentage of viable cells with respect to the untreated cells and represent the mean (±SD) of three biological replicates. The table shows the IC50 and the CI of each condition. Fisher’s exact test was used to calculate the global P value reported in the graph. (H) Western blot evaluating the expression of USP1, KLF4, c-Myc, Snail, and γH2AX in OVCAR-8 USP1 KO cells and in USP1 KO cells re-overexpressing USP1WT or USP1S42A/S331A proteins treated or not with CDDP for 6 hours (t0) and allowed to repair for 24 hours (t24). Densitometric analysis of Snail expression (normalized to GAPDH) is reported under the blot and represents the mean of three independent experiments. (I) Graph (top) and representative phase-contrast images (bottom) showing the ovarysphere-forming ability (mean ± SD of three experiments) of the indicated OVCAR-8 cells. Scale bars, 50 μm. (J) Graph (top) and representative phase-contrast images (bottom) indicating the ovarysphere-forming ability (mean ± SD of three experiments) of OVCAR-8 USP1 KO, USP1 KO cells re-overexpressing USP1WT, and USP1 KO cells re-overexpressing USP1WT and silenced for Snail expression using a specific shRNA (shSnail #4). On the right, the Western blot analysis shows USP1 and Snail expression in the used cells. In (A), (C), (D), (E), (F), (H), and (J), tubulin and GAPDH were used as loading control. In the figure, an asterisk indicates a nonspecific band; Input shows the expression of the indicated proteins or phospho-proteins in the lysates used for IP experiments; IgG represents the control IP using an unrelated antibody. In the figure, data represent the mean (±SD) of three independent experiments, and statistical significance was determined by a two-tailed, unpaired Student’s t test. Error bars denote SD (*P < 0.05, **P < 0.01, ***P < 0.001).
To evaluate whether USP1 phosphorylation after CDDP treatment was directly operated by ATM/ATR, we treated OC cells with CDDP in combination with selective inhibitors of ATM (Ku55933) and/or ATR (VE-821) and performed co-IP experiments. Combined ATM and ATR inhibition strongly reduced S/TQ phosphorylation of endogenous USP1 in OC cells (fig. S4C, top left panels) and prevented Snail/USP1 association (fig. S4C, bottom left panels). Further, USP1 phosphorylation was important for USP1-mediated stabilization of Snail, as inhibition of ATM/ATR reduced CDDP-induced Snail protein up-regulation (Fig. 4D and fig. S4D). Together, these data indicate that phosphorylation of USP1 on S/TQ motifs by ATM and ATR is a critical step for USP1-mediated Snail stabilization.
USP1 phosphorylation on S42 and S331 is necessary for its binding to Snail
We next exploited a mass spectrometry–based proteomic approach to precisely map the USP1 residues phosphorylated in control and CDDP-treated cells. We observed that under basal condition, USP1 is phosphorylated on four serines, but only S42 phosphorylation was strongly increased after CDDP treatment (Fig. 4B, fig. S4E, and table S1). However, because of trypsin digestion sites, peptides containing S331 were lost in this analysis. To confirm these data and to verify whether both USP1 S/TQ motifs on S42 and S331 were phosphorylated after CDDP treatment, we generated anti-pS42 and anti-pS331 phospho-specific antibodies and tested their specificity in OVCAR-8 KO cells transfected with USP1WT or a double phosphomutant, USP1S42A/S331A. Anti-pS42 and anti-pS331 specifically recognized the WT protein but not the USP1S42A/S331A mutant, and the phosphorylation of both S42 and S331 increased in CDDP-treated cells (fig. S4F). The same results were observed on endogenous USP1 in MDAH-2774 and OVCAR-8 cells (Fig. 4C and fig. S4G). In accordance with the above experiments, ATM and, even more, ATR inhibitors strongly reduced CDDP-induced USP1 S42 phosphorylation (Fig. 4D) and eventually reduced Snail half-line in CDDP-treated cells (fig. S4H).
In vitro studies have already demonstrated that phosphorylation on S42 does not modify USP1 catalytic activity (28). We thus hypothesized that S42 and/or S331 phosphorylation could be involved in USP1 interaction with Snail, and we transfected USP1S42A, USP1S331A, or USP1S42A/S331A mutants with Snail and performed co-IP assays. We observed that USP1 binding to Snail was greatly impaired when either one of the two residues was mutated (Fig. 4E). Accordingly, compared to USP1WT, expression of USP1S42A/S331A mutant did not lead to Snail de-ubiquitination (Fig. 4F), and from a biological point of view, it failed to protect OVCAR-8 cells from CDDP-induced cell death (Fig. 4G).
USP1 is necessary for metastatic dissemination of OC cells
Poor prognosis for patients with OC is essentially due to the development of chemoresistant disease that disseminates to the peritoneum and omentum and to the organs located in the peritoneal cavity (4). To verify whether USP1, via stabilization of Snail, played a role in the metastatic dissemination of OC cells, we exploited the OVCAR-8 xenograft model of intraperitoneal spreading that closely resembles the dissemination occurring in patients with OC (29). To this aim, we generated OVCAR-8 USP1 KO cells stably re-expressing USP1WT or USP1S42A/S331A. Re-expression of USP1WT but not of USP1S42A/S331A regulated Snail expression, both under basal conditions and following CDDP treatment, and rescued the ovarysphere-forming ability of OVCAR-8 parental cells (Fig. 4, H and I). Furthermore, Snail silencing in OVCAR-8 USP1 KO cells stably re-expressing USP1WT was sufficient to decrease the ovarysphere-forming ability to the same levels as USP1 KO cells (Fig. 4J), confirming that Snail was the critical downstream target of USP1 in this signaling axis.
To verify whether these in vitro phenotypes translated into different abilities of OC cells to grow in vivo, we intraperitoneally injected OVCAR-8 parental cells, USP1 KO, and USP1 KO stably re-expressing USP1WT or USP1S42A/S331A in female nude mice and evaluated tumor spreading both by macroscopic observation at necroscopy and by pathological analyses. As reported by others (29), in less than 6 weeks, OVCAR-8 cells formed neoplastic foci that grew as disseminated floating cell clusters, with cells diffusely infiltrating the peritoneal wall and the abdominal organs, such as pancreas, small intestine, spleen, uterus and fallopian tubes, and the diaphragm surface. We injected mice with USP1 KO re-expressing or not USP1WT or USP1S42A/S331A and treated or not with low doses of CBDCA, as previously described (30). Macroscopic evaluation of tumor dissemination clearly demonstrated that the tumor burden in mice injected with USP1WT cells was significantly higher than the one observed with USP1 KO cells or with cells re-expressing USP1S42A/S331A, especially under CBDCA treatment (fig. S5A). Combined macroscopic and pathological analyses indicated that USP1 KO OVCAR-8 cells almost completely failed to grow, either as infiltrating cells (Fig. 5, A to C, and fig. S5B) or as disseminated floating cell clusters (fig. S5C), paralleling what was observed in vitro in the ovarysphere and 3D Matrigel invasion assays. USP1 KO cells re-expressing USP1WT efficiently disseminated to the mouse abdomen, while re-expression of USP1S42A/S331A mutant resulted only in a marginal engraftment of the cells to the peritoneal cavity (Fig. 5, A to C, and fig. S5, B and C). When mice were treated with low doses of CBDCA, no residual tumor mass was detectable in mice injected with USP1 KO cells, and mice injected with USP1WT-expressing cells were significantly more resistant to CBDCA than those injected with USP1S42A/S331A-expressing cells (Fig. 5, A to C, and fig. S5, B and C). These phenotypes paralleled the expression of Snail, which was down-modulated in tumors from USP1S42A/S331A-expressing cells, compared to tumors from USP1WT-expressing cells (Fig. 5D), overall confirming in vivo what was observed in vitro. However, Snail expression was only slightly increased in USP1WT tumors treated with CBDCA compared to the untreated ones (Fig. 5D). This was likely due to the timing of the in vivo experiments, in which mice were CBDCA-treated for 10 days and tumors were explanted 36 hours after the last CBDCA administration. This is in line with our in vitro data showing that Snail increases transiently after platinum treatment owing to USP1 activity.
Fig. 5. USP1 is necessary for metastatic dissemination of OC cells via Snail.
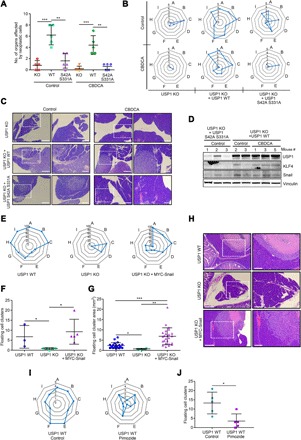
(A and B) Dot (A) and radar (B) plots reporting the total number of organs colonized (A) and the distribution of abdominal metastasis (B) in mice injected intraperitoneally with OVCAR-8 USP1 KO cells or USP1 KO cells re-overexpressing USP1WT or USP1S42A/S331A and treated or not with CBDCA, as determined by macroscopic and pathological analyses. (C) Typical images of hematoxylin and eosin (H&E) analyses of the left hypochondrium area, where the bulk tumor masses were localized in mice injected with OVCAR-8 cells. USP1 KO re-expressing USP1WT cells clearly infiltrate this area in both CBDCA-treated and untreated mice. Minor intrapancreatic tumor localizations were observed in mice injected with USP1S42AS331A-expressing cells, especially after CBDCA treatment. For each mouse, 5× and 10× images of the same field are shown. White dashed boxes represent the areas magnified in the right panels. Scale bars, 500 μm; magnification bars, 200 μm. (D) Western blot analysis of USP1, Snail, and KLF4 expression in the tumor masses formed in the hypochondria of the indicated mice described in (B) and (C). Vinculin was used as loading control. (E to G) Radar plots showing the distribution of abdominal metastasis (E) and dot plots showing the number (± SD) (F) and the area (G) of floating cancer cell clusters in mice injected intraperitoneally with OVCAR-8 USP1 WT (n = 3), USP1 KO cells (n = 5), and USP1 KO cells re-overexpressing Snail (n = 5). (H) Typical images of H&E analyses of pancreas of mice described in (E). Mice injected with USP1 WT or USP1 KO + Snail cells clearly showed an ab extrinseco infiltration of the organ, while intrapancreatic tumor localizations were absent in mice injected with USP1 KO cells. For each mouse, 10× and 20× images of the same field are shown. White dashed boxes represent the areas magnified in the right panels. Scale bars, 500 μm; magnification bars, 200 μm. (I) Radar plots showing the distribution of abdominal metastasis in mice injected intraperitoneally with parental OVCAR-8 cells and treated or not with pimozide. (J) Dot plot reporting the mean (± SD) of floating cancer cell clusters used as a measure of in vivo ovarysphere formation in mice described in (A). In (B), (E), and (I), blue bold lines indicate the number (values 0 to 5) or percentage (E) of mice affected for each district (A, pancreas; B, liver; C, gastrointestinal tract, D, reproductive tract; E, peritoneal wall; F, omentum; G, diaphragm; H, spleen; and I, hypochondrium sx). Statistical significance was determined by a two-tailed, unpaired Student’s t test. Error bars denote SD (*P < 0.05, **P < 0.01, ***P < 0.001).
To verify whether Snail was the key mediator of USP1 activity in vivo, we intraperitoneally injected in female nude mice OVCAR-8 USP1 WT, USP1 KO, and USP1 KO cells stably overexpressing Snail (USP1 KO + Snail). Snail overexpression restored the ability of OVCAR-8 USP1 KO cells to grow and spread to the peritoneal cavity (Fig. 5, E to H), although these cells displayed some differences compared to parental OVCAR-8 cells. While parental cells colonized approximately all structures and organs of the peritoneal cavity, USP1 KO + Snail cells prevalently localized to the pancreas, liver, and reproductive tract (Fig. 5E) and formed a slightly higher number of floating clusters that were significantly bigger in size (Fig. 5, F and G), suggesting that these cells grew much better in suspension than adhered to mesothelium-covered internal organs.
The above results suggested that USP1, by regulating Snail basal and platinum-induced expression, could represent a good therapeutic target to prevent OC spreading. When used in USP1 KO cells, the USP1 inhibitors pimozide and SJB-019A did not significantly affect the expression of Snail and the response to CDDP treatment (fig. S5, D and E), demonstrating that they could be considered quite specific for USP1 in this model. Then, following the workflow depicted in fig. S5F, we treated mice carrying tumors from OVCAR-8 parental cells with pimozide. The treatment with the USP1 inhibitor strongly restrained the metastatic tumor dissemination, reducing the number of peritoneal organs affected by tumor cell infiltration and the number of disseminated floating cell clusters (Fig. 5, I and J, and fig. S5G). In accordance with the results obtained in vitro, these tumor masses displayed lower levels of Snail, compared to the untreated ones (fig. S5H). Overall, these data demonstrate that USP1 expression, activity, and phosphorylation on S42/S331 are all necessary for OC cell growth and spreading, in vivo. Accordingly, we observed that tumors formed by USP1 WT and USP1 KO cells grew similarly when cells were injected subcutaneously, although USP1 KO cells displayed a slightly longer tumor latency (fig. S5, I to K).
High USP1 expression and activity predict poor prognosis in patients eligible for platinum treatment
To verify the role of the USP1/Snail axis in the human pathology, we first interrogated the KM Plotter dataset available online that comprises a very large number of OC samples (n = 1435). The high mRNA level of USP1 predicted low progression-free survival (PFS) and overall survival (OS) in patients with OC [hazard ratio (HR) = 1.33, P = 1.4 × 10−5 and HR = 1.19, P = 0.011, respectively, using the log-rank test] (fig. S6, A and B). In agreement with our observation that USP1 regulates Snail protein stability but not its mRNA expression, Snail mRNA expression did not correlate with patient survival (fig. S6C). Next, we performed immunohistochemistry (IHC) analyses of USP1 in matched primary and synchronous metastatic lesions from patients with OC (n = 10) collected in our institute. USP1 was highly expressed in all specimens, displaying a specific nuclear pattern (fig. S6D). Although three different anti-Snail commercial antibodies were tested, we were not able to obtain specific staining by IHC. To circumvent this limit, we set up an assay of “responsiveness to USP1 inhibition” by treating a panel of OC cell lines with CDDP ± pimozide and then analyzing the expression of Snail protein by Western blot. In 8 of 10 tested cell lines, which we called “responder,” pimozide hampered the up-regulation of Snail expression following CDDP treatment (fig. S6E). The two cell lines that did not respond to the CDDP ± pimozide treatment (“nonresponder”) were also insensitive to USP1 silencing–induced cell death after platinum. We next used the same assay in five primary OC cell lines, recently derived and characterized from high-grade serous OC patients and for which a gene expression profile was available (31). All primary cells expressed USP1, but only three were classified responder according to the above-specified criteria, namely, the 8587, 13699, and 13914 cell lines (Fig. 6A, top panels, and fig. S6F). Accordingly, when we treated the primary OC cell lines with increasing doses of CDDP in combination with pimozide, we found that pharmacological inhibition of USP1 exhibited dose-dependent cytotoxicity only in responder cells (Fig. 6A, bottom panels), confirming that the USP1/Snail axis influenced CDDP response in OC primary cells as well.
Fig. 6. High USP1 expression and activity predict poor prognosis in patients eligible for platinum treatment.

(A) Western blot analyses of USP1 and Snail expression in the indicated primary OC cells, treated with pimozide and/or CDDP. Tubulin and vinculin were used as loading control (an asterisk indicates a nonspecific band). Graphs show cell viability of the same cells treated with increasing doses of CDDP with or without pimozide for 16 hours. Data are expressed as percentage of viable cells with respect to untreated cells and represent the mean (±SD) of three independent experiments. Statistical significance was determined by a two-tailed, unpaired Student’s t test (*P < 0.05; **P < 0.01, ***P < 0.001). (B) Table showing the 10 most up-regulated genes and their fold change in the responder with respect to the nonresponder cells as evaluated by RNA sequencing analyses. (C) Kaplan-Meier survival curve evaluating the PFS of patients with OC, based on the expression of the 10-gene “responder signature” using the KM Plotter online tool on all the available datasets (n = 614). (D and E) Kaplan-Meier survival curves evaluating the PFS based on the 10-gene signature in TNBC (n = 161) (D) and ER+ BC (n = 762) (E) using the online KM Plotter tool on all the available datasets. In (C), (D), and (E), the CIs are enclosed in parentheses. P values were calculated using the log-rank test.
To identify common molecular alterations and/or pathways explaining the phenotypes that we observed, we clustered the responder versus the nonresponder OC primary cells, using the available gene expression profiles (31), and identified ~500 genes commonly up-regulated in the responder primary OC cells (table S2). These genes were then analyzed using the DAVID Bioinformatics Resources online tool (https://david.ncifcrf.gov). Cell plasticity and EMT-related pathways, such as basal cell carcinoma, extracellular matrix–receptor interaction, cell adhesion, and Hippo signaling pathways, were the most significantly enriched in responder cells (table S3), reinforcing our data indicating that activation of the USP1/Snail axis greatly contributed to plasticity and invasion of OC cells.
We next explored whether the mRNA signature associated to USP1 activity could predict the prognosis of patients with OC and selected the 10 most up-regulated genes of this signature, using as cutoff a fold change >100 in responder versus nonresponder primary OC cells (Fig. 6B). We interrogated the KM Plotter dataset and observed that this 10-gene signature, better than USP1 expression alone, was able to segregate OC patients with worse PFS and OS (Fig. 6C and fig. S6G; HR = 1.69, P = 3.5 × 10−7 for PFS and HR = 1.58, P = 8.5 × 10−6 for OS). We also observed that USP1 and Snail expression were increased in two different models of isogenic platinum-resistant OC cells recently established in our laboratory (32), especially under CDDP treatment (fig. S6H), supporting again that this pathway could contribute to the establishment of a platinum-resistant phenotype in OC.
Given the above results, we wondered whether this signature could be used as a more general biomarker of platinum sensitivity, and we chose breast cancer as a model. Among breast cancer subtypes, triple-negative breast cancer (TNBC) is known to be genetically more similar to high-grade OC, and accordingly, it is considered a platinum-sensitive tumor (33, 34). Both USP1 and the 10-gene signature were able to predict PFS in TNBC (Fig. 6D and fig. S7A), but not in estrogen receptor–positive (ER+) BC (Fig. 6E and fig. S7B), confirming the robustness of this signature to detect patients eligible for platinum treatment at higher risk of developing drug resistance and disease progression. Furthermore, in in vitro culture, MDA-MB 468 (TNBC), but not the MCF-7 (ER+) cell line, up-regulated Snail upon CDDP treatment, and USP1 inhibition impaired CDDP-induced overexpression of Snail, demonstrating that the USP1/Snail axis plays a role in drug response and cell plasticity in different platinum-sensitive tumors (fig. S7, C and D). Accordingly, as observed in OC cells, pimozide increased the efficacy of CDDP in MDA-MB 468 cells, but not in MCF-7 cells (fig. S7E). Collectively, these findings strongly support the possibility that, following CDDP treatment, a subpopulation of cancer cells, which could be identified using a specific gene signature, up-regulate Snail in a USP1-dependent manner as an escaping mechanism that leads to platinum resistance and increased cell survival, eventually increasing the metastatic spreading and disease relapse.
DISCUSSION
Resistance to chemotherapies and regulation of cell plasticity have been recently linked to elegant works exploiting in vivo models of EMT (23). However, whether the link between EMT and response to chemotherapy is represented by the activation of DDR, specifically due to chemotherapy-induced DNA damage, has not been clarified.
Here, we fill this gap by showing that USP1 de-ubiquitinase, already known to be involved in the DDR, by regulating the function of FANCD2/I, also regulates the EMT by modulating Snail protein stability, thereby regulating response to chemotherapy, survival, cell plasticity, and metastatic capabilities. By identifying Snail as a new target of USP1 activity, our findings point to USP1 as a promising target for anticancer therapies, especially in combination with platinum-based chemotherapy.
We demonstrate that, following CDDP treatment, USP1 is directly phosphorylated by ATM and ATR on S42 and S331. Once phosphorylated, USP1 is able to bind, de-ubiquitinate, and stabilize Snail, which, in turn, activates a stem cell–like program (fig. S7F). It is known that in the regulation of FANCD2/I proteins, USP1 acts as a heterodimeric complex in association with UAF1 (USP1-associated factor 1). UAF1 increases USP1 catalytic activity but does not increase the affinity for the substrate (13). Whether the UAF1 binding to USP1 is also involved in Snail de-ubiquitination and whether USP1 phosphorylation on S42 and/or S331 affects USP1/UAF interaction have not yet been clarified and certainly need to be addressed in future studies.
Recent studies suggest that the cancer stem cell status is not static but dynamic, that the differentiation into a cancer stem cell is not a monodirectional process, and that bulk cancer cells can also be reprogrammed into stem cells, exploiting plasticity programs (35). As a consequence, the “stemness” is more similar to a cellular state that can be acquired, rather than a cell-intrinsic property (36). The fact that CDDP activates a program leading to the expression of Snail and several other stemness-related genes suggests the possibility that chemotherapy itself could induce the reprogramming of a subpopulation of cancer cells. This possibility has recently been proposed for cancer cells treated with radiotherapy (37).
Furthermore, it has been proposed that any tumor cell can potentially acquire stem cell–like properties if placed in the appropriate niche and/or exposed to the appropriate environmental signals (36). Our observations, linking the activation of ATM/ATR to USP1 phosphorylation and Snail stabilization, again support the possibility that CDDP itself could represent one of these key environmental signals. Since cancer stem cells, for their intrinsic characteristics, are more resistant to chemotherapy (35), this reprogramming could represent an adaptive selection implemented by cancer cells to survive to CDDP.
Our experiments indicated that Snail up-regulation was necessary for ovarysphere formation, in vitro and in vivo. However, in vivo, Snail expression needed at some point to decrease, to allow proper attachment of ovaryspheres to the mesothelium and invade the peritoneal organs. Otherwise, cells continued to grow in suspension, as demonstrated by the significantly bigger size of floating clusters dispersed in the peritoneum when Snail was constitutively overexpressed (Fig. 6E). This result is in line with the hypothesis that, for efficient metastasis formation, EMT must be induced to allow efficient detachment of cells from the primary tumor and their survival in anchorage independence, but then a mesenchymal-to-epithelial transition must also take place to allow proper engraftment and growth of the epithelial cancer cells at distant sites (36). Our data showed a role for Snail also in mediating the DDR after CDDP treatment (Fig. 3, B and D). Whether this effect is due to a decreased proliferation of mesenchymal cells, as previously proposed (23), or to the activation of specific signaling pathways linked to stem cell–like phenotypes, is something that needs to be better addressed in the future. Our data, together with current literature, suggest that platinum-treated cancer cells could use a “tunable” pathway, which starts with the phosphorylation of USP1 by ATM/ATR that induces a mesenchymal status via Snail stabilization necessary for the survival and for initial abdominal colonization. At later times, when DDR is concluded, the decrease of USP1 phosphorylation and of Snail protein levels restores the epithelial status of cancer cells and allows their massive growth at the metastatic sites.
Whether this mechanism is unique for Snail or whether it is shared by other EMT regulators is an open point that may deserve further investigation in the future. However, our data collected in OC models clearly showed that only Snail was consistently up-regulated in a USP1-dependent manner following CDDP treatment, while other EMT-related transcription factors, such as Slug, Twist-1, or Zeb1, were either not up-regulated or not responsive to USP1 inhibition. This finding is in line with recent evidences, demonstrating a specific role for each EMT transcription factor in different phases of tumor progression and in different tissues (38, 39).
Cells that responded with the activation of the USP1/Snail axis following CDDP treatment were enriched in cell invasion– and stemness-related pathways (see table S3 for a complete list) and could be identified by a 10-gene signature. This 10-gene signature can select patients at worse prognosis that might benefit from targeting of USP1, alone or in combination with platinum, supporting the rapid transferability of our research to the clinic toward a more personalized therapy of patients with OC. It is intriguing that this 10-gene signature was able to select not only patients with OC but also patients with TNBC at worse prognosis, suggesting that the findings collected in the context of OC preclinical models could be more widely applicable to improve the platinum sensitivity in different human cancer types. In this regard, a recent manuscript has linked USP1 expression and activity to the metastatic ability of 4T1 murine BC cells (40), further reinforcing the possibility of exploiting USP1 as a therapeutic target.
In conclusion, we show a new role for USP1 in promoting tumor invasion and metastasis mediated by Snail. Although current USP1 inhibitors, as many other targeted therapies, are not highly selective and could thus give rise to unexpected off-target effects, the data that we collected here robustly indicate in animal models that inhibition of USP1, in combination with platinum compounds, could represent a successful way to improve platinum efficacy. Given the compelling clinical need to overcome platinum resistance in patients with OC and in those with other cancers, we believe that future investigations will rapidly lead to clinical testing of this combination therapy, to improve the survival of patients with cancer at high risk of recurrence after platinum-based chemotherapies.
MATERIALS AND METHODS
Cell culture
MDAH-2774 (CRL-10303), TOV-21G (CRL-11730), OV-90 (CRL-11732), SKOV-3 (HTB-77), NIH:OVCAR-3 (HTB-161), and Met5a (CRL-9444) cells were obtained from the American Type Culture Collection (ATCC); OVSAHO (JCRB1046) and KURAMOCHI (JCRB0098) cells were from the JCRB Cell Bank; COV-362 [ECACC (European Collection of Authenticated Cell Cultures) 07071910] cells were from Sigma-Aldrich; and OVCAR-8 and OVCAR-4 cells were from the National Cancer Institute Developmental Therapeutics Program Tumor Repository. All these cell lines were maintained in RPMI 1640 medium (Sigma-Aldrich). Breast cancer cell lines MDA-MB 468 (HTB-132), BT-549 (HTB-122), MCF-7 (HTB-22), and T47D (HTB-133) were obtained from ATCC and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich). Primary epithelial OC cell lines (31) were provided by D.C.C.-T. and maintained in DMEM/F12 (Gibco). Human embryonic kidney (HEK) 293/T17 cells (ATCC, CRL-11268) used for overexpression strategies and 293FT cells (Invitrogen) used for lentivirus production following the manufacturer’s instructions (Clontech) were grown in DMEM high glucose (Sigma-Aldrich). Met5a cells were grown in RPMI 1640 medium (Sigma-Aldrich) with insulin (870 nM), epidermal growth factor (EGF (3.3 nM), and hydrocortisone (400 nM). All media were supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. All cell lines were grown in standard conditions at 37°C and 5% CO2. All cell lines were routinely authenticated in our laboratory using the Cell ID TM System (Promega) protocol and using GeneMapper ID version 3.2.1 to identify DNA short tandem repeat profiles (last authentication was on 29 May 2018).
Compounds and drug treatments
Dose-response curves were performed essentially as described previously (30, 41). Briefly, epithelial OC (EOC) cells were seeded in 96-well culture plates and transduced with lentiviral shRNAs or transfected with overexpressing vectors as indicated. Forty-eight or 72 hours after transfection or transduction, respectively, cells were treated with CDDP (TEVA Italia) for 16 hours at 37°C at the indicated concentrations. Cell viability was determined 24 hours after treatment removal using the CellTiter 96 AQueous cell proliferation assay (MTS) (Promega). For treatment with USP1 inhibitors pimozide (Sigma-Aldrich) and SJB3-019A (MedChem Express), EOC cells were pretreated for 24 hours with inhibitors at the indicated concentrations and then treated or not with CDDP. The specific USP1 inhibitors were dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. The specific ATM inhibitor KU-55933 and ATR inhibitor VE-821 (Selleckchem) were dissolved in DMSO as 10 mM stock and stored at −80°C.
Vectors, transfections, and viruses
pEGFP-C1 USP1WT and pLPC-MYC Snail vectors were provided by R. Bernards and R. Maestro, respectively. pEGFP-C1 USP1 mutants C90S, S42A, S331A, and S42A/S331A were generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene). Plasmids were transfected in HEK293/T17 or EOC cells using FuGENE HD Transfection Reagent (Promega) according to the manufacturer’s indications. Stable expressing clones were selected by culturing cells with G418 (0.5 mg/ml). For lentiviral production, 293FT cells were cotransfected, using a standard calcium phosphate precipitation, with the lentiviral-based shRNA constructs (pLKO) and lentiviral vectors pLP1, pLP2, and pVSV-G (Invitrogen). Seventy-two hours after transfection, medium containing viral particles was used to transduce EOC cell lines. pLKO vectors encoding for control (ctrl), USP1, and Snail shRNAs were purchased from Sigma-Aldrich Stable knock-down clones were selected by culturing cells in puromycin (1 μg/ml)–containing medium.
Preparation of cell lysates, immunoblotting, and immunoprecipitation
Cell lysates were prepared using cold RIPA buffer [150 mM NaCl, 50 mM tris-HCl (pH 8), 0.1% SDS, 1% Igepal, and 0.5% NP-40] containing protease inhibitor cocktail (Roche) phosphatase inhibitors 1 mM Na3VO4 and 10 mM NaF (Sigma-Aldrich) plus 1 mM DTT. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad). Proteins were separated in 4 to 20% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Criterion Precast Gel, Bio-Rad) and blotted onto a nitrocellulose membrane (Amersham, GE Healthcare). Immunoprecipitations were performed using cell lysates in HNTG buffer (20 mM Hepes, 150 mM NaCl, 10% glycerol, and 0.1% Triton X-100) and the indicated primary antibody [anti–green fluorescent protein (GFP) (no. 11814460001, Roche), anti-MYC tag (sc-40 clone 9E10, Santa Cruz Biotechnology), or anti-USP1 (HPA028440, Sigma-Aldrich)] and incubated overnight at 4°C. The immunocomplexes were precipitated by adding protein G or protein A agarose conjugated for an additional 1 hour and 30 min at 4°C. IPs were then washed in HNTG buffer, resuspended in 3× Laemmli Sample Buffer [5× Laemmli buffer composition: 50 mM tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 0.05% bromophenol blue, and 125 mM β-mercaptoethanol], and finally separated on SDS-PAGE for Western blot analysis. Membrane strips were blocked with 5% nonfat dried milk in tris-buffered saline (TBS)–0.1% Tween 20 or in Odyssey Blocking Buffer (LI-COR, Biosciences) and incubated at 4°C overnight with primary antibodies. The following primary antibodies were used: Zeb1 (1:500, #3396 clone D80D3), Slug (1:500, #9585 clone C19G7), pChk1Ser317 (1:500, #12302 clone D12H3), pChk2Thr68 (1:500, #2661), Snail (1:1000, #3879 clone C15D3), pS/TQ (1:1000, #9607 and #2851), p53Ser15 (1:500, #9284), KLF4 (1:500, #4038), c-Myc (1:1000, #5605 clone D84C12), Sox2 (1:1000, #3579 clone D6D9), and β-actin (1:1000, #4970) were from Cell Signaling Technology; Chk1 (1:500, sc-8408 clone G-4), Chk2 (1:500, sc-17747 clone A-11), Twist-1 (1:200, sc-81417 clone Twist2C1a), FANCD2 (1:500, sc-20022 clone FI17), and vinculin (1:5000, sc-7649 clone N-19) were from Santa Cruz Biotechnology; α-tubulin (1:5000, T9026 clone DM1A) and USP1 (1:1000, HPA028440) were from Sigma-Aldrich; and GAPDH (1:1000) and γH2AX (1:500) were from Calbiochem. Antibodies were visualized with appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (GE Healthcare) for chemiluminescent detection (Clarity, Bio-Rad) or with Alexa-conjugated secondary antibodies (Invitrogen) for Odyssey infrared detection (LI-COR Biosciences).
Customized generation of phospho-specific USP1 antibodies
The rabbit polyclonal antibodies specific for USP1 phospho–serine 42 (pS42) and phospho–serine 331 (pS331) were generated by GenScript Inc. Two New Zealand rabbits for each phospho-antibody were immunized with specific keyhole limpet hemocyanin (KHL)–conjugate peptides. The sequences of KHL-conjugate peptides used for rabbit immunization were as follows: USP-1-S42, CTKRALDFTDSQENE; USP-1-pS42, CTKRALDFTD{pSER}QENE; USP-1-S331, CSENESPRPSQKKSR; and USP-1-pS331, CSENESPRP{pSER}QKKSR.
Validation of the phospho-antibodies’ specificity was performed by enzyme-linked immunosorbent assay (ELISA) and approved by the Antibody Department of GenScript Inc. We tested the specificity of the two USP1 phosphorylation-specific antibodies in Western blot analysis as follows. Lysates (400 μg for each sample) from OVCAR-8 USP1 KO cells or USP1 KO cells stably expressing USP1WT or USP1S42A/S331A mutant were immunoprecipitated with an anti-USP1 antibody (HPA028440, Sigma-Aldrich) as described in the “Preparation of cell lysates, immunoblotting, and immunoprecipitation” section. Membranes were incubated with pS42- or pS331-USP1 (1:500) primary antibodies diluted in 5% (w/v) bovine serum albumin (BSA), 1× TBS, and 0.1% Tween 20, at 4°C overnight. Membranes were then probed with anti-rabbit HRP-conjugated secondary antibodies (1:2000) (GE Healthcare) and processed by chemiluminescence.
For detection of endogenous phosphorylation of S42-USP1 or S331-USP1, lysates (600 μg) from MDAH-2774 and OVCAR-8 cells, treated or not with CDDP for the indicated times, were immunoprecipitated as described above, and membranes were incubated with pS42-USP1 (1:500) or pS331-USP1 primary antibodies diluted (1:250) in 5% (w/v) BSA, 1× TBS, and 0.1% Tween 20, at 4°C overnight.
In vivo ubiquitination assay
HEK293/T17 cells were transfected with a His-ubiquitin vector in combination with the indicated constructs. Forty-eight hours after transfection, cells were treated with 10 μM MG132 for 6 hours. Cells were then lysed in buffer A [6 M guanidine-HCl, 0.1 M NaH2HPO4/NaH2PO4, and 10 mM imidazole (pH 8.0)] and sonicated. The lysates were incubated with nickel–nitrilotriacetic acid matrices (Qiagen) overnight at 4°C. The His pull-down products were washed twice with buffer A, twice with buffer A/TI [25 mM tris-HCl and 20 mM imidazole (pH 6.8)] (1 volume buffer A and 3 volumes buffer TI), and once with buffer TI. The pull-down proteins were resolved by SDS-PAGE for immunoblotting.
RNA isolation and real-time polymerase chain reaction
Total RNA was isolated using TriZol reagent (Ambion) following the manufacturer’s instructions. Two micrograms of total RNA was retro-transcribed using random hexamers and the AMV Reverse Transcriptase (Promega). We amplified 1/20 of the obtained cDNAs using primers for human Snail (forward, 5′-AGTGGTTCTTCTGCGCTACT-3′; reverse, 5′-TTAGGCTTCCGATTGGGGTC-3′) and human SDHA (forward, 5′-AGAAGCCCTTTGAGGAGCA-3′; reverse, 5′-CGATTACGGGTCTATATTCCAG-3′). Standard curves (10-fold dilution from 10−1 to 10−4 attomoles) were prepared and analyzed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using the MyiQ2 Two Color Real-Time PCR Detection System (Bio-Rad).
Protein stability with CHX/MG132
Snail protein stability was evaluated by treating (or not) with CDDP (15 μM for 3 hours) COV-362 cells transduced with specific USP1 shRNAs or OVCAR-8 cells stably transfected with GFP-USP1 WT or GFP-USP1C90S and treated with CHX (10 μg/ml) or with MG-132 (10 μM) for 30, 60, 120, and 180 min as indicated.
Mesothelial clearance assay
To form spheroids, COV-362 cells (100 per well) stably transduced with control or USP1 shRNAs were plated in a 96-well plate precoated with Poly(2-hydroxyethyl methacrylate), Poly-HEMA (a polymer that allows the growth in suspension). At the same time, Met5a-GFP cells were plated on a six-well dish precoated with fibronectin to resemble the mesothelium monolayer. After 16 hours, the spheroids were collected and plated on the Met5a-GFP monolayer. Cells were then incubated at 37°C in 5% CO2 atmosphere in the Leica Time Lapse AF6000LX workstation equipped with the Leica DMI 6000 motorized microscope and an environmental chamber for the proper setting of temperature humidity and CO2 concentration. The microscope allows the acquisition of multiple fields on the x, y, and z axes and is computer-assisted. The AF6000 Software (Leica) allows the acquisition of the images on the desiderate frames and periods of time. We collected 20 images for each treatment (one image per treatment every 2 hours) over a period of 8 hours using bright-field and fluorescence filters at ×200 magnification. ImageJ software was used to measure the area of each spheroid at time 0 (t0) and the area of the hole in the Met5a-GFP monolayer after 8 hours (t8). Normalized clearance area is defined as the ratio between t8 and t0 as previously described (20).
Ovarysphere assay
To establish primary ovaryspheres, COV-362 cells stably transduced with control or USP1 shRNAs and OVCAR8 WT USP1 KO and USP1 KO cells modified for USP1 and Snail expression were plated into 35-mm poly-HEMA–coated plates in phenol red–free DMEM/F12 medium (Gibco) containing B27 supplement (no vitamin A; Invitrogen), recombinant EGF (10 ng/ml), bFGF (10 ng/ml) (PeproTech), and 1% penicillin/streptomycin (Sigma-Aldrich). Cells were incubated at 37°C for 10 days, and then the ovaryspheres were photographed and then counted with a Nikon Eclipse TS100 inverted microscope.
Evasion assay
Evasion assays were performed as previously described (19). Briefly, 7.5 × 103 USP1-modified EOC cells were included in Matrigel (Cultrex, BME) drops at a final concentration of 8 mg/ml (12 μl of matrix volume per drop). Matrigel was diluted in RPMI 1640 and 0.1% BSA. The drops, sufficiently spaced from one another, were dispensed in cell culture dishes and maintained for 1 hour at 37°C upside down to jellify. Then, the dishes were turned up, and the drops were incubated in complete medium. The evasion ability was evaluated 6 days after inclusion by measuring the distance covered by crystal violet–stained cells exiting from the drops (five drops/cell lines per experiment). Images were collected using a stereo microscope Leica M205FA.
Generation of USP1 KO with CRISPR technology
The following protocol was adapted from Ran et al. (42). Two guide RNAs (gRNAs) were designed using the CRISPR Design online tool (https://zlab.bio/guide-design-resources) to target the first exon of USP1 (NC_000001.11). Sequences of the two gRNAs are as follows: gRNA1 fw, 5′-CACCGCTTTCACTAGGTATGACACC-3′; gRNA1 rev, 5′-AAACGGTGTCATACCTAGTGAAAGC-3′ and gRNA2 fw, 5′-CACCGGTCATACCTAGTGAAAGTAA-3′; gRNA2 rev, 5′-AAACTTACTTTCACTAGGTATGACC-3′.
The pairs of annealed oligonucleotides were cloned in the gRNA expression vector pSpCas9(BB)-2A-GFP plasmid (pX458, Addgene #48138) (www.addgene.org/). We transfected 2 μg of this plasmid in the OVCAR-8 cell line with Lipofectamine 2000 according to the manufacturer’s protocol. Seventy-two hours after transfection, GFP-positive cells were sorted by flow cytometry (Becton Dickinson) and pooled together. To confirm the presence of mutations surrounding gRNA target sites, we purified and sequenced the genomic DNA from the OVCAR-8 pool cells. By these means, we identified mutations around the predicted point of double-strand breaks. Pooled cells were seeded as single colonies (0.5 cells per well) in ten 96-well plates. After 2 to 3 weeks of expansion, the cells in each colony were divided in half; one half was subjected to propagation and the other half was used to assess the expression of USP1 using Western blot analysis. The genomic DNAs of clones showing a consistent reduction in USP1 expression were analyzed using a MiSeq instrument (Illumina) to confirm the presence and exact allelic fraction of mutations. We only used clones where we detected 100% allelic frequency.
In vivo tumor xenograft
Animal experimentation was reviewed and approved by the Centro di Riferimento Oncologico di Aviano (CRO) Institutional Organism for Animal Wellbeing (OPBA) and by the Italian Ministry of Health (authorization no. 1261/2015-PR, released to G.B.). All animal experiments were conducted in adherence to international and institutional committees’ ethical guidelines. Female athymic (nude) mice (6 to 7 weeks old) were acquired from Charles River Laboratories and xenografted with 5 × 106 USP1 and Snail-modified OVCAR-8 cells intraperitoneally or subcutaneously in sterile phosphate-buffered saline. After 30 days from injection (in accordance with our previous observations of intraperitoneal injection of the same cells), animals were randomly divided into two groups (five mice per group) and treated or not intraperitoneally with CBDCA (10 mg/kg) six times every 2 days or pimozide (15 mg/kg) daily for 10 days. At the time of sacrifice, disease (36 hours after the last treatment) dissemination was recorded and annotated by photographs and charted on the basis of organ dissemination. Tissues were fixed in 4% paraformaldehyde before dehydration in ethanol and xylene before paraffin embedding and then stained with hematoxylin and eosin.
Immunohistochemistry
This work was approved by the Centro di Riferimento Oncologico di Aviano (CRO) Internal Review Board (protocol no. CRO-IRB 05-2014). For IHC on human samples, 4-μm-thick sections from OC specimens were deparaffinized and rehydrated, and antigens were retrieved in target retrieval solution (pH 6.1, DAKO Omnis), using a pressure boiler for 10 min at 110°C. Slides were treated with a 3% solution of hydrogen peroxidase in methanol to block the endogenous peroxidase activity and then washed in a phosphate buffer solution before immunoperoxidase staining. Slides were then incubated at 4°C overnight with the primary antibody against USP-1 (anti-rabbit, HPA028440, Sigma-Aldrich) diluted 1:100 in a phosphate buffer solution. Tissue sections were incubated with biotinylated anti-rabbit immunoglobulin and then stained with streptavidin labeled with peroxidase; the signal was developed by using DAB chromogen as substrate. After chromogen development, slides were counterstained with hematoxylin, washed, dehydrated with alcohol and bioclear, and mounted with coverslips using a permanent mounting medium. Staining was recorded considering both staining intensity and the percentage of positive cells. Briefly, each core biopsy was evaluated for staining intensity (0 = no staining, 1 = faint staining, 2 = moderate staining, and 3 = intense staining) and percentage of positive cells (0 = nonreacting cells, 1 = 1 to 25% reacting cells, 2 = 26 to 50% reacting cells, 3 = 51 to 75% reacting cells, and 4 = 76 to 100% reacting cells). A final IHC score was obtained by adding the value of the intensity to the percentage category.
Phosphosite identification and quantification by nano–liquid chromatography–tandem mass spectrometry analysis
Gel pieces containing immunoprecipitated USP1 were manually excised, and proteins were prepared and in-gel–digested as previously described (43). Phosphopeptides were then enriched from resulting peptides using titanium dioxide beads (TitanSphere, GL Sciences Inc.) adapting a protocol described by others (44). Briefly, peptides were solubilized in 80% AcN, 5% TFA, and 1 M glycolic acid (loading buffer) and incubated in batch mode with 0.6 mg of TiO2 beads for 1 hour under shaking. After centrifugation, the pellet was washed with loading buffer, washing buffer 1 (80% AcN, 1% TFA), and then washing buffer 2 (10% AcN, 0.1 TFA) before elution of phosphopeptides using 1.12% ammonia in H2O. Eluted peptides were then acidified before nano–liquid chromatography–tandem mass spectrometry (LC-MS/MS) analyses using UltiMate 3000 RSLCnano and Q Exactive Plus (Thermo Fisher Scientific). Peptides were sampled on a 300 μm by 5 mm PepMap C18 precolumn and separated on a 75 μm by 250 mm C18 column (PepMap, Dionex). The nano-LC method consisted of a 40-min gradient at a flow rate of 300 nl/min, ranging from 5 to 33% acetonitrile in 0.1% formic acid. Spray voltage was set at 1.6 kV; heated capillary was adjusted to 270°C. Survey full-scan MS spectra (mass/charge ratio = 400 to 1600) were acquired with a resolution of 70,000 after accumulation of 106 ions (maximum filling time, 250 ms). The 10 most intense ions were fragmented by higher-energy collisional dissociation after accumulation of 106 ions (maximum filling time, 250 ms). MS and MS/MS data were acquired using the software Xcalibur (Thermo Fisher Scientific). Peptides and proteins were identified through concomitant searches against UniProt (Homo sapiens taxonomy, May 2017 version), classical contaminants (homemade), and the corresponding reversed databases using Mascot (version 2.5.1). Peptide modifications allowed during the search were carbamidomethyl (C, fixed), acetyl (Protein N-ter, variable), oxidation (M, variable), Phospho (ST, variable), and Phospho (Y, variable). The Proline software (http://proline.profiproteomics.fr) was used to filter the results (conservation of rank 1 peptides, peptide length ≥ 7, minimum peptide-spectrum match (PSM) score of 25, P < 0.01 for query homology threshold, and minimum of one specific peptide per identified protein group). Only phosphopeptides exhibiting a modification site confidence ≥90% [based on Mascot Delta Score (MD-score) (45)] were further considered. The Proline software was also used for label-free quantification of identified peptides. Each phosphosite’s intensity was calculated as the sum of extracted intensities of the different peptides bearing this phosphosite. To counteract for unequal starting quantities before phosphopeptide enrichment, extracted intensities of USP1 phosphosites were normalized by the sum of the extracted intensities of USP1 unphosphorylated peptides. Normalized intensities were used to compare USP1 phosphosite levels in cells treated or not with platinum.
Statistical analysis
Statistical significance (P < 0.05), means, SD, and 95% confidence intervals (CIs) were determined by using GraphPad PRISM software (version 6.0) using the most appropriate test, as specified in each figure. For survival analysis, PFS was identified as the relevant clinical endpoint. PFS curves were reported according to the Kaplan-Meier method and were compared with the log-rank test. Median estimates, with 95% CIs, were also reported. We selected two patients’ cohorts from the KM-Plotter website (http://kmplot.com/). For the OC cohort, histological data were available for 1294 patients (62 endometrioid and 1232 serous OC); data on grade were available for 1405 patients (grade 1: 56; grades 2 and 3: 1349); data on stage were available for 1447 patients (stages 1 and 2: 179; stages 3 and 4: 1268); PFS data were available for 1436 patients, of whom 1259 had a known clinical history of platinum-based therapy. OS data were available for 1656 patients, of whom 1409 had known clinical history of platinum-based therapy. For the BC cohort, we selected 2565 patients classified as ER+ and 255 patients classified as TNBC. Of the ER+ patients with BC, relapse-free (RFS) and OS data were available for 2061 and 548 patients, respectively; and of the patients with TNBC, 255 had RFS data and 15 OS data. For patients with BC, no histological, grade, and stage information could be retrieved and matched with hormonal status.
Supplementary Material
Acknowledgments
We thank R. Bernards for USP1 vector and R. Maestro for Snail vector, A. Polacchini for graphical assistance and helpful discussion, S. Benevol and L. Cesaratto for technical support, and all members of the SCICC laboratory for fruitful scientific discussion. We also thank the discovery platform and informatics group at EDyP proteomic facility at Grenoble University for their support. Funding: This work was supported by grants from Ministero della Salute (RF-2010-2309704 and RF-2016-02361040), Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG 12854), and Regione Friuli Venezia Giulia (RIFT project) to G.B., as well as grants from 5X1000 CRO Intramural Young Investigator and from Ministero della Salute (GR-2016-02361041) to M.So. Proteomic experiments were partially supported by the Proteomics French Infrastructure grant (ANR-10-INBS-08-01). Fondazione Biasotto ONLUS (Pordenone) supported A.C. by funding a PhD fellowship. Author contributions: G.B. and M.So. designed the experiments. M.So. and I.P. performed most of the experiments. A.C., M.C., A.D., and S.D. participated in the experimental work. G.L.R.V. and A.V. performed all pathological analyses. A.K. and Y.C. performed the proteomic MS analyses. D.C.C.-T. provided the primary OC cells and their gene expression profiles. C.D. and S.L. provided the IHC on human paired samples. R.S. contributed to the generation of USP1 KO cells. A.D. and M.Sc. performed the functional genomic screening. G.B., M.So., I.P., and B.B. analyzed the data and wrote the manuscript. B.B. and M.Sc. contributed to analyze the data and provided important intellectual support. All authors read and approved the manuscript. Competing interests: M.So. and G.B. are inventors on a submitted patent related to this work filed by the Centro di Riferimento Oncologico, IRCCS (no. 102019000000130, filed 7 January 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The primary epithelial OC cell lines #13914, #8587, #13699, #12370, and #14433 can be provided by D.C.C.-T. pending scientific review and completed material transfer agreement. Requests for those cell lines should be submitted to D.C.C.-T. (dan.cacsire-castillo@meduniwien.ac.at).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav3235/DC1
Fig. S1. USP1 regulates the response to CDDP in OC cells.
Fig. S2. USP1 binds and regulates Snail protein stability in control and CDDP-treated cells.
Fig. S3. Snail coordinates the USP1-mediated effects on CDDP-induced cell death and stem-like properties.
Fig. S4. USP1 is phosphorylated by ATM and ATR kinases on S42 and S331.
Fig. S5. USP1 is necessary for metastatic dissemination of OC cells.
Fig. S6. High USP1 expression predicts poor prognosis in patients with OC and is associated with acquired CDDP resistance.
Fig. S7. High USP1 expression predicts poor prognosis in patients with TNBC.
Table S1. Identification and quantification of USP1 phosphorylated peptides.
Table S2. Genes differentially expressed in responder versus nonresponder cells.
Table S3. KEGG pathways by the DAVID Bioinformatics tool in responder versus nonresponder cells.
REFERENCES AND NOTES
- 1.Kang X., Xiao H.-H., Song H.-Q., Jing X.-B., Yan L.-S., Qi R.-G., Advances in drug delivery system for platinum agents based combination therapy. Cancer Biol. Med. 12, 362–374 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddik Z. H., Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 22, 7265–7279 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Jayson G. C., Kohn E. C., Kitchener H. C., Ledermann J. A., Ovarian cancer. Lancet 384, 1376–1388 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Thibault B., Castells M., Delord J.-P., Couderc B., Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 33, 17–39 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Polyak K., Weinberg R. A., Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Haslehurst A. M., Koti M., Dharsee M., Nuin P., Evans K., Geraci J., Childs T., Chen J., Li J., Weberpals J., Davey S., Squire J., Park P. C., Feilotter H., EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 12, 91 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurrey N. K., Jalgaonkar S. P., Joglekar A. V., Ghanate A. D., Chaskar P. D., Doiphode R. Y., Bapat S. A., Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 27, 2059–2068 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Viñas-Castells R., Beltran M., Valls G., Gómez I., García J. M., Montserrat-Sentís B., Baulida J., Bonilla F., de Herreros A. G., Díaz V. M., The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J. Biol. Chem. 285, 3794–3805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou B. P., Deng J., Xia W., Xu J., Li Y. M., Gunduz M., Hung M.-C., Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 6, 931–940 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Huang T. T., Nijman S. M. B., Mirchandani K. D., Galardy P. J., Cohn M. A., Haas W., Gygi S. P., Ploegh H. L., Bernards R., D’Andrea A. D., Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 8, 339–347 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Nijman S. M. B., Huang T. T., Dirac A. M. G., Brummelkamp T. R., Kerkhoven R. M., D’Andrea A. D., Bernards R., The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17, 331–339 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Oestergaard V. H., Langevin F., Kuiken H. J., Pace P., Niedzwiedz W., Simpson L. J., Ohzeki M., Takata M., Sale J. E., Patel K. J., Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol. Cell 28, 798–809 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohn M. A., Kowal P., Yang K., Haas W., Huang T. T., Gygi S. P., D’Andrea A. D., A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell 28, 786–797 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Williams S. A., Maecker H. L., French D. M., Liu J., Gregg A., Silverstein L. B., Cao T. C., Carano R. A. D., Dixit V. M., USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 146, 918–930 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Taniguchi T., Tischkowitz M., Ameziane N., Hodgson S. V., Mathew C. G., Joenje H., Mok S. C., D’Andrea A. D., Disruption of the Fanconi anemia–BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 9, 568–574 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Das D. S., Das A., Ray A., Song Y., Samur M. K., Munshi N. C., Chauhan D., Anderson K. C., Blockade of deubiquitylating enzyme USP1 inhibits DNA repair and triggers apoptosis in multiple myeloma cells. Clin. Cancer Res. 23, 4280–4289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J.-K., Chang N., Yoon Y., Yang H., Cho H., Kim E., Shin Y., Kang W., Oh Y. T., Mun G. I., Joo K. M., Nam D.-H., Lee J., USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance. Neuro Oncol. 18, 37–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmed N., Abubaker K., Findlay J., Quinn M., Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets 10, 268–278 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Baldassarre G., Belletti B., Nicoloso M. S., Schiappacassi M., Vecchione A., Spessotto P., Morrione A., Canzonieri V., Colombatti A., p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell 7, 51–63 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Davidowitz R. A., Iwanicki M. P., Brugge J. S., In vitro mesothelial clearance assay that models the early steps of ovarian cancer metastasis. J. Vis. Exp. 2012, e3888 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidowitz R. A., Selfors L. M., Iwanicki M. P., Elias K. M., Karst A., Piao H., Ince T. A., Drage M. G., Dering J., Konecny G. E., Matulonis U., Mills G. B., Slamon D. J., Drapkin R., Brugge J. S., Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Invest. 124, 2611–2625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brozovic A., The relationship between platinum drug resistance and epithelial–mesenchymal transition. Arch. Toxicol. 91, 605–619 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Zheng X., Carstens J. L., Kim J., Scheible M., Kaye J., Sugimoto H., Wu C.-C., LeBleu V. S., Kalluri R., Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu D. S.-S., Lan H.-Y., Huang C.-H., Tai S.-K., Chang S.-Y., Tsai T.-L., Chang C.-C., Tzeng C.-H., Wu K.-J., Kao J.-Y., Yang M.-H., Regulation of excision repair cross-complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin. Cancer Res. 16, 4561–4571 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Díaz V. M., Viñas-Castells R., García de Herreros A., Regulation of the protein stability of EMT transcription factors. Cell Adh. Migr. 8, 418–428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traven A., Heierhorst J., SQ/TQ cluster domains: Concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 27, 397–407 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Sun M., Guo X., Qian X., Wang H., Yang C., Brinkman K. L., Serrano-Gonzalez M., Jope R. S., Zhou B., Engler D. A., Zhan M., Wong S. T. C., Fu L., Xu B., Activation of the ATM-Snail pathway promotes breast cancer metastasis. J. Mol. Cell Biol. 4, 304–315 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villamil M. A., Liang Q., Chen J., Choi Y. S., Hou S., Lee K. H., Zhuang Z., Serine phosphorylation is critical for the activation of ubiquitin-specific protease 1 and its interaction with WD40-repeat protein UAF1. Biochemistry 51, 9112–9123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra A. K., Davis D. A., Tomar S., Roy L., Gurler H., Xie J., Lantvit D. D., Cardenas H., Fang F., Liu Y., Loughran E., Yang J., Sharon Stack M., Emerson R. E., Cowden Dahl K. D., Barbolina M. V., Nephew K. P., Matei D., Burdette J. E., In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol. Oncol. 138, 372–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonego M., Schiappacassi M., Lovisa S., Dall’Acqua A., Bagnoli M., Lovat F., Libra M., D’Andrea S., Canzonieri V., Militello L., Napoli M., Giorda G., Pivetta B., Mezzanzanica D., Barbareschi M., Valeri B., Canevari S., Colombatti A., Belletti B., Del Sal G., Baldassarre G., Stathmin regulates mutant p53 stability and transcriptional activity in ovarian cancer. EMBO Mol. Med. 5, 707–722 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreuzinger C., Gamperl M., Wolf A., Heinze G., Geroldinger A., Lambrechts D., Boeckx B., Smeets D., Horvat R., Aust S., Hamilton G., Zeillinger R., Cacsire Castillo-Tong D., Molecular characterization of 7 new established cell lines from high grade serous ovarian cancer. Cancer Lett. 362, 218–228 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Sonego M., Pellizzari I., Dall’Acqua A., Pivetta E., Lorenzon I., Benevol S., Bomben R., Spessotto P., Sorio R., Gattei V., Belletti B., Schiappacassi M., Baldassarre G., Common biological phenotypes characterize the acquisition of platinum-resistance in epithelial ovarian cancer cells. Sci. Rep. 7, 7104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Network , Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egger S. J., Willson M. L., Morgan J., Walker H. S., Carrick S., Ghersi D., Wilcken N., Platinum-containing regimens for metastatic breast cancer. Cochrane Database Syst. Rev. 6, CD003374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batlle E., Clevers H., Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Varga J., Greten F. R., Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 19, 1133–1141 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Chen X., Liao R., Li D., Sun J., Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget 8, 17301–17312 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye X., Tam W. L., Shibue T., Kaygusuz Y., Reinhardt F., Ng Eaton E., Weinberg R. A., Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525, 256–260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye X., Brabletz T., Kang Y., Longmore G. D., Nieto M. A., Stanger B. Z., Yang J., Weinberg R. A., Upholding a role for EMT in breast cancer metastasis. Nature 547, E1–E3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma A., Tang M., Zhang L., Wang B., Yang Z., Liu Y., Xu G., Wu L., Jing T., Xu X., Yang S., Liu Y., USP1 inhibition destabilizes KPNA2 and suppresses breast cancer metastasis. Oncogene 38, 2405–2419 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Dall'Acqua A., Sonego M., Pellizzari I., Pellarin I., Canzonieri V., D'Andrea S., Benevol S., Sorio R., Giorda G., Califano D., Bagnoli M., Militello L., Mezzanzanica D., Chiappetta G., Armenia J., Belletti B., Schiappacassi M., Baldassarre G., CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol. Med. 9, 1415–1433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casabona M. G., Vandenbrouck Y., Attree I., Couté Y., Proteomic characterization of Pseudomonas aeruginosa PAO1 inner membrane. Proteomics 13, 2419–2423 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Engholm-Keller K., Hansen T. A., Palmisano G., Larsen M. R., Multidimensional strategy for sensitive phosphoproteomics incorporating protein prefractionation combined with SIMAC, HILIC, and TiO2 chromatography applied to proximal EGF signaling. J. Proteome Res. 10, 5383–5397 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Savitski M. M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B., Confident phosphorylation site localization using the Mascot Delta Score. Mol. Cell. Proteomics 10, M110.003830 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav3235/DC1
Fig. S1. USP1 regulates the response to CDDP in OC cells.
Fig. S2. USP1 binds and regulates Snail protein stability in control and CDDP-treated cells.
Fig. S3. Snail coordinates the USP1-mediated effects on CDDP-induced cell death and stem-like properties.
Fig. S4. USP1 is phosphorylated by ATM and ATR kinases on S42 and S331.
Fig. S5. USP1 is necessary for metastatic dissemination of OC cells.
Fig. S6. High USP1 expression predicts poor prognosis in patients with OC and is associated with acquired CDDP resistance.
Fig. S7. High USP1 expression predicts poor prognosis in patients with TNBC.
Table S1. Identification and quantification of USP1 phosphorylated peptides.
Table S2. Genes differentially expressed in responder versus nonresponder cells.
Table S3. KEGG pathways by the DAVID Bioinformatics tool in responder versus nonresponder cells.