

Immunoproteasome subunit β5i regulates cardiac hypertrophy.
Abstract
Pathological cardiac hypertrophy eventually leads to heart failure without adequate treatment. The immunoproteasome is an inducible form of the proteasome that is intimately involved in inflammatory diseases. Here, we found that the expression and activity of immunoproteasome catalytic subunit β5i were significantly up-regulated in angiotensin II (Ang II)–treated cardiomyocytes and in the hypertrophic hearts. Knockout of β5i in cardiomyocytes and mice markedly attenuated the hypertrophic response, and this effect was aggravated by β5i overexpression in cardiomyocytes and transgenic mice. Mechanistically, β5i interacted with and promoted ATG5 degradation thereby leading to inhibition of autophagy and cardiac hypertrophy. Further, knockdown of ATG5 or inhibition of autophagy reversed the β5i knockout-mediated reduction of cardiomyocyte hypertrophy induced by Ang II or pressure overload. Together, this study identifies a novel role for β5i in the regulation of cardiac hypertrophy. The inhibition of β5i activity may provide a new therapeutic approach for hypertrophic diseases.
INTRODUCTION
Cardiac hypertrophy is an important adaptive response to pathological stimuli, including hypertension, myocardial infarction, pressure overload, and the activation of the renin-angiotensin system (1). Hallmarks of cardiac hypertrophy include increased cell size, protein synthesis, actin reorganization, and fetal gene reexpression (1). Although the hypertrophic response may serve to maintain cardiac function for a certain period, prolonged hypertrophy becomes detrimental, resulting in cardiac dysfunction and heart failure (HF) (1). To date, multiple signaling pathways have been demonstrated to positively regulate protein synthesis and cardiac hypertrophy, including the insulin-like growth factor 1 receptor (IGF1R)/phosphatidylinositol 3-kinase (PI3K)/AKT, epidermal growth factor receptor (EGFR)/mitogen-activated protein kinase (MAPK), gp130/Janus kinase (Jak)/signal transducer and activator of transcription 3 (STAT3), and calcineurin/nuclear factor of activated T cells (NFAT) pathways (1). Thus, therapeutic strategies that modulate the expression and activation of these receptors and downstream mediators are promising.
The ubiquitin-proteasome system (UPS) and lysosomal-autophagy pathway are the two most important cellular mechanisms for protein degradation (2). Autophagy is often thought of as a nonspecific process that degrades long-lived proteins and aberrant protein aggregates (3). In contrast to autophagy, the UPS is the major pathway for intracellular protein degradation in eukaryotic cells (4). The standard 20S proteasome comprises three β subunits, namely, β1 (PSMB6), β2 (PSMB7), and β5 (PSMB5), which account for the caspase-like, trypsin-like, and chymotrypsin-like activity of the proteasome, respectively. Upon stimulation with cytokines such as interferon-γ (IFN-γ), three alternative β subunits (also termed immunosubunits)—β1i [large multifunctional peptidase 2 (LMP2) or PSMB9)], β2i (MECL-1 or PSMB10), and β5i (LMP7 or PSMB8)—are induced, which replace their standard subunits to form the 20S immunoproteasome (5). The primary function of the immunoproteasome is to improve major histocompatibility complex-I (MHC-I) antigen presentation (5). The immunoproteasome also has various biological functions, including proinflammatory cytokine production, T cell differentiation and survival, oxidative stress, and muscle mass (5). Previous results have shown that the expression and chymotrypsin-like activity of immunosubunit β5i are markedly increased in the heart under hypertrophic stress (6–8). Recently, our data revealed that β5i is also involved in the regulation of angiotensin II (Ang II)–induced atrial fibrillation in mice (9). However, the functional role of β5i in regulating cardiac hypertrophy remains unknown.
In this study, using primary cardiomyocytes, β5i knockout (KO) mice, and cardiac-specific β5i transgenic (β5i-Tg) mice, we found that β5i acts as a novel positive regulator of cardiac hypertrophy induced by Ang II or pressure overload by targeting the ubiquitination and degradation of autophagy-related 5 (ATG5). Thus, this study identifies a new role of β5i in the development of cardiac hypertrophy and highlights an essential compensatory link between the immunoproteasome and autophagy in the hypertrophic heart.
RESULTS
β5i expression is up-regulated in the hypertrophic heart and patients with HF
To identify the proteasome subunit expression profiles within the injured myocardium, we induced acute cardiac hypertrophy in wild-type (WT) mice by Ang II infusion and analyzed proteasome subunit expression using a microarray assay (10). We found that, among the 47 proteasome subunit genes, immunosubunit β5i (also known as PSMB8 or LMP7) was highly up-regulated in the heart on day 1 of Ang II infusion (P < 0.05 versus saline; Fig. 1A). The increased expression of β5i mRNA was confirmed by quantitative polymerase chain reaction (qPCR) analysis (Fig. 1B). Meanwhile, the mRNA level of β2i (also known as PSMB10) was increased in Ang II–treated heart but was less than that of β5i (P < 0.05; Fig. 1B). However, the expression of standard subunits (β1, β2, and β5) and immunosubunit β1i was not different in heart tissue after Ang II treatment (Fig. 1, A and B). Moreover, the β5i protein level was increased in Ang II–treated neonatal rat cardiomyocytes (NRCMs) (P < 0.05 or 0.01; Fig. 1C). The expression of β5i was also up-regulated in mouse heart after 2 weeks of Ang II infusion (P < 0.01; Fig. 1D) and transverse aortic constriction (TAC) (Fig. 1E). β5i expression was not altered in neonatal rat cardiac fibroblasts after Ang II stimulation (Fig. 1F).
Fig. 1. β5i is increased in hypertrophic hearts and HF patients.
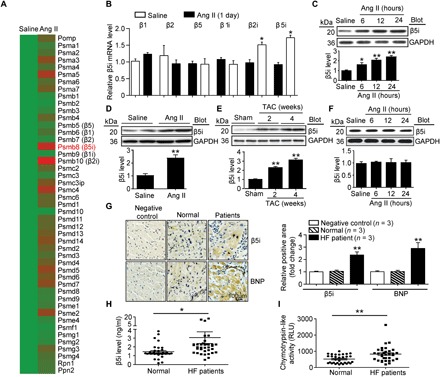
(A) Cluster of the proteasome gene expression profiles in saline- or Ang II–infused mouse heart at day 1 (n = 3 samples per group). (B) qPCR analyses of β1, β2, β5, β1i, β2i, and β5i mRNA expression in saline- or Ang II–infused mouse heart at day 1 and control (n = 3 per group). (C) Protein levels of β5i in NRCMs exposed to Ang II (100 nM) at different time points (top) and quantification (bottom, n = 3). (D and E) Protein levels of β5i in WT hearts after 2 or 4 weeks of Ang II infusion (1000 ng/kg per min) or TAC operation (top) and quantification (bottom, n = 3). (F) Neonatal rat fibroblasts were treated with Ang II for 0 to 24 hours. The β5i protein expression was examined by Western blot analysis (top) and quantification of the relative protein levels (bottom, n = 3). (G) Representative immunohistochemical staining of β5i and BNP proteins in the heart tissues from normal control and HF patients (left). Quantification of β5i- and BNP-positive areas (right, n = 3). (H and I) β5i level and chymotrypsin-like activity in blood samples of normal controls (n = 38) and HF patients (n = 38). Data are presented as means ± SEM, and n represents the number of samples per group. *P < 0.05 and **P < 0.01 versus saline, sham, or normal control. RLU, relative luciferase units.
To examine whether β5i has an important role in human HF, we examined the expression of β5i and B-type natriuretic peptide (BNP; a marker for HF) in heart tissue. Immunohistochemistry revealed that the expression of both β5i and BNP in the failing heart was significantly higher than in normal controls (P < 0.01; Fig. 1G). Moreover, the levels of serum β5i and chymotrypsin-like activity were also increased (P < 0.05 or 0.01) in patients with HF compared with control individuals (P < 0.05 or 0.01; Fig. 1, H and I, and table S1). We then analyzed the relationship of the levels of serum β5i or chymotrypsin-like activity in patients with HF by simple cross-tabulation and the calculations of odds ratios (ORs). Multivariable logistic regression analyses were conducted to test the independent contribution of the levels of serum β5i or chymotrypsin-like activity to HF. After adjusting for sex, age, and cardiovascular risk factors, a significant association was found between the levels of β5i (OR, 12.123) or chymotrypsin-like activity (OR, 1.005) and HF (P < 0.01; table S2). Overall, these results suggest that β5i may play a critical role in the regulation of cardiac hypertrophy.
Knockdown of β5i inhibits cardiomyocyte hypertrophy in vitro
To determine the functional role of β5i in the heart under hypertrophic conditions, we first examined whether β5i exerts a pro- or anti-hypertrophic effect in vitro. NRCMs were infected with an adenovirus vector expressing small interfering RNA (siRNA) against β5i (siRNA-β5i) or scrambled control (siRNA-control). The level of endogenous β5i protein was reduced by approximately 80% upon transfection with siRNA-β5i (P < 0.01; fig. S1A). Notably, knockdown of β5i attenuated the Ang II–induced increase of cardiomyocyte size and the expression of atrial natriuretic factor (ANF) and BNP compared with the siRNA-control (P < 0.05; Fig. 2, A and B). To corroborate these findings, NRCMs were infected with an adenovirus vector overexpressing β5i (Ad-β5i) or green fluorescent protein (Ad-GFP). Infection of NRCMs with Ad-β5i increased the level of β5i 2.3-fold (P < 0.01; fig. S1B) and enhanced cardiomyocyte size and the expression of ANF and BNP compared with the Ad-GFP control after Ang II treatment (P < 0.05; Fig. 2, C and D). In addition, we found that siRNA knockdown of β5i reduced the phosphorylation levels of AKT and extracellular signal–regulated kinase 1/2 (ERK1/2) compared with the siRNA-control after Ang II stimulation (P < 0.05 or 0.01; Fig. 2E). However, knockdown or overexpression of β5i had no effect on cardiomyocytes in the basal condition (Fig. 2, A to D).
Fig. 2. Knockdown of β5i inhibits cardiomyocyte hypertrophy and activates autophagy.
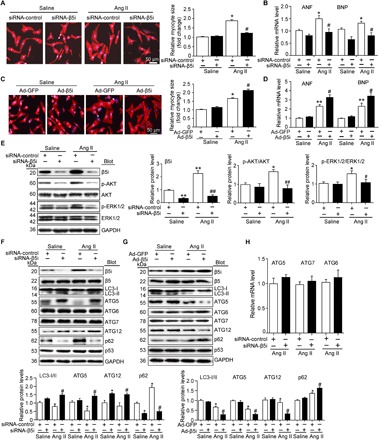
(A) Representative images of double immunostaining [green for α-actinin and blue for 4′,6-diamidino-2-phenylindole (DAPI)] of NRCMs transfected with Ad-siRNA-β5i or siRNA-control after Ang II stimulation. Quantification of cell surface area (at least 150 cells counted per experiment; right). (B) qPCR analyses of ANF and BNP mRNA expression (right, n = 5). (C) Immunostaining of cardiomyocyte size with α-actinin (red) and DAPI (blue) infected with Ad-GFP or Ad-β5i after Ang II stimulation. Quantification of cell surface area (at least 150 cells counted per experiment; right). (D) qPCR analyses of ANF and BNP mRNA expression (right, n = 5). (E) Protein levels of β5i, p-AKT, AKT, p-ERK1/2, and ERK1/2 in NRCMs treated as in (A) and quantification (n = 3). (F) Protein levels of β5i, LC3, ATG5, ATG6, ATG7, ATG12, p62, and p53 in NRCMs infected with scrambled siRNA-control or siRNA-β5i after Ang II treatment (top) and quantification (bottom, n = 3). (G) Protein levels of β5i, LC3, ATG5, ATG6, ATG7, ATG12, p62, and p53 in NRCMs infected with Ad-GFP control or Ad-β5i after Ang II treatment (top) and quantification (bottom, n = 3). (H) qPCR analysis of autophagy-related genes ATG5, ATG7, and ATG6 (Beclin-1) and mRNA levels in primary cardiomyocytes infected with siRNA-control or siRNA-β5i after Ang II treatment (n = 3). Data are presented as means ± SEM, and n represents the number of samples. For (A), (B), (E), and (F), *P < 0.05 and **P < 0.01 versus siRNA-control + saline; #P < 0.05 and ##P < 0.01 versus siRNA-control + Ang II. For (C), (D), and (G), *P < 0.05 and **P < 0.01 versus Ad-GFP + saline; #P < 0.05 versus Ad-GFP + Ang II.
Next, we examined the effect of β5i on phenylephrine-treated cardiomyocytes. Consistent with the data from Ang II–treated cells, knockdown of β5i also reduced phenylephrine-induced cardiomyocyte hypertrophy and the expression of ANF compared with the siRNA-control (P < 0.05; fig. S2, A and B), whereas the increase of cardiomyocyte hypertrophy and the expression of ANF were enhanced in Ad-β5i–infected NRCMs after phenylephrine treatment (P < 0.05; fig. S2, C and D). Collectively, our data indicate that knockdown of β5i inhibits cardiomyocyte hypertrophy in vitro.
β5i regulates the induction of autophagy by targeting ATG5
Growing evidence suggests that inhibition of the proteasome is compensated for by the up-regulation of autophagy (2, 11); thus, we examined the effect of β5i on autophagy. Two reliable markers of autophagy activation, the formation of autophagy flux and the conversion of LC3-I to LC3-II, were examined (12). Fluorescence microscopy revealed that Ang II treatment for 24 hours decreased the number of autophagosomes and autolysosomes, as well as the conversion of LC3-I to LC3-II (P < 0.05; Fig. 2, F and G, and fig. S3, A and B). This decrease was reversed in β5i KO NRCMs compared with the siRNA-control (P < 0.05; Fig. 2F and fig. S3A) but was aggravated in Ad-β5i–infected NRCMs compared with the Ad-GFP control after Ang II treatment (P < 0.05; Fig. 2G and fig. S3B), indicating that knockdown of β5i was sufficient to promote the induction of autophagy upon Ang II treatment.
To determine how β5i regulates the activation of autophagy, we analyzed several key autophagy proteins, including ATG5, ATG6/Beclin-1, ATG7, ATG12, p62, and p53, which play critical roles in the induction of autophagy (2). We found that knockdown of endogenous β5i in NRCMs up-regulated ATG5 and ATG12 protein levels (P < 0.05) but did not affect the levels of the standard proteasome β5 subunit, ATG6, ATG7, and p53 compared with the siRNA-control after saline or Ang II stimulation (Fig. 2F). Conversely, the protein levels of ATG5 and ATG12 were reduced in Ad-β5i–infected cells compared with the Ad-GFP control after Ang II treatment (P < 0.05; Fig. 2G). In addition, p62 expression was also decreased in β5i KO NRCMs after saline or Ang II treatment but increased in Ad-β5i–infected NRCMs (P < 0.05; Fig. 2, F and G). Knockdown of β5i did not alter the expression of ATG5, ATG6, and ATG7 at the transcriptional level after Ang II treatment (Fig. 2H). These results suggest that β5i influences the induction of autophagy by regulating the stability of ATG5 protein.
β5i interacts with ATG5
To test whether β5i mediates its effects on ATG5 through a direct interaction, we performed coimmunoprecipitation assays in primary cardiomyocytes. β5i protein was efficiently precipitated by an antibody against ATG5 in NRCMs but not by a nonspecific immunoglobulin G (IgG) control (Fig. 3A). Moreover, we transfected human embryonic kidney (HEK) 293 cells with Myc-tagged ATG5 and Flag-tagged β5i. An immunoprecipitation assay showed that Flag-β5i was detected in the Myc-ATG5 immune complex, whereas no Flag-β5i was found in the controls (Fig. 3B), indicating that β5i interacts directly with ATG5.
Fig. 3. β5i interacts with ATG5 and promotes its degradation.
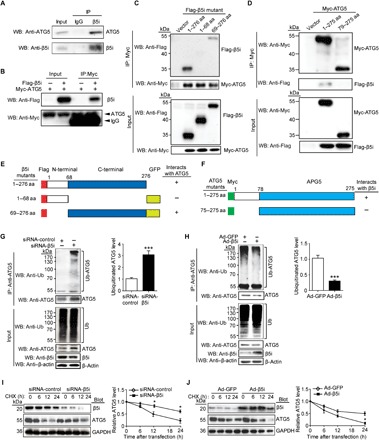
(A) Endogenous protein interactions were examined in cardiomyocyte lysates immunoprecipitated (IP) with rabbit IgG or anti-β5i antibody and analyzed by Western blot (WB) with antibody to detect ATG5 and β5i. (B) HEK293 cells were transfected with the indicated plasmids. Equal amounts of protein lysates were immunoprecipitated with anti-Myc antibody and analyzed by WB with the indicated antibodies. (C) Domains of β5i involved in binding to ATG5. WT β5i and β5i truncations expressed in HEK293 cells were mapped with Flag-GFP tagged. Flag-GFP–tagged β5i proteins were precipitated with anti-Myc antibody and analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) with antibodies against Flag and Myc. aa, amino acids. (D) Interaction domains of ATG5 required for binding to β5i were mapped by coimmunoprecipitation. HEK293 cells were transfected with Flag-GFP-β5i and Myc-ATG5 (full-length and truncations) plasmids. Equal amounts of lysates were prepared for immunoprecipitation with a Myc antibody and immunoblotted with antibodies for Myc or Flag. (E) Deletion constructs of β5i in (C) that bind to ATG5. (F) Deletion constructs of ATG5 in (E) that bind to β5i. (G and H) Lysates from NRCMs infected with siRNA-control, siRNA-β5i, Ad-GFP, or Ad-β5i were immunoprecipitated with anti-ATG5 antibody. Western blot analysis of ubiquitin-conjugated ATG5 with anti-ubiquitin (Ub) and anti-ATG5 (top, n = 3). Input showed the expression of the corresponding proteins in whole-cell lysates (bottom). Quantification of ubiquitinated ATG5 (right). (I and J) NRCMs were infected with siRNA-control, siRNA-β5i, Ad-GFP, or Ad-β5i and then treated with cycloheximide (CHX; 10 μM) for the indicated time periods. Representative Western blot analysis of β5i and ATG5 protein levels for each group (left) and quantification of ATG5 level (right, n = 3). GAPDH as an internal control. *P < 0.05 and ***P < 0.001 versus siRNA-control or Ad-GFP.
To map the sites for the interaction between β5i and ATG5, we tested a full-length Flag-tagged β5i and GFP-Flag–tagged β5i deletion mutants for their ability to bind to full-length Myc-tagged ATG5 expressed in HEK293 cells. Coimmunoprecipitation assays with an anti-Flag antibody revealed that amino acids 69 to 276 of β5i were required for its binding to ATG5 (Fig. 3, C and E). To identify the regions in ATG5 required for its interaction with β5i, a full-length Myc-tagged ATG5 and deletion mutants were used in coimmunoprecipitation assays, with full-length Flag-tagged β5i expressed in HEK293 cells. We found that amino acids 1 to 78 within ATG5 were necessary for its interaction with β5i (Fig. 3, D and F).
β5i promotes the degradation of ATG5
β5i exerts chymotrypsin-like proteolytic activity (5); thus, we studied whether it regulates ATG5 ubiquitination and degradation in cardiomyocytes. Immunoprecipitation analysis revealed that knockdown of endogenous β5i in NRCMs significantly increased ubiquitinated ATG5 and its protein level compared with the siRNA-control (Fig. 3G), whereas overexpression of β5i had the opposite effect (Fig. 3H). To further test whether β5i is involved in the degradation of ubiquitinated ATG5 protein, we performed a pulse-chase assay in NRCMs with cycloheximide. Knockdown of β5i with siRNA significantly prolonged the half-life of ATG5 protein compared with the siRNA-control (Fig. 3I), whereas its half-life was markedly reduced in Ad-β5i–infected NRCMs compared with the Ad-GFP control (Fig. 3J). Together, these results indicate that ATG5 is a previously unrecognized substrate for β5i in cardiomyocytes.
β5i regulates cardiomyocyte size through autophagy
To determine whether β5i regulates cardiomyocyte size via autophagy, we infected cardiomyocytes with siRNAs to reduce the levels of endogenous β5i and ATG5. Western blot analysis showed that the reduction of β5i and ATG5 levels was achieved by siRNA-β5i and siRNA-ATG5, respectively, as compared with the siRNA-control (P < 0.05 or 0.01; Fig. 4C). Moreover, knockdown of β5i attenuated the Ang II–induced increase of cardiomyocyte size, ANF expression, and phosphorylation of AKT and ERK1/2 compared with the siRNA-control (P < 0.05; Fig. 4, A and B, lane 2 versus lane 1), whereas this decrease was markedly or fully reversed by knockdown of ATG5 or both β5i and ATG5 (P < 0.05; Fig. 4, A and B, lane 3 or 4 versus lane 2).
Fig. 4. KO of β5i suppresses cardiomyocyte size through autophagy.
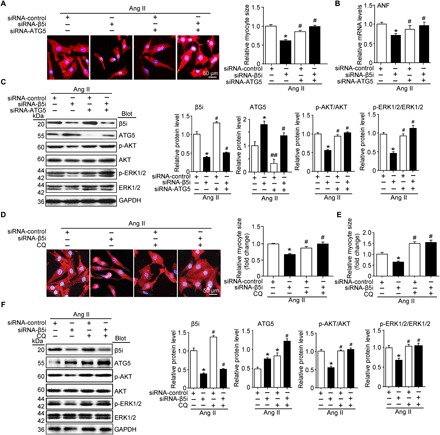
(A) NRCMs were infected with siRNA-control or siRNA-β5i and then treated with Ang II for 24 hours. Representative images of double immunostaining (red: α-actinin for cardiomyocytes; blue: DAPI for nuclei) for measurement of cell size. Quantification of myocyte surface area (right, 150 cells counted per experiment, n = 3). Scale bar, 50 μm. (B) qPCR analysis of ANF mRNA expression in NRCMs treated as in (A) (n = 3). (C) Protein levels of β5i, ATG5, p-AKT, AKT, p-ERK1/2, and ERK1/2 in NRCMs transfected with siRNA-control, siRNA-β5i, or siRNA-ATG5 after 24 hours of saline or Ang II stimulation and quantification of each protein level (n = 3). (D) NRCMs were infected with siRNA-control or Ad-siRNA-β5i and then treated with chloroquine (CQ) (5 μM) and Ang II for 24 hours. Representative images of double immunostaining for measurement of cardiomyocyte size (left). Quantification of cell surface area (150 cells counted per experiment, n = 3). Scale bar, 50 μm. (E) qPCR analysis of ANF mRNA expression in NRCMs treated as in (D) (n = 5). (F) Protein levels of β5i, ATG5, p-AKT, AKT, p-ERK1/2, and ERK1/2 in NRCMs treated as in (D) and quantification of each protein level (n = 3). GAPDH as an internal control. Data are presented as means ± SEM. *P < 0.05 versus siRNA-control + Ang II; #P < 0.05 and ##P < 0.01 versus siRNA-β5i + Ang II.
To further confirmed the effect of autophagy on cardiomyocyte size and related signaling pathways, cardiomyocytes were infected with siRNA-β5i and treated with chloroquine, an inhibitor of autophagy. We found that knockdown of β5i increased ATG5 expression compared with the siRNA-control, and chloroquine treatment further enhanced this effect (P < 0.05; Fig. 4F). Consistent with observations from siRNA-ATG5–infected cells (Fig. 4, A to C), the β5i knockdown-mediated attenuation of the Ang II–induced enlargement of cardiomyocyte size, ANF expression, and phosphorylation of AKT and ERK1/2 were also reversed by chloroquine treatment in siRNA-β5i–infected cells (P < 0.05; Fig. 4, D to F, lane 4 versus lane 2). In addition, the inhibitory effect of β5i knockdown on cardiomyocyte hypertrophy was restored by chloroquine alone (P < 0.05; Fig. 4, D to F, lane 3 versus lane 2). Together, these results indicate that blocking β5i activity is compensated for by the induction of autophagy, and the inhibition of autophagy reverses the protective effect of β5i knockdown.
Deficiency of β5i improves pressure overload–induced cardiac hypertrophy in mice
To verify the effect of β5i on cardiac hypertrophy in vivo, we subjected WT and β5i KO mice to TAC or sham surgery for 6 weeks. KO of β5i reduced TAC-induced chymotrypsin-like activity (P < 0.05; fig. S4A) but did not affect caspase- and trypsin-like activity compared with the WT control after TAC (fig. S4A). Echocardiography showed characteristics of HF reflected by a significant decrease in the left ventricular ejection fraction (EF%) and fractional shortening (FS%) in TAC mice compared with the sham group (P < 0.01), whereas this effect was markedly reversed in β5i KO mice (P < 0.05; Fig. 5A). Furthermore, TAC-induced cardiac hypertrophy and fibrosis of WT mice, as reflected by the increased ratios of heart weight (HW)/body weight (BW) and HW/tibia length (TL), myocyte cross-sectional area, and expression of ANF, BNP, collagen I, and collagen III, were attenuated in β5i KO mice (P < 0.05 or 0.01; Fig. 5, B to D, and fig. S4, B and C). Notably, the expression of tumor necrosis factor–α (TNF-α) mRNA was also increased in WT mice (P < 0.001; fig. S4D) but was reduced in β5i KO mice after TAC (P < 0.01; fig. S4D). There was no notable increase in the expression of markers for cardiac contractile function, hypertrophy, and fibrosis between the two groups after sham surgery (Fig. 5, A to D).
Fig. 5. Deficiency of β5i attenuates pressure overload–induced cardiac hypertrophy in mice.
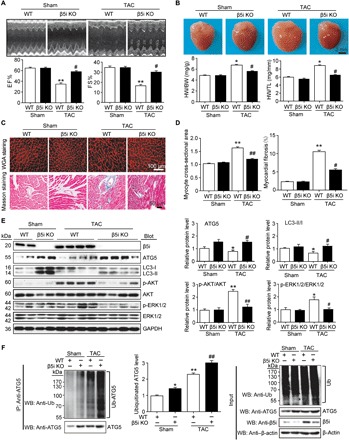
WT and β5i KO mice were subjected to sham or TAC operation for 6 weeks. (A) Representative M-mode echocardiography of the left ventricle (top). Measurement of ejection fraction (EF%) and fractional shortening (FS%) (bottom, n = 7). (B) Representative images of heart size photographed with a stereomicroscope (top) and HW/BW and HW/TL ratios (bottom, n = 7). (C) Cardiac myocyte size and fibrosis were detected by tetramethyl rhodamine isothiocyanate (TRITC)–labeled wheat germ agglutinin (WGA) staining and Masson’s trichrome staining, respectively. Scale bar, 50 μm. (D) Quantification of the relative myocyte cross-sectional area (200 cells counted per heart) and fibrosis (n = 5). (E) Protein levels of β5i, ATG5, LC3, p-AKT, AKT, p-ERK1/2, and ERK1/2 in the heart tissues and quantification (n = 5). GAPDH as an internal control. (F) Lysates from heart tissues of WT β5i KO mice after 6 weeks of sham or TAC were immunoprecipitated with anti-ATG5 antibody. Western blot analysis of ATG5 ubiquitination with anti-ubiquitin or ATG5 antibody (left). Quantification of the relative ubiquitinated ATG5 level (middle, n = 3) and input (Western blot analysis of each protein with corresponding antibody) (right). β-Actin as an internal control. Data are presented as means ± SEM, and n represents the number of animals per group. *P < 0.05 and **P < 0.01 versus WT + sham; ##P < 0.01 versus WT + TAC.
To confirm the in vivo action of β5i on ATG5 stability and the induction of autophagy, we examined the protein levels of ATG5, LC3, and hypertrophic mediators in WT and β5i KO hearts. TAC reduced the level of ATG5 and the LC3-II/LC3-I ratio but increased the phosphorylation of AKT and ERK1/2 in WT mice compared with sham mice (P < 0.05 or 0.01; Fig. 5E), but the reduction of ATG5 expression and the LC3-II/LC3-I ratio and the increase of AKT and ERK1/2 phosphorylation were reversed in β5i KO hearts compared with WT hearts after TAC (P < 0.05 or 0.01; Fig. 5E). In addition, the levels of ubiquitinated ATG5 and ATG5 protein expression were also increased in β5i KO mice compared with WT mice after sham or TAC surgery (P < 0.05 or 0.01; Fig. 5F).
Cardiac-specific β5i overexpression accelerates pressure overload–induced cardiac hypertrophy in mice
To further evaluate whether β5i overexpression influences cardiac hypertrophy in vivo, we generated β5i-Tg mice and confirmed the increased expression (approximately 2.5-fold versus WT; P < 0.01) and specificity of β5i in the heart (fig. S5A). At 3 weeks after TAC surgery, the death rate of β5i-Tg mice was significantly higher than that of WT mice (80% versus 20%, respectively; P < 0.001) (fig. S5B). β5i-Tg mice had more severe left ventricular dilation and cardiac dysfunction, as reflected by a marked decrease in left ventricular EF% and FS%, compared with WT mice (P < 0.001; Fig. 6A). Moreover, TAC-induced cardiac hypertrophy (increased HW/BW and HW/TL ratios and the cross-sectional area of myocytes), left ventricular dilation, and fibrosis were further aggravated in β5i-Tg mice compared with WT control mice (P < 0.05 or 0.01; Fig. 6, B to D). Accordingly, β5i overexpression in mice markedly reduced the protein levels of ATG5 and the LC3-II/I ratio, but enhanced the phosphorylation of AKT and ERK1/2, compared with the WT control after TAC (Fig. 6E). In addition, β5i overexpression in mice also decreased the ubiquitination and protein levels of ATG5 in heart tissue compared with WT mice after sham or TAC surgery (P < 0.05; Fig. 6F).
Fig. 6. Cardiac-specific overexpression of β5i aggravates pressure overload–induced cardiac hypertrophy.
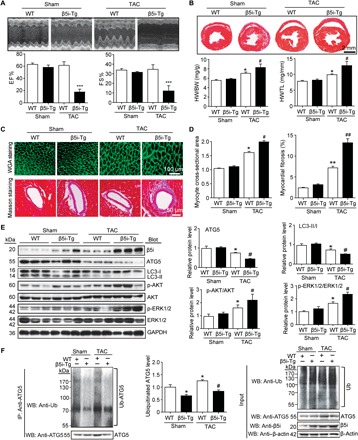
WT and β5i-Tg mice were subjected to sham or TAC operation for 3 weeks. (A) Representative M-mode echocardiography of the left ventricle (top). Measurement of EF% and FS% (bottom, n = 5). (B) Hematoxylin and eosin staining of heart sections and HW/BW and HW/TL ratios (bottom, n = 5). (C) Cardiac myocyte size and fibrosis were detected by FITC-labeled WGA staining (top) and Masson’s trichrome staining (bottom). Scale bar, 100 μm. (D) Quantification of the relative myocyte cross-sectional area (200 cells counted per heart, left) and fibrotic area (right, n = 5). (E) Protein levels of β5i, ATG5, LC3, p-AKT, AKT, p-ERK1/2, and ERK1/2 in the heart tissues (left) and quantification (right, n = 5). (F) Lysates from WT and β5i-Tg mice heart after 3 weeks of sham or TAC were immunoprecipitated with anti-ATG5 antibody. Western blot analysis of ubiquitin-conjugated ATG5 (left). Quantification of the relative ubiquitinated ATG5 level (middle, n = 3) and input of each protein (right). Data are presented as means ± SEM, and n represents the number of animals per group. *P < 0.05 and **P < 0.01 versus WT + sham; #P < 0.05 and ##P < 0.01 versus WT + TAC.
Knockdown of ATG5 abolishes cardioprotection in β5i-deficient mice after pressure overload
To test whether ATG5-dependent autophagy mediates the inhibitory effect of β5i deletion on cardiac hypertrophy, we injected WT or β5i KO mice with rAAV9-siRNA to knock down endogenous ATG5 expression. At 3 weeks after rAAV9-siATG5 injection, the mice were subjected to TAC for an additional 6 weeks. We found that AAV9-siATG5 injection significantly reduced the ATG5 protein level in the heart compared with rAAV9-sicontrol (P < 0.05; fig. S6F). β5i KO mice exhibited improved cardiac function, as reflected by EF% and FS%, as well as reduced left ventricular dilation, HW/BW and HW/TL ratios, myocyte size, fibrosis, and expression of ANF mRNA, compared with rAAV9-sicontrol mice at 6 weeks after TAC (P < 0.05 or 0.01; fig. S6, A to E, lane 3 versus lane 1), whereas these effects were reversed in rAAV9-siATG5–injected β5i KO mice (P < 0.05; fig. S6, A to E, lane 4 versus lane 3). Accordingly, knockdown of ATG5 in β5i KO mice also reduced the LC3-II/I ratio, but increased ERK1/2 activation, compared with rAAV9-sicontrol mice (P < 0.05 or 0.01; fig. S6F, lane 4 versus lane 3). ATG5 knockdown in WT mice had a similar effect compared with rAAV9-sicontrol animals after TAC (fig. S6, A to E, lane 2 versus lane 1). Hence, these in vivo data suggest that β5i KO attenuates cardiac hypertrophy by increasing the level of ATG5.
DISCUSSION
In this study, with the use of loss- and gain-of-function approaches, we identified a novel role for immunosubunit β5i in the regulation of cardiac hypertrophy and dysfunction. KO of β5i in primary cardiomyocytes and mice attenuated cardiac hypertrophy. In contrast, overexpression of β5i aggravated this effect, demonstrating a pro-hypertrophic role for β5i. We further found that ATG5, a key regulator of autophagy, was a direct target of β5i. The overexpression of β5i promoted the degradation of ubiquitinated ATG5, which inhibited the induction of autophagy, thereby leading to cardiac hypertrophy. These results suggest an essential compensatory link between the immunoproteasome and autophagy in the heart and highlight that β5i may be a potential therapeutic target for hypertrophic diseases. These data are summarized in fig. S7.
The immunoproteasome is constitutively expressed in immune cells and is induced in immune and nonimmune cells under conditions of inflammation and other forms of stress through different mechanisms (5). Proinflammatory cytokines, such as IFN-γ, IFN-α/β, and TNF-α, can induce the expression of the β1i, β2i, and β5i immunosubunits by activating the STAT1-IRF1 (interferon regulatory factor 1) pathway (5). Some cytokine-independent regulators, such as nitric oxide, also stimulate immunosubunit expression through the guanosine 3′,5′-monophosphate (cGMP)/adenosine 3′,5′-monophosphate (cAMP)—protein kinase A (PKA)/PKG/cAMP response element–binding protein (CREB) pathway (5). Recently, our data revealed that Ang II increases the expression of the β1i, β2i, and β5i immunosubunits and proteasome activity partially via the AT1R-PKA signaling pathway in Jurkat cells (13). Moreover, the proteolytic activity and expression of the β2i and β5i immunosubunits are up-regulated in primary cardiomyocytes, heart, and atrial tissue after Ang II stimulation (6, 9, 14). In the present study, we found that β5i expression and chymotrypsin-like activity were markedly up-regulated in primary cardiomyocytes and heart tissue from mice after hypertrophic stimuli or in serum from patients with HF (Fig. 1), suggesting that increased β5i expression may regulate cardiac hypertrophy. Moreover, inflammatory cytokines such as TNF-α were markedly up-regulated by Ang II (fig. S4D), which may induce immunosubunit β5i expression in the hypertrophic heart. However, the molecular mechanism by which Ang II up-regulates β5i expression remains to be determined.
The heart is capable of significant remodeling and hypertrophic growth as a means of adapting to injury or altered workloads (1). Cardiac hypertrophy involves alterations in the balance between protein synthesis and degradation, and there have been major advances in the identification of the genes and signaling pathways involved in this process (1). In recent years, the importance of the UPS in the development of cardiac remodeling has been reported. The immunoproteasome has several pathophysiological functions in the regulation of inflammatory diseases (5). Recent studies have indicated that the immunosubunits, such as β2i and β5i, are involved in the control of several cardiovascular diseases, such as enterovirus myocarditis (15), vascular cell apoptosis (16), deoxycortone acetate/salt-induced cardiac hypertrophy (17), and Ang II–induced atrial fibrillation (9, 14), through different mechanisms. However, the functional role of β5i in regulating cardiac hypertrophy remains unknown. The present study extended the previous findings and demonstrated that β5i was also involved in the regulation of cardiac hypertrophy. KO of β5i efficiently attenuated Ang II– or pressure overload–induced cardiomyocyte hypertrophy accompanied with inhibition of autophagy in vitro and in vivo, but these effects were reversed by overexpression of β5i (Figs. 2, 4, 5, and 6). Thus, these data suggest that β5i is a previously unrecognized regulator of cardiac hypertrophy, which may be related to autophagy.
Autophagy is a highly conserved catabolic process that targets proteins and organelles for lysosomal-mediated degradation. The induction of autophagy is controlled by a specific set of autophagy genes including ATG5, ATG6, ATG7, and ATG8/LC3 (18–20). Among them, ATG5, a key protein involved in the extension of the phagophore membrane in autophagic vesicles, is activated by ATG7 and forms a complex with ATG12 and ATG16L1. This complex is necessary for the conjugation of LC3-I to phosphatidylethanolamine to form LC3-II. After formation of the autophagosome, the ATG12-ATG5:ATG16L complex dissociates from the autophagosome (18–20). A wide variety of hypertrophic stimuli can regulate autophagy in cardiomyocytes. Subjection to TAC for 2 to 4 weeks usually induces the expression of ATG5, ATG6/Beclin-1, ATG16L1, and LC3-II, leading to the induction of autophagy in the heart (21, 22). Conversely, exposure to chronic pressure load for 5 to 6 weeks inhibits the conversion of LC3-I to LC3-II and the activation of autophagy (23). Basic autophagy is required for the physiological maintenance of cellular homeostasis, but excessive autophagy or insufficient autophagy may influence cell survival, cardiac hypertrophy, and HF (24, 25). To date, the beneficial role of autophagy in hypertrophic remodeling remains controversial. For example, the heterozygous deletion of ATG6/Beclin-1 reduces pressure overload–induced cardiomyocyte autophagy and pathological remodeling. Conversely, the cardiomyocyte-specific overexpression of ATG6 amplifies this process (22), indicating a maladaptive role of autophagy in the pathogenesis of pressure overload–induced HF. However, completely blocking autophagy induces cardiomyopathy. Cardiac-specific deficiency of ATG5 results in left ventricular hypertrophy, dilatation, and contractile dysfunction and mitochondrial misalignment and aggregation at 1 week after pressure overload (18). However, the role of autophagy in cardiac hypertrophy requires further study. Consistent with the idea outlined by Xie et al. (23), our data revealed that Ang II or TAC treatment markedly reduced the ATG5 protein level, LC3-I to LC3-II conversion, and autophagy, thereby contributing to cardiomyocyte hypertrophy (Figs. 2, F and G, 5E, and 6E). The inhibition of autophagy induced by Ang II or TAC was attenuated by β5i KO but further enhanced by overexpression of β5i in primary cardiomyocytes and in the heart (Figs. 2, F and G, 5E, and 6E). Furthermore, knockdown of ATG5 or inhibition of autophagy with chloroquine abolished the β5i KO-mediated attenuation of cardiac hypertrophy in vitro and in vivo (Fig. 4 and fig. S6), suggesting that ATG5-mediated autophagy is essential for the inhibition of cardiomyocyte hypertrophy. Overall, these results indicate that β5i modulates cardiac hypertrophy likely by targeting ATG5-mediated autophagy.
The UPS and autophagy have been considered two independent protein degradative systems for a long time (3, 5). However, several studies have revealed a link between the UPS and autophagy in neurons and other cells (2, 11, 26, 27). Multiple mechanisms by which the UPS regulates autophagy have been proposed. Following proteasomal inhibition, the accumulation of misfolded proteins leads to the induction of the unfolded protein response, which stabilizes transcription factors such as ATF4, resulting in the up-regulation of ATG5 and ATG7 or LC3 expression (28, 29). Moreover, proteasomal inhibition also increases the level of p53, leading to the induction of autophagy (30). Here, our data extended the mechanisms by which the proteasome alters autophagy, and found that β5i could regulate ATG5 stability, thereby contributing to the induction of autophagy (Fig. 3), suggesting that the immunoproteasome (β5i) is a novel regulator of autophagy in cardiomyocytes.
There are several potential mechanisms involved in the regulation of ATG5 expression and activation. For example, ATG5 is regulated by several transcription factors, including p73, GATA4, E2F1, ATF4, and CHOP (31–34). PDCD4 inhibits ATG5 protein translation by binding to the RNA-helicase EIF4A (35). Moreover, RAB37 and RACK1 interact directly with ATG5 and promote autophagosome formation, but caveolin-1 interacts with the ATG12-ATG5 system to suppress autophagy (36, 37). Protein kinases such as Gadd45ß-MEKK4-p38MAPK can phosphorylate ATG5 at Thr75, leading to its inactivation and the inhibition of autophagy (38). ATG5 expression can be negatively regulated posttranslationally by the microRNA 181A (39). Furthermore, methylation of the ATG5 promoter leads to down-regulation of ATG5 and inhibition of autophagy (40), whereas deacetylation of ATG5 by Sirt1 induces autophagy (41). In addition, cysteine proteases, such as calpain, cleave ATG5, leading to the inhibition of ATG5 (42). Notably, our recent results showed that the E3 ligase CDC20 associates with LC3 and promotes the ubiquitination and degradation of LC3 by the proteasome, thereby attenuating autophagy in cardiomyocytes (23), indicating that ubiquitination is an important mechanism mediating the induction of autophagy in cardiomyocytes. The present study provided further novel evidence that β5i interacts with ATG5 and promotes the degradation of ubiquitinated ATG5, resulting in the inhibition of autophagy and cardiac hypertrophy (Fig. 3). Together, these results suggest that ATG5 is a direct target of β5i in cardiomyocytes, and inhibition of the immunoproteasome (β5i) is compensated for by the up-regulation of autophagy in hypertrophy.
In summary, we have discovered a novel role of immunosubunit β5i as a critical regulator that promotes pathological hypertrophic remodeling. β5i targets ATG5 degradation and inhibits the activation of autophagy, thereby leading to cardiac hypertrophy and dysfunction; this indicates an essential compensatory relationship between the immunoproteasome and the activation of autophagy in the cardiac hypertrophic program. Further studies are needed to identify which E3 ligases promote ATG5 ubiquitination, and to determine whether inhibition of β5i may be a therapeutic strategy for hypertrophic diseases.
METHODS
Antibodies and reagents
The following primary and secondary antibodies were used: anti-sarcomeric α-actinin (EA-53) and anti-Flag (F7425) (Sigma-Aldrich); anti-mouse lgG (sc-2025), anti-rabbit lgG (sc-2027), and β-actin (sc-47778) (Santa Cruz Biotechnology); anti-β5i (ab3329), anti-Myc (ab9106), VeriBlot for IP Detection Reagents (HRP) (ab131366), and anti-β5i (ab180606) (Abcam); anti-BNP (13299-1-AP) (Proteintech); anti-ATG5 (12994), anti–Beclin-1 (3495), anti-ATG7 (8558), anti-ATG12 (4180), anti-p62 (8025), anti-p53 (2524), anti-LC3B (3868), anti–phospho-AKT (9271), anti–phospho-AKT (9272), anti-ERK1/2 (9102), anti–phospho-ERK1/2 (9101), horseradish peroxidase–linked anti-mouse IgG (7074S), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (2118) (Cell Signaling Technology); and anti-ubiquitin (10201-2-AP) (Proteintech). The following reagents were used: Ang II (Aladdin); phenylephrine (Selleck); chloroquine, cycloheximide, and MG132 (Sigma-Aldrich); cell-based proteasome assay (Promega); and human PSMB8 enzyme-linked immunosorbent assay (ELISA) assay kit (Cloud-Clone).
Plasmids
The full-length clone DNA of human proteasome (prosome, macropain) subunit β type 8 (large multifunctional peptidase 7) with pCMV3-N-FLAG–expressing plasmid was purchased from Sino Biological Inc. (Beijing, China). The plasmids pEGFP-CMV-flag-GGGS linker-human β5i-GGGS linker-EGFP mutants (1 to 68) (69 to 276) were constructed by Peking Syngenetech Co. Ltd. (China). The plasmids pCMV-myc-ATG5 (human) (1 to 275) and mutant (79 to 275) were constructed by Peking Syngenetech Co. Ltd. (China).
Primary culture of NRCMs and fibroblasts
The hearts from 1-day-old SD rats were cut for approximately 9 to 10 pieces and dissociated with 0.04% trypsin and 0.07% type II collagenase, as described (43). After dispersed cells were incubated on 100-mm culture dishes for 90 min at 37°C in 5% CO2, nonattached cells were collected and transferred into six-well plates, which were previously treated with laminin (10 μg/ml), and then 0.1 M 5-bromo-2′-deoxyuridine was added. Primary cardiomyocytes were incubated in Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% fetal bovine serum (FBS) for 16 hours and then replaced with serum-free DMEM/F12, which contained appropriate chemicals or adenovirus. Primary fibroblasts, which had attached the culture dishes, continued to grow in DMEM/F12 containing 10% FBS for 24 to 48 hours and then treated with appropriate chemicals or adenovirus.
Adenoviruses and cell infection
Ad-β5i or GFP alone was generated using the pAdEasy system according to the manufacturer’s protocol (HanBio Technology, China). NRCMs were infected with Ad-β5i or Ad-GFP at a multiplicity of infection of 50 and then treated with Ang II (100 nM, Sigma) for different time points. To determine the efficiency of β5i gene infection, immunofluorescence microscopy and Western blot analyses were performed at 24 hours after Ad-β5i or Ad-GFP infection. For the kinetics of autophagic flux, NRCMs were co-infected with adenoviral vector expressing mRFP-GFP-LC3, together with Ad-GFP, Ad-β5i, siRNA-β5i, or Scramble-siRNA (HanBio Technology, Shanghai, China), according to the manufacturer’s instructions. NRCMs were incubated in growth medium with Ad-mRFP-GFP-LC3 for 2 hours at 37°C and were grown in medium containing Ang II (100 nM) for 48 hours. LC3 puncta were examined by fluorescence microscopy (Olympus BX61).
Animals
β5i KO mice (β5i KO, STOCK Psmb8tm1Hif/J, C57BL/6J) were purchased from the Jackson Laboratory. β5i-Tg mice (pRP-ExSi-αMHC-Psmb8-3xFLAG) under α-myosin heavy chain promoter were generated by Cyagen Biosciences Company (China). The DNA of mice was isolated from the tails or fingernails and subjected to PCR analyses to identify mice belonging to which type of gene. β5i KO mice was identified by PCR analysis using the forward (5′-CCGACGGCGAGGATCTCGTCGTGA-3′) and reverse (5′-CTTGTACAGCAGGTCACTGACATCG-3′) primers. For β5i-Tg mice, the forward (5′-CCCCATAAGAGTTTGAGTCG-3′) and reverse (5′-GGGCCATCTCAATTTGAACA-3′) primers as well as the internal forward (5′-ACTCCAAGGCCACTTATCACC-3′) and reverse (5′-ATTGTTACCAACTGGGACGACA-3′) primers were used.
Establishment of cardiac hypertrophy
Mice of 8 to 10 weeks of age were subjected to TAC or sham for 2 to 6 weeks or were infused with Ang II (1000 ng kg−1 day−1) or saline for 2 weeks with osmotic mini-pumps (ALZET, Cupertino, CA), as described in our previous studies (6, 44, 45). Mice were sacrificed, and the heart was harvested to analyze the hypertrophic response. All mice used in this study were kept in a pathogen-free facility, and all procedures were approved by the Institutional Animal Care and Use Committee of Capital Medical University and performed in accordance with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication no. 85–23, 1996).
Echocardiography
Mice were lightly anesthetized with 1.5% isoflurane. Cardiac contractile function and structure were evaluated by M-mode echocardiography at different time points by using a 30-MHz probe (Vevo 770 System, VisualSonics, Toronto, Ontario, Canada). The left ventricular ejection fraction (EF%) and fractional shortening (FS%) were calculated as described (45).
rAAV9-siATG5 treatment
rAAV9-siATG5 and rAAV9-sicontrol were conducted by HanBio Biotechnology (Shanghai, China). Mice were given rAAV9-siATG5 or rAAV9-sicontrol (1.04 × 1012 μg/mg) by intravenous injection of tail, and after 3 weeks, mice were treated with sham or TAC surgery for another 6 weeks.
Histopathology
The heart size was photographed with a stereomicroscope (Olympus SZ61, Japan). The heart samples were fixed in formalin, embedded in paraffin, and then cut into 5-μm serial sections. Hematoxylin and eosin, Masson’s trichrome, and wheat germ agglutinin (WGA) staining were performed as described previously (44, 45). Digital images were taken at ×100 or ×200 magnification of 15 to 20 random fields from each heart sample. The fibrosis areas were analyzed by Image-Pro Plus 3.0 (Nikon, Japan). The cell area was calculated by measuring 150 to 200 cells per slide.
Immunohistochemistry for human hearts
The study included three male patients and three age- and gender-matched controls. Failing heart samples were obtained from patients with end-stage HF (average ejection fraction was 20 ± 5%) at the time of cardiac transplantation (Beijing An Zhen Hospital of the Capital University of Medical Sciences). Nonfailing hearts were obtained from donors who had normal cardiac contractile function by echocardiography and had died from accidents. The procurement of the heart tissues conforms to the principles outlined in the Declaration of Helsinki and was approved by the Institutional Ethics Committee of First Affiliated Hospital of Dalian Medical University. Heart tissues were fixed in neutral buffered formalin solution, embedded in paraffin, and then cut into 5-μm serial sections. Immunohistochemistry staining was performed as described (6). Sections were permeabilized for 15 min at room temperature in 0.1% phosphate-buffered saline (PBS)–Triton X-100 (Sigma-Aldrich). Slides were blocked in 0.1% PBS–Triton X-100 + 3% bovine serum albumin (BSA) (Sigma-Aldrich) for 1 hour at room temperature before being incubated in primary antibodies against monoclonal antibody β5i (1:1000 dilution) and BNP (1:500 dilution) at 4°C for 16 hours. Thereafter, the sections were treated sequentially with ready-to-use biotinylated secondary antibody and ready-to-use streptavidin-peroxidase conjugate, followed by standardized development in 3,3′-diaminobenzidine (DAB) chromogen.
Immunofluorescence
NRCMs were seeded onto laminin-coated coverslips before infection of siRNA-control, siRNA-β5i, Ad-GFP, or Ad-β5i for 24 hours, followed by Ang II stimulation for 24 hours. Immunohistochemistry was performed as described previously (45). Briefly, the cells were washed with PBS and postfixed in 4% paraformaldehyde for 15 min at room temperature. After the cells were blocked in PBS containing 1% BSA and 0.2% Triton X-100 for 10 min, monoclonal antibody against sarcomeric α-actinin was added at a dilution of 1:500 for overnight incubation at 4°C. After washing by PBS three times for 5 min, the cells were then incubated with secondary antibody tetramethyl rhodamine isothiocyanate (TRITC) or fluorescein isothiocyanate (FITC) for 1 hour at room temperature, and the nuclear staining was performed with 4′,6-diamidino-2-phenylindole (DAPI) (100 ng/ml) for 5 min. The cardiomyocyte surface area was depicted by using ImageJ software, and the relative surface area was read with the arbitrary units (the number of pixels) for evaluating hypertrophy.
Measurement of proteasome activity
Proteasome activity in the hearts was measured using fluorogenic substrates, and Z-LLE-AMC (45 μM), Ac-RLR-AMC (40 μM), or Suc-LLVY-AMC (18 μM) were used to detect caspase-like, trypsin-like, or chymotrypsin-like activity, respectively (46). Briefly, proteins of heart tissue were extracted in Hepes buffer (50 mM, pH 7.5) containing 20 mM KCl, 5 mM MgCl2, and 1 mM dithiothreitol. Twenty micrograms of protein was added to 200 μl of the Hepes buffer containing the fluorogenic substrates and incubated at 37°C for 1 hour. The fluorescence intensity was measured with excitation at 380 nm and emission at 460 nm.
Real-time PCR analysis
One microgram of total RNA from each sample was used to generate complementary DNA (cDNA) by using M-MLV reverse transcriptase as per the manufacturer’s specifications (Promega Corporation, USA). Real-time PCR was cycled in 95°C/15 s, 60°C/30 s, and 72°C/30 s for 40 cycles, after an initial denaturation step at 95°C for 10 min using SYBR Green PCR Master Mix purchased from Applied Biosystems (USA). Amplification was performed by using 7500 Fast Real-Time PCR Systems (Applied Biosystems, USA), and the products were routinely checked by using dissociation curve software. Transcript quantities were compared by using the relative quantitative method, where the amount of detected mRNA was normalized to the amount of endogenous control (GAPDH). The relative value to the control sample is given by 2−ΔΔCT.
Western blot analysis
Primary cardiomyocytes or heart tissues were lysed with radioimmunoprecipitation assay lysis buffer (Solarbio). Equal amounts of protein (50 to 60 μg) were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels, transferred to polyvinylidene difluoride membranes, and incubated with primary antibodies, as indicated in each experiment, and then with horseradish peroxidase–conjugated secondary antibodies (1:2500), as described previously (6, 45). Western blot signal intensities were analyzed with a Gel-Pro 4.5 analyzer (Media Cybernetics, USA) and were normalized to controls.
Immunoprecipitation
NRCMs or cardiac tissues were lysed in lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 1% SDS, and 0.5% sodium deoxycholate] plus phenylmethylsulfonyl fluoride (PMSF) (Solarbio) on ice for 20 min. Then, the samples were centrifuged at 12,000 rpm at 4°C for 15 min to obtain the protein extract. After the protein concentration was quantitated by bicinchoninic acid (Thermo Fisher Scientific), 2 μg of appropriate primary antibody and protein A–Sepharose (Amersham Biosciences) were added to the protein samples with gentle shaking at 4°C for 24 hours, followed by centrifugation at 5000 rpm at 4°C for 10 min. The pellet was washed twice with wash buffer I [50 mM tris-HCl (pH 7.5), 500 mM sodium chloride, 0.1% NP-40, and 0.05% sodium deoxycholate] and once with wash buffer II [50 mM tris-HCl (pH 7.5), 0.1% NP-40, and 0.05% sodium deoxycholate]. Bound proteins were eluted by boiling beads in 2× sample buffer, and the precipitated proteins were subjected to SDS-PAGE. Immunoblot data were quantified and analyzed with a Gel-Pro 4.5 analyzer (Media Cybernetics, USA).
Ubiquitylation assays
NRCMs or heart tissues were lysed with lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 1% NP-40, and 0.5% sodium deoxycholate] plus PMSF (Solarbio) and protease inhibitor cocktail (Roche) on ice for 30 min. Immunoprecipitation was performed with ATG5 antibody, and the ubiquitinated ATG5 was detected by Western blot analysis with ubiquitin antibody, as described previously (43, 47, 48).
Pulse-chase analysis
Primary cardiomyocytes were infected with adenovirus expressing GFP alone, β5i, siRNA-control, or siRNA-β5i for 24 hours. Protein lysates were prepared at indicated time points after the addition of cycloheximide (10 μM) or both cycloheximide (10 μM) and MG132 (10 μM). The levels of endogenous ATG5 protein were detected by Western blot analysis with anti-ATG5, quantified, and normalized to GAPDH.
Human subjects and blood samples
Thirty-eight HF patients (24 men and 14 women; 59 to 81 years; mean, 68 years) with left ventricular EF of <40% were selected from the First Affiliated Hospital of Dalian Medical University to collect the blood samples and medical record information. Furthermore, patients associated with infectious disease, immunological disease, pulmonary disease, and malignancy were not included. HF medication consisted of angiotensin converting enzyme (ACE) inhibitors (81%), diuretics (73%), digitalis (68%), and β blockers (42%). Their clinical and hemodynamic situations were stable, with no change in medication in the last month before the study. Controls subjects were 38 donors who received health examination (18 men and 20 women; 58 to 67 years old; mean, 62 years) without any history of cardiac disease. Both serum and plasma were collected to examine biochemical factors. For serum sampling, fresh blood samples without any anticoagulant were immediately immersed in ice for 1 hour before centrifugation (1000g at 4°C for 10 min). For plasma sampling, EDTA was added and samples were immediately centrifuged (1000g at 4°C) for 15 min.
ELISA assay
The blood serum was diluted in saline (1:2) and centrifuged at 1000g for 20 min, and then the β5i protein concentration in supernatants was detected by using an LMP7 ELISA kit (Cloud-Clone Corporation, Houston, TX, USA) according to the manufacturer’s instructions.
Statistical analysis
For multiple group, significance was determined by using one-way analysis of variance (ANOVA) followed by Tukey’s post test or two-way ANOVA followed by Bonferroni corrected post hoc t test. For two-group comparisons, Student’s t test was performed. All data of clinical patients were analyzed by SPSS 16.0 to identify whether they were normally distributed or not. Student’s t test was used to determine the difference between two groups in normally distributed data, and for the data not normally distributed, Mann-Whitney test was performed to determine the difference. Multivariable logistic regression models were used to evaluate the association of HF and blood β5i concentration and its chymotrypsin-like activity and then adjusted for sex, age, and cardiovascular risk factors such as alcohol drinking, smoke, diabetes, dyslipidemia, and obesity. P < 0.05 was considered statistically significant. All statistical analyses were done using SPSS 16.0 software.
Supplementary Material
Acknowledgments
We would like to thank J. Wang and Y. Zhang for helping with mouse colony management and the staff for assistance with animal procedures and experiments. Funding: This work was supported by grants from the National Natural Science Foundation of China (81330003 and 31571170 to H.-H.L., 81600315 to X.X., and 81570207 to H.-X.W.), Dalian high level Talents Innovation and Entrepreneurship Projects (2015R019), and Chang Jiang Scholar Program of China (T2011160 to H.-H.L). Author contributions: H.-H.L. conceived the project. X.X., N.L., S.L., and Y.-L.Z. performed and analyzed in vivo experiments. X.X., H.-L.B., and H.-X.W. performed the in vitro experiments and analyzed the data. X.X., H.-L.B., and S.L. performed and analyzed the biochemical and biophysical experiments. H.-J.C. and L.H. were responsible for human clinical and molecular genetic studies. H.-H.L. and X.X. wrote the paper with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau0495/DC1
Table S1. Clinical characteristics of HF cohort cases.
Table S2. Multivariable logistic regression analysis of β5i and chymotrypsin-like activity associated with HF (n = 38).
Fig. S1. Analysis of the β5i protein levels in rat neonatal cardiomyocytes.
Fig. S2. KO of β5i inhibits phenylephrine (PE)–induced cardiomyocyte hypertrophy.
Fig. S3. Analysis of autophagic influx in rat neonatal cardiomyocytes.
Fig. S4. KO of β5i decreases chymotrypsin-like activity in mice after TAC operation.
Fig. S5. Analysis of the β5i expression and survival rate in WT and β5i-Tg mice.
Fig. S6. Knockdown of ATG5 by rAAV9-siATG5 in mice abolishes the cardioprotective effect of β5i KO after pressure overload.
Fig. S7. The summarized diagram showing that the proposed mechanisms underlying β5i regulate cardiac hypertrophy.
REFERENCES AND NOTES
- 1.Frey N., Olson E. N., Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Korolchuk V. I., Menzies F. M., Rubinsztein D. C., Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N., Komatsu M., Autophagy: Renovation of cells and tissues. Cell 147, 728–741 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Cacciapuoti F., Role of ubiquitin-proteasome system (UPS) in left ventricular hypertrophy (LVH). Am. J. Cardiovasc. Dis. 4, 1–5 (2014). [PMC free article] [PubMed] [Google Scholar]
- 5.Angeles A., Fung G., Luo H., Immune and non-immune functions of the immunoproteasome. Front. Biosci. 17, 1904–1916 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Li N., Wang H. X., Han Q. Y., Li W. J., Zhang Y. L., Du J., Xia Y. L., Li H. H., Activation of the cardiac proteasome promotes angiotension II-induced hypertrophy by down-regulation of ATRAP. J. Mol. Cell. Cardiol. 79, 303–314 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Depre C., Wang Q., Yan L., Hedhli N., Peter P., Chen L., Hong C., Hittinger L., Ghaleh B., Sadoshima J., Vatner D. E., Vatner S. F., Madura K., Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation 114, 1821–1828 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Drews O., Tsukamoto O., Liem D., Streicher J., Wang Y., Ping P., Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ. Res. 107, 1094–1101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J., Wang S., Bai J., Yang X. L., Zhang Y. L., Che Y. L., Li H. H., Yang Y. Z., Novel role for the immunoproteasome subunit PSMB10 in angiotensin II-induced atrial fibrillation in mice. Hypertension 71, 866–876 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Dang M.-Q., Zhao X.-C., Lai S., Wang X., Wang L., Zhang Y.-L., Liu Y., Yu X.-H., Liu Y., Li H.-H., Xia Y.-L., Gene expression profile in the early stage of angiotensin II-induced cardiac remodeling: A time series microarray study in a mouse model. Cell. Physiol. Biochem. 35, 467–476 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Pandey U. B., Nie Z., Batlevi Y., McCray B., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., Yao T.-P., Baehrecke E. H., Taylor J. P., HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Kassiotis C., Ballal K., Wellnitz K., Vela D., Gong M., Salazar R., Frazier O. H., Taegtmeyer H., Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 120, S191–S197 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin X.-Y., Zhang Y.-L., Chi Y.-F., Yan B., Zeng X.-J., Li H.-H., Liu Y., Angiotensin II regulates Th1 T cell differentiation through angiotensin II type 1 receptor-PKA-mediated activation of proteasome. Cell. Physiol. Biochem. 45, 1366–1376 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Li J., Wang S., Zhang Y.-L., Bai J., Lin Q.-Y., Liu R.-S., Yu X.-H., Li H.-H., Immunoproteasome subunit β5i promotes ang II (Angiotensin II)-induced atrial fibrillation by targeting ATRAP (Ang II Type I receptor-associated protein) degradation in mice. Hypertension 73, 92–101 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Opitz E., Koch A., Klingel K., Schmidt F., Prokop S., Rahnefeld A., Sauter M., Heppner F. L., Volker U., Kandolf R., Kuckelkorn U., Stangl K., Krüger E., Kloetzel P. M., Voigt A., Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLOS Pathog. 7, e1002233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z., Gagarin D., St. Laurent G. III, Hammell N., Toma I., Hu C.-a., Iwasa A., McCaffrey T. A., Cardiovascular inflammation and lesion cell apoptosis: A novel connection via the interferon-inducible immunoproteasome. Arterioscler. Thromb. Vasc. Biol. 29, 1213–1219 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan W., Bi H.-L., Liu L.-X., Li N.-N., Liu Y., Du J., Wang H.-X., Li H.-H., Knockout of immunoproteasome subunit β2i ameliorates cardiac fibrosis and inflammation in DOCA/Salt hypertensive mice. Biochem. Biophys. Res. Commun. 490, 84–90 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., Otsu K., The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Cao D. J., Wang Z. V., Battiprolu P. K., Jiang N., Morales C. R., Kong Y., Rothermel B. A., Gillette T. G., Hill J. A., Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. U.S.A. 108, 4123–4128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhuiyan M. S., Pattison J. S., Osinska H., James J., Gulick J., McLendon P. M., Hill J. A., Sadoshima J., Robbins J., Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284–5297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng L.-q., Zhang W.-b., Ye Y., Yin P.-p., Yuan J., Wang X.-x., Kang L., Jiang S.-s., You J.-y., Wu J., Gong H., Ge J.-b., Zou Y.-z., Aliskiren ameliorates pressure overload-induced heart hypertrophy and fibrosis in mice. Acta Pharmacol. Sin. 35, 1005–1014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H., Tannous P., Johnstone J. L., Kong Y., Shelton J. M., Richardson J. A., Le V., Levine B., Rothermel B. A., Hill J. A., Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 117, 1782–1793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Y.-P., Lai S., Lin Q.-Y., Xie X., Liao J.-W., Wang H.-X., Tian C., Li H.-H., CDC20 regulates cardiac hypertrophy via targeting LC3-dependent autophagy. Theranostics 8, 5995–6007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishida K., Yamaguchi O., Otsu K., Crosstalk between autophagy and apoptosis in heart disease. Circ. Res. 103, 343–351 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Wang X., Cui T., Autophagy modulation: A potential therapeutic approach in cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 313, H304–H319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nedelsky N. B., Todd P. K., Taylor J. P., Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan T., Kondo S., Zhu W., Xie W., Jankovic J., Le W., Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol. Dis. 32, 16–25 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Zhu K., Dunner K. Jr., McConkey D. J., Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29, 451–462 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milani M., Rzymski T., Mellor H. R., Pike L., Bottini A., Generali D., Harris A. L., The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 69, 4415–4423 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Du Y., Yang D., Li L., Luo G., Li T., Fan X., Wang Q., Zhang X., Wang Y., Le W., An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 5, 663–675 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Costanzo A., Merlo P., Pediconi N., Fulco M., Sartorelli V., Cole P. A., Fontemaggi G., Fanciulli M., Schiltz L., Blandino G., Balsano C., Levrero M., DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol. Cell 9, 175–186 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi S., Volden P., Timm D., Mao K., Xu X., Liang Q., Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 285, 793–804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Garcia A., Rodriguez-Rocha H., Tseng M. T., de Oca-Luna R. M., Zhou H. S., McMasters K. M., Gomez-Gutierrez J. G., E2F-1 lacking the transcriptional activity domain induces autophagy. Cancer Biol. Ther. 13, 1091–1101 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouschop K. M. A., van den Beucken T., Dubois L., Niessen H., Bussink J., Savelkouls K., Keulers T., Mujcic H., Landuyt W., Voncken J. W., Lambin P., van der Kogel A. J., Koritzinsky M., Wouters B. G., The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest. 120, 127–141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song X., Zhang X., Wang X., Zhu F., Guo C., Wang Q., Shi Y., Wang J., Chen Y., Zhang L., Tumor suppressor gene PDCD4 negatively regulates autophagy by inhibiting the expression of autophagy-related gene ATG5. Autophagy 9, 743–755 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erbil S., Oral O., Mitou G., Kig C., Durmaz-Timucin E., Guven-Maiorov E., Gulacti F., Gokce G., Dengjel J., Sezerman O. U., Gozuacik D., RACK1 Is an Interaction Partner of ATG5 and a Novel Regulator of Autophagy. J. Biol. Chem. 291, 16753–16765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng Y., Song Y., Li Z., Wang Y., Lin H., Cheng H., Zhou R., RAB37 interacts directly with ATG5 and promotes autophagosome formation via regulating ATG5-12-16 complex assembly. Cell Death Differ. 25, 918–934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keil E., Höcker R., Schuster M., Essmann F., Ueffing N., Hoffman B., Liebermann D. A., Pfeffer K., Schulze-Osthoff K., Schmitz I., Phosphorylation of Atg5 by the Gadd45β–MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 20, 321–332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tekirdag K. A., Korkmaz G., Ozturk D. G., Agami R., Gozuacik D., MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy 9, 374–385 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H., He Z., Simon H.-U., Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 10, 372–373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee I. H., Cao L., Mostoslavsky R., Lombard D. B., Liu J., Bruns N. E., Tsokos M., Alt F. W., Finkel T., A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 3374–3379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kroemer G., Mariño G., Levine B., Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H.-H., Kedar V., Zhang C., McDonough H., Arya R., Wang D.-Z., Patterson C., Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Invest. 114, 1058–1071 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L., Li Y.-L., Zhang C.-C., Cui W., Wang X., Xia Y., Du J., Li H.-H., Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc. Res. 101, 383–392 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Wang X., Wang H.-X., Li Y.-L., Zhang C.-C., Zhou C.-Y., Wang L., Xia Y.-L., Du J., Li H.-H., MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension 66, 776–785 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Geserick P., Wang J., Schilling R., Horn S., Harris P. A., Bertin J., Gough P. J., Feoktistova M., Leverkus M., Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 6, e1884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie P., Guo S., Fan Y., Zhang H., Gu D., Li H., Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J. Biol. Chem. 284, 5488–5496 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Li H.-H., Willis M. S., Lockyer P., Miller N., McDonough H., Glass D. J., Patterson C., Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J. Clin. Invest. 117, 3211–3223 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau0495/DC1
Table S1. Clinical characteristics of HF cohort cases.
Table S2. Multivariable logistic regression analysis of β5i and chymotrypsin-like activity associated with HF (n = 38).
Fig. S1. Analysis of the β5i protein levels in rat neonatal cardiomyocytes.
Fig. S2. KO of β5i inhibits phenylephrine (PE)–induced cardiomyocyte hypertrophy.
Fig. S3. Analysis of autophagic influx in rat neonatal cardiomyocytes.
Fig. S4. KO of β5i decreases chymotrypsin-like activity in mice after TAC operation.
Fig. S5. Analysis of the β5i expression and survival rate in WT and β5i-Tg mice.
Fig. S6. Knockdown of ATG5 by rAAV9-siATG5 in mice abolishes the cardioprotective effect of β5i KO after pressure overload.
Fig. S7. The summarized diagram showing that the proposed mechanisms underlying β5i regulate cardiac hypertrophy.