

Measles virus gene expression requires a precise range of cohesiveness and a kink in the coiled coil of its phosphoprotein.
Abstract
The polymerase of negative-stranded RNA viruses consists of the large protein (L) and the phosphoprotein (P), the latter serving both as a chaperon and a cofactor for L. We mapped within measles virus (MeV) P the regions responsible for binding and stabilizing L and showed that the coiled-coil multimerization domain (MD) of P is required for gene expression. MeV MD is kinked as a result of the presence of a stammer. Both restoration of the heptad regularity and displacement of the stammer strongly decrease or abrogate activity in a minigenome assay. By contrast, P activity is rather tolerant of substitutions within the stammer. Single substitutions at the “a” or “d” hydrophobic anchor positions with residues of variable hydrophobicity revealed that P functionality requires a narrow range of cohesiveness of its MD. Results collectively indicate that, beyond merely ensuring P oligomerization, the MD finely tunes viral gene expression through its cohesiveness.
INTRODUCTION
RNA synthesis by nonsegmented negative-stranded RNA viruses (nsNSVs) is ensured by a unique and complex interplay between at least four components: the genomic RNA, the nucleoprotein (N), the phosphoprotein (P), and the polymerase or large protein (L). The genome starts with a promoter region followed by adjacent genes transcribed sequentially via a termination/reinitiation mechanism [see (1) for a review]. The N protein shields the genome and antigenome RNAs within a helical nucleocapsid (NC) away from nucleases and RNA-mediated silencing (2). Atomic structures of NC from several nsNSV show the RNA enwrapped in a homopolymer of N with tight contacts between monomers [for an example, see (3)]. Both transcription and replication are performed by the viral polymerase complex made of the polymerase L associated with its cofactor P.The >2000–amino acid–long L protein, whose structure has been solved for a Rhabdoviridae member (4), contains all the enzymatic activities that are necessary for RNA polymerization, addition of the mRNA cap, and its N- and O-methylation [see (5) for review]. Its cofactor, the P protein, plays a central role in nsNSV RNA synthesis: It binds to and stabilizes both L (6) and N in an RNA-free form (N0) (7, 8); it tethers the polymerase onto the NC (9), thus giving access to the shielded RNA template; it increases the processivity of the polymerase (10, 11); and it regulates RNA synthesis (2, 12, 13).
The P proteins of members of the Paramyxoviridae family are composed of three well-conserved domains [N-terminal domain (NTD), multimerization domain (MD), and X domain (XD)] linked together by long intrinsically disordered regions (Fig. 1A, top). The NTD consists of a molecular recognition element with α-helical propensities (α-MoRE) (14–16) that serves as a chaperon for keeping the neo-synthesized N protein in N0 monomeric and RNA-free form, thus forming the N0P complex (7). An additional short α-MoRE (α4) identified in measles virus (MeV) and located ~160 residues downstream the NTD also participates in N chaperoning (17). The N0P complex is the encapsidation substrate used by the polymerase to encapsidate nascent genomic and antigenomic RNA during replication.
Fig. 1. MeV P organization in conserved modules responsible for binding, stabilizing and supporting L.
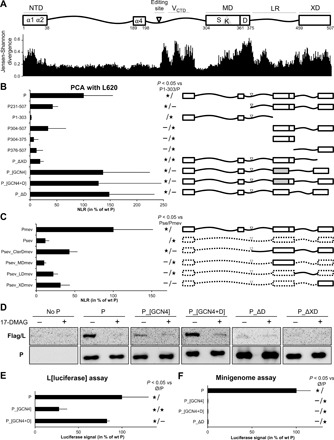
(A) Top: NTD with α1α2 and α4 helix-prone regions involved in N0P complex formation. LR, linker region. MD can be subdivided into two regions: a stable S module (amino acids 304 to 360) and a dynamic D module (amino acids 361 to 375), with former including a kink (K). P mRNA editing site and the region of P overlapping with V protein C-terminal domain (VCTD) are also shown. Bottom: Conservation score for each amino acid of MeV P calculated on the basis of Jensen-Shannon divergence (32) with an alignment of amino acid sequence of P from at least one member of each genus of the Paramyxoviridae family recorded in National Centre for Biotechnology Information (NCBI) databank with the following accession numbers: HQ385703, KF687315, KP717417, KT749883, MF795094, LC000638, EU439428, NC_028362, MG573141, MG516455, KX940962, KY370098, NC_007454, MG573140, KJ641657, KC154054, NC_002199, MF943130, NC_005339, NC_017937, KY971532, JN234009, EU267273, Z47758, MF627832, JN255804, FN869553, NC_025351, NC_025256, NC_025347, NC_025361, KP762799, KX258200, MF033136, JX901129, NC_025349, EU338414, NC_034968, NC_025407, GQ338311, KX119151, NC_025363, NC_025390, MF594598, KY452444, KY452443, KY452442, JN571485, NC_025373, KX953297, NC_003443, NC_006428, AJ749976, NC_021928, KX064232, KT213728, NC_009489, NC_004074, JX112711, NC_025403, NC_025348, NC_025350, NC_025343, NC_025404, NC_025410, KJ641654, JF795583, NC_025360, NC_005084, and MH411104. (B and C) Truncated and chimeric P binding to L620 protein as measured by Gaussia luciferase–derived PCA. P constructs are fusion proteins, with the first domain of the Gaussia luciferase (glu1) fused to their N terminus except for the 1–303 fragment, where glu1 was grafted at the C terminus. L620 was grafted with the second domain of the luciferase (glu2) at its N terminus. 293T cells were transfected to coexpress glu2-L620 with a glu1-P variant. Gaussia luciferase activity was measured 24 hours after transfection. Normalized luminescence ratio (NLR) was calculated for each condition. Results are shown in percentage of wt P NLR. P_[GCN4] and P_[GCN4+D] have the S region of their MD substituted with a tetramerization domain derived from the yeast GCN4 transcription factor. (D) Ability of P variants to assist L folding by the HSP90 chaperon as monitored by SDS–polyacrylamide gel electrophoresis and Western blot in the absence (−) and presence (+) of the HSP90 inhibitor 17-DMAG. Note the very poor expression level of Flag/L in the absence of P (“no P” panel). (E) P variant ability to assist L folding as measured using an L[luciferase] folding assay (30). The principle is that the folding, and consequently the enzymatic activity, of the luciferase embedded in the middle of the L protein relies on the appropriate folding of the upstream L region. Hence, the level of luciferase activity reflects both the amount of L and the quality of its folding in the presence of each P variant. (F) P variant activity in a luciferase-based minigenome assay. BSR-T7 cells were transfected with the mixture of expression vectors encoding the minigenome, N, L, and P variants. The firefly luciferase activity was measured 2 days after transfection. Data in (A), (B), (D), and (E) are means ± SD of at least three independent experiments, with each point done in triplicate. Statistically significant differences (Student’s t test) at P < 0.05 and below, with respect to the negative “Ø” (unless otherwise indicated) and positive “wt” P (unless otherwise indicated) control, are quoted by stars. Although not below the 0.05 threshold, P values for Psev_LDmev versus Psev, Psev_XDmev versus Psev, and Psev_XDmev versus Pmev are 0.096, 0.064, and 0.07, respectively.
The structure of the MD of the P protein of several paramyxoviruses has been solved by x-ray crystallography. It is mainly composed of a long α-helix forming a homotetrameric coiled coil in parallel (18–21) or antiparallel orientation (22). The oligomeric state seems to be context dependent in the case of Henipavirus P proteins: Nipah virus (NiV) MD crystallizes as a tetramer (18), and MD from both NiV and Hendra viruses appears trimeric in solution (23, 24). Coiled coils are made of α-helices interacting with each other via hydrophobic contacts. Coiled coils are low-complexity sequences in which a motif of seven amino acids (heptad) is repeated. The seven residues of the heptad are denoted “a, b, c, d, e, f, g,” with “a” and “d” positions being occupied by hydrophobic residues. Because an α-helix has a pitch of 3.65 residues, heptad repetitions constraint each α-helix to coil over each other to comply with the 7:2 = 3.5 pitch (25). Structural comparisons among the available crystal forms of MeV P MD unveil considerable variations in the quaternary structure and varying degrees of disorder in the C-terminal region (21). MeV P MD can therefore be divided in a 55–amino acid–long stable (S) region followed by a 15–amino acid–long dynamic module (D) (Fig. 1A, top). Furthermore, the heptad regularity of the MeV coiled coil is interrupted by the insertion of a “stammer,” i.e., a three–amino acid motif (L339L340L341), resulting in the formation of a short 310-helix that forms a kink (K). Kinked helices are found in all the structures of MD from Paramyxoviridae members solved so far (18–22). The role of MD remains speculative with the postulate that the oligomerization of P is necessary for the cartwheeling of the P-L complex along the NC with sequential binding of the XD monomers to contiguous N monomers (26).
The C-terminal XD of P folds into a triple α-helical bundle that dynamically accommodates the α-MoRE located within the intrinsically disordered C-terminal domain (NTAIL) of the N protein (27–29). By doing so, it allows the dynamic anchoring of the P-L complex to the NC and tightly regulates the efficiency of the reinitiation of the transcription at each intergenic junction (2, 12, 13).
For paramyxoviruses, the L protein requires both HSP90-mediated chaperoning and P assistance to be properly folded and kept as a soluble P-L complex. In the absence of P, L either is degraded by the proteasome or precipitates into insoluble aggregates (30, 31).
To shed light on the P-L interplay, we carried out a mutational study of the P protein from MeV, a prototypic paramyxovirus, with the specific aim of identifying key structural features and/or motifs of P that mediate binding to L and folding and/or stability assistance to L and those essential to viral gene expression. Results support an overall picture where multiple modules of P cooperate to exert these different functions. In particular, the obtained data strongly suggest that L-P interaction, L folding/stability assistance, and viral gene expression crucially rely on multiple features of its coiled-coil MD.
RESULTS
L-binding site lies in the C terminus of P and requires multimerization
Using algorithms that predict functionally important amino acids based on their conservation (32), an analysis of P proteins from Paramyxoviridae members showed a good conservation of domains required for the formation of the N0P complex (NTD) and for binding to the NC (XD) (Fig. 1A, bottom). The P gene has an editing site where the viral polymerase may add one nontemplated G residue in the mRNA, thus resulting in the production of the nonstructural V protein. A region with high sequence conservation occurs downstream the P mRNA editing site and is a secondary consequence of the overlap with the C-terminal cysteine-rich domain of the V protein. Together with the high conservation of the P mRNA editing site, it likely reflects the crucial role played by paramyxoviral V proteins in counteracting the host innate immune response, with V C terminus being critically involved in binding to DDB1 (33) and in inhibiting MDA5 (34). The amino acid sequence of the MD is also well conserved, with the D module being the most conserved P sequence, suggesting that it plays a crucial functional role. The linker region between MD and XD also bears highly conserved sequences for which functions have not been assigned yet. The L-P interaction being essential for the virus, it is tempting to suggest that L may bind to one or several of these highly conserved sequences.
The L-binding site within P was mapped using a protein complementation assay (PCA) based on split-luciferase reassembly (35). The instability of MeV L protein when expressed alone (30), however, prevents the use of the full-length L protein as reliable bait for mapping the L-binding site using truncated forms of P. Results could be biased by the inability of some P deletion variants to support the folding of L that would result in its aggregation and degradation. Because the P-binding site on L has been previously mapped to its N-terminal L1-408 region (36), we used the N-terminal L1-620 (L620) fragment as the bait. L620, which encompasses both CRI and CRII polymerase motifs, is expressed as a soluble protein requiring neither HSP90 activity nor P (fig. S1). In a Gaussia luciferase–based PCA, L620 interacts with the P protein as efficiently as the entire L, and the interaction is lost when L620 contains four substitutions (201VELLI > 201AEAAA, L640_4A) reported to abolish the P-L interaction (fig. S1) (36).
A significant binding of L620 could be detected with P231-507, in agreement with data obtained using yeast two-hybrid assay (Fig. 1B) (37). The shorter P304-507 construct interacts as well, while no significant PCA signal was detected with the N-terminal 1–303 fragment. The MD (304–376) and the C-terminal 376–507 fragment led on their own to no significant PCA signal (Fig. 1B). This suggests that the L-binding site may encompass both fragments or that stable binding requires the multimerization of P376-507. To discriminate between these two hypotheses, the MD was swapped with the minimal yeast-derived GCN4pLI tetramerization domain (P_[GCN4]) (38). The strong PCA signal generated by P_[GCN4] (Fig. 1B) suggests that the main L-binding site resides in the C-terminal region of P (amino acids 376 to 507) and that its multimerization may be required for binding to L. Furthermore, the deletion of the XD strongly decreases the binding to L620, revealing its involvement in P interaction with L620 (Fig. 1B).
To avoid possible adverse effects of truncation and to keep the different regions of MeV P in a more relevant context, we generated chimeric constructs by introducing different MeV P regions in the Sendai virus (SeV) P protein that shares a similar architecture and does not bind MeV L620 in PCA (Fig. 1C). The insertion of MeV P231-507 in SeV P backbone partially restores interaction with L620 similarly to what was observed with P231-507 alone (Fig. 1B). Replacement of SeV MD by MeV MD does not increase SeV P binding to L620, whereas swapping of the linker region and the XD improves binding (Fig. 1C). Overall, the data obtained in PCA support the conclusion that XD and the linker region contain a major and possibly a weaker L-binding site and that the MD plays a role in the P-L interaction via the multimerization of these domains.
The D module of the MD is required for P to assist L folding
The ability of P_[GCN4] chimeric protein to provide assistance to the stable folding of L by HSP90 was then evaluated. In the absence of P or in the presence of an inhibitor of HSP90 activity (17-DMAG), L is unstable and degraded, and very low amounts are detectable by Western blot (Fig. 1D), as reported previously (30). When coexpressed with P_[GCN4], the expression of L is poor compared to the coexpression with wild-type (wt) P indicating L instability (Fig. 1D). However, the addition of the short dynamic region downstream to the GCN4 coiled coil (P_[GCN4+D]) restores the expression of L. Reciprocally, the deletion of D from P (P_ΔD) results in the impairment of the L folding/stability assistance. As expected from the loss of binding to L620 (Fig. 1B), P_∆XD is also unable to stabilize L (Fig. 1D).
To confirm these biochemical data, the GCN4 constructs were also analyzed using an L[luciferase] folding assay (30). In this assay, the coding sequence of the Renilla luciferase is inserted in a loop in L, and the Renilla luciferase activity is detected only if L is stable and correctly folded. The L[Renilla] is functional because a virus expressing this chimeric polymerase was rescued, reaching a titer of 1.6 × 106 TCID50 (median tissue culture infective dose)/ml, a value comparable to that obtained with a virus expressing wt L. As previously reported for a recombinant virus expressing an L[GFP] construct, the polymerase was, however, somehow debilitated because the peak of virus production was delayed by 2 days (39). The Renilla activity of this virus reached a value of ~4 × 107 relative luminescence units (RLU) 1 day after infection at a multiplicity of infection (MOI) of 1 compared to the ~2 × 105 RLU background obtained with wt L. Results confirm the requirement of the dynamic region of P for L stability and/or folding (Fig. 1E). P_ΔD, P_[GCN4], and P_[GCN4+D] all bind similarly to L620 as judged from their PCA signals (Fig. 1B). Therefore, although not required for binding to L620, the D module is required for P to stabilize L.
In addition to P multimerization and L stabilization, MD plays a critical role in viral gene expression
The ability of P variants to support viral gene expression was then tested with a minigenome assay (Fig. 1F). As expected from proteins unable to stabilize L, P_[GCN4] and P_∆D were found to be not functional (Fig. 1F). However, although P_[GCN4+D] binds and assists HSP90-mediated folding of L, it does not support gene expression (Fig. 1F). This was further verified in the viral context by introducing P_ΔD, P_ΔS (lacking the 304–360 S region), and P_[GCN4+D] as a second P gene (referred to as HA-P) in biG-biS recombinant viruses. These viruses allow either the expression of the two duplicated genes (Flag-P and HA-P) or the selective expression of only one of them owing to the use of specific host cells expressing small interfering RNAs (siRNAs) targeting each of the transcript from the other gene copy (see scheme, Fig. 2A) (2, 12). In cells preventing the expression of the mutated P gene, thus allowing only wt Flag-P to be expressed, all viruses grow well, as measured from the expression of the F protein overtime, although the growth of P¦P_[GCN4+D] and P¦P_ΔS is somewhat delayed when compared to the virus with duplicated wt P gene (Fig. 2, B to E, compare green dashed lines). For all P constructs, virus growth was abrogated when only the mutated P gene is expressed, whereas the P¦P control grow well, as previously reported (Fig. 2, B to E, compare red dashed lines). Furthermore, when the mutated P constructs are expressed together with wt P, while the expression of P_ΔS is neutral, virus growth is severely limited in the case of P_ΔD and P_[GCN4+D], revealing that they exert a dominant negative effect over their wt P counterpart (Fig. 2, B to E, compare differences between green and black lines). To test whether the dominant negative effect of P_ΔD and P_[GCN4+D] could reflect their hetero-oligomerization with wt P, we measured such interactions by PCA (Fig. 2F). P_∆D conserves its ability to oligomerize and can hetero-oligomerize with wt P (Fig. 2F). P_[GCN4+D] can also hetero-oligomerize with wt P and P_∆D, although the signal intensities are reduced to about half of the wt P homo-oligomerization (Fig. 2F). On the other hand, the deletion of the S module abolishes the ability of P to hetero-oligomerize with wt P or P_[GCN4+D] (Fig. 2F). These data support the conclusion that the dominant negative effect of P_∆D and P_[GCN4+D] is mediated by their hetero-oligomerization with wt P and that the heterocomplexes are not functional. Together, these data indicate that, in addition to P tetramerization, the S region of the MD plays a crucial role in viral growth.
Fig. 2. Virus production in the context of biG-biS viruses encoding P constructs with modified MD.
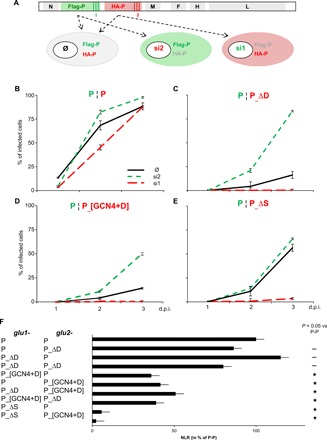
(A) Scheme of the biG-biS system. BiG-biS viruses bear duplicated P genes coding for wt Flag-P and HA-P variants, the expression of which can be independently silenced with two distinct small interfering RNAs (siRNAs). Infection of cells, expressing or not one siRNA, results in the production of wt P, P variant, or both. (B to E) P variant activity as measured with the biG-biS system. Vero cells (black lines) and Vero cells constitutively expressing either si1 (red dashed lines) or si2 RNA (green dashed lines) were infected at an MOI of 0.1, with biG-biS viruses encoding either two wt P proteins (B) or one wt P and either P_ΔD (C), or P_[GCN4+D] (D) or P_ΔS (E) protein construct. The percentage of cells expressing MeV F was determined by flow cytometry after immunolabeling with anti-F monoclonal antibody. (F) Hetero-oligomerization of different P variants. P constructs are fusion proteins with the first or second domain of the Gaussia luciferase fused to their N terminus. 293T cells were transfected to coexpress glu1- and glu2-P variants. Gaussia luciferase activity was measured 24 hours after transfection. NLR was calculated for each condition. Results are shown in percentage of wt P NLR. Statistically significant differences (Student’s t test) at P < 0.05 and below, with respect to the negative “Ø” and positive “wt” control, are quoted by stars.
P MD kink is required for P function in gene expression
Unlike the GCN4pLI coiled coil, the regularity of the MeV MD coiled-coil structure is broken in the S region because of the presence of a stammer (Fig. 3, A and B). Such a break in heptad regularity imposes a conformational constraint to the α-helices of the tetrameric coiled coil, thereby generating a 310-helix that kinks the three-dimensional (3D) structure (Fig. 3A). NiV and mumps virus (MuV) MDs show similar kinked structures and heptad regularity interruptions (Fig. 3A). The presence of kinks in MDs is a hallmark within at least four genera of the Paramyxoviridae family, including the Respirovirus. Although in SeV (a Respirovirus member) the MD sequence does not have any stammer, its structure is locally distorted because of the insertion of two stutters, i.e., a four–amino acid motif (a tetrad). To assess the importance of the kink for P function, we restored the heptad regularity either by deleting the residues of the kink (LLL), which belongs to the LLLL motif, or by adding an amino acid tetrad upstream the kink (fig. S2A). P deleted of the LLL triad is poorly (if at all) able to support gene expression from a minigenome (P_ΔK1; fig. S2B). The presence of the LLL triad in the context of a regular α-helix is not sufficient to support gene expression because the abrogation of the local 310-helix by addition of a tetrad (LATV, LGHG, or LEKE) upstream to the stammer, which should restore a regular heptad repeat, abolishes the polymerase activity (P_ΔK2, P_ΔK3, and P_ΔK4; fig. S2B). Displacing the LLL triad either upstream of preceding triad (P_K-3) or downstream the following tetrad (P_K+3) or the preceding heptad (P_K-7) or the following heptad (P_K + 7) is also not functionally tolerated (fig. S2C). Similarly, the displacement of the whole 336–345 triad-triad-tetrad upstream or downstream the preceding or following heptad (P_K*-H, P_K*+H) is not tolerated either (fig. S2C). Likewise, the LLL stammer by itself does not confer functionality when the flanking heptads were substituted with those from the GCN4 tetramerization domain despite the overall similar length of the two tetramerization domains and the conservation of the D module (P_[GCN4+K+GCN4+D]; fig. S2B). Last, replacement of the S region of MeV P MD with the corresponding S region from NiV P MD (P_[Sniv+D]), which also contains a kink, does not support cofactor function (fig. S2B). These data suggest that the kink is required but not sufficient for the MD to support viral gene expression.
Fig. 3. Conservation of deformations in the coiled coil of P MD within paramyxoviruses and functional impact of substitutions within the stammer of MeV P.
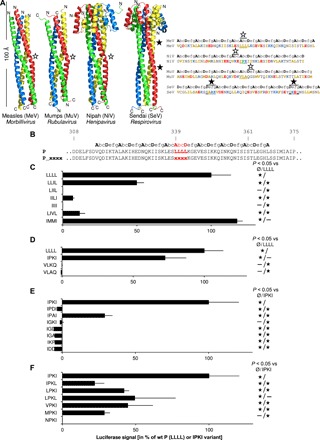
(A) Ribbon representation of the crystal structures of the P MDs from MeV (PDB code 3ZDO), MuV (PDB code 4EIJ) (note its particular antiparallel orientation), NiV (PDB code 4GJW), and SeV (PDB code 1EZJ), four Paramyxoviridae members representative of four genera. Kinks induced by insertion of stammers (i.e., insertion of three extra amino acids) or stutters (i.e., insertion of four extra amino acids) that disrupt the heptad regularity of the coiled coil are indicated by empty and full stars, respectively. (B) Amino acid sequences and register of the P MDs from MeV, NiV, MuV, and SeV. For the amino acids out of register, a register was manually assigned according to the expected register resulting from the insertion of either a stammer or a stutter (letters in italics). Stammers and stutters are underlined, and the residues of the kinks are highlighted by empty or full stars for stammer- or stutter-induced kinks, respectively. (C) Sequences of kink variants with the expected heptad register. (D to F) Activity in a minigenome assay of P stammer variants bearing mutations with hydrophobic residues (C), replacement with other paramyxovirus stammers (D), and mutations of exposed (E) or buried residues (F) of IPKI. BSR-T7 cells were transfected with the mixture of expression vectors encoding the minigenome, N, L, and P variants. The firefly luciferase activity was measured 2 days after transfection. Data are means ± SD of at least three independent experiments, with each point done in triplicate. Statistically significant differences (Student’s t test) at P < 0.05 and below with respect to the negative “Ø” and positive LLLL or IPKI controls are quoted by stars.
The sequence of the stammer is relatively tolerant of substitutions
To evaluate the importance of the sequence of the stammer, we introduced substitutions in the LLLL motif (Fig. 3B). When residues of the LLLL motif were replaced with other hydrophobic amino acids, the gene expression was found to be strongly affected, with some mutants being inactive in a minigenome assay (Fig. 3C). Although all leucines can be substituted without affecting activity (see IMMI mutant), progressive replacement of leucines by isoleucines decreases the activity.
Next, we assessed whether the stammer of MeV can be replaced with the sequence of the stammers from other Paramyxoviridae members (NiV, IPKI; MuV, VLAQ; Newcastle disease virus, VLKQ). Only the IPKI motif from NiV was found to be well tolerated without significant change in gene expression (Fig. 3D). Other variants inspired from IPKI were also tested and only IPAI retains some activity (Fig. 3E). Change of the charge (IPDI), shift of the proline (IKPI), and substitution of the proline with a glycine, another amino acid known to disfavor the formation of α-helices, all abrogate the ability of P to support viral gene expression (Fig. 3E). Conservation of the exposed residues of IPKI (P and K) and substitution of the buried isoleucine residues produce a set of mutants whose activity ranges between 0 and 36% of that of wt P (Fig. 3F).
Together these data show that the sequence of the stammer is constrained but not by the chemical properties of the amino acids, as judged based on the fact that the IPKI variant is functional and the LIIL variant (that is chemically much closer to wt than IPKI) is not. This suggests that specific structural properties of the MD may be critical to support viral gene expression.
P tolerates variations in the position and the dynamics of the 310-helices
The presence of prolines in coiled coils has a well-known impact on the structure of coiled coils in that they induce a kink similar to that induced by a stammer (40). To quantify the structural changes induced by the IPKI substitution (where a proline occurs in a stammer), we solved the structure of the IPKI variant of MeV MD by x-ray crystallography (Fig. 4A and table S1). The data were initially indexed using XDS programme package with decent statistics in space group I422 (table S1) and scaled with AIMLESS. The crystal gave rise to an anisotropic diffraction pattern characterized by a maximum resolution of 2.64 Å on the h k plane and 2.20 Å resolution on the l plane assuming a signal-to-noise ratio greater than 1.5. Note that, in the l plane, the crystal likely diffracted to a higher resolution, but the experimental setup did not allow us to measure reflections at a resolution higher than 2.2. Å. An anisotropic cutoff and correction was applied using the STARANISO server. Phases were obtained using molecular replacement and the structure of wt P MD [Protein Data Bank (PDB) code 3ZDO] as the search model (with one chain in the asymmetric unit). The 2.2 to 2.3 Å resolution shell was found to be characterized by an average I/σ(I) lower than 1.5 (table S1). Because of the poor signal-to-noise ratio of the highest-resolution shell, and of the anisotropic nature of the diffraction, the inclusion of the last resolution shell in the refinement led to poor statistics. We therefore choose to cut the data at 2.3 Å and to refine the model at this resolution to improve model statistics. This resolution limit is still good enough to appreciate the impact of substitutions on the geometry of the coiled coil. The structure was solved for residues 306 to 368 and was deposited in the PDB (PDB code 6HTL) with acceptable statistics (Rwork/Rfree = 0.254/0.285).
Fig. 4. Structural comparison between wt and IPKI MDs and molecular dynamic simulations.
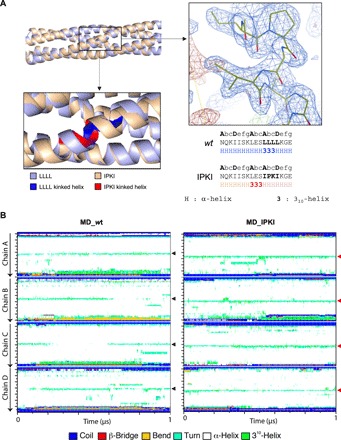
(A) Structural superimposition between the crystal structure of wt MD (PDB code 3ZDO) (light blue) and of the IPKI variant (PDB code 6HTL) (beige) both in ribbon representation. The zoomed-in region below the superimposed models displays the kink that is shown either in red (IPKI) or in dark blue (wt). The zoomed-in region on the left shows the 2Fo-Fc (contoured at 1σ) (blue) and Fo-Fc (contoured at 3σ) (red) electron density maps centered on the IPKI motif. Below are the amino acid sequences of the two proteins in the region encompassing the kink with the register and the secondary structure shown above and beneath the sequence, respectively. (B) Secondary structure timeline analysis of wt (left) and IPKI (right) MDs, as obtained using the GROMACS dssp routine utility (91, 94, 95), during a 1-μs molecular dynamic simulation. Regions adopting a 310-helical conformation are shown in green, and the central kinks are pinpointed by arrowheads. Note the N-terminal shift of the kink in the IPKI variant (see also fig. S3B).
The structure of IPKI is almost perfectly superimposable onto that of wt MD with the only exception of the kink region (Fig. 4A). Although IPKI MD and wt MD are structurally very close to each other and have the same coiled-coil parameters (table S2), the presence of the proline triggers a shift of the kink three residues upstream the IPKI stammer motif, i.e., on L336E337S338 residues, in the same way as it does in its natural P NiV context (PDB code 4GJW; see also Fig. 3).
Solved crystal structures of MeV wt P MD (with the wt LLLL motif) have revealed subtle differences in the coiled coil, with the latter being either asymmetrical (i.e., three chains with a 310-helix plus one chain fully α-helical) [3ZDO (19)] or symmetrical (i.e., four chains with a 310-helix) [4C5Q and 4BHV (21)]. A 1-μs molecular dynamics simulation of wt MeV MD starting with a symmetrical model that was prolonged to encompass the entire MD (amino acids 304 to 375) (fig. S3A) revealed that the symmetrical model readily converts to an asymmetrical one (three 310-helices plus one α-helix), with the α-helical arrangement being randomly distributed among the four chains over time (Fig. 4B, left panels). When zooming over a 5-ns simulation, very short-lived α-helices in one of the four chains can be observed (fig. S3B, left panel). If the kink disappears over time in one of the four chains to reappear in another chain, it never shifts to adjacent residues of the same chain [see arrowheads in Fig. 4B (left panels) and black arrows in fig. S3B (left panels)].
In the case of IPKI, the four chains of the tetramer adopt a 310-helical conformation encompassing residues L336E337S338, and the simulation shows the persistence of the four 310-helices over time [Fig. 4B (right panel) and fig. S3B (right panel, red arrows)]. This was expected considering the α-helix breaking properties of proline. Accordingly, the root mean square fluctuation (RMSF) of the residues surrounding the stammer of each of the four chains is higher for wt MD (fig. S4, A, B, and I, compare red and black lines). However, and as observed for wt MD, when zooming over a 5-ns simulation, very short-lived α-helices occurring in one of the four chains can be observed (fig. S3B, right panel). This suggests a constant equilibrium favoring the 310-helical conformation over the α-helical one.
Analysis of the RMSFs of wt and IPKI MDs also revealed that the N- and C-terminal regions are the most dynamic ones (fig. S4, A, B, and I). Unexpectedly, however, the D region does not appear to be more dynamic than the N-terminal region. The discrepancy between structural data and data from molecular dynamic simulations might reflect static rather than dynamic disorder in the crystal (i.e., the D region may adopt specific and distinct conformations within different asymmetric units, thereby leading to a lack of coherence in diffraction data).
Because the IPKI variant was found to be efficient in a minigenome assay (Fig. 3E), these results support the conclusion that neither the precise location of the kink nor a continuous maintenance of the symmetry of the coiled coil would be crucial for P ability to support viral gene expression. The 310-helix can occur either at residues L339L340L341 on three of four chains (wt MD) or at residues L336E337S338 on the four chains (IPKI MD).
P ability to support viral gene expression relies on a restricted range of MD coiled-coil stabilities
Because substitutions of the buried “a” and “d” positions of the LLLL or IPKI motif can abrogate gene expression (Fig. 3, C and E), features other than the nature of surface-exposed residues within the S region of P MD are likely to be critical for the P cofactor activity. The cohesiveness of a coiled coil mainly relies on the regular distribution and nature of hydrophobic residues at positions “a” and “d” within repeated “a, b, c, d, e, f, g” heptads [see (25) for review]. MD variants, in which anchor residues (i.e., residues at “a” or “d” positions) were substituted with residues with varying degree of hydrophobicity, were generated (Fig. 5A) and characterized biochemically and biophysically (Fig. 5, B and C, and figs. S5, A and B, and S6).
Fig. 5. Mutation of buried residues within the S region affects MD stability and P cofactor function.
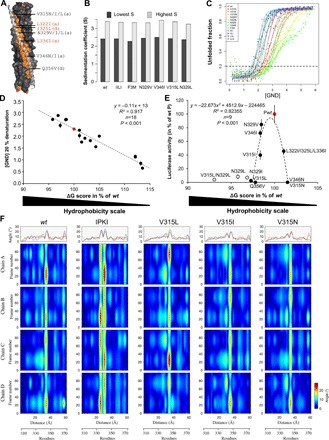
(A) Structure of wt MD (PDB code 3DZO), with one chain shown in ribbon representation with side chains shown as sticks (light gray) and the other chains in surface representation (dark gray). Hydrophobic residues are shown in orange. Residues in “a” and “d” positions targeted for substitutions are highlighted. The precise nature of the substitution in single-site variants is indicated. Substitutions introduced in the triple variant [referred to as F3M in (B) and (C)] are indicated in red. (B) Sedimentation coefficients, as obtained by AUC, of the various MD variants. (C) Normalized denaturation profiles of the MD variants as a function of GND concentration. Denaturation was estimated by CD by monitoring the ellipticity at 222 nm. Experimental points (dots) were fitted to a sigmoidal curve (continuous line). Each point is the mean of 20 measurements at 222 nm. For each variant, the data obtained for two different protein samples (shown with the same color) are shown. The horizontal dashed line corresponds to an unfolded ratio of 20%. (D) Correlation between the stability of MD variants (wt, V315L, V315I, V315N, N329L, N329I, N329V, V346I, L322I/I325L/L336I, LLIL, LIIL, IILI, IIII, IMMI, IPKI, LPKI, MPKI, and IPKL) as defined on the basis of the concentration of GND leading to 20% of denaturation and their overall hydrophobicity corresponding to the sum of the free energies of transfer ΔG (kcal/mol) from water to palmitoyloleoylphosphatidylcholine interface of each residue (42). P wt is shown in red. (E) Functionality of the MD variants bearing substitutions in “a” and “d” positions, as measured in a minigenome assay, as a function of their hydrophobicity level. BSR-T7 cells were transfected with the mixture of expression vectors encoding the minigenome, N, L, and P variants. The firefly luciferase activity was measured 2 days after transfection. Data in (D) and (E) are means ± SD of at least three independent experiments, with each point done in triplicate. Statistical analysis was performed using Pearson’s r correlation test after best-fit regression curve of full dots. (F) Analysis of helix axis curvature. The different panels presented on the first line show the maximum curvature angle seen for each chain versus residue numbers. The chains are colored according to blue (chain A), red (chain B), orange (chain C), and purple (chain D). The isosurface angle curvatures over 100 generated frames versus the distance along the helix or residue number are shown for wt MD, IPKI, V315L, V315I, and V315N. The angle curvature is represented by a color gradient, which varies from 0° (dark blue) to 28° (dark red).
Purification of MD variants by size exclusion chromatography (SEC) led to a typical profile with three peaks (at 45, 50, and 60 ml) (fig. S5A), of which the first one corresponds to the void volume as already described in previous studies (19, 21). The peak at 60 ml corresponds to the elution of a protein with an apparent mass of 39 kDa, a value in close agreement with the expected mass for a tetrameric form (36 kDa). On the other hand, the peak at 50 ml could correspond to an oligomeric state of higher order or to a more extended conformation of the MD. The similarity of the SEC elution profiles of the different variants suggests that they likely share the same oligomeric state. To confirm this, and to avoid possible pitfalls related to the poor suitability of SEC to infer information about the molecular mass of very elongated macromolecules, we also used analytical ultracentrifugation (AUC). In particular, wt MD and a subset of six variants mutated at anchor positions were studied by AUC. In agreement with the SEC profile, sedimentation data could not be fitted assuming a monodisperse single-specie model. Thus, they were fitted assuming a continuous distribution of sedimentation coefficient (S) (see fig. S6 for the quality of the fits). The best fit led to a distribution similar to that of the SEC elution profile characterized by two distinct species, with a mean sedimentation coefficient of 2.5S and 3.5S compatible, respectively, with a tetramer and an octamer (Fig. 5B). Comparison of those values with the theoretical S computed using HYDROPRO (41) and the MeV MD crystal structure (PDB code 3ZDO) confirms that the species with the lowest S correspond to the tetrameric MD as observed in the crystal. Several computations were performed on dimers of tetrameric MDs modeled by assembling two adjacent tetramers, as observed in the crystal packing of 3ZDO. While a complex between two tetrameric MDs interacting side to side led to a theoretical sedimentation coefficient of 4.34 S, assuming an apical interaction between two tetrameric MDs led to a calculated sedimentation coefficient of 3.38 S, a value quite close to the highest S estimated from sedimentation fitting. This suggests that dimers likely arise from apical interactions between tetramers, with this type of interactions being a notorious source of artifacts in coiled coils. Sedimentation data could also be fitted using a polydisperse discrete species model of two components using the sedimentation coefficients calculated from HYDROPRO for the tetrameric MD and for the dimer of tetrameric MDs. Discrete and continuous models were found to fit the experimental data equally well.
Together, SEC and AUC studies indicate that the substitutions introduced at anchor positions have impact neither on the oligomeric state of the protein nor on the relative amounts of tetramers versus dimers of tetramers (fig. S5, A and B).
The secondary structure content of the variants was estimated using far-ultraviolet circular dichroism (CD). Measured spectra are all typical of proteins, with a predominant α-helical content characterized by maximal amplitudes at 222 and 209 nm (fig. S5B). Furthermore, the ellipticity ratio at 222 and 209 nm (θ222/θ209) is greater than one for all variants, a hallmark of proteins composed of α-helices interacting with each other’s. Last, all spectra are nicely superimposable and almost identical, indicating that the substitutions introduced in the MD have no impact on the secondary structure content of the protein (fig. S5B).
However, the variants significantly differ in their resistance to chemical denaturation induced by increasing concentration of guanidinium chloride (GND) (Fig. 5C). Most of the variants show a cooperative denaturation according to a two-state model, as expected for a folded single-domain protein. Unexpectedly, substituting the asparagine-329 with either a leucine or an isoleucine led to a denaturation profile showing two transitions, while substituting N329 by a valine led to a single but very long transition.
From the experimentally determined free energy of transfer from water to a bilayer interface (ΔG score) of each residue, the overall hydrophobicity of each MD variant could be estimated (42). A linear correlation was obtained when the GND concentration inducing 20% denaturation was plotted against the overall hydrophobicity, indicating that increasing the hydrophobicity of the coiled-coil core increases the overall MD stability (Fig. 5D). Only a small subset of MD variants bearing substitutions at anchor positions displays activity in a minigenome assay (Fig. 5E). In particular, N329V and V346I variants exhibit the highest activity, close to that of wt P; V315I and the triple variant (F3M) bearing the substitutions L322I/I325L/L336I exhibit an intermediate activity. All other variants (V346N, N329I, Q356V, N329L, V315L/N329L, I353D, and L322D) were found to be inactive (Fig. 5E). When plotted against the overall hydrophobicity, the activity appears to be restricted to a narrow range of hydrophobicity levels comprised between >98 and <102% of the ΔG score of wt P MD (Fig. 5E). Only P variants bearing substitutions at anchor (i.e., buried) residues were included in this analysis to avoid possible changes in the interaction network involving surface-exposed residues that may jeopardize the analysis.
These results support a scenario where either decreasing or increasing the MD stability reduces gene expression. That is, the ability of P to support viral gene expression appears to rely on a small range of MD coiled-coil cohesiveness.
The possibility that the observed variations in minigenome activity could arise from differences in the accessibility of exposed residues induced by the substitution of anchor residues was checked and ruled out. The RMSFs of variants bearing a substitution at position 315, chosen because they subtly vary in their hydrophobicity and in their activity in a minigenome assay (i.e. V315I, V315N, and V315L), were determined from a 1-μs molecular dynamic simulation and were found to be not significantly different neither among each other nor with respect to wt and IPKI (fig. S4). Notably, the lack of structural variations also concerns residues in the vicinity of the position targeted for substitution (i.e., position 315), as well as the basic patch K350/K351/N354 that might be involved in binding to L by analogy with SeV (fig. S4I, insets) (43). Likewise, the observed differences in activity cannot be ascribed to possible differences in the dynamics of the kink. The active V315I variant and the inactive V315L and V315N variants all display a similar overall constant composition of three 310-helices plus one α-helix (with each monomer equally sampling the two conformations over time as observed with wt MD) (fig. S7).
Normal mode analysis suggests that the bending of the coiled coil is not critical for P activity
While molecular dynamics simulations are particularly well suited to study local motions and secondary structure changes within short time scales, events like folding, helix bending, and deformation involve larger molecular displacements and become computationally very expensive. To study large structure rearrangements or deformations, normal mode analysis is better suited and can be carried out on a single structure (44). Normal mode analysis is based on the fact that the vibrational normal modes exhibiting the lowest frequencies are associated with the largest movements in proteins and are the ones that are functionally relevant. From the single structure used as starting model in molecular dynamic simulations (fig. S3A), the five most collective motions (i.e., motions showing the five highest eigenvalues) were computed. From these modes, a collection of 100 PDB files was generated and assembled to represent the change of secondary structure arising from normal mode calculation (see movie S1, A and B). The 100 assembled structures allowed us to analyze the curvature of the four helices of the wt MD and IPKI, which are shown in Fig. 5F and can be appreciated from movie S2 (A and B). The analysis was performed along the helix axis to obtain local angles along the length of the helix versus frame number (100 in total). The helix distortion distribution is represented across residues by an iso-contour map with color code according to local angle magnitude (44). In wt MD, a pronounced curvature that equally involves the four chains and encompasses residues L341 to V346 is observed, and the bending can be assimilated to a hinge effect around residue L340. In the IPKI variant, the curvature encompasses two sets of residues, S334 to L336 and S338 to I342, with the bending axis centered on E337. In addition, in this case, the four chains do not appear to contribute equally to the bending in the two sets of residues, i.e., the coiled coil likely bends in a less coherent manner compared to wt MD (Fig. 5F, compare upper curves). IPKI is characterized by a marked twisting movement (compare movie S1A with movie S1B), possibly resulting from the presence of two consecutive hinges.
Normal mode analysis of the helix curvature of variants V315I, V315L, and V315N reveals subtle variations between these three variants and wt MD in the maximal curvature amplitude (highest for V315L) and/or degree of homogeneity in curvature variations of each chain with higher symmetry for wt and V315I (Fig. 5F, compare upper curves). When considering, in addition, the helix curvature of IPKI, no obvious feature in the bending of the coiled coil that allows discrimination of active versus inactive variants could be found.
Another observation from the normal mode is the presence of another peak of angular mobility centered on L367 at the C terminus, i.e., in the D subdomain, a feature absent in the N terminus. This is indicative of a local increase in chain mobility as previously speculated (21).
DISCUSSION
With the goal of investigating the L-P complex, for which molecular data were scarce, several P variants have been designed, generated, and characterized in their structural and functional properties.
Results bring new insights into the MeV P regions required for binding to L and for the formation of a stable and active polymerase complex (Fig. 6A). In particular, we found subtle modifications of the P MD to strongly affect the ability of P to support viral gene expression despite conservation of its tetrameric coiled-coil structure, thus supporting a critical role played by the MD beyond mere P tetramerization. Notably, although the expression levels of the P MD variants that were tested in a minigenome assay vary to some extent (fig. S8), no correlation was found between expression levels and activity (fig. S8).
Fig. 6. Map of P functions and model of tight regulation exerted by MD on RNA synthesis activity by the polymerase.
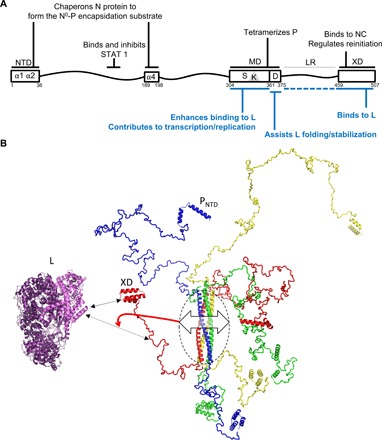
(A) P functions previously described and functions characterized in this study are written in black and blue, respectively. (B) Cartoon representation of a model of MeV P generated using RANCH (96). The crystal structures of MeV P MD (PDB code 3ZDO) (21), MeV XD (PDB code 1OKS) (28), and the PNTD region (residues 1 to 37 of P) as observed in the MeV N0-PNTD complex (PDB code 5E4V) (15) were used to build up the model. Chains are colored in blue, green, red, and yellow. The cryo-electron microscopy structure of the L protein from VSV (PDB code 5A22) (4) is shown in purple and pink. The L region shown in pink corresponds to conserved regions CRI and CRII, i.e., the structural counterpart of the MeV L region spanning residues 1 to 620. The L protein is shown oriented with its CRI and CRII facing XD and the linker. The continuous and dashed double-headed black arrows indicate the major (XD) and secondary (linker) L-binding sites within P. The double-headed arrow on P MD (circled) illustrates variations in MD cohesiveness that, in turn, may affect the conformational landscape of the linker region (see the red arrow), thus modulating binding to L.
The main MeV L-binding site lies within P C-terminal region and seems conserved within the Paramyxoviridae family
Previous attempts to map the reciprocal L- and P-binding sites in various Paramyxoviridae members have located the P-binding site on the L N-terminal region and the L-binding site on the P C-terminal region downstream the MD and/or possibly including it (22, 45–51). The instability of the L protein from Mononegavirales when expressed in the absence of the homologous P protein has been a potential source of confusing data including repeated reporting of L-L interactions via its NTD that was likely due to nonspecific aggregation of collapsed protein species (52). By using a rationally designed truncated N-terminal L protein construct that is stably expressed in the absence of P and does not require the HSP90 chaperon apparatus, the main L-binding site within MeV P was mapped to the XD, with a possible weaker interaction in the linker region. In addition, P oligomerization was required for an efficient binding to L of the C-terminal region of P. Although previous work reported that the deletion of the XD of SeV did not abrogate L binding to P (49, 50), the XD of rinderpest virus, a virus closely related to MeV, is required for interaction with L (47). The enhancing effect of the MD on the P-L interaction has also been reported in the case of hPIV3 and MuV (22, 53). This enhancement may be due to the binding of more than one P to a single L protein because of two independent P-binding sites on L and/or to the increase of P avidity for L because of the fourfold increase in the local concentration of the monomeric L-binding site located on each chain of the tetrameric P. In the absence of the MD, the C-terminal region could also adopt a structure inappropriate for binding to L.
The dual binding properties of the C-terminal domain of P toward both L and the NC have also been reported for respiratory syncytial virus, a member of the Pneumoviridae family (54, 55). This raises the question as to whether NTAIL (or N) and L may bind simultaneously to P or rather whether their binding is mutually exclusive with possible functional impact on the regulation of the transcription at intergenic junctions (12, 13).
Beyond NTAIL and L, MeV XD also binds and stabilizes the E3 ubiquitin ligase Pirh2 by preventing self-ubiquitination of the latter (56). Exploratory experiments aimed at unraveling possible links between the recruitment of Pirh2 and the role of P in assisting L folding/stabilization were nonconclusive, thus setting the scene for future studies.
The C-terminal end of the MD is required for P to assist L folding/stability
Although P MD is not strictly required for binding to L, the last two heptads of the coiled coil, referred to as the D region, are critically required to allow proper folding of L. The corresponding region of SeV P has previously been linked to L binding but only as part of larger deletions encompassing downstream regions (49, 50). In addition, the use of full-length L in these studies precludes the distinction between the lack of L binding and the loss of L stability. Moreover, mutations of hydrophobic residues within this region on SeV MD abrogate RNA synthesis without affecting P oligomerization or binding to L (43). How MeV D region controls L stability and/or folding is yet to be determined more precisely. Similarly to the low affinity N0-binding site recently mapped on P N-terminal region (17), the D region may contain an additional weak L-binding site that may be required for maintaining L in a stable form (Fig. 6B).
Unlike paramyxoviruses, the L-binding site on vesicular stomatitis virus (VSV) and rabies virus (RabV) P proteins, two Rhabdoviridae family members, has been mapped to the N-terminal region of P, upstream the oligomerization domain (10, 57–59). Although a peptide encompassing P residues 41 to 61 is required for VSV P to bind L by coprecipitation, the region P81-106 is sufficient for stimulation of L processivity on a naked RNA (59). Similarly, RabV P1-19 is sufficient to bind L but P40-50 is required for stimulation of L processivity on naked RNA (10). Therefore, a main L-binding site associated with more distant regions with lower affinity and implicated in other functions such as L stability or processivity may be a common feature of mononegavirus P proteins.
Structural features of the MD are required for the activity of the polymerase complex
The insertion of a stammer or a stutter in the MD sequence seems to be a hallmark of Paramyxoviral MDs. Substitution of the kinked LLLL motif by the IPKI motif from the cognate NiV causes the kink to shift upstream the stammer and induces the formation of a 310-helix on the four chains but does not abrogate the viral gene expression. By contrast, all our attempts at removing the kink by either insertion or deletion led to a nonfunctional or poorly efficient polymerase complex. Therefore, although the sequence and position of the kink can be modified to some extent, the kink appears to be a required feature of MeV MD. Whether it is the kink itself that is crucial or its effects on the MD cohesiveness, overall stability, and/or dynamics remains to be determined. That the kink itself may establish a weak contact with L looks unlikely because the substitution of the two solvent-exposed residues of the stammer (i.e., L340L341) with residues with different physicochemical properties (i.e., PK in IPKI, MM in IMMI, and PA in IPAI) and the shifting of the kink location (in IPKI) are tolerated. To the best of our knowledge, such a crucial functional role of a kink within a coiled coil had not been reported so far.
The oligomerization domain of the MuV P protein was also shown to be required for viral gene expression in a cell-based assay, and the alanine substitution of arginine and lysine of the coiled coil was found to be deleterious (22). The substituted positively charged residues are all located in either “e” or “g” position. Those positions, when occupied by charged residues, are known to be involved in both coiled-coil cohesion and parallelism through electrostatic interactions, and alanine substitutions might affect these parameters, although this was not documented in that study (22). Here, modification of the coiled-coil cohesion through substitution of buried hydrophobic residues in either “a” or “d” positions is reported to affect viral gene expression, with the least tolerated substitutions being those inducing the highest variations in stability with respect to wt MD. Because substitutions are introduced in the hydrophobic core of the protein and have no detectable impact on oligomeric state, α-helical content, and RMSF of MD residues, one can conclude that the diminution of the activity does not result from a modification of the protein-protein interaction network in which MeV P MD could be involved through its exposed residues. Rather, subtle variations in the hydrophobicity of buried residues are associated with a marked impact on the ability of P to sustain viral gene expression, although those substitutions result only in negligible changes in the overall dynamics of MD, as determined from RMSF calculation in molecular dynamic simulations, and in coiled-coil curvature angle, as judged from normal mode analysis. The requirement of a kink and the need for a restricted range of coiled-coil cohesiveness are not of straightforward interpretation and raise the puzzling question of which is the modulatory signal intrinsic to MD and how it is delivered to the polymerase machinery.
The minigenome assay does not allow to look specifically at RNA synthesis, and a loss of luciferase signal could result from several mechanisms such as a default in the initial encapsidation of the minigenome or the inability of the P variants to undergo a liquid-liquid phase separation such as the one required for the formation of viral factories in RabV (60) and in VSV (61). However, because of the role of MD in the L-P binding, its proximity with the L-binding site(s), and the requirement of the D module to form stable, folded L protein, it is highly probable that the loss of signal is linked to a default in the formation, or the activity, of the L-P polymerase complex.
The need for a kinked coiled coil with a precise cohesiveness might reflect a role of P as a dynamic (mobile) scaffold to enable accommodation of L, which is a very large protein, likely adopting different conformations according to its multiple enzymatic activities and/or depending on whether it is in the transcriptase versus replicase mode. The conformational entropy of P MD may, for instance, affect the conformational landscape of the linker region that possibly contains a second weaker L-binding site (Fig. 6B). It can also, although to a lower extent, have an impact on the conformational sampling of the D region that is responsible for enabling L folding into a mature, functional form.
It is also tempting to speculate that the MD entropy might mechanically be transduced along the P protein to affect the interactions that P establishes with the monomeric (N0) and assembled form of N through its N- and C-terminal domains (PNTD and XD), respectively. These interactions need to be highly coordinated and are probably subjected to spatial constraints (for example, to allow the concomitant N encapsidation of the nascent genomic RNA), as well as to affinity constraints [as illustrated in the case of the XD-NTAIL interaction strength (12)]. However, this scenario seems to be unlikely, in light of the following considerations. First, although the MD can sample more than one conformational state, its conformational entropy, and hence ability to exert long-range effects on distant domains, is by far much lower compared to that of intrinsically disordered regions able to tune enzymatic function (62). Second, the possible impact of MD motions on the relative spatial position of PNTD and XD is likely “entropically rectified” (62) by the large intrinsically disordered regions that flank P MD.
Last, in light of the ability of coiled coils to both homo- and hetero-oligomerize [see (63) and Fig. 2F], substitutions can also possibly affect the ability of P to form putative functional hetero-oligomers with host cell proteins. Substitutions introduced in P MD, irrespective of whether they stabilize or destabilize the homotetrameric MD, will expectedly have an impact on hetero-oligomerization. If those hetero-oligomers are required for P to be able to sustain transcription and replication, then substitutions will have an impact on viral gene expression, with the native MD sequence being the one sustaining the highest activity. This hypothesis would also be consistent with the experimentally observed bell-shape dependency of P function on coiled-coil cohesiveness (Fig. 5E).
Even if the correlation between protein stability and the ability of P to support viral gene expression requires additional studies to be fully understood, the present results indicate that P MD is much more than a mere oligomerization device. Rather, it plays a crucial role in transcription and/or replication and requires a very specific set of physical properties governing its cohesiveness to lead to a fully functional system.
Coiled-coil structures are also involved in the oligomerization of several other nsNSV phosphoproteins. The atomic structures of P MDs from members of the Filoviridae family (Ebola virus and Marburg virus) (64, 65) and the Pneumoviridae family (66) are made of trimeric or tetrameric coiled coils with evidence of interconversion between trimers and tetramers for Ebola virus. Whether a narrow range of cohesiveness and dynamics is also essential for these coiled coils to support viral gene expression is presently unknown and worth to be studied.
From a broader point of view, the present work describes an additional mechanism by which a coiled coil can sustain an enzymatic cofactor function. A coiled coil can mediate oligomerization and hence increases the avidity by multiplying valence of appended binding sites, a process that can be itself regulated by the interconversion between monomers and multimers. The dynamic association between coils supports the cytoskeleton dynamics and the regulation of transcription factors or can act as a spring loaded hinge in fusion proteins (67). The surface of a coiled coil can constitute a specific binding site for a protein partner. It can bring together adjacent enzymatic domains or acts as a scissors grip to bind to DNA. Last, coiled coils can also serve as pH or temperature sensors and “rigid,” elastic, or spacer scaffolds [see (68) for review].
The discovery that the presence of a kink and the cohesiveness of the MD ensure a tight regulation of P functionality in viral gene expression enlarges the set of mechanisms by which coiled coils can rely on to mediate a broad range of different functions. The present work appears in line with the evolutionary conservation of destabilizing residues in Streptococcus M protein that enables the capture of the fibrinogen (69).
From a more practical perspective, the present results shed light onto the multiple P regions that could be targeted in those approaches. Drugs that interfere with P homo-oligomerization and/or with the P MD cohesiveness are likely to impair viral gene expression, thereby designating the structure of P MD as an asset for the design of novel antiviral agents.
Last, the discovery that efficient viral gene expression relies on a highly restricted range of P MD cohesiveness may explain the high conservation level of this P region within the Paramyxoviridae family. The corollary of this hypothesis is that paramyxovirus members would share a unique common mechanism to finely tune viral gene expression.
MATERIALS AND METHODS
Eukaryotic cell lines and biG-biS viruses
BSR-T7 (70), 293T (CelluloNet BioBank BB-0033-00072; SFR BioSciences, Lyon, France), 293-3-46 (71), Vero-hSLAM (72), Vero (si2), and Vero-hSLAM (si1) (2) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (30 min at 56°C), 1% l-glutamine, and gentamicin (10 μg/ml) at 37°C and with 5% CO2. Medium of 293-3-46 helper cells was supplemented with G418 (1.2 mg/ml). To rescue recombinant viruses, the helper cell line 293-3-46, stably expressing T7 polymerase, MeV N, and P, was transfected using the ProFection Kit with two plasmids coding for the MeV genome and MeV-L protein (pEMC-La) (71). Three days after transfection, the cells were overlaid on Vero-si2 cells. Upon appearance, isolated syncytia were picked and individually propagated on Vero-si2 cells. Virus stock was produced after a second passage at an MOI of 0.03 in the Vero-si2 cell line. This stock was checked to rule out mycoplasma contamination; had its N, P1, P2, M, and L genes sequenced; and was titrated on the relevant host cell before use.
Plasmids
The coding sequence of wt, truncated, and/or substituted L and/or P proteins from MeV (Moraten strain) was subcloned in frame either downstream or upstream of the Gaussia luciferase glu1 and glu2 domains under a cytomegalovirus promoter in the pCi vector (2) and under the T7 promoter in the pEMC vector (30). Some P variants were subcloned into the duplicated P2 gene position in the biG-biS–modified p(+)MVNSe vector (Fig. 2A) (2). The constructs for the eukaryotic expression of the P variants were generated by polymerase chain reaction (PCR) using dedicated oligonucleotides, and the PCR products were introduced into pCi, pEMC, and p(+)MVNSe by recombination using the InFusion Kit (Clontech). Whenever possible, new restriction sites with silent mutations were also introduced in the oligonucleotides to facilitate the screening of Escherichia coli–transformed clones. Plasmids used in the minigenome assay have been already described (12).
The bacterial constructs encoding variants of the P(304-375) MD with a C-terminal hexahistidine tag were subcloned in the pDEST14 vector (Invitrogen) by Gateway recombination (Invitrogen) of PCR products amplified from the corresponding pEMC constructs. All insert sequences were verified and validated by sequencing.
Protein expression in eukaryotic cells
The expression of P protein from every plasmid construct was verified by SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot. 293T or BSR-T7 cells were plated in a 12-well plate at 2 × 105 cells per well in DMEM supplemented with 10% of FBS. The next day, cells were transfected with 2 μg of plasmid with jetPRIME (Polyplus). For the assessment of L stability, some cells were also treated 6 hours after transfection with 2 μM 17-DMAG. Twenty-four hours after transfection, cells were trypsinized, washed with phosphate-buffered saline (PBS), and lysed for 30 min on ice. Cells expressing P only were lysed with a urea-free lysis buffer [50 mM tris-HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 0.1% NP-40, 5% glycerol, 1 mM dithiothreitol, and cOmplete protease inhibitor (Roche)], and cells expressing L were lysed with the same buffer supplemented with 8 M urea. Proteins were separated by SDS-PAGE and electrotransferred onto polyvinylidene difluoride membranes (Hybond-P, Amersham Biosciences). After saturation for 1 hour in tris-buffered saline (TBS) containing 0.05% Tween 20 (TBS-Tween) and 5% fat-free milk, the membranes were incubated for 2 hours with primary antibodies specific for P [either the mouse monoclonal antibody 49.21 (56) or a rabbit polyclonal antibody (37, 56) diluted 1:3000 and 1:10,000 in TBS–Tween–5% milk, respectively] or Flag/L (1:2000; mouse anti-Flag monoclonal antibody, Sigma), washed with TBS-Tween, and incubated with peroxidase-conjugated sheep anti-mouse or anti-rabbit immunoglobulin G (IgG) antibody (Promega) for 1 hour at room temperature. The membranes were then washed and incubated in Covalight reagent (Covalab), and chemiluminescent signals were measured with the VersaDoc Imaging System (Bio-Rad). All P variants migrated at their expected molecular weight. Expression levels of the P MD variants that were tested in the minigenome assay were assessed using a semiquantitative dot blot assay as described previously (73) using the two anti-P antibodies described above.
Protein complementation assays
Gaussia princeps luciferase-based complementation assay and data analysis [normalized luminescence ratio (NLR)] were performed according to Cassonnet et al. (74). Human 293T cells were plated in a 96-well plate with 2 × 104 cells per well and transfected the next day with 100 ng of each plasmid using the jetPRIME reagent (Polyplus-transfection). Luciferase signals were read 24 hours after transfection. NLR was calculated by dividing the luciferase value of the two chimeric partners by the sum of the luciferase values of each chimeric partner mixed with the other free Gaussia luciferase domain. Results were expressed as fold increase with respect to the reference (wt full-length P), which was set to 1.
L folding assays
BSR-T7 cells were plated into a 96-well plate at 2 × 104 cells per well and transfected the next day with plasmids coding for Flag/L[Renilla] and a P variant with jetPRIME (Polyplus). Luciferase signals were measured 24 hours later according to the manufacturer’s protocol (Renilla Luciferase Assay System, Promega).
Analysis of viral growth
Parental Vero si1 and si2 cells were infected at an MOI of 0.1 with recombinant viruses with the addition of the fusion inhibitor peptide z-fFG (10 μg/ml) to prevent syncytium formation. At indicated times after infection, cells were trypsinized, stained with an anti-F monoclonal antibody (Y503) (73) for 30 min at 4°C, centrifuged, washed, stained with the Alexa Fluor 647 anti-mouse IgG antibody (1:700; Invitrogen) for 30 min at 4°C, centrifuged, washed, fixed in 1% paraformaldehyde for 15 min at room temperature, centrifuged, and resuspended in PBS. Fluorescence signals were quantified with Accuri C6 (BD Biosciences).
Minigenome assays
BSR-T7 cells were plated in 96-well plates at 2 × 104 cells per well and transfected the day after with 66 ng of pEMC-N, 44 ng of pEMC-(Flag/L), 28 ng of pEMC-P construct (encoding either wt P or the individual P variants), and 90 ng of plasmid encoding for the minigenome, using jetPRIME (Polyplus). Two days after transfection, the firefly luciferase activity was measured using the Nano-Glo Dual-Luciferase Reporter Assay (Promega). The background luciferase activity observed in the absence of active L protein was subtracted from the signal measured in the presence of L, and data obtained from three independent experiments were normalized to each other using the mean signal observed for all combinations tested at the same time (12).
Expression and purification of MD variants
The E. coli strain T7 pLysS (New England Biolabs) was used as the host for heterologous expression of all MD variants. E. coli cells were transformed with pDEST14 derivatives encoding P MD variants with a C-terminal hexahistidine tag. Transformed cells were grown overnight at 37°C in Luria-Bertani broth medium supplemented with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) and then diluted 1:40 in 1 liter of Super Broth medium supplemented with ampicillin and chloramphenicol. Cultures were incubated at 37°C until the OD600 (optical density at 600 nm) reached 0.7, and then protein expression was induced by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside. After induction, cultures were incubated for 4 hours at 37°C. Bacteria were harvested by centrifugation at 4000g for 20 min and then resuspended in 30 ml per liter of culture of buffer A [50 mM tris-HCl (pH 8), 1 M NaCl, and 10 mM imidazole] supplemented with lysozyme (0.1 mg/ml), deoxyribonuclease (10 μg/ml), and 20 mM MgSO4 and stored at −20°C. The cells were disrupted by sonication, and the lysate was clarified by centrifugation at 12,000g for 45 min at 4°C. The soluble fraction was loaded onto 2 ml of Sepharose Fast Flow resin preloaded with nickel and pre-equilibrated with buffer A. The resin was then washed successively with 20 column volumes of buffer A and then with 20 column volumes of buffer A containing 20 mM imidazole. Proteins were eluted with 5 column volumes of buffer B [50 mM tris-HCl (pH 8), 1 M NaCl, and 500 mM imidazole]. Proteins that were soluble at pH 8 were directly concentrated on a 10-kDa concentrator (Ultracel, Amicon), then loaded onto a S75 16:60 SEC column, and eluted in 10 mM tris-HCl (pH 8) and 150 mM NaCl. Proteins that were soluble at acidic pH were dialyzed against 50 mM sodium acetate (pH 4.5) and 150 mM NaCl and then concentrated and purified by SEC using this buffer. The fractions were analyzed by 18% SDS-PAGE, and those containing the protein of interest were pooled, concentrated as described above, and stored at 4°C.
Far-UV CD studies
CD spectra were recorded using a JASCO 810 dichrograph flushed with N2 and equipped with a Peltier thermoregulation system. Spectra were recorded between 190 and 250 nm with scanning speed of 20 nm/min and data pitch of 0.2 nm. All measurements were carried out at 20°C on samples at 0.1 mg/ml in 10 mM phosphate buffer (pH 8) using a 1-mm-thick quartz cell. Each spectrum is the average of three acquisitions. The spectrum of buffer was subtracted from the protein spectrum. Spectra were smoothed using the “means-movement” smoothing procedure implemented in the Spectra Manager package. Mean molar residue ellipticity (MRE) values per residue were calculated as [θ] = 3300 m∆A/lcn, where l is the path length in centimeters, n is the number of residues, m is the molecular mass in daltons, and c is the concentration of the protein in milligrams per milliliter. CD spectra were also recorded in the presence of increasing GND concentrations. To this end, protein samples at 10 mg/ml were diluted to 0.1 mg/ml into 40 different GND solutions, ranging from 0 to 6 M, in 10 mM tris-HCl (pH 8). Chemical denaturation was monitored by measuring the ellipticity values at 222 nm. For each GND concentration, 20 measurements were performed and averaged. The denaturation midpoints were derived upon fitting the data to a sigmoid curve using the nonlinear least-squared R routine (Gauss-Newton).
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out at 42,000 rpm and 20°C in a ProteomeLab XL-L analytical ultracentrifuge, using 12-mm aluminum-Epon double-sector centerpieces in an An55Ti rotor. Protein concentrations were recorded in continuous mode using absorbance at 280 nm and interferometry. P MD variants were studied at three different concentrations, namely, 0.8, 1.3, and 5 mg/ml. The partial specific volume of wt P MD, solvent density, and viscosity, as calculated with SEDNTER (75), were 0.741 ml/g according to P MD structure (PDB code 3ZDO), 0.99821 g/cm3, and 0.01002 poise, respectively. The partial specific volume of MD variants was assumed to be identical to that of wt MD. The data recorded from moving boundaries were analyzed in terms of both discrete species and continuous size distribution function of sedimentation coefficient C(S) using the program SEDFIT (76).
Crystallization and x-ray data collection of the IPKI variant
Crystallization experiments were performed immediately after protein purification. The IPKI variant was dissolved in 50 mM sodium acetate (pH 4.5) and 150 mM NaCl. Protein crystallization was carried out at 20°C using the sitting-drop vapor diffusion method in 288-well crystallization plates (Swissi 3-wells). Wells were filled using a nanodrop-dispensing robot (Mosquito Crystal, TTP Labtech) and various commercial crystallization kits [JCSG (Molecular Dimensions) and PEGs Suite & PEGs II Suite (Qiagen)]. Reservoir solutions were 40 μl in volume, and crystallization drops were composed of 100 nl of reservoir solution and 100, 200, and 300 nl of protein solution at 5 mg/ml. Crystallization plates were sealed with transparent film after setup of the drops and were transferred to a storage cabinet at 20°C. Crystallization was followed using a Rock Imager system (Formulatrix). The IPKI variant was crystallized in 25% polyethylene glycol 3350, 200 mM CaCl2, and 100 mM tris-HCl (pH 8) in 5 days. A single crystal was fished out, washed in the same solution, and flash-cooled in liquid nitrogen. X-ray diffraction was carried out using synchrotron light on the PROXIMA1 beamline at SOLEIL, Gif-sur-Yvette, France. Three thousand six hundred images were collected over 360° and recorded using an Eiger X Dectris detector, with a sample to detector distance yielding a resolution limit of 2.2 Å.
Structure determination, refinement, and analysis
Diffraction data were integrated by XDS (77) and scaled with AIMLESS (78). Phases were obtained by molecular replacement using the crystal structure of MeV wt P MD (PDB code 3ZDO) (19) using MOLREP (79). Data analysis by the STARANISO server (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi) revealed an anisotropic diffraction with resolution limits of 2.5 Å along the a* and b* axes and 1.9 Å on the c* axis. To take into account this anisotropy, an ellipsoidal resolution limit mask was used and rescaling was performed by STARANISO. Model building was manually done with COOT (80), and refinement was performed using BUSTER (81) and REFMAC (82). PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC) was used to visualize and draw the models. The quality of the final model was evaluated using MolProbity (http://molprobity.biochem.duke.edu/). The server PDBeFold from EMBL-EBI (http://www.ebi.ac.uk/msd-srv/ssm/ssmstart.html) was used to compute root mean square deviations (RMSDs) between P MD structures. Coiled-coil analysis was carried out using TWISTER (83).
Structure modeling and molecular dynamics simulations of MDs
In view of performing molecular dynamic simulations, we first generated structural models of wt and IPKI MDs prolonged to cover the entire MD (amino acids 304 to 375) and hence to include the dynamic domain (D) to be able to monitor over time the possible structural changes occurring within this region. In addition, the starting model of wt P MD was chosen to be symmetric, i.e., to have a kink on each of the four chains. The rationale for this choice resides in the fact that the asymmetry of the 3ZDO structure likely arises from crystal packing artifacts (19). To generate the prolonged wt and IPKI MD models for molecular dynamic simulations, the structures of wt P MD (PDB code 4C5Q) and IPKI variant (PDB code 6HTL) were used as starting models complemented by the C terminus of the 3ZDO structure (residues 357 to 370). The alignment of the query and template sequences was first performed by means of the MUSCLE web server (84) with the Pearson output file format. The resulting target alignment file was used as input to generate a 3D structural model of MD using MODELLER (85). The models of MD variants bearing a substitution at position 315 were built in the same way by means of the MODELLER software as described above.
Molecular dynamics simulations were carried out by means of the ACEMD software (86) with the AMBER99SB-ILDN force field (87) and with the TIP3P water model (88). The systems were minimized and equilibrated under constant pressure (1 atm) and temperature (300 K) [constant number of particles (N), pressure (P) and temperature (T), NPT] conditions using a time step of 4 fs [owing to the use of the hydrogen mass repartitioning scheme (89) implemented in ACEMD], a nonbonded cutoff of 9 Å, and particle-mesh Ewald long-range electrostatics with a grid of 70 × 74 × 138 with spacing of 1 Å. The systems were equilibrated first using 2000 steps of steepest-descent minimization, followed by running 0.12 ns of the isothermal NVT [constant number of particles (N), volume (V), and temperature (T)] ensemble, using a Langevin thermostat set at 300 K, followed by 2.8 ns of the isothermal-isobaric NPT ensemble using a Langevin thermostat at the same temperature and the Berendsen barostat of ACEMD. During minimization and equilibration, the heavy atoms were restrained spatially using a spring constant (1 kcal mol−1 Å−2). Then, a production run of 1 μs in the NVT ensemble was performed. The processing of the different trajectories and the secondary structure timeline analysis were performed by using VMD (90) and the GROMACS dssp utility (91). Normal mode analysis was performed by means of the NOLB software (92) that is able to generate structures with a fewer number of geometric distortions compared to the linear approach. To represent the amplitude of the normal modes, we generated 100 frames for wt P MD and for the IPKI, V315I, V315L, and V315N variants. The amplitude of the different curvatures was then analyzed by means of the Bendix module (93) of VMD, while the isosurface curvature map was displayed within MATLAB (MathWorks Inc).
Statistical analyses
Data from PCA and minigenome assays were analyzed statistically by comparing data from at least three independent experiments (with each data point performed in triplicate) with a negative “Ø” and a positive “wt” control (unless otherwise indicated in the figures) using Student’s t test. Statistically significant differences at P < 0.05 and below with respect to the negative and positive control are quoted by stars. Correlation studies were performed using Pearson’s r correlation test.
Supplementary Material
Acknowledgments
We acknowledge the contribution of SFR Biosciences (UMS3444/CNRS, US8/Inserm, ENS de Lyon, UCBL) flow cytometry facilities (T. Andrieu and S. Dussurgey). We thank C. Mathieu for critical reading of the manuscript. We also wish to thank G. Sulzenbacher, J. Roche, and F. Fadel (from the AFMB lab) and P. Legrand (beamline Proxima-1, Soleil synchrotron, Gif-sur-Yvette, France) for their help and useful advice on the resolution of the crystal structure. G. Sulzenbacher is also acknowledged for efficient managing of the AFMB BAG. Funding: This work was supported, in part, by French ANR NITRODEP (project ANR-13-PDOC-0010-01) (www.agence-nationale-recherche.fr) and LABEX ECOFECT (ANR-11-LABX-0048) of Lyon University, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR). The studies described here were carried out with the financial support of the CNRS. A.S. is a recipient of a Ph.D. fellowship from the French Ministry of National Education, Research and Technology allotted to the Ecole Doctorale des Sciences de la Vie et de la Santé (Aix-Marseille Université). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Author contributions: L.-M.B. and A.S. designed and performed experiments, analyzed data, and wrote part of the manuscript; C.L. performed and analyzed data; B.R. designed and analyzed experiments; M.H. designed, performed and analyzed data, and wrote part of the manuscript. O.W. designed and analyzed data. S.L. supervised the project, analyzed data, and wrote the manuscript. D.G. designed and performed experiments, supervised the project, and wrote the manuscript. All authors approved the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The structure of the IPKI MD variant has been deposited in the PDB under PDB code 6HTL. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw3702/DC1
Fig. S1. Stability of L620 fragment in the absence of P.
Fig. S2. Impact of kink abrogation and displacement on P functionality.
Fig. S3. Workflow of molecular dynamics simulation versus normal mode analysis and results of molecular dynamics simulations on wt and IPKI MD variants.
Fig. S4. Comparison of RMSF per residue for each chain and average over the four chains.
Fig. S5. Biochemical analysis of MD variants.
Fig. S6. AUC analysis of MD variants.
Fig. S7. Molecular dynamics simulations of V315N, V315I, and V315L MD variants.
Fig. S8. Lack of relationships between expression levels of the variants and their ability to support viral gene expression.
Table S1. Data collection and refinement statistics for the IPKI MD variant.
Table S2. Values for the main structural coiled-coil parameters of wt MD and of the IPKI variant as determined by TWISTER and RMSDs (Å) among the IPKI and the wt MD structures.
Movie S1 (A and B). Animations showing the low frequency collective motions [also known as normal modes (NM)] computed to extract large structure rearrangements of the wt and IPKI.
Movie S2 (A and B). Analysis of the curvature of the four helices along the different frames used to compute NM. The color gradient used for the animation is similar to the one used in Fig. 5C.
REFERENCES AND NOTES
- 1.Ortin J., Martin-Benito J., The RNA synthesis machinery of negative-stranded RNA viruses. Virology 479-480, 532–544 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Brunel J., Chopy D., Dosnon M., Bloyet L.-M., Devaux P., Urzua E., Cattaneo R., Longhi S., Gerlier D., Sequence of events in measles virus replication: Role of phosphoprotein-nucleocapsid interactions. J. Virol. 88, 10851–10863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutsche I., Desfosses A., Effantin G., Ling W. L., Haupt M., Ruigrok R. W. H., Sachse C., Schoehn G., Structural virology. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 348, 704–707 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Liang B., Li Z., Jenni S., Rahmeh A. A., Morin B. M., Grant T., Grigorieff N., Harrison S. C., Whelan S. P. J., Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162, 314–327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morin B., Kranzusch P. J., Rahmeh A. A., Whelan S. P. J., The polymerase of negative-stranded RNA viruses. Curr. Opin. Virol. 3, 103–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canter D. M., Perrault J., Stabilization of vesicular stomatitis virus L polymerase protein by P protein binding: A small deletion in the C-terminal domain of L abrogates binding. Virology 219, 376–386 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Curran J., Marq J. B., Kolakofsky D., An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol. 69, 849–855 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masters P. S., Banerjee A. K., Complex formation with vesicular stomatitis virus phosphoprotein NS prevents binding of nucleocapsid protein N to nonspecific RNA. J. Virol. 62, 2658–2664 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellon M. G., Emerson S. U., Rebinding of transcriptase components (L and NS proteins) to the nucleocapsid template of vesicular stomatitis virus. J. Virol. 27, 560–567 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morin B., Liang B., Gardner E., Ross R. A., Whelan S. P. J., An in vitro RNA synthesis assay for rabies virus defines ribonucleoprotein interactions critical for polymerase activity. J. Virol. 91, e01508-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin B., Rahmeh A. A., Whelan S. P. J., Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 31, 1320–1329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bloyet L.-M., Brunel J., Dosnon M., Hamon V., Erales J., Gruet A., Lazert C., Bignon C., Roche P., Longhi S., Gerlier D., Modulation of re-initiation of measles virus transcription at intergenic regions by PXD to NTAIL binding strength. PLOS Pathog. 12, e1006058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox R. M., Krumm S. A., Thakkar V. D., Sohn M., Plemper R. K., The structurally disordered paramyxovirus nucleocapsid protein tail domain is a regulator of the mRNA transcription gradient. Sci. Adv. 3, e1602350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggarwal M., Leser G. P., Kors C. A., Lamb R. A., Structure of the paramyxovirus PIV5 nucleoprotein in complex with an amino-terminal peptide of the phosphoprotein. J. Virol. 92, e01304-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guryanov S. G., Liljeroos L., Kasaragod P., Kajander T., Butcher S. J., Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J. Virol. 90, 2849–2857 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yabukarski F., Lawrence P., Tarbouriech N., Bourhis J.-M., Delaforge E., Jensen M. R., Ruigrok R. W. H., Blackledge M., Volchkov V., Jamin M., Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat. Struct. Mol. Biol. 21, 754–759 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Milles S., Jensen M. R., Lazert C., Guseva S., Ivashchenko S., Communie G., Maurin D., Gerlier D., Ruigrok R. W. H., Blackledge M., An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci. Adv. 4, eaat7778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruhn J. F., Barnett K. C., Bibby J., Thomas J. M. H., Keegan R. M., Rigden D. J., Bornholdt Z. A., Saphire E. O., Crystal structure of the nipah virus phosphoprotein tetramerization domain. J. Virol. 88, 758–762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Communie G., Crepin T., Maurin D., Jensen M. R., Blackledge M., Ruigrok R. W. H., Structure of the tetramerization domain of measles virus phosphoprotein. J. Virol. 87, 7166–7169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarbouriech N., Curran J., Ruigrok R. W. H., Burmeister W. P., Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 7, 777–781 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Blocquel D., Habchi J., Durand E., Sevajol M., Ferron F., Erales J., Papageorgiou N., Longhi S., Coiled-coil deformations in crystal structures: The measles virus phosphoprotein multimerization domain as an illustrative example. Acta Crystallogr. D Biol. Crystallogr. 70, 1589–1603 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Pickar A., Elson A., Yang Y., Xu P., Luo M., He B., Oligomerization of mumps virus phosphoprotein. J. Virol. 89, 11002–11010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blocquel D., Beltrandi M., Erales J., Barbier P., Longhi S., Biochemical and structural studies of the oligomerization domain of the Nipah virus phosphoprotein: Evidence for an elongated coiled-coil homotrimer. Virology 446, 162–172 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Beltrandi M., Blocquel D., Erales J., Barbier P., Cavalli A., Longhi S., Insights into the coiled-coil organization of the Hendra virus phosphoprotein from combined biochemical and SAXS studies. Virology 477, 42–55 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Lupas A. N., Bassler J., Coiled coils - a model system for the 21st Century. Trends Biochem. Sci. 42, 130–140 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kolakofsky D., Le Mercier P., Iseni F., Garcin D., Viral RNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 318, 463–473 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Longhi S., Receveur-Bréchot V., Karlin D., Johansson K., Darbon H., Bhella D., Yeo R., Finet S., Canard B., The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 278, 18638–18648 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Johansson K., Bourhis J.-M., Campanacci V., Cambillau C., Canard B., Longhi S., Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 278, 44567–44573 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Kingston R. L., Hamel D. J., Gay L. S., Dahlquist F. W., Matthews B. W., Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. U.S.A. 101, 8301–8306 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bloyet L.-M., Welsch J., Enchery F., Mathieu C., de Breyne S., Horvat B., Grigorov B., Gerlier D., HSP90 chaperoning in addition to phosphoprotein required for folding but not for supporting enzymatic activities of measles and nipah virus L polymerases. J. Virol. 90, 6642–6656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katoh H., Kubota T., Nakatsu Y., Tahara M., Kidokoro M., Takeda M., Heat shock protein 90 ensures efficient mumps virus replication by assisting with viral polymerase complex formation. J. Virol. 91, e02220-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capra J. A., Singh M., Predicting functionally important residues from sequence conservation. Bioinformatics 23, 1875–1882 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Salladini E., Delauzun V., Longhi S., The Henipavirus V protein is a prevalently unfolded protein with a zinc-finger domain involved in binding to DDB1. Mol. BioSyst. 13, 2254–2267 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez K. R., Horvath C. M., Amino acid requirements for MDA5 and LGP2 recognition by paramyxovirus V proteins: A single arginine distinguishes MDA5 from RIG-I. J. Virol. 87, 2974–2978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Remy I., Michnick S. W., A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 3, 977–979 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Cevik B., Holmes D. E., Vrotsos E., Feller J. A., Smallwood S., Moyer S. A., The phosphoprotein (P) and L binding sites reside in the N-terminus of the L subunit of the measles virus RNA polymerase. Virology 327, 297–306 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Chen M., Cortay J. C., Gerlier D., Measles virus protein interactions in yeast: New findings and caveats. Virus Res. 98, 123–129 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Harbury P. B., Zhang T., Kim P. S., Alber T., A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262, 1401–1407 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Plumet S., Duprex W. P., Gerlier D., Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 79, 6900–6908 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deiss S., Hernandez Alvarez B., Bär K., Ewers C. P., Coles M., Albrecht R., Hartmann M. D., Your personalized protein structure: Andrei N. Lupas fused to GCN4 adaptors. J. Struct. Biol. 186, 380–385 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Ortega A., Amorós D., García de la Torre J., Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White S. H., Wimley W. C., Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta 1376, 339–352 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Bowman M. C., Smallwood S., Moyer S. A., Dissection of individual functions of the Sendai virus phosphoprotein in transcription. J. Virol. 73, 6474–6483 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skjaerven L., Hollup S. M., Reuter N., Normal mode analysis for proteins. Theochem 298, 42–48 (2009). [Google Scholar]
- 45.Horikami S. M., Smallwood S., Bankamp B., Moyer S. A., An amino-proximal domain of the L protein binds to the P protein in the measles virus RNA polymerase complex. Virology 205, 540–545 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Bousse T., Takimoto T., Matrosovich T., Portner A., Two regions of the P protein are required to be active with the L protein for human parainfluenza virus type 1 RNA polymerase activity. Virology 283, 306–314 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Chattopadhyay A., Shaila M. S., Rinderpest virus RNA polymerase subunits: Mapping of mutual interacting domains on the large protein L and phosphoprotein p. Virus Genes 28, 169–178 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Parks G. D., Mapping of a region of the paramyxovirus L protein required for the formation of a stable complex with the viral phosphoprotein P. J. Virol. 68, 4862–4872 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curran J., Pelet T., Kolakofsky D., An acidic activation-like domain of the Sendai virus P protein is required for RNA synthesis and encapsidation. Virology 202, 875–884 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Smallwood S., Ryan K. W., Moyer S. A., Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202, 154–163 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Cevik B., Smallwood S., Moyer S. A., The L-L oligomerization domain resides at the very N-terminus of the sendai virus L RNA polymerase protein. Virology 313, 525–536 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Chattopadhyay S., Banerjee A. K., Phosphoprotein, P of human parainfluenza virus type 3 prevents self-association of RNA-dependent RNA polymerase, L. Virology 383, 226–236 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choudhary S. K., Malur A. G., Huo Y., De B. P., Banerjee A. K., Characterization of the oligomerization domain of the phosphoprotein of human parainfluenza virus type 3. Virology 302, 373–382 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Tran T.-L., Castagné N., Bhella D., Varela P. F., Bernard J., Chilmonczyk S., Berkenkamp S., Benhamo V., Grznarova K., Grosclaude J., Nespoulos C., Rey F. A., Eléouët J.-F., The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J. Gen. Virol. 88, 196–206 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Sourimant J., Rameix-Welti M.-A., Gaillard A.-L., Chevret D., Galloux M., Gault E., Eléouët J.-F., Fine mapping and characterization of the L-polymerase-binding domain of the respiratory syncytial virus phosphoprotein. J. Virol. 89, 4421–4433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen M., Cortay J.-C., Logan I. R., Sapountzi V., Robson C. N., Gerlier D., Inhibition of ubiquitination and stabilization of human ubiquitin E3 ligase PIRH2 by measles virus phosphoprotein. J. Virol. 79, 11824–11836 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chenik M., Schnell M., Conzelmann K. K., Blondel D., Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J. Virol. 72, 1925–1930 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emerson S. U., Schubert M., Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 84, 5655–5659 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rahmeh A. A., Morin B., Schenk A. D., Liang B., Heinrich B. S., Brusic V., Walz T., Whelan S. P. J., Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 109, 14628–14633 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nikolic J., le Bars R., Lama Z., Scrima N., Lagaudrière-Gesbert C., Gaudin Y., Blondel D., Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 8, 58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinrich B. S., Maliga Z., Stein D. A., Hyman A. A., Whelan S. P. J., Phase transitions drive the formation of vesicular stomatitis virus replication compartments. MBio 9, e02290-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keul N. D., Oruganty K., Schaper Bergman E. T., Beattie N. R., McDonald W. E., Kadirvelraj R., Gross M. L., Phillips R. S., Harvey S. C., Wood Z. A., The entropic force generated by intrinsically disordered segments tunes protein function. Nature 563, 584–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White J. T., Toptygin D., Cohen R., Murphy N., Hilser V. J., Structural stability of the coiled-coil domain of tumor susceptibility gene (TSG)-101. Biochemistry 56, 4646–4655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bruhn J. F., Kirchdoerfer R. N., Urata S. M., Li S., Tickle I. J., Bricogne G., Saphire E. O., Crystal structure of the marburg virus VP35 oligomerization domain. J. Virol. 91, e01085-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zinzula L., Nagy I., Orsini M., Weyher-Stingl E., Bracher A., Baumeister W., Structures of ebola and reston virus VP35 oligomerization domains and comparative biophysical characterization in all ebolavirus species. Structure 27, 39–54.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Leyrat C., Renner M., Harlos K., Grimes J. M., Solution and crystallographic structures of the central region of the phosphoprotein from human metapneumovirus. PLOS ONE 8, e80371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harrison S. C., Viral membrane fusion. Virology 479-480, 498–507 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hartmann M. D., Functional and structural roles of coiled coils. Subcell. Biochem. 82, 63–93 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Stewart C. M., Buffalo C. Z., Valderrama J. A., Henningham A., Cole J. N., Nizet V., Ghosh P., Coiled-coil destabilizing residues in the group A Streptococcus M1 protein are required for functional interaction. Proc. Natl. Acad. Sci. U.S.A. 113, 9515–9520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conzelmann K. K., Schnell M., Rescue of synthetic genomic RNA analogs of rabies virus by plasmid-encoded proteins. J. Virol. 68, 713–719 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Radecke F., Spielhofer P., Schneider H., Kaelin K., Huber M., Dötsch C., Christiansen G., Billeter M. A., Rescue of measles viruses from cloned DNA. EMBO J. 14, 5773–5784 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tatsuo H., Ono N., Tanaka K., Yanagi Y., SLAM (CDw150) is a cellular receptor for measles virus. Nature 406, 893–897 (2000). [DOI] [PubMed] [Google Scholar]
- 73.Vincent S., Spehner D., Manié S., Delorme R., Drillien R., Gerlier D., Inefficient measles virus budding in murine L.CD46 fibroblasts. Virology 265, 185–195 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Cassonnet P., Rolloy C., Neveu G., Vidalain P.-O., Chantier T., Pellet J., Jones L., Muller M., Demeret C., Gaud G., Vuillier F., Lotteau V., Tangy F., Favre M., Jacob Y., Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Methods 8, 990–992 (2011). [DOI] [PubMed] [Google Scholar]
- 75.D. Hayes, T. Laue, J. Philo, Program Sednterp: Sedimentation Interpretation Program (University of New Hampshire, 1995).
- 76.Schuck P., Rossmanith P., Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 54, 328–341 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Evans P. R., An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vagin A., Teplyakov A., Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G., Refinement of severely incomplete structures with maximum likelihood inBUSTER–TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strelkov S. V., Burkhard P., Analysis of alpha-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 137, 54–64 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sali A., Blundell T. L., Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 86.Harvey M. J., Giupponi G., Fabritiis G. D., ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 5, 1632–1639 (2009). [DOI] [PubMed] [Google Scholar]
- 87.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E., Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 89.Feenstra K. A., Hess B., Berendsen H. J. C., Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798 (1999). [DOI] [PubMed] [Google Scholar]
- 90.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 91.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015). [Google Scholar]
- 92.Hoffmann A., Grudinin S., NOLB: Nonlinear rigid block normal-mode analysis method. J. Chem. Theory Comput. 13, 2123–2134 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Dahl A. C. E., Chavent M., Sansom M. S. P., Bendix: Intuitive helix geometry analysis and abstraction. Bioinformatics 28, 2193–2194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Joosten R. P., te Beek T. A. H., Krieger E., Hekkelman M. L., Hooft R. W. W., Schneider R., Sander C., Vriend G., A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–D419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kabsch W., Sander C., Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- 96.Tria G., Mertens H. D. T., Kachala M., Svergun D. I., Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw3702/DC1
Fig. S1. Stability of L620 fragment in the absence of P.
Fig. S2. Impact of kink abrogation and displacement on P functionality.
Fig. S3. Workflow of molecular dynamics simulation versus normal mode analysis and results of molecular dynamics simulations on wt and IPKI MD variants.
Fig. S4. Comparison of RMSF per residue for each chain and average over the four chains.
Fig. S5. Biochemical analysis of MD variants.
Fig. S6. AUC analysis of MD variants.
Fig. S7. Molecular dynamics simulations of V315N, V315I, and V315L MD variants.
Fig. S8. Lack of relationships between expression levels of the variants and their ability to support viral gene expression.
Table S1. Data collection and refinement statistics for the IPKI MD variant.
Table S2. Values for the main structural coiled-coil parameters of wt MD and of the IPKI variant as determined by TWISTER and RMSDs (Å) among the IPKI and the wt MD structures.
Movie S1 (A and B). Animations showing the low frequency collective motions [also known as normal modes (NM)] computed to extract large structure rearrangements of the wt and IPKI.
Movie S2 (A and B). Analysis of the curvature of the four helices along the different frames used to compute NM. The color gradient used for the animation is similar to the one used in Fig. 5C.