

Abstract
Background/Aim: Retinoblastoma (RB) is the most common primary intraocular malignancy. Carboplatin (CPt) is a DNA damage-inducing agent that is widely used for the treatment of RB. Unfortunately, this drug also activates the transcription factornuclear factor-kappa B (NF-ĸB), leading to promotion of tumor survival. Pentoxifylline (PTX) is a drug that inhibits the phosphorylation of I kappa B-alpha (IĸBα) in serines 32 and 36, and this disrupts NF-ĸB activity that promotes tumor survival. The goal of this study was to evaluate the effect of the PTX on the antitumor activity of CPt. Materials and Methods: Y79 RB cells were treated with CPt, PTX, or both. Cell viability, apoptosis, loss of mitochondrial membrane potential, the activity of caspase-9, -8, and -3, cytochrome c release, cell-cycle progression, p53, and phosphorylation of IĸBα, and pro- and anti-apoptotic genes were evaluated. Results: Both drugs significantly affected the viability of the Y79 RB cells in a time- and dose-dependent manner. The PTX+CPt combination exhibited the highest rate of apoptosis, a decrease in cell viability and significant caspase activation, as well as loss of mitochondrial membrane potential, release of cytochrome c, and increased p53 protein levels. Cells treated with PTX alone displayed decreased I kappa B-alpha phosphorylation, compared to the CPt treated group. In addition, the PTX+CPt combination treatment induced up-regulation of the proapoptotic genes Bax, Bad, Bak, and caspases- 3, -8, and -9, compared to the CPt and PTX individual treated groups. Conclusion: PTX induces apoptosis per se and increases the CPt-induced apoptosis, augmenting its antitumor effectiveness.
Keywords: Pentoxifylline, Y79 cells, carboplatin, apoptosis, NF-ĸB
Retinoblastoma (RB) is a genetically determined tumor that represents the most common intraocular malignancy of infancy and early childhood (1). Untreated RB is always fatal, with patients dying of intracranial extension and metastasis within 2 years (2). Traditionally, RB has been treated by surgical removal of the whole eye (enucleation), cryotherapy, and radiotherapy, whereas chemotherapy is used to improve the prognosis of patients with RB (3). In this regard, carboplatin (CPt) is a chemotherapeutic agent based on platinum that has been well-documented in clinical and experimental protocols against RB and other tumors (4). Unfortunately, RB is either intrinsically resistant to systemic therapy or acquires resistance at some point during multiple courses of therapy (5). In an attempt to increase the efficiency of RB treatments higher doses of the cytotoxic agents have been used either as monotherapy or in different combinations. In the majority of cases where higher doses have been put into effect, these did not demonstrate good results, but also led to increased adverse effects (6). These reasons necessitate the development of novel therapeutic modalities. Pharmacological induction of apoptosis is a preferred approach in targeting malignant cells and can now provide with new directions in cancer therapy based on the concept of “chemotherapy with a rational molecular basis” (7). Chemotherapy acts mainly on induction of apoptosis through regulation of a wide variety of signals, resulting in chromatin condensation, internucleosomal fragmentation of DNA, followed by nuclear and cellular damages, loss of the membrane integrity and formation of apoptotic bodies (8), all of these processes are mediated by caspases; the primary enzymes that act as apoptotic initiators and effectors (9). However, it is noteworthy that upon CPt stimulation not only there is induction of apoptosis but also activation of an anti-apoptotic mechanism, probably providing a defense mechanism to tumor cells (10,11). In this regard, CPt has a dual role; i) inducing apoptosis in tumor cells and, paradoxically, ii) activating Nuclear Factor-kappa B (NF-ĸB) transcription factor, which promotes the tumor cell proliferation and survival (12). Under normal conditions, NF-ĸB exists in an inactive form, bound to its inhibitor I-kappa B (IĸB) in the cytoplasm, which prevents it from entering the nucleus and activating target genes such as the anti-apoptotic Bcl-2 and Bcl-XL. Thus, NF-ĸB can affect the efficiency of chemotherapy (13). Nonetheless, it is noteworthy that the chemotherapy itself can activate the NF-ĸB pathway (14,15). On the other hand, the drug pentoxifylline (PTX), is a xanthine and a non-specific phosphodiesterase (PDE) inhibitor that can act as a potent tumor necrosis factor alpha (TNF-α) inhibitor and reduce leukotriene synthesis, and inflammation, through the inhibition of IĸB phosphorylation on serines 32 and 36 (7,16). We have previously shown that PTX in combination with antitumoral drugs such as adriamycin, cisplatin, MG132, and perillyl alcohol significantly increases the levels of apoptosis in vivo and in vitro studies, on L5178Y (mouse lymphoma), U937 (human leukemia), and HeLa and SiHa (human cervical cancer cells) (7,17-19). Finally, in the clinical setting, it has been demonstrated that PTX can induce cancer remission by increasing apoptosis in children with acute lymphoblastic leukemia during the steroid-window phase (20,21). Similar effects of PTX in other types of cancers have confirmed the potency of this drug (16,22-25).
The work presented here aimed to study the antitumor effect of PTX either alone or in combination with CPt in human retinoblastoma Y79 cells.
Materials and Methods
The protocol was approved by the Committee of Research, Ethics, and Biosafety of the Western Biomedical Research Center (CIBO), Mexican Institute of Social Insurance (IMSS), 2016-1305-1.
Cell culture. The cell line Y79 (ATCC® HTB-18™ Manassas, VA, USA), derived from human retinoblastoma, was cultured in RPMI-1640 medium supplemented with 15% (v/v) heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, and antibiotic/antimycotic (Penicillin-Streptomycin-Neomycin); which will be designated RPMI-S. Cells were maintained at 37˚C in a humidified atmosphere containing 5% CO2 and 95% of air. Media was changed every 48 h. All of the previously mentioned products were obtained from the GIBCO™ Invitrogen Corporation (Carlsbad, CA, USA).
Drugs. CPt was obtained from PISA Laboratories (Guadalajara, Mexico) as a crystalline powder, and was dissolved in sterile saline solution at a concentration of 6000 μg/ml and stored at –4˚C for <4 days. PTX (Sigma Aldrich, St. Louis, MO, USA) was dissolved in sterile saline solution at a concentration of 200 mM and stored at –4˚C for less than 4 days (7).
Cell culture and experimental conditions. Y79 cells (2.5×105 cells/ml in T75 flasks) were grown in RPMI-S for 24 h and collected by centrifugation. The cells were then reseeded in 6-well plates (1×106 cells/well) or in 96-well plates (2×104 cells/well) and were treated with CPt (30 μg/ml), PTX (4 mM), or PTX (4 mM)+ CPt (30 μg/ml). In the latter group, PTX was added to the culture 1 h prior to the addition of CPt. Cells treated with RPMI-S alone were used as control, referred as Untreated Control Group (UCG).
Concentration of the treatments employed in this study were obtained from the dose-response curve. Y79 cells were treated with CPt at concentrations of 5, 15, 30, 40, 50, 80, and 150 μg/ml or with PTX at 2, 4, 8, 10, and 12 mM. Cell viability was assessed at 24, 48 and 72 h after treatment. Before all experiments were initiated, cell viability was determined by trypan blue (Sigma Aldrich, St Louis, MO, USA) exclusion (>95%).
Cell viability. Y79 cells were seeded in 96-well plates (2×104 cells/well) and were treated with RPMI-S, PTX, CPt or PTX+CPt for 24 h. Cell viability was assessed using the cell proliferation reagent WST-1 (Commercial Kit, Sigma Aldrich), according to the manufacturer’s instructions. After incubation in WST-1, an electron coupling reagent (ECS) was added, and the Y79 cells were incubated for another 3 h. Absorbance was measured in a microtiter plate reader (Synergy™ HT Multi-Mode Microplate Reader; Biotek, Winooski, VT, USA) at 450 nm with a reading reference wavelength at 690 nm. Data are reported as a percentage of cell viability compared to the respective percentage in UCG cells considered as 100%.
Assessment of Annexin-V, mitochondrial membrane potential loss (ΔΨm), and DNA fragmentation. Y79 cells seeded in 6-well plates were treated with the appropriate drug, drug combination, or medium (control) for 24 h; apoptosis was evaluated by different methods.
Early detection of apoptosis was performed using the Annexin-V-FLUOS staining Kit (Sigma Aldrich, St Louis, MO, USA) according to the manufacturer’s protocol. Briefly, 1×106 cells were collected and resuspended in 500 μl 1x Annexin-V binding buffer. Afterward, cells were incubated with FITC-conjugated Annexin-V FLUOS for 15 min and were analyzed by flow cytometry.
For mitochondrial membrane potential assays, 1×106 cells/ml were collected and stained for 20min with MitoCapture™ staining solution (MitoCapture™ Mitochondrial Apoptosis Detection Kit, BioVision Research, Mountain View, CA, USA) followed by two washes with PBS prior to analysis by flow cytometry. As an internal positive control for the ΔΨm loss, cells were treated for 4 h with 150 μM of protonophore Carbonyl Cyanide m-ChloroPhenylhydrazone (CCCP, Sigma Aldrich), which induces mitochondrial depolarization (26). The percentage of cells with ΔΨm loss was analyzed by flow cytometry using an Attune™ flow cytometer (Life Technologies, Carlsbad, CA, USA); at least 20,000 events were acquired for each sample and were analyzed using Attune software version 2.1 (Life Technologies). Results are represented as the percentage of Annexin-V and ΔΨm loss.
Apoptotic DNA fragmentation is a crucial feature of apoptosis (27); for this reason, internucleosomal DNA fragmentation was quantitatively assayed by antibody-mediated capture and detection of cytoplasmic mononucleosome-and-oligonucleosome-associated histone-DNA complexes (Cell Death Detection ELISAPLUS Kit; Sigma Aldrich). Briefly, Y79 cells were cultured in 96-well plates at 2×104 cells/well and treated with CPt 30 μg/ml, PTX 4 mM, or combined PTX 4 mM+CPt 30 μg/ml, for 24 h. The cell culture supernatants were removed, then the cells were resuspended in 200 μl of lysis buffer™ and lysed directly in the well, centrifugated (1,200 rpm, 10 min), and 20 μl of the cytoplasmic fraction was used to determinate DNA fragmentation according to the manufacturer’s standard protocol. Subsequently, absorbance was measured in a microplate reader (Synergy™ HT Multi-Mode Microplate Reader; Biotek, Winooski, VT, USA) at 405 nm. In the DNA fragmentation test, the rate of apoptosis is reflected by the enrichment (fold increase) of mono- and oligonucleosomes accumulated in the cytoplasm and was calculated according to the following formula:
Rate of Apoptosis=Absorbance of Sample cells/Absorbance of Control cells.
Pan-caspase activity by flow cytometry and colorimetric assay for determining the specific caspase -9, -8, and-3 activity. Determination of activated caspases was performed using the Generic Caspase Activity Assay FITC staining kit for use in flow cytometry (Abcam, Cambridge, UK). This assay can detect activated caspases-1, -3, -4, -5, -6, -7, -8, and -9 in cells undergoing apoptosis. After seeded in 6-well plated and treated with the appropriate drug, drug combination or medium (control) for 24 h, cells were incubated with 1 μl/ml of 500× TF2-VAD-FMK for 1 h at 37˚C in 5% CO2. Finally, cells (1×106 cells) were harvested, washed twice with PBS, and were resuspended in PBS before being analyzed by flow cytometry. Flow cytometry experiments were carried out as before.
In a second step, caspase-9, -8, and -3 activities were determined using colorimetric kits (Abcam, Cambridge, UK). Y79 cells (10×106 cells) were treated for 24 h with the set of drugs mentioned previously. Subsequently, the cells were washed twice with PBS, were resuspended in 300 μl of cell lysis buffer™ containing protease inhibitor (cOmplete™, Mini, EDTA-Free, Roche-Sigma Aldrich) and were incubated on ice for 10 min. Crude lysates were centrifuged for 10 min at 12,000 rpm. Protein concentrations were determined by the Bradford assay using the Dc Protein Kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA), according to the manufacturer’s instructions. Finally, absorbance was measured in a microplate reader (Synergy™ HT Multi-Mode Microplate Reader; Biotek, Winooski, VT, USA) at 405 nm. Results are represented as a percentage of pancaspase activity, and the mean±standard deviation of the optical density (OD) values obtained in each group.
Protein extraction for Cytochrome C level measurement. Cytochrome C (CytC) was determined using the Cytochrome C Profiling ELISA kit (Abcam, Cambridge, UK). Y79 cells (10×106 cells) were treated with CPt, PTX, or both drugs for 24 h. The cells were then harvested, washed twice with PBS, and were resuspended in lysis buffer (Standard Cell Fractionation buffer, Abcam, Cambridge, UK) containing a cocktail of protein inhibitors (cOmplete™, Mini, EDTA-Free, Roche-Sigma Aldrich), according to the manufacturer’s instructions, so as to obtain the cytosolic and mitochondrial fractions. Protein concentrations were determined by the Bradford assay as before. For CytC profiling of subcellular fractions, equal amount of protein (60 μg for cytosolic or mitochondrial fractions) from each sample was added and was analyzed according to the manufacturer’s instructions. The absorbance of cytosolic fractions and mitochondrial were measured at 660 nm using a plate, and the values for each standard concentration were plotted on the vertical (Y) axis versus the corresponding CytC concentration on the horizontal (X) axis, using the Gen5™ software (BioTek Instruments, Inc., Winooski, VT, USA). The results are expressed in μg/ml.
Cell-cycle distribution analysis. For the cell-cycle analysis, the Y79 cells were initially synchronized. In brief, cells were cultured in RPMI-1640 containing 5% FBS for 12 h. Subsequently, the cells were washed and cultured in RPMI-1640 containing 1% FBS. After incubation in this medium overnight, the cells were washed with PBS and were incubated in serum-free medium for 18 h. Finally, the cells were split and were released into cell cycle by the addition of 15% FBS in RPMI-1640 culture medium. A total of 2×106 cells were treated with each drug alone or with both drugs for 24 h. The BD Cycletest™ Plus DNA Reagent kit was used according to the manufacturer’s instructions (BD Biosciences, San Jose, CA, USA). DNA quality control (QC) Particles (BD Biosciences) were used for verification of the instrument performance and quality control of the Attune™ flow cytometer used for DNA analysis. For each sample, at least 30,000 events were acquired, and data were processed with Kaluza v2.0 software (Beckman Coulter) (18).
Western blot analysis of p53 protein levels. Y79 cells (10×106) were treated with CPt, PTX, and PTX+CPt for 18, 24, and 48 h. After each treatment, cell were harvested, washed twice with PBS and were lysed with RIPA buffer (0.5% deoxycholate, 1% NP-40, 0.1% SDS, 50 mM Tris-HCl pH 8.0, and 150 mM NaCl) containing a protein inhibitor cocktail (cOmplete™, Mini, EDTA-Free Roche-Sigma Aldrich) for 30 min on ice. Following sonication (15 pulses, 50% amplitude), protein extracts were centrifuged for 12 min at 12,000 rpm, 4˚C. Protein concentrations were determined using the Dc Protein Kit (Bio-Rad Laboratories, Inc., CA, USA). Equal protein amount (50 μg) from each sample was subjected to electrophoresis using a 10% SDS/polyacrylamide gel. Subsequently, proteins were transferred to Immobilon-P PVDF membranes (Millipore, Bedford, MA, USA) and were incubated with the Odyssey® Blocking Buffer (PBS) reagent for 2 h. Immunodetection of p53 was performed using a mouse monoclonal anti-p53 antibody (DO-1 Abcam Cambridge, UK, diluted at 1:1,000 in PBS+0.1% Tween-20) at 4˚C overnight. After incubation with a fluorescently-labeled secondary antibody (IRDye 680 Donkey Anti-Mouse IgG, LI-COR Biosciences, NE, USA) diluted at 1:10,000 in PBS+0.1% Tween-20 and SDS (0.1%), p53 protein was visualized using the Odyssey™ infrared Imaging System (LI-COR Biotechnology, Nebraska, USA). Anti-actin-β antibody (Abcam Cambridge, UK) was used as loading control diluted at 1:1000 in PBS+0.1% Tween-20. Results were normalized for all experiments by the mean optical density of control Y79 cells (time=18, 24 and 48 h) and were expressed as a fold-change over untreated basal cells. Protein levels of p53 in the Western blot were quantified using the ImageJ v1.51 software package (National Institute of Health, Bethesda, MD, USA www.rsbweb.nih.gov/ij/index.html).
Detection of total-IĸBa and IĸBα (pS32/36) proteins by ELISA. Y79 cells (10×106) were treated with CPt, PTX, and PTX+CPt for 24 h. After treatment, cells were harvested, and cell protein extracts were prepared by lysing the cells using buffer lysis™ provided in the Cell Extraction/Enhancer Buffer PTR kit (Abcam, Cambridge, UK), according to the manufacturer’s instructions. Protein concentrations were determined by the Bradford assay as before. Total and phosphorylated I-kappa B-alpha (IĸBα) protein levels from the cell extracts were measured using the IĸBα (pS32/36)+Total IĸBα SimpleStep ELISA kit (Abcam, Cambridge, UK), according to the manufacturer’s protocol. Equal amounts of protein (50 μg) were used in each experiment, and the absorbance (OD) was measured at 450 nm with a microtiter plate reader. The values of absorbance at 450 nm minus the values for each standard concentration were plotted on the vertical (Y) axis versus the corresponding IĸBα (pS32/36) and Total IĸBα concentrations on the horizontal (X) axis using the Gen5™ software. The results are expressed in μg/ml.
RNA extraction and quantitative real time-PCR. Total RNA of Y79 cells (10×106 cell) was obtained after 3 h of incubating the cells with the different treatments, using the column-based RNA purification kit (GeneJET™ RNA purification kit, Thermo Scientific Waltham, MA, USA), following the manufacturer’ s instructions, The cDNA was synthesized starting from 5 μg of total RNA using the Transcript First Strand cDNA (Roche). Real-Time PCR was carried out with the System Light Cycler® 2.0 apparatus (Roche Applied Science, Mannheim, Germany), using DNA Master plus SYBR Green I (Roche Applied Science). The final concentration of primers in a real time-PCR reaction was 5 pg. The PCR program consisted of an initial 10 min step at 95˚C, and 40 cycles of 15-sec at 95˚C, 5-sec at 60˚C, and 15-sec cycles at 72˚C. Analysis of PCR products was performed using LightCycler® software (Roche Applied Science). Data are expressed as relative normalized quantities employing L32 Ribosomal Protein (RPL32) gene expression to verify the specificity of the amplified reaction, which was nearly 100%. Each sample was processed in triplicates to verify the specificity of the amplification reaction. The oligonucleotides (Invitrogen Corporation, Carlsbad, CA, USA) used to amplify human Bad, Bak, Bax, caspase-3, -8, and -9, Bcl-XL, Bcl-2, and RPL32 are shown in Table I. The oligonucleotides were designed using Oligo software (version 6). Gene sequences were obtained from the GenBank Nucleotide Database of the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov) (19).
Table I. Primer pairs used in real-time quantitative PCR.

Statistical analysis. All experimental procedures were performed in triplicates and were repeated three times. The values represent the mean±standard deviation from the actual values obtained. Statistical analysis was performed using the non-parametric Mann-Whitney U test to compare two groups. Considering values of p<0.05 as significant. In some experiments, we calculated the Δ%, which represents the percentage of increase or decrease in relation to the corresponding UCG. For the determination of significant variation in gene expression levels, this was set at a fold-change≥1.3 between the experimental and control groups.
Results
Dose- and time-dependent effects of CPt and PTX treatments on the viability of Y79 cells. First, we analyzed and compared the effects exerted on Y79 cells after their treatment with CPt or PTX alone. For that purpose, Y79 cell cultures were treated for 24, 48 and 72 h with various doses of either CPt (5-150 μg/ml) or PTX (1-10 mM), and the cell viability was assessed using the WST-1 assay. The cell viability decreased over time in a dose-dependent manner in the CPt or PTX alone groups, and a significant cytotoxic effect was observed after 24 h of incubation with 30 μg/ml of CPt or PTX 4 mM (45% inhibition of cell viability, p<0.05) in comparison to UCG (Figure 1A and B). Cell viability diminished progressively after 48 h. These results show that the CPt and PTX alone exhibit important antitumoral activity. We then performed experiments to verify whether the combination of the two drugs at doses close to IC50 may be more efficient than the CPt alone. Figure 1C illustrates that the combined treatment with PTX (4 mM)+CPt (30 μg/ml) for 24 h decreased cell viability to 87.0±5.0 %, p<0.01 in comparison with UCG, CPt, and PTX treated groups. Likewise, PTX per se decreased cell viability in a similar way to CPt alone group. Taken together, these results demonstrate a cytotoxic effect of PTX and a synergistic action with CPt.
Figure 1. Cell viability of Y79 RB cells treated with carboplatin (CPt)(A) and pentoxifylline (PTX) (B) at different doses and for different amounts of time. The effects of CPt, PTX, and PTX and CPt combined on cell viability (C) are expressed as a percentage of non-viable cells in comparison with the untreated control group (UCG) considered as 100%. The results are presented as means±standard deviation. **p<0.001 (PTX+CPt compared to the UCG), *p<0.05 (CPt and PTX alone groups compared to the UCG, and PTX+CPt treated group compared to the CPt and PTX groups).
Apoptosis induction by PTX+CPt. Since chemotherapy is known to induce apoptosis, we were interested in determining the apoptotic activity of the Y79 cells in our experimental model. Results from the Annexin-V and the ΔΨm tests are expressed in percentage whereas the DNA fragmentation in fold-change (Table II). In the case of the annexin-V determination, the percentage of apoptosis was similar between the CPt (19.9±5.0) and PTX group (18.6±5.0), with an increment of Δ%>100% over UCG (9.1±3.1% of apoptosis, p<0.05). The determination of apoptosis by DNA-fragmentation showed similar results, since the fold-change for CPt- or PTX-treated groups were 4.8±0.3 and 4.1±0.3, respectively, representing an increment of Δ%=310% (p<0.05 compared to UCG). Finally, the combined treatment of combined CPt and PTX showed the highest level of DNA fragmentation with an 8.7±0.3 fold-change and with Δ%=770% higher than that of the groups treated exclusively with either CPt or with PTX alone (p<0.001 vs. all groups). However, the ΔΨm was not affected in the same way, the percentage of ΔΨm CPt was similar to UCG, that is, 31.2±6.1% and 29.7±4.1%, respectively, and lower in comparison to the PTX (37.1±1.7%) alone and after combination treatment with PTX and CPt (41.1±3.7%). Statistically significant differences were observed between the CPt and UCG ΔΨm when they were compared to the PTX and PTX/CPt ΔΨm with p<0.05.
Table II. Assessment of apoptosis induced by Pentoxifylline (PTX), Carboplatin (CPt), and PTX combined with CPt in Y79 cells.

SD, Standard deviation; UCG, untreated control group. *p<0.05, compared to the corresponding UCG.
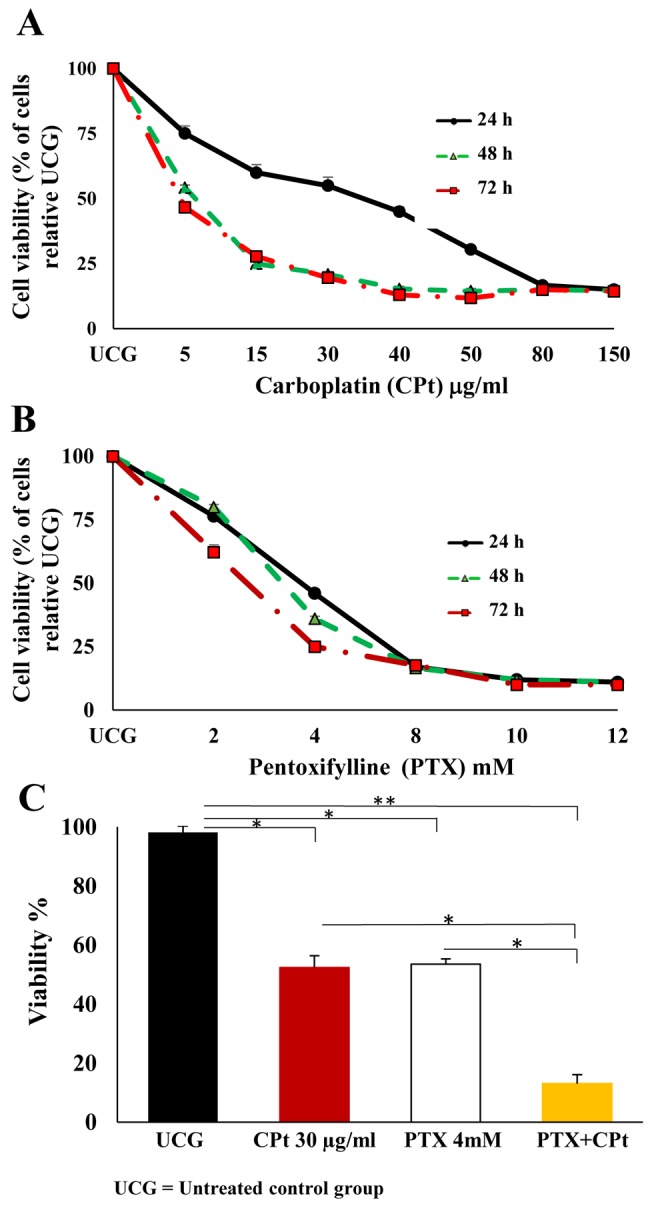
PTX combined with CPt increased caspase activity in Y79 RB cells. Due to the importance of the participation of caspases in apoptosis, as a first step, we analyzed the pan-caspase activity in Y79 cells. Our results, shown in Figure 2A, demonstrate that the minimal pan-caspase activity in UCG was 3.0±1.0%, while the activity of the cells incubated either exclusively with CPt or PTX alone, reached 12.3±3.2% and 13.4±3.0%, respectively. Cells incubated with combined PTX+CPt, showed the highest values for pan-caspase activity, at 15.3±2.8%. All values obtained from treated cells were statistically different from the ones of the UCG (p<0.001), but comparable to the two treatments using a single drug. These results demonstrate the general activity of caspases in our experimental models. For this reason, the activities of caspase-9, -8, and -3 in the different experimental groups were further evaluated (Figure 2B). Analysis revealed that the combination treatment with PTX+CPt significantly increased caspase-9, and -3 activity (Δ%=42.1% and Δ%=30.9%, respectively; p<0.001 compared to the UCG). Caspase-8 activity also exhibited significant changes following treatment with PTX alone and CPt alone (Δ%=48.1%, Δ%=54.3%, respectively; p<0.007, compared to UCG), while the highest change was observed in the PTX+CPt-treated group (Δ%=79.0%, p<0.007) compared to the UCG. Taken together, these results suggest that in treated groups, caspase-9, -3 and -8 play an important role in the apoptosis observed in Y79 cells.
Figure 2. Analysis of caspase activity. Pancaspase activity (A) and caspases 9, 8 and 3 activity (B) in Y79 cells treated with pentoxifylline (PTX; 4 mM), carboplatin (CPt; 30 μg/ml) or their combination for 24 h. The results are presented as means±standard deviation. **p<0.007 [CPt, PTX or PTX+CPt groups compared to untreated control group (UCG)] *p<0.001(CPt, PTX or PTX+CPt compared to the UCG).
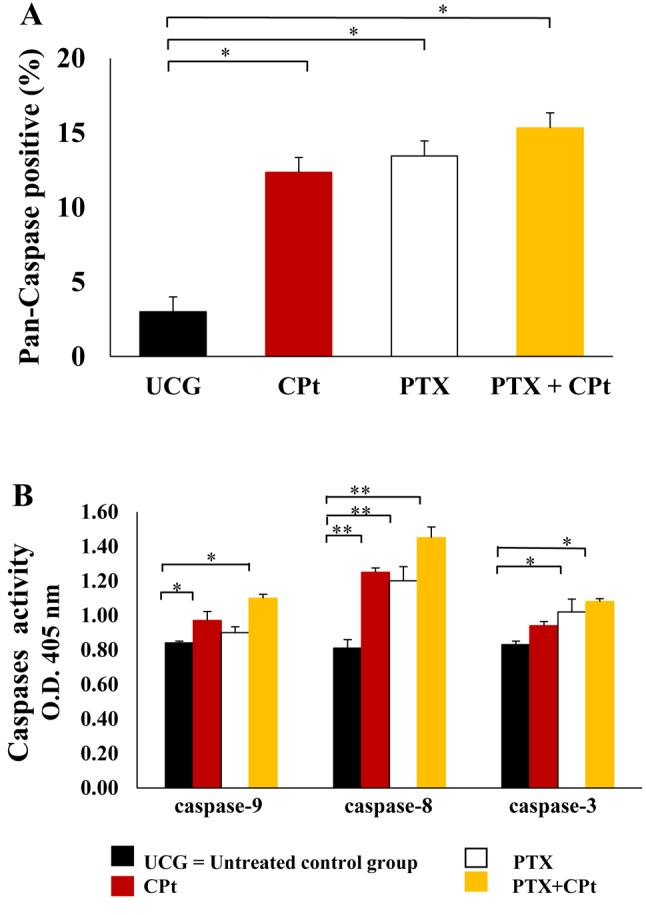
PTX+CPt increased CytC release. The release of CytC in cells treated with PTX, CPt or with both drugs combined was determined, as this comprises a central event in apoptosis induced by the mitochondrial/caspase-9 pathway. As shown in Figure 3A, the cells treated with PTX or the PTX+CPt displayed decreased levels of mitochondrial CytC release (p<0.05) in comparison with the levels in UCG (Δ%=–23.7% and Δ%=–25.4% respectively). Therefore, the PTX and the PTX+CPt groups demonstrated important increase of cytosolic CytC with a Δ%=1,250% and Δ%=2,350%, respectively (p<0.001) (Figure 3B), compared to the UCG. No significant changes were observed in the CPt-treated group. These results reveal the role of CytC in the induction of cell death by PTX alone or when combined with CPt and suggest that mitochondria play an important role in the CytC-induced apoptosis.
Figure 3. Pentoxifylline (PTX) alone and combined with carboplatin (CPt) decrease mitochondrial and increase cytosolic levels of cytochrome C (CytC) in Y79 cells. Mitochondrial (A) and cytosolic (B) CytC levels in Y79 cells treated with PTX, CPt or PTX and CPt combined. The results are presented as means±standard deviation. *p<0.05, [PTX and PTX+CPt groups compared to the untreated control group (UCG) and CPt treated group] in the mitochondrial CytC analysis. **p<0.001, (PTX and PTX+CPt groups compared to the UCG and CPt alone) in the cytosolic CytC analysis.
Effect of PTX on Y79 cell-cycle progression. We were next interested in elucidating whether the aforementioned treatments modulate the cell cycle progression in Y79 cancer cells. The cells were, as previously, treated with PTX or CPt alone or their combination for 24 h, and subsequently, the cell cycle was determined by flow cytometry. As shown in Figure 4, 61.8% of UCG cells were in G1 phase, while the cells in the PTX and PTX+CPt treated groups exhibited similar G1 levels, with a variation at Δ%=4.6% and Δ%=–2.2% respectively. Interestingly, 54.5%, of cells treated with CPt alone were in G1 phase (Δ%=–11.8% as compared to UCG, p<0.05). Opposite results were obtained for the S phase, with the lowest percentage at 15.6% corresponding to the UCG group of cells. Among the cells treated with CPt 26.9% were in phase S (p<0.01 compared to the UCG); while 23.5% of the cells treated with PTX+CPt and 21.1% of the cells treated PTX alone were in the phase S in comparison with UCG. Finally, the percentage of cells in the G2 phase, decreased from 22.5% in the UCG group to 6.4% for the PTX+CPt treated group, to 8.2% for the PTX alone group and 4.0% for the CPt alone group.
Figure 4. Pentoxifylline (PTX) and carboplatin (CPt) modulate the cell cycle checkpoints of Y79 cells. Data are representative of 3 independent experiments, which yielded a similar result *p<0.05; **p<0.01 compared to the untreated control group (UCG).

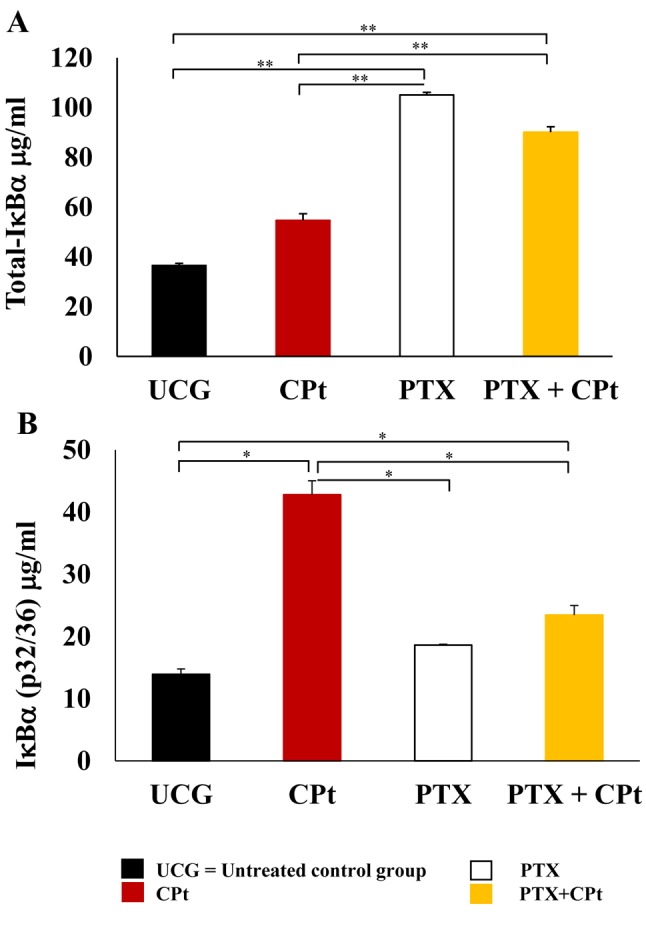
Determination of total-IĸBα and phosphorylated IĸBα (pIĸBα) protein by ELISA. At 24 h post-treatment with the after-mentioned drugs, the total-IĸBα was quantified in Y79 cells. In Figure 5A, an important increase of total Iĸβα was observed in the groups treated with PTX and PTX+CPt (105.0±1.0 μg/ml and 90.1±2.1 μg/ml, respectively). These values represent an increment of Δ%=184.86% and Δ%=144.49%, respectively in comparison with the UCG (p<0.001) and CPt-treated group (p<0.001). In contrast, the pIĸBα increased after the CPt-treatment Δ%=221.02% (p<0.05) compared to the untreated group (Figure 5B). In the case of PTX or PTX+CPt groups, a reduction of the pIĸBα was observed; Δ%=–56.45% and Δ%=–45.16%, respectively (p<0.05) compared to CPt treated group. These experiments strongly suggest that the PTX treatment decrease phosphorylation of the IĸBα induced by CPt, and confirm precedents observations (19).
Figure 5. Determination of Total-IĸBa and phosphorylated IĸBα (pS32/36) and in Y79 cells treated with carboplatin (CPt), pentoxifylline (PTX), and PTX+CPt. The results are presented as means±standard deviation. **p<0.001, [PTX and PTX+CPt treated groups compared to the untreated control group (UCG) and CPt alone group]. *p<0.05 (PTX and PTX+CPt treated groups compared to UCG and CPt group, and CPt alone group compared to the UCG).
PTX+CPt increase p53 protein in Y79 cells. Western blotting analyses allowed us to assess the effects of CPt, PTX, and PTX+CPt on the expression of the p53 protein. Figure 6 shows that untreated Y79 cells express the p53 protein constitutively. However, treatment with CPt increased the level of p53 expression in a time-dependent response after 24 h by 1.70-fold and at 48 h by 2.5-fold (p<0.05) compared to the PTX group and UCG. On the contrary, PTX treatment of Y79 cells resulted in slight increased the p53 levels at 24 h by 1.15-fold as compared to the UCG. When the two drugs were combined (PTX+CPt), the levels of p53 after 24 h increased by 1.5-fold, compared to the UCG (p<0.05). These results further support the role of these two drugs in increasing the levels of apoptosis in Y79 cells.
Figure 6. Protein levels of p53 in Y79 cells treated with pentoxifylline (PTX), carboplatin (CPt), or PTX+CPt for 18, 24 or 48 h were analyzed by western blot. The results in the charts are presented as means±standard deviation. *p<0.05 [CPt and PTX+CPt groups, compared to the untreated control group (UCG), and CPt alone group compared to PTX and PTX+CPt treated groups].

Changes in the expression of proapoptotic and anti-apoptotic-related genes. Real-time PCR was employed to determine the relative changes in the expression of apoptosis-related genes mRNA (Figure 7). Upregulation or downregulation of gene expression was considered when fold-change in mRNA levels compared to the UCG was ≥1.3. PCR assay revealed that CPt treatment did not cause any significant changes in the expression levels of the examined genes. On the contrary, when Y79 RB cells were treated with PTX, mRNA levels of Bak, Bax, Bad, caspase-9, caspase-8, and caspase-3 were significantly increased; however, fold-change was <1.3. When Y79 RB cells were treated with the combination of PTX+CPt, the proapoptotic genes Bax, Bad, and caspase 8 had significantly increased mRNA levels (p<0.05; fold change <1.3), while the highest increase was observed in Bak, caspase 9, and caspase 3 expression (3.0-, 2.5-, and 1.8-fold change, respectively; p<0.05). In general, the data obtained suggested that only the PTX+CPt treatment of Y79 RB cells favored the activation of genes with proapoptotic activity.
Figure 7. Expression of pro- and anti-apoptotic genes in Y79 cells treated with pentoxifylline (PTX), carboplatin (CPt), and PTX combined with CPt. The data are expressed as fold change of mRNA expression versus untreated control group (UCG). *p<0.05 (PTX and PTX+CPt treated groups compared to CPt group and UCG).

Discussion
Our study has focused on assessing the ability of PTX, CPt, and their combination to induce cell death in Y79 retinoblastoma cells. The Y79 RB cell line represents one of the most useful in vitro model systems for investigating human retinoblastoma (28). The potential of PTX as an antitumor agent is great, and successful combinations of PTX with antitumor treatments, including Adriamycin, Cisplatin, MG132, Perillyl alcohol, and radiotherapy produce a synergistic more robust antitumoral response (16,17,19,29). In this study, we observed that the PTX and the CPt treatments alone exhibited an important antitumoral activity per se in the Y79 RB cells; this effect was stronger when the drugs were combined, suggesting a possible synergistic effect. This was supported by the evaluation of cell viability and apoptosis, and DNA fragmentation assays.
In response to DNA damage, cell cycle arrest in G1 phase allows cells to undergo apoptosis instead of progressing to S phase (30). Interestingly, in our study, the used treatments caused significant changes in the control of Y79 cell cycle. The highest amount of cells in S phase was under the effect of CPt, whereas slightly fewer cells were in S phase after treatment with PTX+CPt. It has been previously reported that CPt induces cell-cycle arrest in S-phase and directs tumor cells towards to senescence and drug resistance (31). These results are in agreement with our previously reported evidence that treatment of U937 cells with PTX alone or in combination with the proteasome inhibitor MG132 induces cell arrest in G1 phase (18). Moreover, it has been demonstrated that PTX induces cell cycle arrest at G0/G1 phase and apoptosis in HepG2 cells (32). Therefore, it is possible that the increment in the percentages of Y79 cells in G1 phase may be induced by PTX. These observations suggest different mechanisms action of the two drugs in the cell cycle progression.
By combining different assays, we observed that the disruption of ΔΨm by PTX or PTX+CPt appears to precede the activation of caspase-3, -8 and -9. Following the apoptosis-induced permeabilization of the mitochondrial membrane and CytC release, other proapoptotic proteins are released into the cytosol. Stronger caspase-8 activation was observed when Y79 cells were treated with PTX and CPt alone or with the combination of the two drugs. A similar report has shown that the use of PTX alone or in combination with other chemotherapeutic agents induces the activation of caspase-8 in cervical tumor cells (19). CPt exhibited a different behavior from that of PTX, as it did not induce a ΔΨm loss or CytC release. These results provide evidence that CPt has a different function from PTX.
In RB cells, it has been observed that involvement of the p53 can mediate cell death (33,34). In the present study, the magnitude and kinetics of the changes in p53 caused by CPt alone were quite different from that caused by PTX alone or by the combined actions of PTX and CPt. The accumulation of p53 in cells incubated with CPt for 24-48 h was considerably increased as compared to the PTX, PTX+CPt treated groups and UCG. It has been well documented that cisplatin and CPt increase p53 levels and facilitate the apoptotic response in these cells (35). Contrary to the CPt, treatment with PTX alone did not change p53 levels in Y79 RB cells. In agreement with this finding, other investigators have reported that PTX sensitizes and enhanced antitumor effect of cisplatin in tumor cells independently of p53 status (19,36). In addition, we previously showed that PTX sensitizes p53-null cells to perillyl alcohol-induced apoptosis (17). Taken together, these data suggest that PTX and CPt induce apoptosis in retinoblastoma Y79 cells via different mechanisms.
On the other hand, NF-ĸB is constitutively activated in many types of cancer and can prevent the apoptosis induced by antitumor drugs (37). It is well documented that CPt can induce NF-ĸB activation in cervical and ovarian cancer (38). In addition, NF-ĸB plays a fundamental role in the survival of tumor cells and occasionally confers significant resistance of cancer cells to the different antitumoral therapies (39). We found a great increase in the levels of IĸBα phosphorylation in Y79 cells treated with CPt compared to UCG (p<0.05), which, in turn, translates to an increased activity of NF-ĸB. At the same time, PTX prevented the activation of NF-ĸB by avoiding the breakdown of its inhibitory molecule, IĸBα. When PTX plus CPt was used, the IĸBα phosphorylation was decreased compared to the CPt-alone treatment. This suggested that diminution of phosphorylation of IĸBα keeps NF-ĸB inactive in the cytoplasm, thus inhibiting tumor resistance and survival of these cancer cells. Our findings are in agreement with observations from other groups, who have reported that inhibition of the translocation of NF-ĸB to the nucleus significantly reduces retinoblastoma-cell invasion, migration, and viability, and decreases the resistance of tumor cells to different chemotherapy drugs (37).
Tumor cells develop resistance to apoptosis through multiple mechanisms, including the expression of anti-apoptotic genes (10). NF-ĸB activation induces the modulation of several genes related to inhibition of apoptosis. Our results demonstrate that the Y79 cells exposed to both PTX and CPt show an up-regulation of proapoptotic genes (Bak, Bax, Bad, caspases-9 -8 and -3). Among the proapoptotic genes analyzed, we found that the highest upregulation occurred in the Bak and caspase-9 genes (18). In contraposition, PTX alone induced the overexpression of Bak, caspase-3 and -9 genes. The latter suggests that there is a gene balance that favors apoptosis in the PTX-treated cells, and differences in the expression of these genes strongly suggest alternative mechanisms of gene regulation. Taken together, our results point to a general mechanism of apoptosis in retinoblastoma cells induced by PTX or PTX and CPt combined, that involves parallel or overlapping cascades of events that lead to the disruption of NF-ĸB, cell-cycle arrest, and caspase activation with consequent apoptotic cell death. All these data demonstrate that PTX in combination with CPt can favor a proapoptotic machinery in Y79 retinoblastoma cells, further supporting previous observations from our group (7). Importantly, PTX is a strong inhibitor of PDE activity with PDE1 and PDE4 expression predominating in Y79 cells. It may be possible that PTX may exhibit its proapoptotic activity in these cells through inhibition of PDE 4, as it has been reported to be the case in other types of cells (40).
Even though PTX is experimentally used to sensitize tumor cells to chemotherapy and can exhibit similar or, in some cases, better efficacy against tumor cell survival, it is not classified as an antitumor drug. In our study, we generated a considerable amount of in vitro evidence of the antitumoral role of PTX, similar to other studies performed in lymphoblastic-leukemia and cervical cancer cells (20,23,24). The importance of this lies in the necessity to find chemotherapeutic agents with fewer adverse effects and robust antitumoral activity. With more than 30 years of clinical use and fewer adverse effects, PTX (41,42) can provide protection against the adverse symptoms from chemotherapy (43,44). In addition, PTX is not as expensive as other antitumor drugs, and it displays excellent tolerance even in children with cancer (20,21). Due to these characteristics, PTX may represent a new alternative for the treatment of retinoblastoma. Thus, it would be interesting to include more specific studies on the function of PTX and evaluate it as an antitumoral drug and not only as an adjuvant agent.
Conclusion
PTX and CPt showed antitumor activity in the in vitro model with human Y79 RB cells, while the two drugs combined had an additive effect on apoptosis. These findings suggest that the combination of PTX and CPt may comprise a promising strategy for the treatment of retinoblastoma tumors.
Conflicts of Interests
The Authors declare that they have no conflicting interests.
Acknowledgements
This work was supported by Grant (FIS/IMSS/PROT/MD16/1567) from the Instituto Mexicano del Seguro Social (IMSS), Red de Inmunología del Cáncer y Enfermedades Infecciosas-CONACYT-253053 (INMUNOCANEI) and CONACYT-382889.
References
- 1.Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, Brisse H, Doz F, Desjardins L. Retinoblastoma. Orphanet J Rare Dis. 2006;1(1):1–31. doi: 10.1186/1750-1172-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chawla B, Jain A, Azad R. Conservative treatment modalities in retinoblastoma. Indian J Ophthalmol. 2013;61(9):479–485. doi: 10.4103/0301-4738.119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartuma K, Pal N, Kosek S, Holm S, All-Ericsson C. A 10-year experience of outcome in chemotherapy-treated hereditary retinoblastoma. Acta Ophthalmol. 2014;92(5):404–411. doi: 10.1111/aos.12282. [DOI] [PubMed] [Google Scholar]
- 4.Kim H, Lee JW, Kang HJ, Park HJ, Kim YY, Shin HY, Yu YS, Kim IH, Ahn HS. Clinical results of chemotherapy based treatment in retinoblastoma patients: A single center experience. Cancer Res Treat. 2008;40(4):164–171. doi: 10.4143/crt.2008.40.4.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85(9):1219–1226. doi: 10.1016/j.bcp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 6.Ruggiero A, Trombatore G, Triarico S, Arena R, Ferrara P, Scalzone M, Pierri F, Riccardi R. Platinum compounds in children with cancer: Toxicity and clinical management. Anticancer Drugs. 2013;24(10):1007–1019. doi: 10.1097/CAD.0b013e3283650bda. [DOI] [PubMed] [Google Scholar]
- 7.Lerma-Diaz JM, Hernandez-Flores G, Dominguez-Rodriguez JR, Ortiz-Lazareno PC, Gomez-Contreras P, Cervantes-Munguia R, Scott-Algara D, Aguilar-Lemarroy A, Jave-Suarez LF, Bravo-Cuellar A. In vivo and in vitro sensitization of leukemic cells to adriamycin-induced apoptosis by pentoxifylline. Involvement of caspase cascades and ikappabalpha phosphorylation. Immunol Lett. 2006;103(2):149–158. doi: 10.1016/j.imlet.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63(1):12–31. doi: 10.1016/j.critrevonc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Florea AM, Busselberg D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011;3(1):1351–1371. doi: 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor nf-kappab. J Clin Invest. 2001;107(3):241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J-L, Kamata H, Karin M. IKK/NF-kappab signaling: Balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115(10):2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greten FR, Karin M. The ikk/nf-kappab activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206(2):193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 15.Montagut C, Tusquets I, Ferrer B, Corominas JM, Bellosillo B, Campas C, Suarez M, Fabregat X, Campo E, Gascon P, Serrano S, Fernandez PL, Rovira A, Albanell J. Activation of nuclear factor-kappa b is linked to resistance to neoadjuvant chemotherapy in breast cancer patients. Endocr Relat Cancer. 2006;13(2):607–616. doi: 10.1677/erc.1.01171. [DOI] [PubMed] [Google Scholar]
- 16.Bohm L, Roos WP, Serafin AM. Inhibition of DNA repair by pentoxifylline and related methylxanthine derivatives. Toxicology. 2003;193(1-2):153–160. doi: 10.1016/s0300-483x(03)00294-4. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Contreras PC, Hernandez-Flores G, Ortiz-Lazareno PC, Del Toro-Arreola S, Delgado-Rizo V, Lerma-Diaz JM, Barba-Barajas M, Dominguez-Rodriguez JR, Bravo Cuellar A. In vitro induction of apoptosis in u937 cells by perillyl alcohol with sensitization by pentoxifylline: Increased bcl-2 and bax protein expression. Chemotherapy. 2006;52(6):308–315. doi: 10.1159/000096003. [DOI] [PubMed] [Google Scholar]
- 18.Bravo-Cuellar A, Hernandez-Flores G, Lerma-Diaz JM, Dominguez-Rodriguez JR, Jave-Suarez LF, De Celis-Carrillo R, Aguilar-Lemarroy A, Gomez-Lomeli P, Ortiz-Lazareno PC. Pentoxifylline and the proteasome inhibitor mg132 induce apoptosis in human leukemia u937 cells through a decrease in the expression of bcl-2 and bcl-xl and phosphorylation of p65. J Biomed Sci. 2013;20(1):13. doi: 10.1186/1423-0127-20-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez-Flores G, Ortiz-Lazareno PC, Lerma-Diaz JM, Dominguez-Rodriguez JR, Jave-Suarez LF, Aguilar-Lemarroy Adel C, de Celis-Carrillo R, del Toro-Arreola S, Castellanos-Esparza YC, Bravo-Cuellar A. Pentoxifylline sensitizes human cervical tumor cells to cisplatin-induced apoptosis by suppressing nf-kappa b and decreased cell senescence. BMC Cancer. 2011;11(1):483. doi: 10.1186/1471-2407-11-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Ramella O, Ortiz-Lazareno PC, Jimenez-Lopez X, Gallegos-Castorena S, Hernandez-Flores G, Medina-Barajas F, Meza-Arroyo J, Jave-Suarez LF, Lerma-Diaz JM, Sanchez-Zubieta F, Bravo-Cuellar A. Pentoxifylline during steroid window phase at induction to remission increases apoptosis in childhood with acute lymphoblastic leukemia. Clin Transl Oncol. 2016;18(4):369–374. doi: 10.1007/s12094-015-1376-x. [DOI] [PubMed] [Google Scholar]
- 21.Meza-Arroyo J, Bravo-Cuellar A, Jave-Suarez LF, Hernandez-Flores G, Ortiz-Lazareno P, Aguilar-Lemarroy A, Padilla-Corona M, Sanchez-Zubieta F, Gonzalez-Ramella O. Pentoxifylline added to steroid window treatment phase modified apoptotic gene expression in pediatric patients with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2018;40(5):360–367. doi: 10.1097/MPH.0000000000001152. [DOI] [PubMed] [Google Scholar]
- 22.Barancik M, Bohacova V, Gibalova L, Sedlak J, Sulova Z, Breier A. Potentiation of anticancer drugs: Effects of pentoxifylline on neoplastic cells. Int J Mol Sci. 2012;13(1):369–382. doi: 10.3390/ijms13010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohsaki Y, Ishida S, Fujikane T, Kikuchi K. Pentoxifylline potentiates the antitumor effect of cisplatin and etoposide on human lung cancer cell lines. Oncology. 1996;53(4):327–333. doi: 10.1159/000227582. [DOI] [PubMed] [Google Scholar]
- 24.Schiano MA, Sevin BU, Perras J, Ramos R, Wolloch EH, Averette HE. In vitro enhancement of cis-platinum antitumor activity by caffeine and pentoxifylline in a human ovarian cell line. Gynecol Oncol. 1991;43(1):37–45. doi: 10.1016/0090-8258(91)90006-q. [DOI] [PubMed] [Google Scholar]
- 25.Jimenez-Luevano MA, Rodriguez-Chavez JL, Ramirez-Flores S, Rodriguez-Villa P, Jimenez-Partida MA, Cervantes-Rodriguez G, Hernandez-Flores G, Solis-Martinez R, Bravo-Cuellar A. Treatment of hepatocarcinoma with celecoxib and pentoxifylline: A case report. Rev Med Inst Mex Seguro Soc. 2018;56(3):309–315. [PubMed] [Google Scholar]
- 26.Hernandez-Flores G, Gomez-Contreras PC, Dominguez-Rodriguez JR, Lerma-Diaz JM, Ortiz-Lazareno PC, Cervantes-Munguia R, Sahagun-Flores JE, Orbach-Arbouys S, Scott-Algara D, Bravo-Cuellar A. Gamma-irradiation induced apoptosis in peritoneal macrophages by oxidative stress. Implications of antioxidants in caspase mitochondrial pathway. Anticancer Res. 2005;25(6B):4091–4100. [PubMed] [Google Scholar]
- 27.Collins JA, Schandi CA, Young KK, Vesely J, Willingham MC. Major DNA fragmentation is a late event in apoptosis. J Histochem Cytochem. 1997;45(7):923–934. doi: 10.1177/002215549704500702. [DOI] [PubMed] [Google Scholar]
- 28.Chévez-Barrios P, Hurwitz MY, Louie K, Marcus KT, Holcombe VN, Schafer P, Aguilar-Cordova CE, Hurwitz RL. Metastatic and nonmetastatic models of retinoblastoma. Am J Pathol. 2000;157(4):1405–1412. doi: 10.1016/S0002-9440(10)64653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bravo-Cuellar A, Ortiz-Lazareno PC, Lerma-Diaz JM, Dominguez-Rodriguez JR, Jave-Suarez LF, Aguilar-Lemarroy A, del Toro-Arreola S, de Celis-Carrillo R, Sahagun-Flores JE, de Alba-Garcia JE, Hernandez-Flores G. Sensitization of cervix cancer cells to adriamycin by pentoxifylline induces an increase in apoptosis and decrease senescence. Mol Cancer. 2010;9(1):114. doi: 10.1186/1476-4598-9-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spoerri L, Oo Z, Larsen J, Haass N, Gabrielli B, Pavey S. Cell cycle checkpoint and DNA damage response defects as anticancer targets: From molecular mechanisms to therapeutic opportunities. In: Stress response pathways in cancer: From molecular targets to novel therapeutics. Wondrak GT (ed.). Dordrecht, Springer Netherlands. 2015:29–49. [Google Scholar]
- 31.Cruet-Hennequart S, Villalan S, Kaczmarczyk A, O’Meara E, Sokol AM, Carty MP. Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle. 2009;8(18):3039–3050. [PubMed] [Google Scholar]
- 32.Wang Y, Dong L, Li J, Luo M, Shang B. Pentoxifylline induces apoptosis of hepg2 cells by reducing reactive oxygen species production and activating the mapk signaling. Life Sci. 2017;183(15):60–68. doi: 10.1016/j.lfs.2017.05.029. [DOI] [PubMed] [Google Scholar]
- 33.Hsiao WT, Tsai MD, Jow GM, Tien LT, Lee YJ. Involvement of Smac, p53, and caspase pathways in induction of apoptosis by gossypol in human retinoblastoma cells. Mol Vis. 2012;18:2033–2042. [PMC free article] [PubMed] [Google Scholar]
- 34.Dyer MA. Lessons from retinoblastoma: Implications for cancer, development, evolution, and regenerative medicine. Trends Mol Med. 2016;22(10):863–876. doi: 10.1016/j.molmed.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Felice V, Lauricella M, Giuliano M, Emanuele S, Vento R, Tesoriere G. The apoptotic effects of cisplatin and carboplatin in retinoblastoma y79 cells. Int J Oncol. 1998;13(2):225–232. doi: 10.3892/ijo.13.2.225. [DOI] [PubMed] [Google Scholar]
- 36.Fan S, Smith ML, Rivet DJ, Duba D, Zhan Q, Kohn KW, Fornace AJ Jr., O’Connor PM. Disruption of p53 function sensitizes breast cancer mcf-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995;55(8):1649–1654. [PubMed] [Google Scholar]
- 37.Poulaki V, Mitsiades CS, Joussen AM, Lappas A, Kirchhof B, Mitsiades N. Constitutive nuclear factor-ĸb activity is crucial for human retinoblastoma cell viability. Am J Pathol. 2002;161(6):2229–2240. doi: 10.1016/s0002-9440(10)64499-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li F, Sethi G. Targeting transcription factor nf-ĸb to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. 2010;1805(2):167–180. doi: 10.1016/j.bbcan.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Lagunas VM, Meléndez-Zajgla J. Nuclear factor-kappa B as a resistance factor to platinum-based antineoplasic drugs. Met Based Drugs. 2008;2008:576104–576104. doi: 10.1155/2008/576104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White JB, Thompson WJ, Pittler SJ. Characterization of 3’,5’ cyclic nucleotide phosphodiesterase activity in y79 retinoblastoma cells: Absence of functional PDE6. Mol Vis. 2004;10:738–749. [PubMed] [Google Scholar]
- 41.Chua DT, Lo C, Yuen J, Foo YC. A pilot study of pentoxifylline in the treatment of radiation-induced trismus. Am J Clin Oncol. 2001;24(4):366–369. doi: 10.1097/00000421-200108000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Ward A, Clissold SP. Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs. 1987;34(1):50–97. doi: 10.2165/00003495-198734010-00003. [DOI] [PubMed] [Google Scholar]
- 43.Moss RW. Do antioxidants interfere with radiation therapy for cancer. Integr Cancer Ther. 2007;6(3):281–292. doi: 10.1177/1534735407305655. [DOI] [PubMed] [Google Scholar]
- 44.Letur-Konirsch H, Guis F, Delanian S. Uterine restoration by radiation sequelae regression with combined pentoxifylline-tocopherol: A phase ii study. Fertil Steril. 2002;77(6):1219–1226. doi: 10.1016/s0015-0282(02)03120-5. [DOI] [PubMed] [Google Scholar]