

Abstract
Our laboratory has developed chemically self-assembled nanorings (CSANs) as prosthetic antigen receptors (PARs) for the non-genetic modification of T-cell surfaces. PARs have been successfully employed in vitro to activate T cells for the selective killing of leukemia cells. However, PAR efficacy has yet to be evaluated in vivo or against solid tumors. Therefore, we developed bispecific PARs that selectively target the human CD3 receptor and human Epithelial Cell Adhesion Molecule (EpCAM), which is overexpressed on multiple carcinomas and cancer stem cells. The αEpCAM/αCD3 PARs were found to stably bind T cells for >4 days, and treating EpCAM+ MCF-7 breast cancer cells with αEpCAM/αCD3 PAR-functionalized T cells resulted in the induction of IL-2, IFN-γ and MCF-7 cytotoxicity. Furthermore, an orthotopic breast cancer model validated the ability of αEpCAM/αCD3 PAR therapy to direct T cell lytic activity towards EpCAM+ breast cancer cells in vivo leading to tumor eradication. In vivo biodistribution studies demonstrated that PAR-T cells were formed in vivo and persist for over 48 hours with rapid accumulation in tumor tissue. Following PAR treatment, the production of IL-2, IFN-γ, IL-6 and TNF-α could be significantly reduced by an infusion of clinically-relevant concentrations of the FDA-approved antibiotic, trimethoprim, signaling pharmacologic PAR deactivation. Importantly, CSANs did not induce naïve T cell activation and thus exhibit a limited potential to induce naïve T cell anergy. In addition, murine immunogenicity studies demonstrated that CSANs do not induce a significant antibody response, nor do they activate splenic cells. Collectively, our results demonstrate that bispecific CSANs are able to non-genetically generate reversibly modified T cells that are capable of eradicating targeted solid tumors.
Keywords: Immunotherapy, Nanotechnology, Bispecific, anti-cancer, T cell, EpCAM
The past decade has produced several promising immunotherapeutic strategies for redirecting the patient’s own immune system to selectively target and kill cancer cells. Of these, chimeric antigen receptors (CARs) have demonstrated significant early success and clinical promise.1–6 However, despite the significant strides achieved in CAR therapy there are a number of treatment-associated toxicities resulting from the uncontrolled release of cytokines following the activation and rapid proliferation of CAR T cells.7–11 Additionally, their application to solid tumors has been challenging as antigens associated with solid tumors can also be expressed on normal tissues.2, 12–14 To minimize these toxicities, a number of promising techniques have been developed to either enhance tumor specificity or regulate their persistence.15–18 In practice, the use of bispecific molecules has also demonstrated significant potential to harness T cells for specific targeted killing of cancer cells for immunotherapy, without eliciting the drawbacks associated with genetically engineering T cells.19 While Bispecific T cell Engagers (BiTEs) have achieved excellent success, they typically exhibit limited avidity for binding both the T cell and target cancer cell, which when combined with their small size results in their rapid in vivo elimination (within minutes), thus requiring continuous infusion.20–22 There have been advances to increase the circulating half-life of BiTEs (e.g., pegylation); however, the potential to elicit a cytokine storm and other off target toxicity is still a possibility with the increased circulation time,20, 23 causing significant concern and hesitation for therapeutic use.
As an alternative approach to current CAR and bispecific antibody agents, we explored the use of prosthetic antigen receptors (PARs) as a non-genetic system for directing selective cell-cell interactions. PARs are formed by fusing a single-chain variable fragment (scFv) to two E. Coli dihydrofolate reductase (DHFR2) molecules that spontaneously assemble into octomeric chemically self-assembled nanorings (CSANs) upon the addition of a chemical dimerizer, bis-methotrexate (bisMTX).24, 25 The combination of an αCD3 fusion protein with a tumor targeting fusion protein results in the formation of bispecific, multivalent, CSANs that stably bind to CD3+ T cell surfaces, generating PARs, and selectively target tumor cells (Figure 1).26 We have previously incorporated a variety of scFvs and small peptide sequences onto the C-termini of our DHFR2 proteins (DHFR2-scFv), thereby enabling the CSANs to target specific overexpressed receptors.27–30 Additionally, CSAN size, and therefore overall scFv valency, can be varied by modifying the length of the peptide linker between the two DHFR proteins.31 PAR therapy has several innovations, such as the capability of quickly labeling T cell membranes in a matter of minutes and is stable on the cell surface for multiple days.26 Importantly, our approach has the capability of removing PARs from T cells by incubation with the FDA approved antibiotic trimethoprim, at clinically relevant concentrations, allowing pharmacological deactivation of T cells in the event of any unseen adverse or off target effects.28 Due to these innovations, the application of PAR therapy to solid tumors cells offers significant potential.
Figure 1. Prosthetic Antigen Receptor (PAR) Schematic.

Bispecific CSANs are constructed from αCD3 DHFR2, αEpCAM DHFR2, and BisMTX to form multivalent nanorings. Effector T cells are engaged with bispecific CSANs which tightly bind, due to their high avidity, and generate PAR T cells that facilitate the cell lysis of target cells. Additionally, the use of clinically relevant concentrations of Trimethoprim disassembles CSANs on the cell surface.
The epithelial cell adhesion molecule (EpCAM, CD326) is a viable target for anti-cancer bispecific antibodies,32–34 as the EpCAM receptor is overexpressed on a variety of different tumor cell types including breast, ovarian, colon, pancreatic, prostate, and lung cancer.35–37 Even more attractive is the presence of EpCAM receptor expression on cancer stem cells (CSCs), which are thought to be responsible for the reemergence of tumors in patients that have been in remission.38 While the EpCAM receptor is expressed on selective normal tissue, recent data suggests these receptors are mostly found within intracellular boundaries and are inaccessible to EpCAM targeting drugs.36, 39, 40 In contrast to normal tissues, significant amounts of EpCAM can be found on the cell surface of tumor tissue, making this an excellent target.41
The trifunctional αEpCAM/αCD3 antibody, catumaxomab, has shown clinical efficacy against ovarian ascites and has recently received European approval.34 Nevertheless, the Fc domain of catumaxomab has been associated with liver toxicity by activation of Kupfer cells.42 Amgen, Inc. (Thousand Oaks, CA) has also advanced an αEpCAM/αCD3 BiTE, Solitomab, into the clinic for solid tumors.43–45 Unfortunately, while tumor growth was suppressed, there were associated dose-limiting toxicities that prevented drug concentrations from approaching the IC90 values during continuous infusion.21 To address these issues, αEpCAM CAR T cells prepared by transient RNA transfection have been generated.46 While αEpCAM CAR T cells have successfully demonstrated the suppression of tumor growth they’ve been unable to completely eradicate tumors; resulting in tumor growth rebounding when dosing with the αEpCAM RNA transfected T cells was stopped.46 On-going clinical trials with αEpCAM CAR T cell therapy are underway in order to address the efficacy of this approach. Nevertheless, the development of a less evasive, more efficacious and safe alternative for the targeting of EpCAM-expressing cancer cells by T cells could significantly advance solid tumor immunotherapy.
Previous studies performed in our lab demonstrated that CSANs displaying single chain antibodies can bind to both the CD3ε subunit of the T cell-receptor/CD3 complex and CD22 on malignant B cells.26 We demonstrated the multivalent and bispecific format allows the αCD3/αCD22 CSANs to stably bind to T cell surfaces for greater than four days in vitro, while being easily disassembled on the cell membrane by treatment with the non-toxic FDA approved drug, trimethoprim. In the presence of CD22+ Raji cells, T cells modified with αCD3/αCD22 CSANs were shown to selectively increase the production of interleukin-2 (IL-2) and interferon-γ (IFN-γ) and initiate cytotoxicity. Herein, we have characterized the use of PARs T cells to target solid tumors expressing the EpCAM receptor for anti-cancer cell directed immunotherapy using an orthotopic xenograft model.
RESULTS AND DISCUSSION
Preparation of αEpCAM/αCD3 Bispecific CSANs
To extend the potential of our PAR T cell approach, we have now investigated the ability of bispecific CSANs to direct T cell killing of solid tumor tissue both in vitro and in vivo. Consequently, we prepared αEpCAM/αCD3 CSANs and characterized their ability to bind and activate T cells in the presence of the breast cancer cell line, MCF-7 cells, which like many epithelial cancers express EpCAM on their membrane surface.32–34
The development and preparation of DHFR2-αEpCAM and DHFR2-αCD3 proteins have been previously described.25, 28 Once prepared the purified DHFR2-αEpCAM and DHFR2-αCD3 fusion proteins were incubated with bisMTX dimerizer (1:1:2.2 equivalents) for 30 minutes to form αEpCAM/αCD3 CSANs, which were characterized by size exclusion chromatography (Figure 2A). These octameric rings are generated by random combinations, and statistically calculated to be greater than ninety-nine percent bispecific in makeup.26 As expected the bispecific CSANs generated larger, octameric species, which eluted at 18.5 minutes with nearly 100% oligomerization whereas the smaller DHFR2-αEpCAM and DHFR2-αCD3 monomer proteins eluted at 28.5 minutes. Using cryo-transmission electron microscopy (Cryo-TEM) we were able to directly visualize CSAN ring formation and confirmed an average mean diameter of 19.8 ± 4.3 nM (Figure 2B-C). Similar to previously prepared CSANs, the bispecific CSANs were found by dynamic light scattering (DLS) to exhibit a higher hydrodynamic radius, 16.1 ± 0.1 nM (Figure 2D), compared to those of the monomeric proteins, DHFR2-αEpCAM and DHFR2-αCD3, at 9.7 ± 0.1 nM (Figure 2E) and 9.6 ± 0.1 nM (Figure 2F), respectively.26, 30 Importantly, the self-assembled αEpCAM/αCD3 CSANs exhibited the same size dimensions (20–25 nM diameter) and thus polyvalency (7–10) found for previous bispecific CSANs.25, 28
Figure 2. Bispecific CSAN Characterization.

(A) The assembly of bispecific CSANs was characterized by size exclusion chromatography: Black curve, αEpCAM/αCD3 Bispecific CSANs; blue curve, αCD3 DHFR2 monomer; red curve, αEpCAM DHFR2 monomer. Ring formation was confirmed by Cryo-Transmission Electron Microscopy (Cryo-TEM): (B) size distribution analysis and (C) the corresponding Cryo-TEM image. The hydrodynamic size of bispecific CSANs was confirmed by dynamic light scattering: (D) αEpCAM/αCD3 bispecific CSANSs (16.1 ± 0.1 nM), (E) αEpCAM DHFR2 monomer (9.7 ± 0.1 nM) and (F) αCD3 DHFR2 monomer (9.6 ± 0.1 nM).
Once we confirmed ring formation, binding studies were performed using flow cytometry to ensure the functionality of CSAN antigen targeting. Anti-EpCAM/αCD3 CSANs, αCD3 monospecific CSANs, and αEpCAM monospecific CSANs were generated using a previously described fluorescein-labeled bisMTX (FITC-bisMTX) dimerizer,27 and incubated with a breast cancer cell line, MCF-7 cells (confirmed EpCAM positive, Figure S1A), and peripheral blood mononuclear cells (PBMCs) which are primarily comprised of CD3 expressing T cells (confirmed CD3 positive, Figure S1B). Binding of the bispecific CSANs to both MCF-7 cells and PBMCs was observed (Figure S2). Additionally, since EpCAM is known to undergo little or no internalization, we utilized confocal microscopy to confirm the lack of αEpCAM CSAN internalization by MCF-7 cells (Figure S3). Similar to previous studies with αCD22/αCD3 CSANs, αEpCAM/αCD3 CSANs stably bound to the surface of PBMCs, with only a 16% loss of nanorings from the cell surface observed after three days (Figure S4A).
As previously discussed, an important feature of the PAR T cell approach is our ability to pharmacologically disassemble the rings in the presence of the FDA approved drug, trimethoprim.26, 28 To characterize the ability of trimethoprim to disassemble αEpCAM/αCD3 CSANs bound to the T cells, we incubated PBMCs with the bispecific CSANs (100 nM) prepared with FITC-bisMTX. After washing the cells, the αEpCAM/αCD3 CSANs modified T cells were incubated with variable concentrations of trimethoprim for 30 min. The percentage of CSANs remaining on the cell surface was then monitored by flow cytometry. Similar to previous studies, at a trimethoprim concentration of 3.9 µM, approximately 50% of the CSANs were removed from the surface, while at a concentration of 60 µM approximately 90% of the αEpCAM/αCD3 CSANs were removed from the cell surface (Figure S4B).26
In Vitro T cell Activation and Tumor Cell Cytotoxicity
Cytotoxicity studies were performed with unactivated T cells treated with increasing αEpCAM/αCD3 concentrations for 30 minutes, and then incubated with target MCF-7 cells for 24 hours at an effector-to-target ratio of 10:1 (Figure 3A). Maximal cytotoxicity was observed for cells incubated with 50 nM of the αEpCAM/αCD3 CSANs, with significant cytotoxicity observed at a concentration of 10 nM. At a fixed CSAN concentration of 50 nM, a 4-fold specific lysis was achieved at an effector to target ratio of 10:1 and 20:1 using bispecific CSANs (Figure 3B).
Figure 3. αEpCAM/αCD3 CSAN Labeled T Cells Selectively Activate and Kill Target EpCAM+ Cells.

(A) Target cell lysis was evaluated at a set 10:1 E:T ratio with increasing concentrations of CSANs as well as (B) at a set concentration of CSANs (50 nM) with increasing E:T ratio. Unactivated PBMCs were co-cultured with media, monospecific CSANs or bispecific CSANs in the presence or absence of target MCF-7 cells. CD69 expression levels was measured on (C) CD4+ T cells and (D) CD8+ T cells following a 12-hour incubation while CD25 expression was measured on (E) CD4+ Tells and (F) CD8+ T cells following a 24-hour incubation. CD4+ and CD8+ T cells were gated on the lymphocyte population as determined by the forward and side scatter as well as live/dead cell dye. Following incubation, the media was analyzed for (G) IL-2 and (H) IFN-γ by ELISA. Data shown was obtained from one donor (n=3), but representative of three donors (Figure S16 & S17). *P<0.05 with respect to αEpCAM CSAN and no treatment controls, by 2-tailed Student’s t test.
To further confirm the specificity of αEpCAM/αCD3 CSAN targeting, we carried out competition studies using the parental αEpCAM mAb, Moc31. PBMCs were treated with increasing αEpCAM/αCD3 CSAN concentrations and then incubated with MCF-7 cells at an effector-to-target ratio of 10:1 for 24 hours with or without Moc31 (Figure S5). Inclusion of Moc31 significantly decreased target cell killing to a level comparable to the non-targeted control, αCD3 monospecific CSANs. These data suggest the αEpCAM scFv of the αEpCAM/αCD3 CSANs selectively targets the EpCAM receptor and is required to bind for target cell killing.
Time-lapse microscopy was performed using live cell imaging methods to observe selective cell killing events in the presence of EpCAM+ MCF-7 cells co-cultured with the EpCAM negative (confirmed EpCAM negative in figure S6) U87 glioblastoma cells (Figure S7). In addition to observing the interactions between the target and effector cells, we wanted to observe the selective cytotoxicity of the bispecific CSAN-treated T cells against EpCAM+ cells without affecting the growth and proliferation of an EpCAM negative cell line. Over the course of 25 hours, the redirected T cells were selectively attached to and interacted with the EpCAM+ cell line leading to cell death while leaving EpCAM negative cells unharmed. Similarly, when in the presence of untreated T cells neither cell line was compromised but instead continued to grow and divide normally (Figure S7).
As observed with αCD22/αCD3 PAR T cells, full activation of the αEpCAM/αCD3 PAR T cells was dependent on the presence of antigen-positive tumor cells. PBMC T cell signaling activation was monitored by measuring CD69 and CD25 upregulation following cytotoxicity assays, as well as the amount of IL-2 and IFN-γ produced. Both CD8+ and CD4+ T cells increased CD69 (Figure 3C & 3D) and CD25 expression (Figure 3E & 3F) in the presence of either αCD3 or αEpCAM/αCD3 CSANs. However, for both CD4+ and CD8+ T cells labeled with αEpCAM/αCD3 CSANs, the presence of EpCAM+ MCF-7 cells led to significantly higher levels of CD69+CD25+ populations within CD4+ and CD8+ T cells. Similarly, when PBMCs were treated with αEpCAM/αCD3 CSANs in the presence of MCF-7 cells, the production of IL-2 (Figure 3G) and IFN-γ (Figure 3H) was enhanced by 7.0- and 2.7-fold, respectively. This observation is consistent with previous studies of the parental monoclonal antibody, UCHT1, which, unlike other αCD3 antibodies (e.g. OKT-3), is unable to induce IL-2 production directly.47, 48 Additionally, IL-2 and IFN-γ production was shown to be dose dependent on the concentration of αEpCAM/αCD3 CSANs (Figure S8A & B) and correlates well with the selective cytotoxicity of the T cells toward MCF-7 cells when in the presence of αEpCAM/αCD3 CSANs (vide supra). Of particular note, the level of increased CD69, CD25, and IFN-γ expression was comparable to αCD22/αCD3 CSANs.26 Therefore, even though the T cell donor was different and the targeting scFv for the bispecific CSANs was different, the effect of the incorporated αCD3 svFv when interacting with the T cells remained the same. Thus, the degree of scFv polyvalent display by the two bispecific CSANs appears to be proportional.
Our results are also comparable to the αEpCAM/αCD3 BiTE, MT110. For instance, the level of CD69 expression (40%) at non-toxic doses was similar. Efficient cell killing over 24 hours was also observed for both αEpCAM/αCD3 PARs and MT110, resulting in CD69 expression levels of 80%, and IL-2 production increase of 3.5-fold.45, 49 However, the total amount of IFN-γ and IL-2 produced by the PBMCs in the presence of MT-110 and a gastric cell carcinoma was found to be 5- to 10-fold greater than αEpCAM/αCD3 PARs.49 Additionally, unlike MT100, αEpCAM/αCD3 PAR T cells produced significantly greater amounts of IL-2 (1.4-fold) than IFN-γ, in vitro, when in the presence of the target tissue.
A significant obstacle in solid tumor immunotherapy is the exhaustion of T cells as a result of the immune suppressive milieu within the tumor microenvironment.50 Specifically, T-regulator cells (Treg) have demonstrated significant immunosuppressive capabilities and correlated strongly with poor prognosis and metastasis in cancer patients when high numbers of activated, tumor resident, Treg cells are found.51 Furthermore, as Treg cells express the CD3 receptor they can theoretically undergo activation in response to any CD3 directed bispecific construct. To evaluate the potential for Treg activation we evaluated the activation levels of CD25+FOXP3+CD127- Tregs in the presence of bispecific CSAN therapy.52 In brief, previously unactivated PBMCs were incubated with target MCF-7 cells and αEpCAM/αCD3 bispecific CSANs followed by monitoring of the CD4 T cell population for the upregulation of FOXP3 by flow cytometry. Our results indicated a 0.7% and 1.6% increase in the Treg population in response to PBMCs treated with either bispecific CSANs and bispecific CSANs in the presence of the target tumor cells, respectively (Figure S9). While this increase was statistically significant the total percentage of Treg cells remained within the standard range for human PBMCs (4–10%),53, 54 and significantly lower than the range typically found within the tumor mass (20–30%).55, 56 Additionally, the PAR T cells remained capable of eliciting cytotoxic effects both in vitro and in vivo; indicating that if Treg activation did occur, it was insufficient to elicit a significant immunosuppressive effect.
Strong, and prolonged, stimulation of the TCR has been shown to result in T cell anergy.57–59 For this reason our laboratory decided to evaluate the affect CSANs have on the TCR receptor to determine whether there was any subsequent activation. There are multiple phosphorylation events downstream of TCR signaling which have been shown to determine the fate of T cells upon recognition of their cognate antigen. For example, engagement of the TCR stimulates activation of Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and protein kinase B (AKt), which lead to the activation of mammalian target rapamycin (mTOR) and phosphorylation of pS6.60 We chose to monitor the biochemical signal pS6 as a representative readout of TCR stimulation. When in the presence of CSANs alone there was no significant increase in the levels of pS6 for both CD4+ and CD8+ naïve T cells and only a 1.5-fold increase in the expression levels for CD4+ and CD8+ memory T cells (Figure 4). Importantly, while there was an increase in memory cell activation, these same culture conditions elicited no increase in IL-2 release (Figure 3G) over 24 hours and only a partial increase in IFN-γ release (Figure 3H) production. In contrast to CSANs alone, incubation with both bispecific CSANs and target MCF-7 cells resulted in a significant 2-fold increase in pS6 levels for CD4+ and CD8+ memory T cells over naïve T cells (Figure 4). These results are similar to those observed for activation of the T cells by the CD3/CD28/CD2 activation complex, this is mirrored by both the significantly increased IL-2 and IFN-γ production under these same conditions (Figure 3G & H). Additionally, CD8+ memory T cells were shown to display significantly higher levels of the degranulation marker CD107a and intracellular IFN-γ compared to naïve T cells upon stimulation with CSANs (Figure S10). In fact, even in the presence of both target cells and CSANs, the naïve CD8+ T cells elicited no upregulation of CD107a or IFN-γ. These results suggest that engagement of naïve T cells by the αEpCAM/αCD3 bispecific CSANs in the presence of the target cells does not appear to significantly stimulate the TCRs of naïve T cells, but preferentially activates CD8+ memory T cells. Taken together, these results suggest that treatment of naïve T cells with bispecific αEpCAM/αCD3 CSANs has a low probability of inducing T cell anergy.
Figure 4. Monitoring pS6 Levels in T Cells Labelled With αEpCAM/αCD3 CSANs.
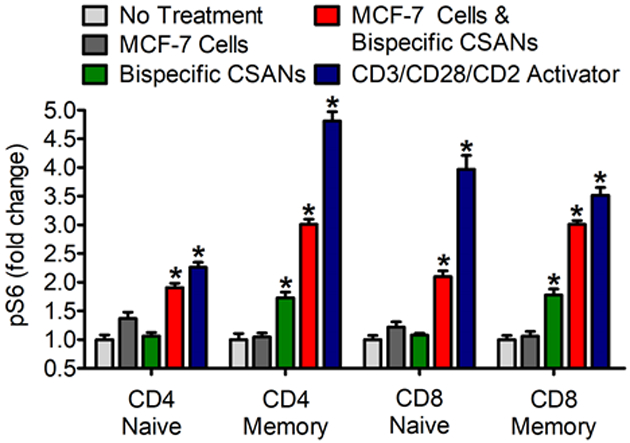
Unactivated PBMCs were co-cultured with media, MCF-7 cells, bispecific CSANs, both bispecific CSANs and target MCF-7 cells or CD3/CD28/CD2 activator complex. PBMCs were incubated for 3 hours before measuring internal pS6 levels by flow cytometry. Lymphocytes were determined by forward and side scatter as well as removal of CD20, CDllb/c positive cells. CD4+ and CD8+ cells were gated for further categorization as naïve (CD45RA+CD45RO−) or memory cells (CD45RA−CD45RO+). *P<0.05 with respect to non-treated controls, by one-way ANOVA analysis.
Since both CD4+ and CD8+ T cells can have effector function, we characterized the activation and cytotoxicity of isolated CD4+ and CD8+ T cells in the presence αEpCAM/αCD3 CSANs. Over the course of 24 hours, the greatest level of targeted cytotoxicity resulted from CD8+ T cells (55%) treated with αEpCAM/αCD3 CSANs and the least from treated CD4+ cells (10%; Figure 5A). The presence of CD4+ T cells was not required for CD8+ T cell targeted cytotoxicity in vitro. Indeed, as the ratio of CD4+ to CD8+ T cells decreased from 4:1 to 1:4, as targeted cellular cytotoxity increased. Further analysis revealed the bispecific CSAN-modified CD8+ T cells in the presence of the target cells were fully capable of producing IFN-γ (Figure 5B) and IL-2 (Figure 5C). Interestingly, both CD4+ and CD8+ T cells in the presence of either αCD3 CSANs or αEpCAM/αCD3 CSANs and target MCF-7 cells expressed the same amount of CD69 and CD25 (Figure S11). However, the CD4+ and CD8+ T cells treated with the bispecific CSANs produced 10- to 12-fold, respectively, more IL-2 than cells treated with αCD3 CSANs. Similarly, a 2- and 3-fold increase in the amount of IFN- γ released by CD4+ and CD8+ T cells, respectively, was found for cells treated with αEpCAM/αCD3 CSANs than αCD3 CSANs in the presence of MCF-7 cells. Thus, while an increase in the amount of CD69 and CD25 is observed for CD4+ and CD8+ T cells treated with αEpCAM/αCD3 CSANs, the magnitude of cytotoxicity correlated with the amount of IFN-γ and, to an even greater extent, IL-2 produced. When the time of incubation was extended to 36 hours, cellular toxicity induced by CD4+ T cells treated αEpCAM/αCD3 CSANs was similar to that mediated by CD8+ T cells (Figure S12).
Figure 5. Isolated CD8+ and CD4+ T Cells are Still Capable of Selective Activation and Target Cell Killing.

(A) CD8+ and CD4+ T cells were isolated separately by negative selection and used for targeting cell killing with 50 nM Bispecific CSANs and a 10:1 E:T ratio at various CD4:CD8 ratios. Cytokine production produced from CD4+ and CD8+ T cells isolated by negative selection. Unactivated PBMCs were co-cultured with media, monospecific CSANs or bispecific CSANs in the presence or absence of target MCF-7 cells for 24 hours at various CD4:CD8 ratios. Following incubation, the media was analyzed for (B) IFN-γ and (C) IL-2 by ELISA. Data shown was obtained from one donor (n=3), but representative of three donors (Figure S18 & S19). *P<0.05 with respect to antiEpCAM CSAN and no treatment controls, by 2-tailed Student’s t test.
In Vivo anti-Tumor Activity
To characterize the in vivo anti-tumor activity of the αEpCAM/αCD3 PAR T cells, we used an orthotopic NSG mouse model amenable to human xenograft receptor targeting. Since the bispecific CSANs were found to not cross-react with either murine CD3 or murine EpCAM; a xenograft model reliant on the engraftment of human cancer cells and PBMCs into an immunodeficient NSG mice was necessary for assessing in vivo anti-tumor efficacy. Specifically, MCF-7-Luc breast cancer cells were unilaterally injected into the fourth mammary fat pad of NOD.Cg-Prkdcscid Il2rγtm1Wjl/SzJ (NSG) mice. Ten days after tumor implantation, mice were separated into control and treatment groups and dosed intravenously with either PBS, PBMCs (20×106), αEpCAM/αCD3 CSANs (1 mg/kg), PBMCs treated with αCD3 CSANs (50 nM), or PBMCs treated with αEpCAM/αCD3 CSANs (50 nM). The αEpCAM/αCD3 CSAN-treated PBMC group was subsequently dosed intravenously with injections of αEpCAM/αCD3 CSANs (1 mg/kg) every 2 days following the initial infusion of αEpCAM/αCD3 CSAN modified PBMCs on day 10, thus re-arming the engrafted T cells in vivo. Similarly, the αCD3 CSAN-treated PBMC group was given additional injections of αCD3 CSANs (1 mg/kg) every 2 days following the initial infusion of αCD3 CSAN modified PBMCs on day 10. Over the course of the next eight days full tumor eradication was observed for the αEpCAM/αCD3 CSAN-treated PBMC group (Figure 6A). The response was found to be durable, with no tumor re-emergence observed over the next two and half weeks. Unfortunately, due to the emergence of graft-vs-host (GVH) disease, it was necessary to sacrifice the study mice two and half weeks after implantation of human PBMCs, thus hindering our ability to assess tumor reemergence. In contrast to EpCAM expressing tumors, when NSG mice were implanted with EpCAM negative U-87 MG xenograft tumors and subsequently dosed with αEpCAM/αCD3 CSAN modified PBMCs, followed by dosing every two days with the CSANs, tumors progressed at the same rate as control groups (Figure S13).
Figure 6. In Vivo Efficacy Study of Bispecific PARs in an Orthotopic NSG Mouse Model.

NSG mice were inoculated in the mammary fat pad with 1.0×106 MCF-7 Luciferase cells. (A) Ten days following tumor implantation mice were IV injected with either PBS, 20×106 unstimulated human PBMCs, 1 mg/kg αEpCAM/αCD3 bispecific CSANs, 20×106 unstimulated PBMCs modified with 50 nM αCD3 monospecific CSANs, and the treatment group of 20×106 unstimulated PBMCs modified with 50 nM αEpCAM/αCD3 bispecific CSANs (n=5). Each cohort was given additional booster injections of the relevant CSAN construct at 1 mg/kg, or PBS, every two days following the initial infusion on day 10. Tumor growth was monitored every by caliper and recorded as mm3. (B) The same NSG model was applied with booster injections provided at varying time intervals, including: daily, every two days, every four days and every 8 days. (C) Body weight was monitored and recorded. (D-G) On days 1, 5 and 9 following treatment the level of IFN-γ (D), IL-2 (E), IL-6 (F) and TNF-α (G) was quantified. All in vivo experiments were performed independently and at least twice. *P<0.05 with respect to readings statistically significant from the PBS control group, by 2-tailed Student’s t test.
To assess the importance of the αEpCAM/αCD3 CSAN dosing schedule on tumor regression, or how often T cells required re-arming, αEpCAM/αCD3 CSAN labeled PBMC were administered prior to intravenously injections of the bispecific CSANs either every day, every two day, every four days or every eight days. One, two, and four-day dosing resulted in full tumor eradication following the initial infusion of αEpCAM/αCD3 CSAN modified PBMC. While complete tumor regression was not observed when dosing occurred every eight days, the rate of tumor growth was significantly reduced by 72% (Figure 6B). Over the course of the anti-tumor studies, no significant weight loss was observed, regardless of dosing schedule (Figure 6C & Figure S14). However, due to tumor burden, as well as GVH disease, a small decrease was observed by the end of the study for the animals treated with the PBS and PBMC only controls (Figure 6C).
An explanation for the potent anti-tumor activity of αEpCAM/αCD3 PAR T cells may rest on the fact that MCF-7 cells, and their associated tumorigenic stem cells, express EpCAM.38 This conclusion is consistent with the observation that tumor re-emergence is not observed for over two weeks following both the 24 and 48 hour dosing schedules of αEpCAM/αCD3 CSANs (Figure 6). Nevertheless, the inability to significantly reduce tumor size for the 8-day dosing schedule implies that once tumors have reached a certain size, it may be difficult to achieve the necessary infiltration of αEpCAM/αCD3 PAR T cells. Future studies will examine the impact that tumor type, tumor size, and CSANs dosing levels have on tumor progression or eradication, which will provide important insights into both the potential and limitations of PAR T cell anti-tumor activity. Additionally, future studies will explore the possibility of a CSAN scaffold targeting mouse EpCAM and CD3 to allow the evaluation of CSANs with a fully immunocompetent model.
Targeting EpCAM+ tumors has also been attempted with an αEpCAM/αCD3 BiTE, MT110.49 Dosing of mice with established tumors, although not orthotopic, did lead to tumor eradication if MT110 was given daily.49 By comparison, tumor eradication with the αEpCAM/αCD3 PAR T cells could be achieved by dosing every four days with a comparable amount of the bispecific CSANs. This difference likely results from, the longer half-life of the αEpCAM/αCD3 CSANs on the T cells is likely responsible for the decreased dosing schedule compared to the BiTEs, which require daily infusion to achieve clinical efficacy. Additionally, similar to MT110, no significant toxicities or activity toward EpCAM negative tumors was observed following administration of the αEpCAM/αCD3 PAR T cells, suggesting specific interaction with the EpCAM positive tumor cells, further suggesting that the observed anti-tumor effect of the PAR T cells is specific.
Effect of CSANs on In Vivo Cytokine Release
Since IL-2, IFN-γ, TNF-α, IL-6 and IL-10 production is hallmark of T cell activation, the plasma cytokine levels of NSG mice bearing orthotopic MCF-7-Luc tumors treated with αEpCAM/αCD3 CSANs was determined by ELISA and compared to non-treated groups. The cytokine analysis indicated IL-2 and IFN-γ production was detectable within one day of treatment and rose slowly over the course of the next eight days (Figure 6D-E). In contrast, neither IL-2 nor IFN-γ production was observed in the plasma for the non-treated PBS control group. By day five, both IL-2 and IFN-γ could be observed in the plasma for the non-treated PBMC control group suggesting the emergence of GVH disease. In addition, IL-6 and TNF-α production by the treatment group and not the PBS control group was also observed after one day (Figure 6F-G). The amount of IL-6 in the plasma doubled over the next four days, but was unchanged by day eight, while the amount of TNF-α rose by 25% by day four and day eight. Thus, T cell activation by the αEpCAM/αCD3 CSANs could be observed by at least the first day of treatment. Interestingly, within the first 24 h 6.5-fold more IFN-γ was produced than IL-2 which is in contrast to the in vitro results where IL-2 production was slightly higher than that of IFN-γ. Additionally, compared to previous studies with CAR T cells and BiTEs, similar or lower levels of cytokine production by αEpCAM/αCD3 CSANs were observed.
Effect of CSANs on In Vivo T Cell Populations
Persistent effector and central memory T cell phenotypes correlate with total remissions in clinical trials for CAR T cell therapy.61, 62 To evaluate the effect αEpCAM/αCD3 CSANs were having on T cell populations we dosed NSG mice bearing the MCF-7-Luc tumors as previously discussed. Mice were dosed with either PBMCs or PBMCs treated with αEpCAM/αCD3 CSANs and redosing was performed every 4 days. CD4+ and CD8+ T cells in the mouse peripheral blood were analyzed for CD45RO and CCR7 on day 0 and 4. The αEpCAM/αCD3 CSAN treatment demonstrated significant expansion of CD45RO+CCR7- (effector memory T cells) for both the CD4+ and CD8+ cells compared to the non-treated PBMC population at day 4 (Figure 7). The bispecific treatment additionally showed a greater decrease in the CD45RO-CCR7+ (naïve T cells) population compared to the non-treated control. In addition, the increased levels of both IL-2 and IFN-γ appear to correlate with both the rise in number of CD4+ and CD8+ T effector memory cells (Tem) and decrease in the number of CD4+ and CD8+ naïve T cells (Tn) relative to the non-PAR T cell treated mice.
Figure 7. CD4+ and CD8+ Memory Cell Formation.
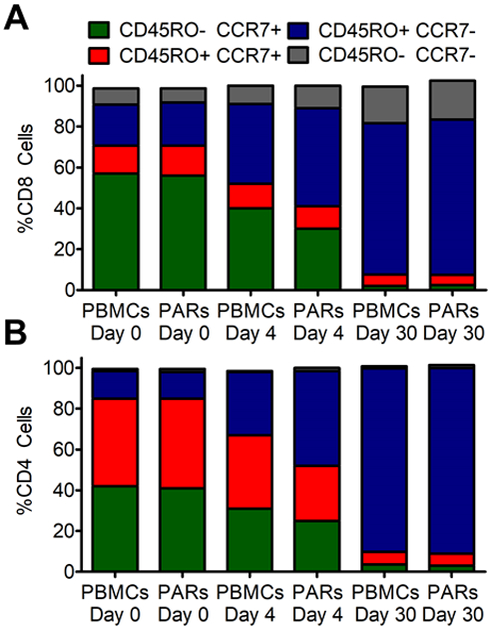
The phenotype of T cells in the peripheral blood from mice treated with PBMCs and PARs formed by modifying PBMCs with αEpCAM/αCD3 bispecific CSANs was analyzed on day 0, 4 and 30. They are displayed as the proportion of (A) CD8+ cells and (B) CD4+ cells by flow cytometry with staining for CCR7and CD45RO (n = 5). All SD < 1.9%.
Previously, we have demonstrated that surface bound CSANs can be removed from T cells by the addition of trimethoprim. Next, we wanted to evaluate whether this ability extended to in vivo applications as well. To determine the ability of trimethoprim to affect the activation of T cells bound to αEpCAM/αCD3 CSANs, we prepared NSG mice bearing orthotopic MCF-7-Luc tumors and treated them with PBMCs and αEpCAM/αCD3 CSANs. One day after treatment, the amount of IFN-γ, IL-2, IL-6 or TNF-α in the blood was determined and compared to the amounts before treatment, as well as to the non-treatment controls. In each case, the amount of each cytokine significantly increased above the background amounts and the control groups, indicating T cell activation. The mice were then dosed intravenously (2 mg/kg) with clinically used trimethoprim/sulfamethoxazole or PBS. Twenty-four hours later, plasma samples were taken and cytokine levels determined. The amount of IL-2, IL-6, IFN-γ or TNF-α remained unchanged for the trimethoprim/sulfamethoxazole treated group, while the levels of each cytokine significantly increased for the mice not treated with trimethoprim/sulfamethoxazole (Figure 8). This result is consistent with removal of the bispecific rings from the surface of the cells, since the production of the cytokines is dependent on the targeted interaction of the T cells with tumor tissue. Whether a higher dose or more frequent dosing would result in a more profound affect remains to be determined. Regardless, these results suggest the toxic side-effects resulting from T cell activating immunotherapies may be addressable pharmacologically for PAR T cells with an FDA approved drug.
Figure 8. Trimethoprim Mediated Dissociation of CSAN Therapy In Vivo.

Three groups of NSG Mice (n=5) previously engrafted with MCF-7 tumors were treated with either PBS, 20 million unstimulated PBMCs or 20 million unstimulated PBMCs functionalized with 50nM αEpCAM/αCD3 CSANs by IV. One day following therapy the experimental group receiving trimethoprim/sulfamethoxazole (TMP) was dosed with 2 mg/kg of TMP. Each day their levels of IFN-γ (A), IL-2 (B), IL-6 (C) and TNF-α (D) was quantified (n=5). *P<0.05 with respect to readings that were statistically significant from bispecific PAR therapy, by 2-tailed Student’s t test.
In Vivo Biodistribution of CSANs
To characterize the in vivo biodistribution of PAR therapy we generated radiolabeled αEpCAM/αCD3 bispecific CSANs and αCD3 non-targeted CSANs based on our previously developed methodology.30 Monomeric DHFR2-αEpCAM and DHFR2-αCD3 were incubated with three equivalents of bis-MTX-DOTA[64Cu] at room temperature to form the relevant 64Cu labeled CSAN constructs as previously described, followed by purification with a Bio-Spin6 SEC column resulting in labelling efficiencies of greater than 95% for all constructs. NSG mice (n = 4) bearing orthotopic MCF-7-Luc tumors were engrafted with 20 million PBMCs by IV infusion followed 4 days later by IV injection of 180 to 200 µCi/µmol of purified 64Cu-αEpCAM/αCD3 CSANs and 64Cu-αCD3 CSANs. Additionally, a blocking study was performed by IV infusion of the parental αEpCAM mAb, Moc31, 10 minutes prior to the injection of CSANs. The mice were subsequently euthanized at 1, 6, 24 and 48 hours post injection (p.i.) and organs were harvested and analyzed for residual radioactivity using a single-tube well counter.
Similar to previous studies, a significant proportion of CSANs accumulated in the major clearance organs, with approximately 25% and 17% of each construct accumulating in the liver and kidneys, respectively (Figure 9A).30 In contrast to previous results, αCD3 containing CSANs exhibit increased targeting to major T cell reservoirs (e.g. spleen and lungs). However, regardless of the construct used in this study, the only significant difference in tissue accumulation was observed in EpCAM overexpressing tumors. The peak tumor accumulation (12.1 ± 1.6% ID/g) for targeted 64Cu-αEpCAM/αCD3 CSANs was observed 24 hours post injection, with only a minor decrease in activity detected 48 hours post injection (10.9 ± 1.1% ID/g) (Figure 9B). Both the non-targeted 64Cu-αCD3 CSANs and blocked 64Cu-αEpCAM/αCD3 CSANs displayed a significantly decreased peak tumor accumulation compared to the targeted construct, with an observed accumulation of 4.7 ± 2.1% and 5.9 ± 1.8% ID/g at 24 hours, respectively. The targeted 64Cu-αEpCAM/αCD3 CSANs accumulated rapidly in the tumor with 4.1 ± 0.9% ID/g observed one-hour post injection and 11.3 ± 1.3% ID/g six hours post injection. Thus, accumulation of the targeted CSANs appeared to be specific for the EpCAM expressing tumor tissue with a life time of greater than 48 hours. (Figure 9B). Moreover, CSANs were observed to exhibit a markedly increased circulation time compared to that of BiTEs, which are rapidly eliminated within minutes in vivo.20, 21 In fact, the increased half-life of CSANs likely contributed to the induced tumor clearance that was seen even when dosed as infrequently as every four days; in contrast, BiTEs required daily dosing to achieve these same results.49
Figure 9. Biodistribution and Tumor Targeting of Bispecific PARs.

NSG mice were inoculated in the mammary fat pad with 1.0×106 MCF-7 cells. Tumors were allowed to grow to 300 mm3 prior to infusing mice with 20×106 PBMCs to allow for uman T cell engraftment. Four days following PBMC infusion mice were injected with 64Cu-radiolabeled αEpCAM/αCD3 bispecific CSANs, 64Cu-radiolabeled αEpCAM/αCD3 bispecific CSANs ten minutes following the infusion of 10 mg/kg αEpCAM mAb (moc31), and 64Cu-radiolabeled non-targeted αCD3 CSANs. Mice were sacrificed and organs were harvested to determine a (A) full biodistribution analysis at 24 hours, (B) the tumor uptake over time, and (C) ratio of CSANs that are bound to T cells or in free plasma circulation. Results are reported as (A & B) percentage injected dose per gram of tissue and (C) ratio of T cell:Plasma (n = 4), representing approximately 87.5 ± 2.4 % percent of radioactivity in the isolatiod blood samples. Statistical significance, *p < 0.05 with respect to block and αCD3 CSAN controls (two tailed T-test).
To ascertain the ability of the injected CSANs to bind the surface of T cells in vivo, blood samples were collected at 1, 6, 24 and 48 hours post injection and the T cell component and the CD4+ and CD8+ positive T cells isolated and the ratio of the T cell bound CSANs to free plasma CSANs was determined. After one hour, only 4% of the radioactive 64Cu-αEpCAM/αCD3 CSANs in the blood was found to be T cell bound; however, by 24 hours and 48 hours the total T cell bound radioactivity was shown to have risen to 42% and 44%, respectively (Figure 9C). These results indicate that the CSANs stably bound the T cells for over 48 hours, while the percentage of unbound CSANs in plasma was decreased by roughly 50% in the first 24 hours. A slightly greater percentage of T-cells (51% vs 46%) was found to be bound to the monospecific αCD3 CSANs was observed, possibly due to the high αCD3 polyvalency of αCD3 CSANs, relative to the αEpCAM/αCD3 CSANs. Importantly, dosing with the αEpCAM MAB, Moc31, did not block the labeling of the T cells in vivo, thus further validating the specificity of the bispecific and monospecific CSANs for T cells.
While the majority of the radioactivity in the blood was located in the plasma and T cell populations, approximately 12% was unaccounted for. To determine the cause of this loss we dosed PBMCs in vitro for 24 hours with 50 nM of both FITC labelled αEpCAM/αCD3 CSANs and non-targeted CSANs. PBMCs were subsequently analyzed by flow cytometry to determine what cell populations demonstrated CSAN binding. As expected the αEpCAM/αCD3 CSANs demonstrated strong binding to the CD4+ and CD8+ T cells and none to the B cell and NK cell populations (Figure S15). However, moderate binding was seen in the monocyte population, by both the targeted and non-targeted constructs, which is validated by our previous results indicating macrophages slowly take up CSANs over time.30
CSANs Immunogenicity
As with all biologics and especially with those derived from foreign sources, the development of immunogenicity may be a clinically important issue that limits efficacy. This is of particular concern for CSANs, since they are composed of E. coli DHFR. Consequently, we evaluated the inherent immunogenicity of our CSAN based therapy. When either CSANs or free DHFR2 were incubated with fresh splenocytes from untreated BALB/c mice for 24 h at 37°C no significant increase in the expression levels of CD40 or CD86 was observed (Figure 10A). Furthermore, no significant increase in the production of IL-6 was observed, indicating that CSANs do not activate B-cells (Figure 10B). Additionally, this indicates that while CSANs alone elicit a mild activation in memory cells, there is no B-cell activation as a result of CD4+ memory cells. To further address the potential immunoreactivity of CSANs we injected BALB/c mice with 30 µg of the CSANs on day 1 and day 13 and then measured the amount of DHFR specific IgG antibody ELISA on days 13, 21 and 30. As can be seen in Figure 10C, over the course of the 30 days, no significant difference in the production of anti-DHFR IgG was observed in the plasma of the CSANs treated mice compared to the untreated PBS control. Thus, CSANs exhibit negligible immunogenicity when mice were dosed intravenously. Although speculative, the apparent inability of the DHFR2 CSANs to display innate immune response pathogen associated molecular patterns could be a factor in limiting potential immune cell activation. In addition, the high stability of the CSANs, even when endocytosed, may also limit their ability to undergo antigen presentation and thus generation of an adaptive immune response. Nevertheless, while the inability to elicit an immune response in mice does not preclude the possibility they may be immunogenic in humans, the development of an adaptive immune response in mice would predict a likely potent immune response in humans.63
Figure 10. Murine αDHFR2 Immune Response.

Freshly isolated spleen cells from naïve mice were incubated with DHFR2 protein, non-targeted CSANs and CpG 1826 (positive control) for 24 hours and monitored for (A) CD40 and CD86 expression as well as (B) IL-6 release. Mice were injected with non-targeted CSANs and (C) monitored on day 13, 21 and 30 since the first immunization. Sera were collected for estimation of αDHFR IgG antibodies by ELISA. Data were shown as absorbance values at 450 nm wavelength at appropriate dilutions. Each bar represents the mean value of absorbance value OD450 ± SD for each group (n=4). Values of p < 0.05 were considered significant and were indicated as follows: *p < 0.05. Statistical differences between the groups were determined by one-way ANOVA analysis and Student t test.
CONCLUSIONS
Our results demonstrate that αEpCAM/αCD3 bispecific CSANs can be used for the rapid, stable and reversible modification of T cells resulting in potent anti-tumor responses. Evidently, the high avidity of the CSANs to central memory and effector memory T cells enables the PAR T cells to be functional over several days, unless removed by treatment with trimethoprim. Thus, compared to existing T-cell engaging and CAR T-cell based immunotherapies, multispecific and polyvalent CSANs have the potential to be an alternative and complementary T-cell targeting approach. In addition, the demonstration that bispecific and polyvalent CSANs can guide and reversibly control cell-cell interactions, both in vitro and in vivo, supports their on-going development as a synthetic biological tool.
MATERIALS AND METHODS
Cell Lines, Culture Conditions and T cell isolation
MCF-7 breast cancer cells and U87 MG human glioblastoma cells were obtained from American Type Culture Collection (ATCC, Rockville, MD). MCF-7 Luciferase cells were kindly provided by Dr. Daniel Vallera (University of Minnesota, Minneapolis, MN). Human cancer lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS, 100 U/mL penicillin, 100 µg/mL streptomycin, and L-glutamine at 37°C in 5% CO2. Human PBMCs were isolated from buffy coats of healthy donor blood samples (obtained from Memorial Blood Centers, St. Paul, MN) by Ficoll density gradient centrifugation. Unactivated CD8+ or CD4+ T cells were isolated from PBMCs by positive selection using CD8+ or CD4+ T cell isolation kit (Invitrogen Life Technologies, Grand Island, NY). PBMCs were cultured in complete RPMI 1640 medium (Lonza) supplemented with 10% (v/v) fetal bovine serum, L-glutamine (final concentration of 2mM), Penicillin (100 units/mL), and Streptomycin (100 µg/mL) in a humidified incubator with 5% CO2 at 37 ˚C.
DHFR2 protein expression and purification
The development and preparation of DHFR2-αEpCAM and DHFR2-αCD3 proteins have been previously described 24, 25, 28. Briefly, E. coli BL21-DE3 OneShot Ultracompetent cells were purchased from Invitrogen and transformed with either DHFR2-αEpCAM and DHFR2-αCD3 plasmids per Invitrogen specifications. Following transformation, individual starting cultures were grown in 4L of LB media containing 0.1 μg/mL ampicillin in 4 separate 2L flasks. The cultures were shaken at 250 RPM at 37°C until the OD600 was within the ranges of 0.4–0.8 and subsequently induced with isopropyl β-D-1-thiogalactopyranoside (IPTG; Sigma Aldrich, St. Louis, MO) at a final concentration of 0.3 mM. Cultures were shaken for an additional 3 hours following induction and centrifuged to obtain a bacterial pellet. The cell pellet was suspended in a 50mM Tris-HCl buffer containing 50mM NaCl, 5mM EDTA, 1mg/mL lysozyme, and a protease inhibitor cocktail (phenylmethanesulfonyl fluoride, Pepstatin A, and Leupeptin). All insoluble material was isolated by centrifugation and further washed using a detergent-based buffer containing 1% sodium deoxycholate, Triton X-100, and glycerin to isolate the inclusion bodies (PPIB). The inclusion bodies were solubilized in 2.5% sodium N-lauroyl-sarcosine (SLS) buffer and air oxidized using 50 μM CuSO4 for 20 hours. All detergent in the SLS buffer was removed by treatment with Dowex 10% 1-X8 anion-exchange resin and 6M urea. Oxidized inclusion bodies were diluted 40-fold into refolding buffer containing 50mM Tris, 0.5 M L-arginine, and 20% glycerol (pH 8) and allowed to sit for 48 hours. Refolded protein was dialyzed against 20mM Tris buffer, and loaded onto a Fast Flow Q-sepharose (FFQ) anion exchange column prior to elution with 720mL of Tris buffer containing 1M sodium chloride (NaCl).
CSAN Oligomerization and Characterization
To create bispecific CSANs, equal parts of purified DHFR2-αEpCAM and DHFR2-αCD3 proteins (2–8 µM) were combined with 2.2 equivalents (1:1:2.2 ratio) of either C9-bisMTX dimerizer or FITC-conjugated bisMTX trilinker in phosphate-buffered saline (PBS). For monospecific CSANs, single purified fusion proteins were incubated with 2.2 equivalents (1:2.2 ratio) of either C9-bisMTX dimerizer or FITC-conjugated bisMTX trilinker in PBS. CSANS were allowed to incubate at room temperature for 30 minutes, in the absence of light. All proteins and CSAN constructs were analyzed by size exclusion chromatography (SEC) to observe the average size of ring formation, by injection onto a Superdex G200 column connected to a Beckman Coulter HPLC equipped with a diode array detector and using PBS as a mobile phase. Elution times were monitored at 280 nm to observe the change in hydrodynamic radius compared to monomeric species. DLS measurements were performed on a Brook haven 90 Plus Particle Analyzer (Holtzville, NY) with a 35 mW red diode laser. Samples (1.5 mL) were measured at room temperature in suspensions of PBS at (1.5 mg/mL). Cryo-TEM samples were prepared using an FEI Vitrobot Mk IV (Hillsboro, OR). Briefly, glow discharged lacey formvar/carbon, 300 mesh, copper grids (Ted Pella Inc., Redding, CA, cat: 01883) were loaded with 3 μL of a 6.6 μM solution of CSANs in PBS within a 95% humidity chamber at 25˚C. After blotting for 5.5 seconds, grids were plunge frozen in liquid ethane, and kept under liquid nitrogen until loading into a cryo-transfer holder (Gatan Inc., Pleasanton, CA, cat: 626). Imaging occurred on an FEI Tecnai G2 Spirit BioTWIN transmission electron microscope with a LaB6 emitter and FEI Eagle 2k CCD detector.
Binding Assays
Protein samples (4–20 µM) were incubated with 1.1 equivalent of bis-MTX-FITC in PBS for 1 hour at room temperature to generate the following three constructs: αEpCAM CSANs, αCD3 CSANs and αEpCAM/αCD3 Bispecific CSANs. All three CSAN constructs (1 μM) were incubated with 1 million EpCAM+ MCF-7 cells at 37°C for 1 hour and subsequently washed three times with PBS supplemented with 1% BSA and 0.1% sodium azide (FACS buffer). Samples were filtered to remove cell aggregate prior to analysis by flow cytometry using a BD LSR II flow cytometer. The mean fluorescence intensity (MFI) was monitored and compared to unstained PBMCs as a background control. To monitor the specific binding to each PBMC cell subtype, either FITC labelled αEpCAM/αCD3 Bispecific CSANs and FITC labelled non-targeted CSANs were incubated with PBMCs for 24 hours. Following treatment PBMCs were harvested and washed with FACs buffer prior to staining with CD4-PE, CD8a-BV421, CD20-BV605, CD11b-APC-ef780, CD11c-APC-ef780, and CD16-AF488 before analysis with an LSRFortessa H0081. To monitor cell surface stability of CSANs, 5×105 PBMCs were incubated with saturating concentrations (1 µM) of either αEpCAM/αCD3 bispecific CSANs or αCD3 DHFR2 monomer for 1 hour at room temperature. Cells were subsequently washed and plated in 48-well plates. For the following 5 days cells were washed with FACs buffer and then replated into the 48-well plate. Each day one of each sample was washed with FACs buffer and treated with saturating concentrations of αCD3 mAb-FITC and analyzed for FITC fluorescence. To determine the maximum fluorescence, the same number of PBMCs not previously stained with bispecific CSANs or αCD3 DHFR2 monomer were additionally stained with αCD3 mAb-FITC. This experiment was run in triplicate for each time point. Percent fluorescence inhibition was determined by subtracting the mean fluorescence at each time point from the maximum 100% cell binding which was set with the absence of competition.
Cytotoxicity Assays
Measurement of cell lysis was evaluated by measuring LDH (lactate dehydrogenase) release from cells with the non-radioactive cytotoxicity assay (CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega). The day previous to the experiment 5×103 target MCF-7 cells or non-target U87-MG cells were seeded into a 96-well plate in 200 μL of RPMI-1640 media per well. The following day the appropriate number of resting PBMCs (according the requested effector (E) to target (T) cell (E:T) ratio as indicated in the respective experiment) were counted and incubated with 0.01–1 μM of the αEpCAM/αCD3 bispecific CSANs for one hour at 37°C with 5% C02. Following the initial incubation, PBMCs were washed and resuspended in RPMI before addition to 96-well plate containing target MCF-7 or U87-MG cells and incubated for 24 hours under standard conditions. MOC-31 (Abcam), which competitively binds the same epitope as αEpCAM DHFR2,28 was co-incubated when indicated to test target receptor specificity. Lysis buffer (provided in assay kit) was added to control wells with only MCF-7 cells to estimate the maximum LDH release. The absorbance at 490 nm was measured and recorded using a Synergy H1 Multi-Mode Reader (Biotek). Data was corrected for media absorbance, and values were determined per the following equation: ((LDH release sample – SReffector – SRtarget)(MRtarget – SRtarget)) x 100. SR: spontaneous release; MR: maximum release.
Immunostaining and Cytokine Analysis
IL-2 and IFN-γ measurements in the cytotoxicity assay supernatants were analyzed using ELISA per the conditions provided by IFN-γ ELISA kit (Invitrogen) and IL-2 ELISA kit (Invitrogen). In brief, following the incubation period the supernatant was removed and 10-fold diluted into ELISA buffer (provided in the kit) prior to placing 50 µL into the respective well of the included ELISA 96-well plate. IFN-γ and IL-2 production of experimental wells was determined through a standard curve generated from known control sample concentrations. T cell activation was measured using the cells from the cytotoxicity assay, which were stained with anti-human CD4 (FITC), anti-human CD8 (PE) and either anti-human CD25 (APC) or anti-human CD69 (APC) (Biolegend) and analyzed by flow cytometry to determine the proportion of CD4+ and CD8+ T cells activated (CD25+CD69+). Cells analyzed for CD69+ expression were incubated in the cytotoxicity assay for 12 hours while those analyzed for CD25+ expression were incubated for 24 hours. In vitro immunogenicity assays were performed with freshly isolated spleen cells from naïve Balb/c mice plated into 96-well plates with the indicated treatments. Cells were stained with anti-mouse CD40-PE-Cy5 (clone: 3/23) (BioLegend) and anti-mouse CD86 (clone:GL1). Cells were analyzed on the BD LSR II flow cytometer. To measure pS6 upregulation PBMCs were incubated for 3 hours at 37°C with either PBS, target MCF-7 cells, 50 nM αEpCAM/αCD3 bispecific CSANs, 50 nM αEpCAM/αCD3 bispecific CSANs which were pre-plated overnight at 37°C, 50 nM αEpCAM/αCD3 bispecific CSANs and target MCF-7 cells, or CD3/CD28/CD2 activator complex. PBMCs were harvested and washed once with PBS before resuspending in 1 ml of 1.6% Paraformaldehyde/PBS. Cells were incubated at RT for 15 min before the addition of 4 ml of 100% methanol at −80°C. Cells were incubated for an additional 15 minutes, spun down at 4°C and washed with FACs buffer. Cells were stained with CD4-PE, CD8a-BV421, CD45RO-BV605, CD45RA-APC, CD11b-APC-ef780, CD11c-APC-ef780, CD20-APC-ef780 and pS6-AF488 before analysis with an LSRFortessa H0081.
MCF-7 Orthotopic Breast Cancer Model
All protocols conformed to institutional regulations. Cohorts of five NSG mice were used. Six- to 8-wk-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were injected unilaterally into the fourth mammary fat pad with 1.0×106 MCF-7 cells. The injection was performed in 30 µL of 50:50 Matrigel/PBS directly through the nipple. Four days prior to tumor implant and every subsequent four days throughout the remainder of the study mice were SQ injected with 1 µg/kg of 17β-estradiol valerate. Ten days following tumor implantation mice were separated into treatment groups: a control group injected with PBS, a control group infused with 20×106 unmodified peripheral Blood Mononuclear Cells (PBMCs) by IV injection, a control group infused with 1 mg/kg αEpCAM/αCD3 CSANs by IV injection, a control group infused with 20×106 αCD3 monospecific PARs modified with 50 nM αCD3 CSANs, and a treatment group infused with 20×106 αEpCAM/αCD3 bispecific PARs modified with 50 nM αEpCAM/αCD3 CSANs. The treatment group was given additional booster injections of αEpCAM/αCD3 CSANs at 1 mg/kg dosage every 2 days following the initial PAR infusion on day 10. The αCD3 PAR group was given additional booster injections of αCD3 CSANs at 1 mg/kg dosage every 2 days following the initial PAR infusion on day 10. PBS and PBMC control groups were dosed every two days with PBS while the αEpCAM/αCD3 CSANs control group was dosed every two days with 1 mg/kg of additional αEpCAM/αCD3 CSANs. When indicated, additional booster injections were provided at alternate time intervals including: daily dosing, every 2 days, every 4 days and every 8 days. Body weight was monitored daily, and tumor growth was monitored daily by caliper to measure the height x width x length and recorded as mm3. Blood was obtained by facial vein bleeds and spun down at 400g for ten minutes to obtain the plasma. The plasma was analyzed by ELISA assay kits (AbCAM) as previously described to quantify the amount of IL-2, IFN-γ gamma, TNF-α, and IL-6. Mice were sacrificed at 35 days and the blood and spleen were immediately harvested for FACs analysis. PBMCs from the blood were obtained by spinning at 400g for 10 minutes in lymphocyte separation media and subsequently washed with FACs buffer. PBMCs from the spleen were obtained by maceration with a wire filter and RPMI media. Red blood cells were lysed using FACs lysing solution and the solution was then spun down and the pellet was washed with FACs buffer to isolate viable PBMCs. These were then stained with CD4 (FITC) (eBioscience), CD8 (PE) (eBioscience), CD20 (APC-ef780) (eBioscience), CD11b (APC-ef780) (eBioscience), CD11c (APC-ef780) (eBioscience), Live/Dead ghost red (Tonbo), CD45RO (BV605) (Biolegend), CCR7 (Percp Cy5.5) (Biolegend) for memory cell analysis. Cells were analyzed using an BD LSR II flow cytometer. In Vivo disassembly of PARs was evaluated with an IV infusion of 2 mg/kg trimethoprim/sulfamethoxazole (Sigma Aldrich) 1 day following the initial infusion of PBMCs or treated PARs.
In Vivo Biodistribution Analysis
64Cu was obtained from Department of Medical Physics, University of Wisconsin - Madison, WI, USA. Preparation of 64Cu labeled CSANs has been previously described.30 In brief, 64Cu Labeling of bis-MTX-DOTA was accomplished by the addition of 50 µL of bis-MTX-DOTA to approximately 10 mCi of 64CuCl2 that had been neutralized in 1M sodium acetate buffer [pH 7.0]. The reaction mixture was incubated for 30 minutes at 50° C, following which the extent of 64Cu chelation was determined by iTLC. The resulting excess of bis-MTX-DOTA[64Cu] was incubated with 1.0 mg of αCD3 DHFR2 to generate αCD3 CSANs and with both .5 mg of αCD3 DHFR2 and .5 mg of αEpCAM DHFR2 to generate bispecific CSANs. The mixture was incubated at room temperature for 30 minutes to prepare each respective CSAN construct. Excess 64Cu was bound by addition of 10 mM ethylenediaminetetraacetic acid for 10 minutes and removed, along with excess bis-MTX-DOTA[64Cu], by Bio-spin 6 Tris column. The extent of labeling was once again determined by iTLC and found to be greater than 95% for both constructs. As previously described, NSG mice were orthotopically grafted with EpCAM expressing MCF-7 tumors for further biodistribution analysis. 14 days following implantation (~125 mm3), mice were IV injected with 20×106 unstimulated human PBMCs to allow the engraftment of a human T cells for four days prior to the biodistribution study. Following engraftment mice (n = 4) were IV injected with 100 to 120 µCi of either αEpCAM/αCD3 Bispecific CSANs or αCD3 monospecific CSANs. An additional group was previously IV injected (10 minutes prior) with the αEpCAM mAb, Moc31, before the injection of αEpCAM/αCD3 Bispecific CSANs to perform a blocking study. Mice were sacrificed at 1, 6, 24 and 48 hours post injection (n =4). The blood, liver, spleen, pancreas, heart, kidney, lung, bone, brain, tail, intestines, skin and tumor were collected and weighed prior to measurement with a gamma ray counter. Decay correction was performed to allow for injected dose per gram of tissue calculation. The percent of CSANs bound to T cells or in free plasma was determined by isolation of the blood and incubation with excess CD4+ and CD8+ T cell positive isolation kits for 30 minutes at 4 degrees. The CD4+ and CD8+ captured T cells were removed by magnetic separation and the radioactive signal was calculated. The remaining blood sample was centrifugated to isolate the plasma. Non-CD4 and non-CD8 cells obtained in the cellular pellet were quantified to ensure the lack of radioactivity.
DHFR2 In Vivo Immunogenicity Analysis
Female BALB/c mice (7 weeks old), stock #01B05, were purchased from the National Cancer Institute (Frederick, MD, USA). Four mice per group were immunized intravenously with 30 µg (834.7 pmol) of CSANs in PBS in a total volume of 300 µl/animal. A booster injection with the same concentration and volume was administered on day 14. Blood samples were taken on day 0, 13, 21 and 30 by retro-orbital bleeding. Mice were sacrificed by cervical dislocation. Single cell suspension was prepared aseptically from the spleens. Mouse splenocytes were grown in RPMI media for FACS and in vitro activation assay. Levels of antibodies binding to DHFR were measured by enzyme-linked immunosorbent assay (ELISA). Briefly, Maxisorp 96-well plates (Thermo scientific) were coated first with DHFR2 (1 µg/well) in ELISA diluent (PBS containing 10% FBS) overnight at 4˚C. Following 1 h blocking with ELISA diluent, diluted sera (1:10) of immunized mice were transferred to the coated plates and incubated for 2 h at room temperature. Horseradish peroxidase-conjugated rabbit anti-mouse IgG-HRP (Sigma–Aldrich) diluted 1:80,000, or goat anti-mouse IgG1-HRP (Santa Cruz) diluted 1:4,000, or goat anti-mouse IgG2a-HRP (Santa Cruz) diluted 1:4,000, in the same diluent were incubated for 1 h at room temperature. The amount of bound peroxidase was visualized by incubation with tetramethylbenzidine and hydrogen peroxide (BD Bioscience). After 15 min, the reaction was stopped with 0.2 M H2SO4 and A450 was measured with a microplate reader (Biotek).
Statistical Analysis
All data analysis was analyzed using PRISM (v4.0; GraphPad Inc.). Data was analyzed using a 2-tailed, equal variance Student’s t test and ANOVA analysis with Bonferroni’s correction for multi group comparisons. Data acquired from in vitro assays using experimental replicates are presented as the mean ± SD and data acquired in vitro or in vivo using biological replicates are presented as the mean ± SEM. Data analyses were not blinded. Outliers were not excluded. Statistical significant threshold was set at P < 0.05.
Study Approval
All in vivo animal experiments were performed under a protocol approved by the University of Minnesota Institutional Animal Care and Use Committees in accordance with both federal and institutional regulations for humane treatment of animals. Human blood samples were obtained from the Memorial Blood Centers which maintains an IRB-approved protocol for use of deidentified human donor specimens.
Supplementary Material
Acknowledgments
We thank Dr. Daniel Vallera for use of the luciferized MCF-7 cell line. This work was supported by NCI R21-CA185627(CRW), NIH/NCI F30-CA210345 (CC), NIH R01-AI106791 (BTF), NIH U24-AI118635 (BTF), the Diabetes Research Foundation 3–2014-215 (JAS) and Tychon Bioscience, LLC. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.
Footnotes
Associated Content:
The supporting information is available on the ACS Publication Website at DOI:10.1021/acsnano.8b01308.
Characterization of cell types used in this manuscript, binding of CSANs, CSAN internalization evaluation by confocal microscopy, CSAN stability study on T-cell membranes, Moc31 competition demonstrating killing is EpCAM mediated, confirmation U87-MG cells are EpCAM negative, cell killing visualized by time lapse microscopy, PARs dose dependent cytokine production, regulatory T cell response to CSAN therapy, memory T cells are primary responders to CSAN therapy, CD69 and CD25 expressed on purified CD4+ and CD8+ t cells, CD4+ T cells exhibit delayed cell killing compared to CD8+ T cells, in vivo efficacy study of bispecific PARs with EpCAM negative U87-MG flank tumors, variable dosing schedule does not exhibit toxicity, CSAN binding profile in PBMC populations after 24 hours. additional donors and corresponding data.
References and Notes
- 1.Lee DW; Barrett DM; Mackall C; Orentas R; Grupp SA, The Future is Now: Chimeric Antigen Receptors as New Targeted Therapies for Childhood Cancer. Clin. Cancer. Res. 2012, 18, 2780–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran KJ; Pegram HJ; Brentjens RJ, Chimeric Antigen Receptors for T Cell Immunotherapy: Current Understanding and Future Directions. J. Gene Med. 2012, 14, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brentjens RJ; Davila ML; Riviere I; Park J; Wang X; Cowell LG; Bartido S; Stefanski J; Taylor C; Olszewska M; Borquez-Ojeda O; Qu J; Wasielewska T; He Q; Bernal Y; Rijo IV; Hedvat C; Kobos R; Curran K; Steinherz P; Jurcic J; Rosenblat T; Maslak P; Frattini M; Sadelain M, CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maus MV; Grupp SA; Porter DL; June CH, Antibody-Modified T Cells: CARs Take the Front Seat for Hematologic Malignancies. Blood 2014, 123, 2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett DM; Singh N; Porter DL; Grupp SA; June CH, Chimeric Antigen Receptor Therapy for Cancer. Annu. Rev. Med. 2014, 65, 333–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grupp SA; Kalos M; Barrett D; Aplenc R; Porter DL; Rheingold SR; Teachey DT; Chew A; Hauck B; Wright JF; Milone MC; Levine BL; June CH, Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DW; Gardner R; Porter DL; Louis CU; Ahmed N; Jensen M; Grupp SA; Mackall CL, Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL; Barrett D; Teachey DT; Grupp SA, Managing Cytokine Release Syndrome Associated with Novel T Cell-Engaging Therapies. Cancer J 2014, 20, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonifant CL; Jackson HJ; Brentjens RJ; Curran KJ, Toxicity and Management in CAR T-Cell Therapy. Mol. Ther. Oncolytics. 2016, 3, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davila ML; Riviere I; Wang X; Bartido S; Park J; Curran K; Chung SS; Stefanski J; Borquez-Ojeda O; Olszewska M; Qu J; Wasielewska T; He Q; Fink M; Shinglot H; Youssif M; Satter M; Wang Y; Hosey J; Quintanilla H; Halton E; Bernal Y; Bouhassira DC; Arcila ME; Gonen M; Roboz GJ; Maslak P; Douer D; Frattini MG; Giralt S; Sadelain M; Brentjens R, Efficacy and Toxicity Management of 19–28z CAR T Cell Therapy in B cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochenderfer JN; Wilson WH; Janik JE; Dudley ME; Stetler-Stevenson M; Feldman SA; Maric I; Raffeld M; Nathan DA; Lanier BJ; Morgan RA; Rosenberg SA, Eradication of B-lineage Cells and Regression of Lymphoma in a Patient Treated with Autologous T Cells Genetically Engineered to Recognize CD19. Blood 2010, 116, 4099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan RA; Yang JC; Kitano M; Dudley ME; Laurencot CM; Rosenberg SA, Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamers CH; Sleijfer S; van Steenbergen S; van Elzakker P; van Krimpen B; Groot C; Vulto A; den Bakker M; Oosterwijk E; Debets R; Gratama JW, Treatment of Metastatic Renal Cell Carcinoma with CAIX CAR-engineered T Cells: Clinical Evaluation and Management of On-Target Toxicity. Mol. Ther. 2013, 21, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhurst MR; Yang JC; Langan RC; Dudley ME; Nathan DA; Feldman SA; Davis JL; Morgan RA; Merino MJ; Sherry RM; Hughes MS; Kammula US; Phan GQ; Lim RM; Wank SA; Restifo NP; Robbins PF; Laurencot CM; Rosenberg SA, T Cells Targeting Carcinoembryonic Antigen can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Stasi A; Tey SK; Dotti G; Fujita Y; Kennedy-Nasser A; Martinez C; Straathof K; Liu E; Durett AG; Grilley B; Liu H; Cruz CR; Savoldo B; Gee AP; Schindler J; Krance RA; Heslop HE; Spencer DM; Rooney CM; Brenner MK, Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y; Moon E; Carpenito C; Paulos CM; Liu X; Brennan AL; Chew A; Carroll RG; Scholler J; Levine BL; Albelda SM; June CH, Multiple Injections of Electroporated Autologous T Cells Expressing a Chimeric Antigen Receptor Mediate Regression of Human Disseminated Tumor. Cancer Re.s 2010, 70, 9053–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CY; Roybal KT; Puchner EM; Onuffer J; Lim WA, Remote Control of Therapeutic T Cells Through a Small Molecule-Gated Chimeric Receptor. Science 2015, 350, aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma JS; Kim JY; Kazane SA; Choi SH; Yun HY; Kim MS; Rodgers DT; Pugh HM; Singer O; Sun SB; Fonslow BR; Kochenderfer JN; Wright TM; Schultz PG; Young TS; Kim CH; Cao Y, Versatile Strategy for Controlling the Specificity and Activity of Engineered T Cells. Proc. Natl. Acad. Sci. U S A. 2016, 113, E450–E458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner J; Schneider SM, Blinatumomab: A New Treatment for Adults With Relapsed Acute Lymphocytic Leukemia. Clin. J. Oncol. Nurs. 2016, 20, 165–168. [DOI] [PubMed] [Google Scholar]
- 20.Kontermann RE; Brinkmann U, Bispecific Antibodies. Drug. Discov. Today. 2015, 20, 838–847. [DOI] [PubMed] [Google Scholar]
- 21.Klinger M; Benjamin J; Kischel R; Stienen S; Zugmaier G, Harnessing T Cells to Fight Cancer with BiTE® Antibody Constructs-Past Developments and Future Directions. Immunol. Rev. 2016, 270, 193–208. [DOI] [PubMed] [Google Scholar]
- 22.Cochlovius B; Kipriyanov SM; Stassar MJ; Christ O; Schuhmacher J; Strauss G; Moldenhauer G; Little M, Treatment of Human B Cell Lymphoma Xenografts with a CD3 x CD19 Diabody and T Cells. J. Immunol. 2000, 165, 888–895. [DOI] [PubMed] [Google Scholar]
- 23.Stork R; Campigna E; Robert B; Müller D; Kontermann RE, Biodistribution of a Bispecific Single-Chain Diabody and its Half-Life Extended Derivatives. J. Biol. Chem. 2009, 284, 25612–25619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q; Hapka D; Chen H; Vallera DA; Wagner CR, Self-Assembly of Antibodies by Chemical Induction. Angew. Chem. Int. Ed. Engl. 2008, 47, 10179–10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q; So CR; Fegan A; Cody V; Sarikaya M; Vallera DA; Wagner CR, Chemically Self-Assembled Antibody Nanorings (CSANs): Design and Characterization of an anti-CD3 IgM Biomimetic. J. Am. Chem. Soc. 2010, 132, 17247–17257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen JJ; Vallera DA; Wagner CR, Prosthetic Antigen Receptors. J. Amer. Chem. Soc. 2015, 137, 10108–10111. [DOI] [PubMed] [Google Scholar]
- 27.Fegan A; Kumarapperuma SC; Wagner CR, Chemically Self-Assembled Antibody Nanostructures as Potential Drug Carriers. Mol. Pharm. 2012, 9, 3218–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabrielse K; Gangar A; Kumar N; Lee JC; Fegan A; Shen JJ; Li Q; Vallera D; Wagner CR, Reversible Re-Programing of Cell-Cell Interactions. Angew. Chem. Int. Ed. Engl. 2014, 53, 5112–5116. [DOI] [PubMed] [Google Scholar]
- 29.Gangar A; Fegan A; Kumarapperuma SC; Wagner CR, Programmable Self-Assembly of Antibody-Oligonucleotide Conjugates as Small Molecule and Protein Carriers. J. Am. Chem. Soc. 2012, 134, 2895–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah R; Petersburg J; Gangar AC; Fegan A; Wagner CR; Kumarapperuma SC, In Vivo Evaluation of Site-Specifically PEGylated Chemically Self-Assembled Protein Nanostructures. Mol. Pharm. 2016, 13, 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson JC; Jena SS; Flenniken M; Chou TF; Siegel RA; Wagner CR, Chemically Controlled Self-Assembly of Protein Nanorings. J. Am. Chem. Soc. 2006, 128, 7630–7638. [DOI] [PubMed] [Google Scholar]
- 32.Fiedler W; Honemann D; Ritter B; Bokemeyer C; Fettes P; Klinger M; Reinhardt C; Zugmaier G; Kaubitzsch S; Wolf M, Safety and Pharmacology of the EpCAM/CD3-Bispecific BiTE Antibody MT110 in Patients with Metastatic Colorectal, Gastric or Lung Cancer. Ejc. Supplements. 2009, 7, 136–137. [Google Scholar]
- 33.Fiedler W; Wolf M; Kebenko M; Goebeler M; Ritter B; Quaas A; Vieser E; Hijazi Y; Patzak I; Friedrich M; Kufer P; Frankel S; Seggewiss-Bernhardt R; Kaubitzsch S, A Phase I Study of EpCAM/CD3-Bispecific Antibody (MT110) in Patients with Advanced Solid Tumors. J. of. Clin. Onc. 2012, 30. [Google Scholar]
- 34.Mau-Sorensen M; Dittrich C; Dienstmann R; Lassen U; Buchler W; Martinius H; Tabernero J, A Phase I Trial of Intravenous Catumaxomab: a Bispecific Monoclonal Antibody Targeting EpCAM and the T cell Coreceptor CD3. Cancer Chemotherapy and Pharmacology 2015, 75, 1065–1073. [DOI] [PubMed] [Google Scholar]
- 35.Balzar M; Winter MJ; de Boer CJ; Litvinov SV, The Biology of the 17–1A Antigen (Ep-CAM). J. Mol. Med. (Berl). 1999, 77, 699–712. [DOI] [PubMed] [Google Scholar]
- 36.El-Sahwi K; Bellone S; Cocco E; Casagrande F; Bellone M; Abu-Khalaf M; Buza N; Tavassoli FA; Hui P; Rüttinger D; Silasi DA; Azodi M; Schwartz PE; Rutherford TJ; Pecorelli S; Santin AD, Overexpression of EpCAM in Uterine Serous Papillary Carcinoma: Implications for EpCAM-Specific Immunotherapy with Human Monoclonal Antibody Adecatumumab (MT201). Mol. Cancer. Ther. 2010, 9, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stefan N; Martin-Killias P; Wyss-Stoeckle S; Honegger A; Zangemeister-Wittke U; Plückthun A, DARPins Recognizing the Tumor-Associated Antigen EpCAM Selected by Phage and Ribosome Display and Engineered for Multivalency. J. Mol. Biol. 2011, 413, 826–843. [DOI] [PubMed] [Google Scholar]
- 38.Jahchan NS; Lim JS; Bola B; Morris K; Seitz G; Tran KQ; Xu L; Trapani F; Morrow CJ; Cristea S; Coles GL; Yang D; Vaka D; Kareta MS; George J; Mazur PK; Nguyen T; Anderson WC; Dylla SJ; Blackhall F; Peifer M; Dive C; Sage J, Identification and Targeting of Long-Term Tumor-Propagating Cells in Small Cell Lung Cancer. Cell. Rep. 2016, 16, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gostner JM; Fong D; Wrulich OA; Lehne F; Zitt M; Hermann M; Krobitsch S; Martowicz A; Gastl G; Spizzo G, Effects of EpCAM Overexpression on Human Breast Cancer Cell Lines. BMC. Cancer. 2011, 11, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winkler J; Martin-Killias P; Plückthun A; Zangemeister-Wittke U, EpCAM-Targeted Delivery of Nanocomplexed siRNA to Tumor Cells with Designed Ankyrin Repeat Proteins. Mol. Cancer. Ther. 2009, 8, 2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linke R; Klein A; Seimetz D, Catumaxomab: Clinical Development and Future Directions. MAbs. 2010, 2, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borlak J; Langer F; Spanel R; Schondorfer G; Dittrich C, Immune-Mediated Liver Injury of the Cancer Therapeutic Antibody Catumaxomab Targeting EpCAM, CD3 and Fc Gamma Receptors. Oncotarget. 2016, 7, 28059–28074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baeuerle PA; Reinhardt C, Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer. Res. 2009, 69, 4941–4944. [DOI] [PubMed] [Google Scholar]
- 44.Cioffi M; Dorado J; Baeuerle PA; Heeschen C, EpCAM/CD3-Bispecific T-cell Engaging Antibody MT110 Eliminates Primary Human Pancreatic Cancer Stem Cells. Clin. Cancer. Res. 2012, 18, 465–474. [DOI] [PubMed] [Google Scholar]
- 45.Haas C; Krinner E; Brischwein K; Hoffmann P; Lutterbüse R; Schlereth B; Kufer P; Baeuerle PA, Mode of Cytotoxic Action of T Cell-Engaging BiTE Antibody MT110. Immunobiology. 2009, 214, 441–453. [DOI] [PubMed] [Google Scholar]
- 46.Ang WX; Li ZD; Chi ZX; Du SH; Chen C; Tay JCK; Toh HC; Connolly JE; Xu XH; Wang S, Intraperitoneal Immunotherapy with T Cells Stably and Transiently Expressing Anti-EpCAM CAR in Xenograft Models of Peritoneal Carcinomatosis. Oncotarget. 2017, 8, 13545–13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwab R; Crow MK; Russo C; Weksler ME, Requirements for T Cell Activation by OKT3 Monoclonal Antibody: Role of Modulation of T3 Molecules and Interleukin 1. J. Immunol. 1985, 135, 1714–1718. [PubMed] [Google Scholar]
- 48.Van Wauwe JP; Goossens JG; Beverley PC, Human T Lymphocyte Activation by Monoclonal Antibodies; OKT3, but Not UCHT1, Triggers Mitogenesis via an Interleukin 2-Dependent Mechanism. J. Immunol. 1984, 133, 129–132. [PubMed] [Google Scholar]
- 49.Brischwein K; Schlereth B; Guller B; Steiger C; Wolf A; Lutterbuese R; Offner S; Locher M; Urbig T; Raum T; Kleindienst P; Wimberger P; Kimmig R; Fichtner I; Kufer P; Hofmeister R; da Silva AJ; Baeuerle PA, MT110: A Novel Bispecific Single-Chain Antibody Construct with High Efficacy in Eradicating Established Tumors. Mol. Immunol. 2006, 43, 1129–1143. [DOI] [PubMed] [Google Scholar]
- 50.Baitsch L; Baumgaertner P; Devêvre E; Raghav SK; Legat A; Barba L; Wieckowski S; Bouzourene H; Deplancke B; Romero P; Rufer N; Speiser DE, Exhaustion of Tumor-Specific CD8⁺ T Cells in Metastases from Melanoma Patients. J. Clin. Invest. 2011, 121, 2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao E; Wang L; Dai J; Kryczek I; Wei S; Vatan L; Altuwaijri S; Sparwasser T; Wang G; Keller ET; Zou W, Regulatory T Cells in the Bone Marrow Microenvironment in Patients with Prostate Cancer. Oncoimmunology. 2012, 1, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drake CG; Jaffee E; Pardoll DM, Mechanisms of Immune Evasion by Tumors. Adv. Immunol. 2006, 90, 51–81. [DOI] [PubMed] [Google Scholar]
- 53.Sakaguchi S, Naturally Arising CD4+ Regulatory T Cells for Immunologic Self-Tolerance and Negative Control of Immune Responses. Annu. Rev. Immunol. 2004, 22, 531–562. [DOI] [PubMed] [Google Scholar]
- 54.Nizar S; Copier J; Meyer B; Bodman-Smith M; Galustian C; Kumar D; Dalgleish A, T-Regulatory Cell Modulation: The Future of Cancer Immunotherapy? Br. J. Cancer. 2009, 100, 1697–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zou W, Regulatory T cells, Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [DOI] [PubMed] [Google Scholar]
- 56.Valzasina B; Piconese S; Guiducci C; Colombo MP, Tumor-Induced Expansion of Regulatory T Cells by Conversion of CD4+CD25- Lymphocytes is Thymus and Proliferation Independent. Cancer. Res. 2006, 66, 4488–4495. [DOI] [PubMed] [Google Scholar]
- 57.Zheng Y; Collins SL; Lutz MA; Allen AN; Kole TP; Zarek PE; Powell JD, A Role for Mammalian Target of Rapamycin in Regulating T Cell Activation Versus Anergy. J. Immunol. 2007, 178, 2163–2170. [DOI] [PubMed] [Google Scholar]
- 58.Zheng Y; Delgoffe GM; Meyer CF; Chan W; Powell JD, Anergic T Cells are Metabolically Anergic. J. Immunol. 2009, 183, 6095–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarbassov DD; Ali SM; Sabatini DM, Growing Roles for the mTOR Pathway. Curr. Opin. Cell. Biol. 2005, 17, 596–603. [DOI] [PubMed] [Google Scholar]
- 60.Salmond RJ; Emery J; Okkenhaug K; Zamoyska R, MAPK, Phosphatidylinositol 3-kinase, and Mammalian Target of Rapamycin Pathways Converge at the Level of Ribosomal Protein S6 Phosphorylation to Control Metabolic Signaling in CD8 T cells. J. Immunol. 2009, 183, 7388–7397. [DOI] [PubMed] [Google Scholar]
- 61.Rosenberg SA; Restifo NP; Yang JC; Morgan RA; Dudley ME, Adoptive Cell Transfer: a Clinical Path to Effective Cancer Immunotherapy. Nat. Rev. Cancer. 2008, 8, 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Terakura S; Yamamoto TN; Gardner RA; Turtle CJ; Jensen MC; Riddell SR, Generation of CD19-Chimeric Antigen Receptor Modified CD8+ T Cells Derived from Virus-Specific Central Memory T Cells. Blood. 2012, 119, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sauerborn M; Brinks V; Jiskoot W; Schellekens H, Immunological Mechanism Underlying the Immune Response to Recombinant Human Protein Therapeutics. Trends. Pharmacol. Sci. 2010, 31, 53–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.