

Summary
Based on their lobule location, hepatocytes display differential gene expression, including pericentral hepatocytes that surround the central vein, which are marked by Wnt-β-catenin signaling. Activating β-catenin mutations occur in a variety of liver tumors, including hepatocellular carcinoma (HCC), but no specific therapies are available to treat these tumor subsets. Here, we identify a positive relationship between β-catenin activation, its transcriptional target glutamine synthetase (GS) and p-mTOR-S2448, an indicator of mTORC1 activation. In normal livers of mice and humans pericentral hepatocytes were simultaneously GS- and p-mTOR-S2448-positive, as were β-catenin-mutated liver tumors. Genetic disruption of β-catenin signaling or GS prevented p-mTOR-S2448 expression, while their forced expression in β-catenin-deficient livers led to ectopic p-mTOR-S2448 expression. Further we found notable therapeutic benefit of mTORC1 inhibition in mutant-β-catenin-driven HCC through suppression of cell proliferation and survival. Thus, mTORC1 inhibitors could be highly relevant in the treatment of liver tumors that are β-catenin-mutated and GS-positive.
One Sentence Summary:
Wnt-β-catenin-GS axis and mTORC1 activation in HCC
Graphical Abstract
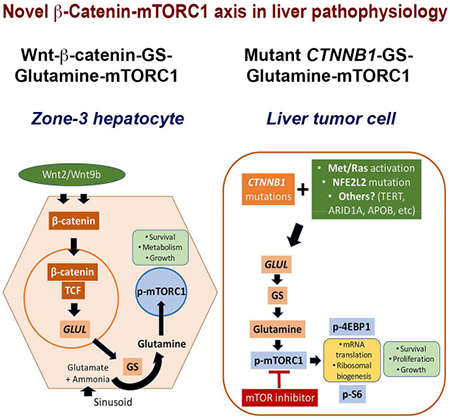
eTOC Blurb
Michael, Ko et al show that β-catenin activation in zone 3 hepatocytes leads to high mTORC1 activity downstream of elevated glutamine synthetase expression and intracellular glutamine. Due to the same reason, liver tumors harboring mutated, hyperactive β-catenin also show mTORC1 activation, making them susceptible to mTOR inhibitors.
Introduction
The relevance of Wnt signaling in development and tissue homeostasis is well appreciated (Steinhart and Angers, 2018). From its fundamental contributions in gastrulation to more specialized roles in organogenesis, and homeostasis in adult tissues via stem cell renewal in organs such as skin and gut, the Wnt pathway is indispensable to normal growth and development. β-Catenin, the chief downstream effector of canonical Wnt signaling, acts as a co-factor for the T cell factor family of transcription factors to regulate tissue-specific target gene expression (Clevers and Nusse, 2012). It is through such targets that the Wnt-β-catenin signaling contributes to specific biological functions such as cell proliferation, survival, migration and others to eventually regulate tissue regeneration and homeostasis. However, aberrations in the various components of the pathway can lead to incessant signaling, anomalous gene expression, dysregulated growth and ultimately neoplasia (Nusse and Clevers, 2017).
The Wnt-β-catenin signaling has also been shown to regulate key biological functions innate to the liver including regeneration, development and metabolic zonation (Monga, 2015; Russell and Monga, 2018). Histologically, an adult liver is divided into hepatic lobules. Hepatocytes are organized within a lobule along sinusoids, which carry blood from the portal vein and hepatic artery to the central vein. The hepatocytes are partitioned into three metabolic zones based on their function and location within the lobule. The Wnt-β-catenin pathway is active in the pericentral or zone-3 hepatocytes owing to both the continuous Wnt2 and Wnt9b expression in the endothelial cells lining central veins, and high levels of adenomatous polyposis coli gene product (APC), an inhibitor of Wnt pathway, in the periportal (zone-1) and midzonal (zone-2) hepatocytes (Benhamouche et al., 2006; Wang et al., 2015). Active β-catenin in zone-3 hepatocytes regulates expression of tissue-specific target genes encoding for glutamine synthetase (GS) and others (Sekine et al., 2006; Tan et al., 2006). Glutamine metabolism is a well-known function of the Wnt-β-catenin pathway (Cadoret et al., 2002). Stabilizing missense mutations or deletions in CTNNB1, the gene encoding β-catenin, are observed in notable subsets of hepatocellular carcinomas (HCCs), hepatoblastomas (HBs) and hepatocellular adenomas (HCAs) (Monga, 2015). Such tumors harboring CTNNB1 mutations, are uniformly positive for GS, which has been touted as their biomarker (Cieply et al., 2009; Zucman-Rossi et al., 2007). The exact mechanism by which β-catenin activation contributes to liver tumors remains unknown
In our current study, we identify a novel cell-intrinsic regulation of mTORC1 by the Wnt-β-catenin pathway. Using multiple genetic mouse models, we identify presence of phospho-mTOR-Serine2448 (p-mTOR-S2448), an indicator of active mTORC1, in zone-3 hepatocytes as a function of GS and high intracellular glutamine (Gebhardt and Coffer, 2013), which can directly phosphorylate mTOR in lysosomes (Jewell et al., 2015). We show several hepatic tumors with active β-catenin and high GS levels, to be simultaneously positive for p-mTOR-S2448. Using previously published clinically relevant HCC models (Patil et al., 2009; Tao et al., 2016; Tao et al., 2017), we demonstrate addiction of β-catenin mutated HCCs to mTOR thus identifying a novel therapeutic strategy to disrupt tumor metabolism and combat β-catenin-mutated hepatic tumors with existing approved pharmacological agents.
Results
Increased expression of GS and active mTORC1 in hepatic tumors harboring β-catenin gene mutations
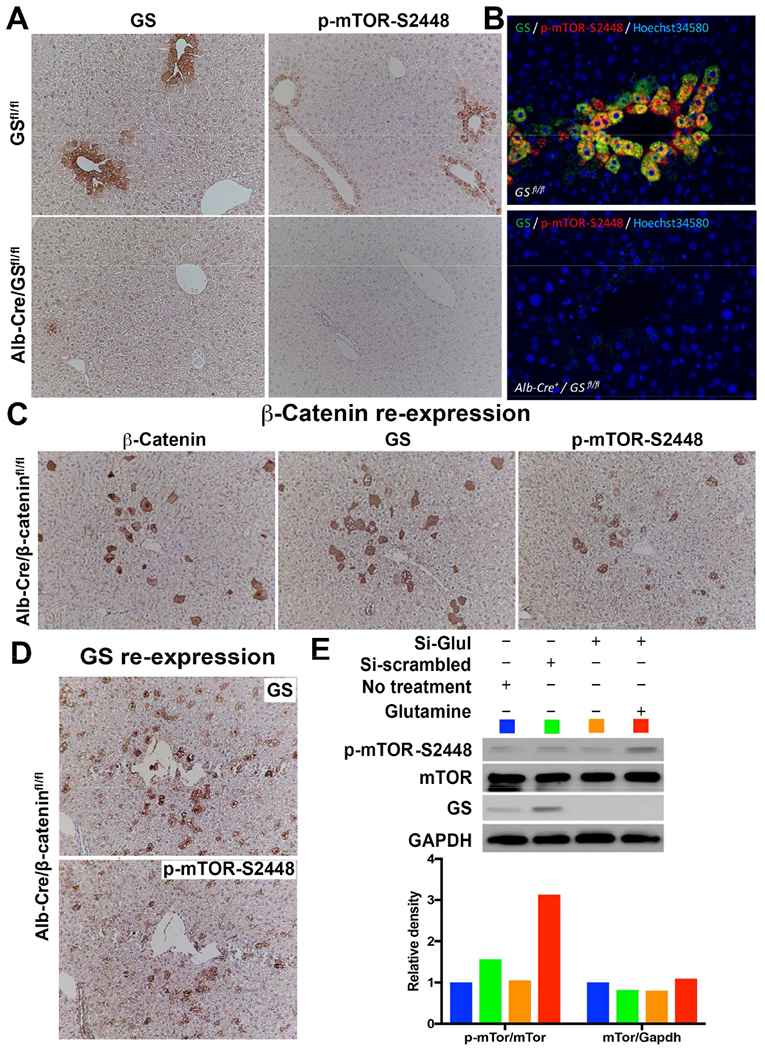
In previous studies, we observed that mice harboring mutant β-catenin (S45Y, S33Y, Δ90), which lead to β-catenin activation, and displaying c-Met co-expression (Met-β-catenin model) using sleeping beauty transposon/transposase and hydrodynamic tail vein injection (SB-HTVI) led to HCC (Patil et al., 2009; Tao et al., 2016; Tao et al., 2017). The HCC in these models showed clear evidence of mTORC1 activation. In a similar HCC mouse model, which was driven by the combination of S45Y- or S33Y-β-catenin and Ras activation downstream of c-Met (Ras-β-catenin model), suppression of β-catenin led to a complete response, which was associated with decreased levels of p-mTOR-S2448 (Tao et al., 2016; Tao et al., 2017). To verify this relationship between β-catenin and mTOR, we examined serial sections from various preclinical models of hepatic tumors including HCC and HB, for GS and p-mTOR-S2448 by immunohistochemistry (IHC). All HCCs occurring in the Met-β-catenin and Ras-β-catenin model that were GS-positive were also positive for p-mTOR-S2448 (Fig. 1A). In these models, occasionally, cholangiocarcinomas (CCAs) are also observed (Tao et al., 2016; Tao et al., 2017), but CCA did not show such concordance in GS and p-mTOR-S2448 by IHC (Fig. 1A). Since activating mutations in NFE2L2, which encodes for Nrf2, can also co-occur with CTNNB1 mutations in a HCC subset (Schulze et al., 2015), we also co-expressed G31A-NFE2L2 and T41A-β-catenin in murine livers which led to HCC. HCC in the Nrf2-β-catenin model were also positive for GS and p-mTOR-S2448 by IHC (Fig. 1A). Co-expression of an active mutant-Yap1 (S127A) and Δ90-β-catenin leads to development of HB (Tao et al., 2014). Co-expression of S45Y-S33Y-β-catenin and S127A-Yap (Yap-β-catenin model) also led to HB and these tumors were simultaneously positive for GS and p-mTOR-S2448 by IHC (Fig. 1A). Lastly, we examined Met-WT-β-catenin model where HCC occurs by co-expressing non-mutant wild-type β-catenin (WT-β-catenin) along with c-Met. IHC showed most HCCs in this model to be GS-negative. Notably, these tumors were also negative for p-mTOR-S2448 (Fig. 1B).
Figure 1. Mouse models of HCC with CTNNB1 mutations display simultaneous positivity for GS and p-mTOR-S2448.
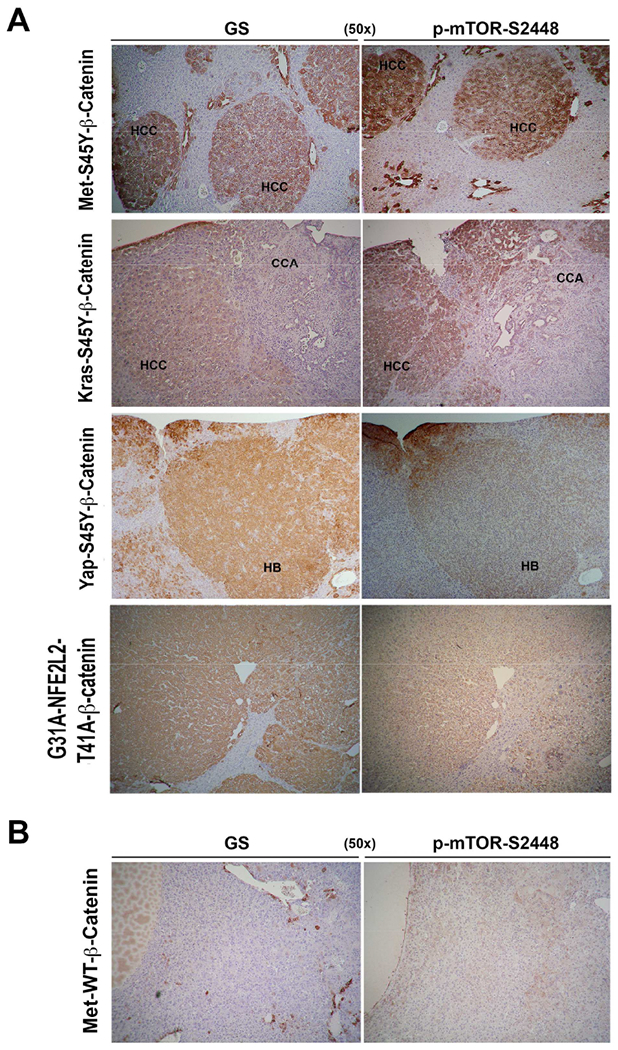
A. IHC on serial liver sections shows same tumor foci to be positive for GS and p-mTOR-S2448 in the Met-β-catenin, Kras-β-catenin and Nrf2-β-catenin models (50×). Cholangiocarcinoma (CCA) occurring in Kras-β-catenin model are negative. IHC on serial sections also shows hepatoblastoma (HB) to be positive for GS and p-mTOR-S2448 in Yap-β-catenin model (50×).
B. IHC on serial liver sections shows same tumor foci to be negative for GS and p-mTOR-S2448 in the Met-WT-β-catenin HCC model generated by co-expressing wild-type non-mutant β-catenin and c-Met (50×).
Next, we determined if a correlation existed between GS and mTORC1 activation in clinical samples. We assessed GS and p-mTOR-S2448 levels in an array of liver tumors. We first assessed HCAs, a notable subset of whom carry CTNNB1 mutations, which can be identified by increased GS expression (Zucman-Rossi et al., 2006). Of the 16 HCAs, all 8 tumors harboring diverse but activating CTNNB1 mutations located in exon-3, showed 6-45-fold increase in the mRNA expression of GLUL, gene encoding GS, as compared to normal adjacent livers for available HCA (n=5) or HCAs with non-mutated CTNNB1 (Fig.2A). All tumors with increased GS expression, showed notable increase in p-mTOR-S2448 by Western blots (WBs) (Fig. 2A). Upon quantification, a significant increase (p=0.0002) in the relative levels of p-mTOR-S2448 were evident in the CTNNB1-mutated HCAs as compared to other groups (Fig.2B)
Figure 2: Human liver tumors with CTNNB1 mutations and/or GS upregulation show significant increase in p-mTOR-S2448.
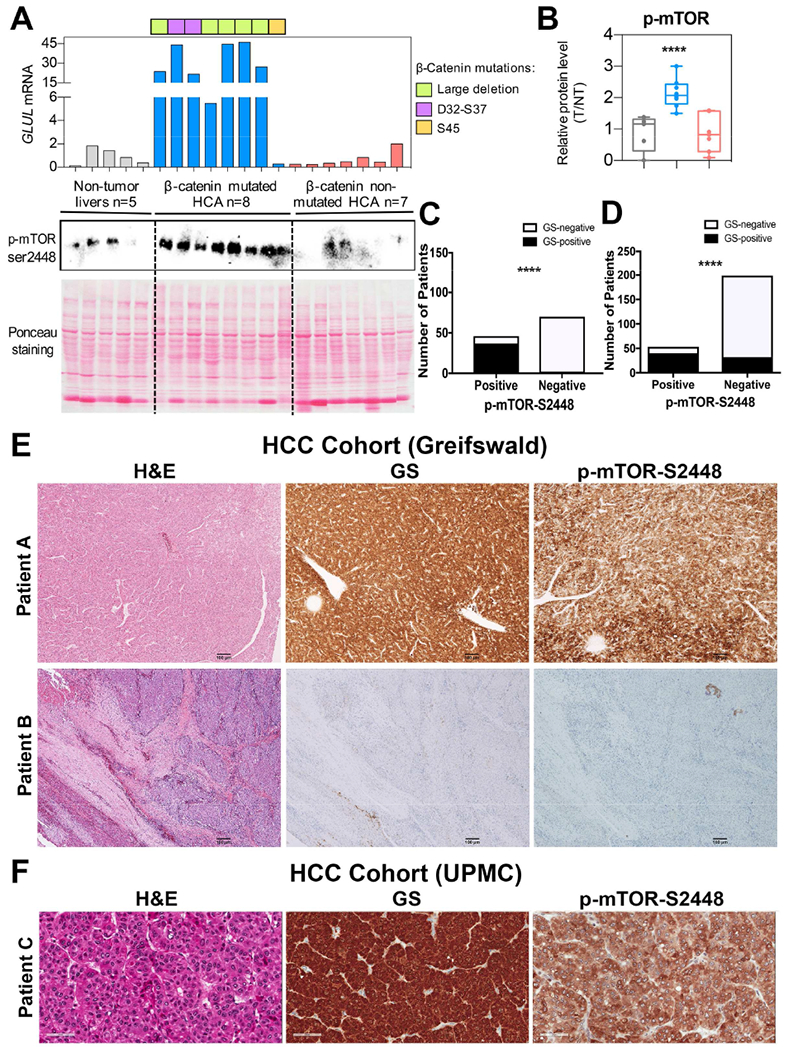
A. Levels of p-mTOR-S2448 were dramatically increased in CTNNB1-mutated HCA (blue) as compared to adjacent non-tumor livers (gray) and CTNNB1 non-mutated HCA (red) by WB. Ponceau staining confirmed comparable protein loading. GLUL mRNA expression was assessed by qRT-PCR and showed increased expression in CTNNB1-mutated HCA with different mutations/deletions in CTNNB1 noted with different colors.
B. For each sample, expression level of each protein was quantified using Image Lab software (Bio-Rad). Kruskall-Wallis and Mann-Whitney test were used to assess differences between groups and showed significant increase in p-mTOR-S2448 in CTNNB1-mutated HCA as compared to other groups (****p=0.0002).
C. 32% (n=37) of all HCC cases (n=116) at the University of Greifswald cohort were simultaneously positive for GS and p-mTOR-S2448. Bar graph representing Fisher’s exact test showed a significant correlation between GS and p-mTOR-S2448 staining in these samples (****p=2.34E-19; 2-sided test).
D. 16% (n=40) of the 252 usable cases represented on 6 TMAs representing the UPMC cohort, were simultaneously positive for GS and p-mTOR-S2448 while 169 cases were negative for both these markers. Fisher’s exact test showed a significant correlation between GS and p-mTOR-S2448 (****p=4.26E-17, 2-sided test).
E. Representative IHC of HCC samples from the University of Greifswald cohort showing simultaneous positivity (Patient A) or negativity (Patient B) for GS and p-mTOR-S2448 (50×).
F. Representative IHC of HCC samples from the UPMC cohort TMA showing simultaneous positivity (Patient C) for GS and p-mTOR-S2448 (100×).
See also Figures S1, S2, S3 and S7 and Tables S1, S2 and S3.
We next tested 55 HB cases on 3 tissue microarrays (TMAs) (Table S1 and Fig.S1). Since each HB can have multiple histological components (fetal, crowded fetal, embryonal, others), a total of 113 components were identified in the 55 cases (Table S1). These numbers represented good quality tissues for which data was obtainable and interpretable for both GS and p-mTOR-S2448 by IHC by a qualified pathologist, and scored as described in Methods. In the component-wide analysis, of the 113 total components, 36 (~32%) were simultaneously positive for both markers and 37 (~33%) were negative for both (Fig.S2A). Fisher’s exact test showed a significant correlation between GS and p-mTOR-S2448 (***p=4.52E-4, 2-sided test) (Fig.S2A). Restricting analysis only to the epithelial components within HB (n=95), the correlation by Fisher’s exact test was also highly significant (***p=2.36E-4, 2-sided test), with 36 of 95 cases being simultaneously positive and 25 cases being simultaneously negative for both markers (Fig.S2B). Representative images from IHC are included (Fig.S2C).
Lastly, we tested two major cohorts of HCC. The first analysis was performed on full liver sections available from 116 HCC cases at the University of Greifswald, Germany (Table S2). Status of CTNNB1 mutations was known in these cases and 28 of the 116 cases (24.1%) displayed missense mutations in the exon-3 of CTNNB1 (Table S2). Twenty-seven of these 28 (96.4%) cases showed strong GS staining while 1 sample showed none. Additional 12 of 116 cases (10.3%) were GS-positive despite absence of obvious CTNNB1 mutations. Overall, 39 of 116 HCC cases were strongly GS-positive (33.6%) and 77 were GS-negative (66.4%). Notably, 37 of 39 GS-positive cases (94.9%) showed strong staining for p-mTOR-S2448, while 2 (5.1%) were negative.
Additional 9 cases were p-mTOR-S2448-positive despite being GS-negative, while 68 of 116 cases (58.6%) were negative for both. Overall, around 32% of HCCs were simultaneously GS and p-mTOR-S2448 positive. Fisher’s exact test showed a significant correlation between GS and p-mTOR-S2448 (p=2.34E-19, 2-sided test) (Fig.2C). We also assessed correlation between CTNNB1 mutations and IHC for GS or p-mTOR-S2448. Of the 116 cases, 27 with CTNNB1 mutations were GS-positive and 1 was negative, while 76 of the non-mutated were also GS-negative (p=5.05E-16, 2-sided test) (Fig.S3A). Likewise, 27 cases with CTNNB1 mutations were p-mTOR-S2448-positive and only 1 was negative, while 69 of the non-mutated were also p-mTOR-S2448-negative (p=4.89E-13, 2-sided test) (Fig.S3B). Six additional TMAs representing 252 usable HCC cases from the University of Pittsburgh Medical Center (UPMC), Pittsburgh were also assessed for GS and p-mTOR-S2448 by IHC (Table S3 and Fig.S3C). We were aware of the caveat that TMAs represent only a small core of the HCC, unlike a larger tissue section, and hence may not be as sensitive in detecting concordance between stains. No genomic data on CTNNB1 mutations was available for these cases. Of the 252 cases, 72 were GS-positive (~29%) of which 40 were simultaneously positive for p-mTOR-S2448 (55.5%) while 32 were negative (44.4%). Additionally, 11 cases that were GS-negative were p-mTOR-S2448-positive. Overall 169 cases were negative for both these markers (67.1%). Fisher’s exact test showed a significant correlation between GS and p-mTOR-S2448 (p=4.26E-17, 2-sided test) (Fig.2D). Representative images of tumors stained for GS and p-mTOR-S2448 from the German HCC cohort (Fig.2E) and UPMC HCC cohort (Fig.2F) are included.
P-mTOR-S2448 is zonated in adult baseline livers and localizes to pericentral hepatocytes
IHC for GS and p-mTOR-S2448 was next performed on normal human and mouse livers to investigate any relationship. As expected, in normal human livers, GS was localized to zone-3 hepatocytes (Cadoret et al., 2002) (Fig.3A). Intriguingly, p-mTOR-S2448 was localized to the same hepatocytes as well (Fig.3A). Normal murine livers showed exactly the same localization for both these proteins in zone-3 (Fig.3A).
Figure 3: Pericentral expression of GS and p-mTOR-S2448 in normal human and mouse liver is β-catenin-dependent.
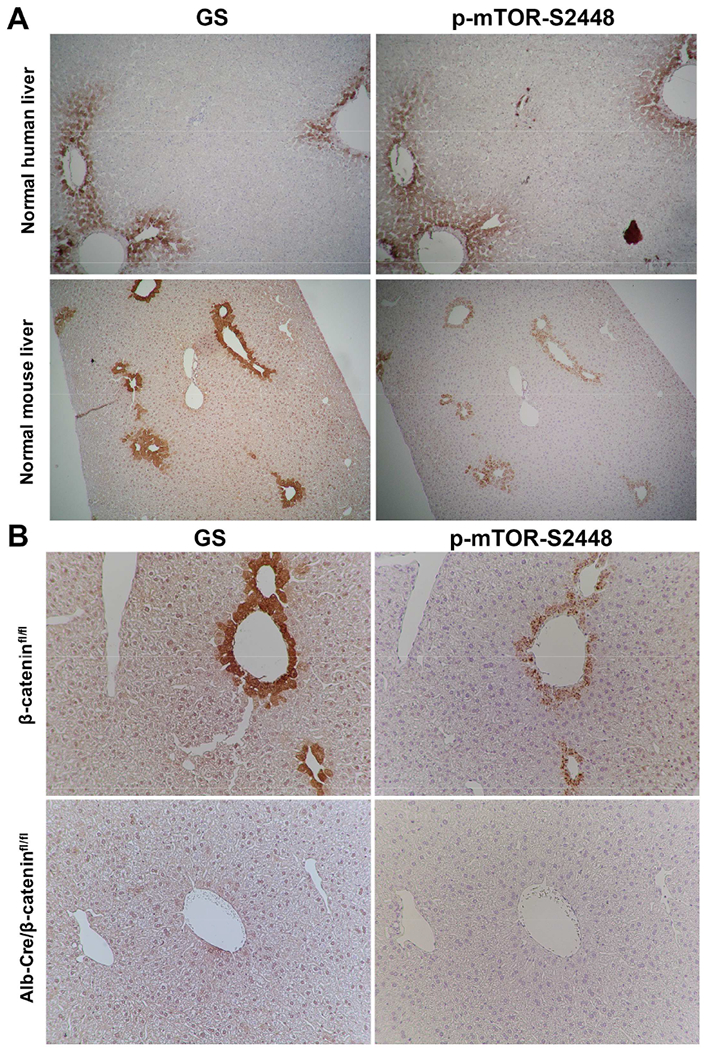
A. Serial sections from a normal human (top) and mouse (bottom) liver stained for GS and p-mTOR-S2448 show positive staining for both only in the zone 3 hepatocytes (50×).
B. Representative IHC shows staining in zone-3 hepatocytes of GS and p-mTOR-S2448 in β-cateninfl/fl mice (top), which was absent in Alb-Cre/β-cateninfl/fl (bottom) (100×).
See also Figures S4 and S7
Conditional disruption of Wnt-β-catenin signaling in vivo or in vitro, disrupts pericentral localization or levels of p-mTOR-S2448, respectively
Wnt-β-catenin signaling regulates pericentral gene expression in an adult liver (Benhamouche et al., 2006). This localized regulation is due to basal Wnt2 and Wnt9b release from the central venous endothelial cells (Preziosi et al., 2018; Wang et al., 2016), which act in a paracrine manner via the redundant Wnt-co-receptors LRP5/6 on hepatocytes (Yang et al., 2014) to activate β-catenin-TCF signaling and regulate target gene expression including Glul (Sekine et al., 2006; Tan et al., 2006). To determine if Wnt-β-catenin signaling regulated p-mTOR-S2448 in adult liver, we assessed its localization in the various genetic knockout mice exhibiting disruption of the Wnt-β-catenin axis in the liver. Liver-specific β-catenin KO (Alb-Cre/β-cateninfl/fl) mice as expected lacked GS in zone-3 hepatocytes (Sekine et al., 2006; Tan et al., 2006) (Fig.3B). Intriguingly, these livers showed absence of p-mTOR-S2448 (Fig.3B). Downstream of mTORC1, we observed decreases in both p-4E-BP1-Threonine37/46 (p-4E-BP1-T37/46) and pS6-S240/244 in whole cell lysates derived from KO livers when compared to controls (not shown).
To validate in vivo findings, we cultured Hep3B cells, a human hepatoma cell line, in the presence of pre-validated siRNA targeting the mRNA of CTNNB1 or a scrambled sequence for 48h and assessed the cells for Wnt signaling and mTORC1 signaling. Knockdown of β-catenin led to decreased GS, and simultaneously, decreased p-mTOR-S2448, p-4E-BP1-T37/46 and pS6-S240/244 by WBs (Fig.S4A).
Liver-specific Wnt co-receptor LRP5-6 double KO (Alb-Cre/LRP5/6fl/fl) also lacked GS as expected (Yang et al., 2014), and these livers also showed absence of p-mTOR-S2448 (Fig.S4B). Like β-catenin KO, the liver lysates from LRP5/6 KO also showed decreased mTORC1 signaling as reflected by decreased p-4E-BP1-T37/46 and pS6-S240/244 levels (not shown).
Recently, we showed Lyve1-cre driven Wntless KO, which are incapable of Wnt secretion from endothelial cells including those lining central veins, also lacked pericentral hepatocyte β-catenin activation (Preziosi et al., 2018). By IHC, livers from these KOs showed not only absence of GS, but also loss of p-mTOR-S2448 (Fig.S4C).
Conditional deletion of GS disrupts p-mTOR-S2448 localization without affecting Wnt-β-catenin signaling
To investigate the basis of p-mTOR-S2448 loss following disruption of Wnt-β-catenin signaling in the liver, we first assessed levels of total mTOR in β-catenin KO and LRP5-6 KO livers. No changes in total mTOR levels were observed by WB, suggesting it to not be a Wnt-β-catenin transcriptional target (Fig.S4D).
Recently, free amino acids such as glutamine were shown to induce lysosomal activation of mTORC1 through direct phosphorylation of mTOR at S2448 in a Rag GTPase-independent but v-ATPase- and adenosine diphosphate ribosylation factor-1 GTPase-dependent manner (Jewell et al., 2015). Since GS catalyzes the condensation of glutamate with ammonia to yield glutamine in an ATP-dependent manner and hence rids remnant ammonia from sinusoidal blood before it leaves liver (Liaw et al., 1995), the highest intracellular levels of glutamine in the liver are observed in the zone-3 hepatocytes (Gebhardt and Coffer, 2013). Based on these observations, we next investigated if baseline p-mTOR-S2448 in zone-3 hepatocytes is a direct consequence of the presence of GS rather than of Wnt-β-catenin signaling. Livers from Alb-Cre/GSfl/fl mice (Qvartskhava et al., 2015), showed complete loss of GS in zone-3 hepatocytes (Fig.4A). Intriguingly, there was a complete absence of p-mTOR-S2448 in pericentral hepatocytes in these livers (Fig.4A). This was verified by double immunofluorescence for GS and p-mTOR-S2448, which co-localized in littermate controls but was absent in Alb-Cre/GSfl/fl livers (Fig.4B;Fig.S5A,B). The expression of β-catenin targets cyp2e1 and cyp1a2 (Loeppen et al., 2005; Sekine et al., 2006; Tan et al., 2006) were unaffected in the Alb-Cre/GSfl/fl livers indicating an intact Wnt-β-catenin signaling (Fig.S5C).
Figure 4: Pericentral expression of p-mTOR-S2448 is the function of GS and in turn Glutamine, downstream of the Wnt-β-catenin pathway.
A. Conditional deletion of Glul in Alb-Cre/GSfl/fl mice leads to loss of GS and p-mTOR-S2448 in zone-3 hepatocytes (bottom), which was intact in GSfl/fl littermates (top) (100×).
B. Double immunofluorescence validates colocalization of GS and p-mTOR-S2448 in the zone-3 hepatocytes in GSfl/fl mice (top), while no staining for either was seen in the liver sections from Alb-Cre/GSfl/fl mice (bottom) (200×).
C. IHC on serial sections from livers of Alb-Cre/β-cateninfl/fl mice following forced expression of S45Y-CTNNB1 by SB-HTVI in a subset of zone-3 hepatocytes shows ectopic expression of β-catenin, GS and p-mTOR-S2448 in the same hepatocytes (100×).
D. IHC on serial sections from livers of Alb-Cre/GSfl/fl mice following forced expression of GLUL by SB-HTVI in a subset of zone-3 hepatocytes shows ectopic expression of GS and p-mTOR-S2448 in the same hepatocytes (100×).
E. GS was silenced in Hep3B cells using validated siRNA against GLUL as compared to siRNA against non-specific scrambled sequence for 24 hours, followed by supplementation with 4mM of Glutamine for another 24 hours. WB shows that GLUL siRNA successfully decreased GS as well as p-mTOR-S2448, which was restored by Glutamine supplementation. Equivalent protein loading was confirmed by GAPDH. Densitometry analysis (color-coded) on normalized sampled showed Glutamine supplementation dramatically increasing p-mTOR-S2448 after GS knockdown.
See also Figure S5.
To further validate these in vivo findings, GS was knocked down in Hep3B cells using a validated siRNA against the mRNA of GLUL versus a scrambled sequence. 48h after transfection, a notable decrease in total GS levels was evident (Fig.S5D). There was no change in total mTOR but a corresponding decrease in p-mTOR-S2448 and pS6-S240/244 levels by WB (Fig.S5D). mTOR knockdown for 48 hours in Hep3B cells did not affect β-catenin or GS levels by WB (data not shown).
Genetic re-expression of CTNNB1 or GLUL or glutamine repletion rescues loss of p-mTOR-S2448 expression
To further validate the relationship of β-catenin, GS and p-mTOR-S2448, we forced expression of β-catenin in the livers of Alb-Cre/β-cateninfl/fl mice using SB-HTVI. Ten days after delivery of CTNNB1 plasmid via SB-HTVI, IHC showed many isolated β-catenin-positive hepatocytes in zone-3 (Fig.4C). Analysis of serial sections showed the same cells to be positive for GS and p-mTOR-S2448 (Fig.4C). We next re-expressed GLUL in the Alb-Cre/β-cateninfl/fl mice via SB-HTVI. Ten days post HTVI, we found the appearance of isolated GS-positive hepatocytes randomly in the pericentral area (Fig.4D). Staining for p-mTOR-S2448 in serial sections showed reappearance of this protein in the same cells (Fig.4D).
To further corroborate this observation in vitro, we knocked down GLUL in Hep3B cells using validated siRNA and 24 hours after transfection, cells were supplemented with 4mM of Glutamine for 24 more hours. Cells were washed and WB using whole cell lysates showed successful knockdown of GS in GLUL-siRNA transfected samples, along with decrease in p-mTOR-S2448 (Fig.4E). Glutamine supplementation induced p-mTOR-S2448 levels despite GS knockdown (Fig.4E)
Preclinical HCC models with increased GS and mTORC1 have increased hepatic glutamine content
We next investigated if β-catenin-mutated HCC, which have high GS levels (Cieply et al., 2009; Zucman-Rossi et al., 2007) (Fig.1A) also have high glutamine levels. This is highly relevant since glutamine can be a major source of energy for cancer cells (Altman et al., 2016) and the primary role of GS is to synthesize glutamine in a cell. Total glutamine levels per milligram protein in the normal and the tumor-bearing livers from various β-catenin-mutant HCC models with high GS levels was determined. Significantly higher levels of glutamine were evident in the Met-β-catenin, Kras-β-catenin and Yap-β-catenin models as compared to the normal livers (Fig.5A).
Figure 5: Increased glutamine levels in β-catenin mutant HCC makes them susceptible to mTORC1 loss or inhibition alone or in combination with GC1.
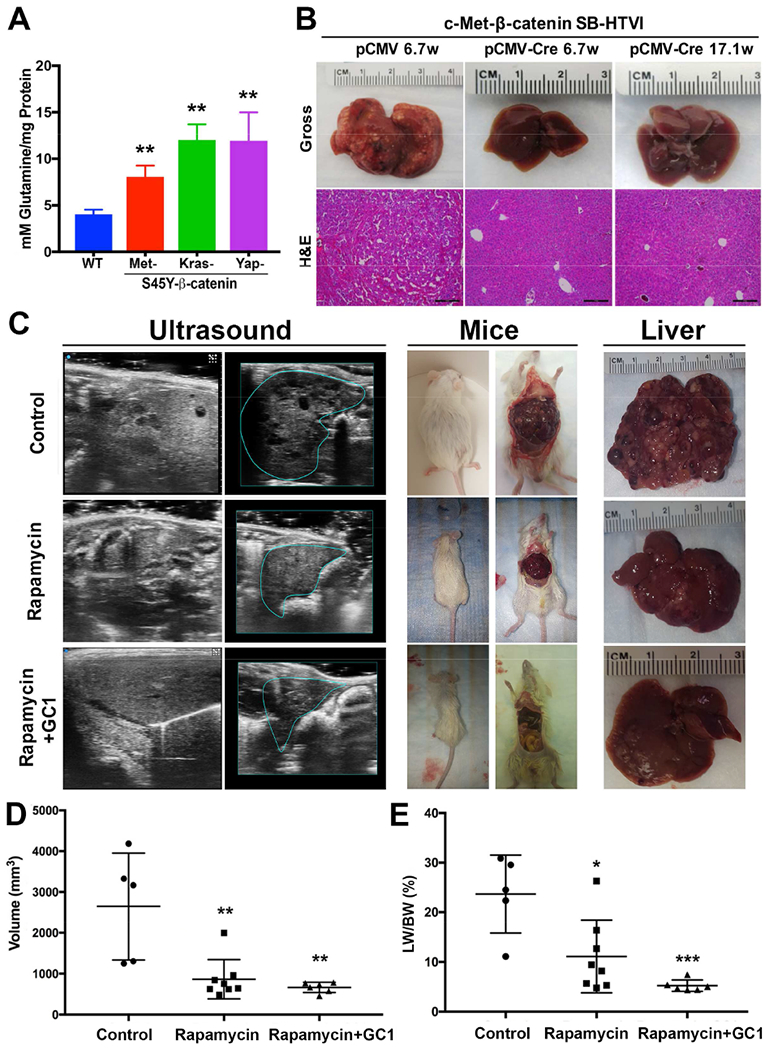
A. Significantly higher glutamine levels were observed in the tumor bearing livers from the S45Y-CTNNB1-Met, S45Y-CTNNB1-G12D-Kras and S45Y-CTNNB1-S127A-Yap models as compared to normal FVB livers. (**p<0.01)
B. Gross (top) and H&E staining from the representative livers (bottom) from the Raptorfl/fl mice injected for 6.7 weeks with the Met-β-catenin-pCMV or Raptorfl/fl mice injected for 6.7 or 17.1 weeks with the Met-β-catenin-pCMV-Cre. Macroscopic and microscopic HCC is visible in the pCMV but not in pCMV-Cre group. (100×; scale bar=200μm)
C. 3D-US identified focal round lesions that were both hypo-echoic and hyper-echoic in the basal diet control group, which were notably decreased in the Rapamycin only group, with even a more profound decrease in the Rapamycin+GC1 group. Gross images of livers in mice confirmed the relative decrease in disease in Rapamycin only and complete absence of disease in combination group as compared to the control.
D. Significant decrease in tumor volume calculated based on 3D-US was evident in both Rapamycin and Rapamycin+GC1 group as compared to basal diet (**p<0.01).
E. Significant decrease in the liver weight to body weight ratio (LW/BW), an indicator of tumor burden was observed after treatment for 5 weeks with either Rapamycin alone (*p<0.05) or Rapamycin+GC1 (***p<0.005)
See also Figures S6 and S7.
Absence of Raptor, which disrupts mTOR signaling, prevents development of HCC in the Met-β-catenin model
Co-expression of dominant-negative TCF4 in Met-β-catenin and Kras-β-catenin models prevented GS expression and HCC development (Tao et al., 2016; Tao et al., 2017). To address if disruption of mTORC1 impacts HCC development, we used SB-HTVI to deliver Met-β-catenin plasmids along with pCMV or pCMV-Cre to the Raptor-floxed mice as described in the methods (Sengupta et al., 2010) (Fig.S6A). While a notable tumor burden was evident in the Met-β-catenin mice injected with pCMV, a complete absence of tumors was noted in the PCMV-Cre injected group, which led to deletion of Raptor in the Met-β-catenin-transfected hepatocytes. A significant decrease in tumor incidence, liver weight (LW) and LW/body weight (BW), an indicator of tumor burden was also evident in this group (Fig.S6B-D). While clear microscopic foci were evident in the Met-β-catenin-pCMV group at 6.7 weeks after SB-HTVI, no tumor foci were evident at either 6.7 or 17.1 weeks after SB-HTVI in the Met-β-catenin-pCMV-Cre group (Fig.5B).
mTOR inhibition alone or in combination with Met inhibition reduces HCC burden
Since HCCs developing in the Met-β-catenin model resemble 11% of all human HCC based on gene expression, and show increased GS and mTOR activation (Tao et al., 2016; Tao et al., 2017), we investigated the impact of mTOR inhibition in this model. Once HCCs were established at 5 weeks after SB-HTVI, the mice were randomized into 3 groups for 5 weeks of therapy (Fig.S6E). Group A was fed basal diet (n=5); Group B was fed diet containing 18 mg/kg of Rapamycin, an mTORC1 inhibitor (n=8); and group C was fed diet containing both Rapamycin (18 mg/kg) and 5 mg/kg of GC1 (Sobiterome) (n=8). GC1 is a thyromimetic with partial Met-inhibitory activity and modestly impacts HCC in the Met-β-catenin model (Puliga et al., 2017).
Three-dimensional ultrasonography (3D-US) was used to visualize tumors in all mice. 3D-US images showed HCC in mice on control diet as focal round lesions that were an admix of well-circumscribed, homogeneous or heterogeneous, hypo-echoic and hyper-echoic, with larger lesions showing pressure necrosis observed as non-echoic, acoustic halo (Fig.5C). After 5 weeks, Rapamycin-treated group showed a decrease in HCC foci whereas Rapamycin+GC1 group exhibited an even more profound absence of tumors (Fig.5C). Next, analytic software from the ultrasound provider (VisualSonics Vivo LAB 3.0.0, Fuji film, Canada) was used to measure liver volume and tumor diameter, which was performed using multi-slice method. The boundary of liver on each sectional slice were delineated by analyzer, then 3D liver image was reconstructed to calculate liver volume (Fig.5C). Liver tumors were recognized on sequential sectional images frame-by-frame and tumor diameter measured on the maximum sectional slice of targeted lesion. Tumor volume was calculated mathematically [4/3 ×π×r3(r=tumor diameter/2)]. A significant decrease in tumor volume was observed in Rapamycin-treated group with an even greater response observed in the combination group when compared to the controls (Fig.5D).
Mice from all 3 groups were sacrificed after 5 weeks of treatment for gross and microscopic assessment of liver. Treatment with Rapamycin alone led to a significant decrease in the LW/BW, which was even more profoundly reduced in the Rapamycin+GC1 treatment group (Fig.5E).
To further validate these findings, gross images of individual representative livers from the three groups were assessed for macroscopic disease. A visible decrease in macroscopic tumor nodules in the Rapamycin and even more pronounced decrease in Rapamycin+GC1 group was evident (Fig.6A). Histological analysis was performed next on livers from each group. Representative analysis shown confirmed the presence of several microscopic tumor foci in the control group by H&E, which were strongly positive for GS and p-mTOR-S2448 (Fig.6B). However, a notable reduction in tumor size and numbers was evident after Rapamycin treatment and these small remnant foci were positive for GS and p-mTOR-S2448 (Fig.6B). Treatment with Rapamycin+GC1 had a more dramatic effect with loss of tumors in majority of the cases with almost normalization of the hepatic architecture (Fig.6C).
Figure 6. Decreased tumor burden seen by macroscopic and histological analysis after 5 weeks of mTORC1 Inhibition alone but more profoundly in combination with GC1.
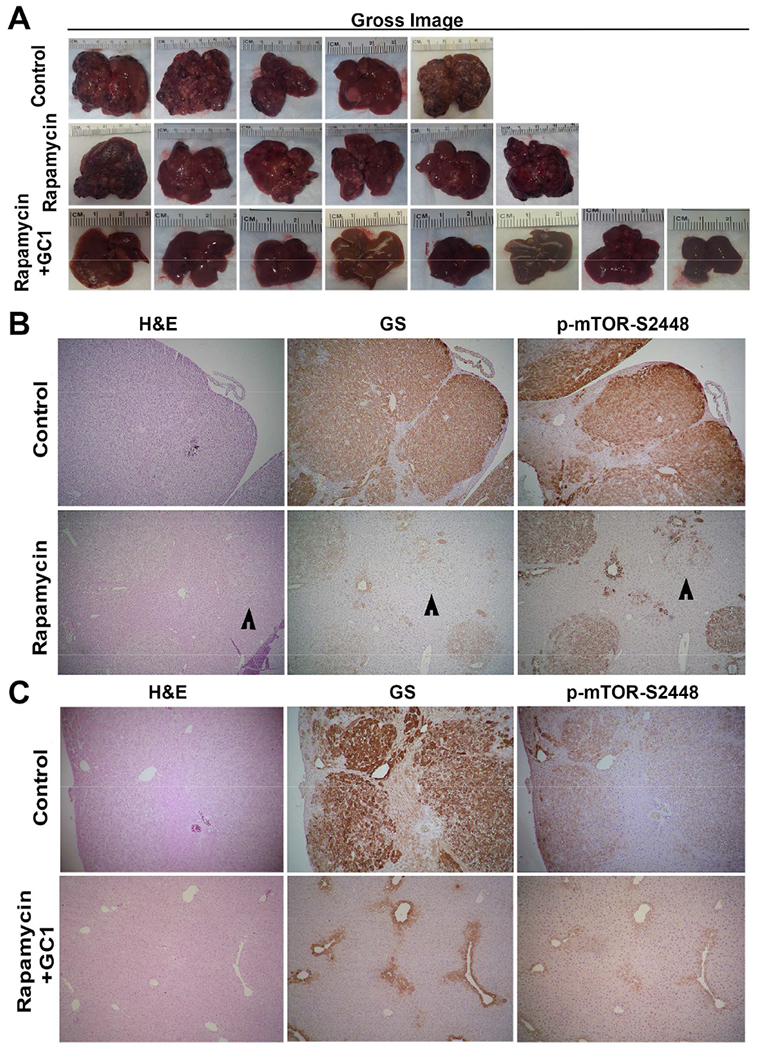
A. Gross images of individual representative livers from the 3 groups showing decreased macroscopic tumor burden in Rapamycin alone and more so in the Rapamycin+GC1 group as compared to the controls.
B. H&E staining and IHC for GS and p-mTOR-S2448 on serial sections shows large tumor foci staining positive for the two markers in basal diet group and notably smaller nodules in Rapamycin treatment only (50×).
C. H&E staining and IHC for GS and p-mTOR-S2448 on serial sections shows large tumor foci staining positive for the two markers in basal diet group and lack of any nodules in the Rapamycin+GC1 treatment with normal zonated appearance for the two markers (50×).
See also Figure S6.
Since the combination therapy showed the most profound therapeutic effect, we performed a more comprehensive analysis on this group along with the controls. First, we performed a more rigorous microscopic analysis utilizing Myc-tag (to detect mutant-β-catenin) and V5-tag (to detect c-Met) as indicators of tumors. While most of the hepatic tissue consisted of tumors which were universally positive for Myc-tag and V5-tag in the control group, the Rapamycin+GC1 showed only occasional foci or a few isolated positive cells (Fig.7A). This profound response to Rapamycin+GC-1 as compared to the control was better seen in representative tiled images of an entire liver section stained for Myc-tag (Fig.7B). Decrease in Myc-tag was also confirmed by WB (Fig.7B). IHC showed large nodules in the control livers to be strongly positive for Ribosomal pS6-Ser235/236 and pS6-Ser240/244, downstream effectors of mTORC1 signaling, while smaller and fewer foci stained positive for these markers in the Rapamycin+GC1 group (Fig.7C)
Figure 7. Five-week treatment with Rapamycin+GC1 combats Met-β-catenin HCC and is superior to treatment with Rapamycin alone.
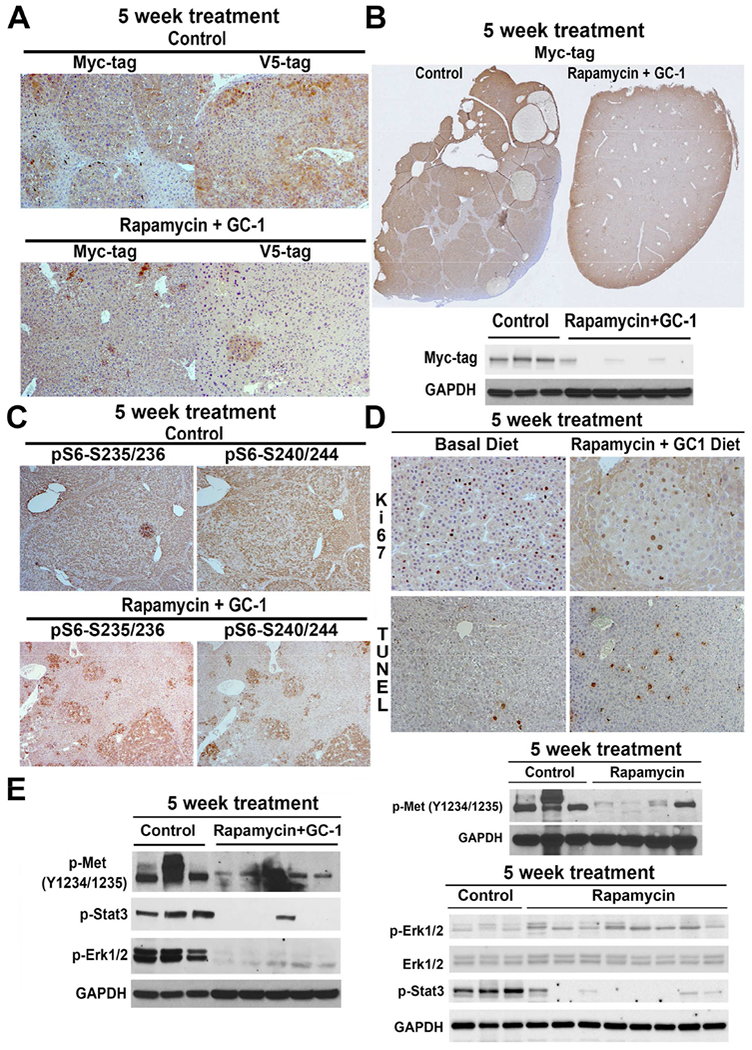
A. IHC for Myc tag representing mutant-β-catenin and V5-tag representing c-Met, shows all tumors in basal diet group to be positive and thus derived from the injected plasmids. A notable decrease in IHC for both markers indicates a complete response to the combination therapy (50×).
B. A representative tiled image from IHC for Myc-tag from the basal diet-fed versus Rapamycin+GC1 diet-fed Met-β-catenin mice shows a dramatic difference in overall histologic tumor burden which was also confirmed by WB for Myc-tag. GAPDH confirmed equal loading.
C. IHC for p-S6-S235/236 and p-S6-240/244, indicators of mTORC1 activity, showed notably smaller positive nodules in the combination treatment as compared to basal diet in the Met-β-catenin model (50×).
D. Decreased number of tumor cells were PCNA-positive in the Rapamycin+GC1 group versus basal diet (100×), and increased number of TUNEL-positive cells were evident in the combination treatment than controls (100×).
E. Representative WB using lysates from tumor-bearing livers from Met-p-catenin HCC model comparing Met signaling in controls versus Rapamycin alone and controls versus Rapamycin+GC1. P-Met- and p-Stat3 were comparably downregulated in both treatment groups as compared to the controls, however p-Erk1/2 was decreased in only the Rapamycin+GC1 group. GAPDH verified comparable loading in both sets of analyses.
See also Figure S6.
Consistent with a decrease in mTORC1 signaling, there was reduced cell proliferation as indicated by decreased number of tumor cells in S-phase of cell cycle detected by IHC for Ki-67 as well as increased cell death detected by TUNEL staining in the treatment group (Fig.7D).
Differences in effects of Rapamycin alone or in combination with GC1 on downstream signaling
To mechanistically address superiority of Rapamycin+GC1 combination versus Rapamycin alone, we performed analysis on c-Met and associated downstream signaling based on our previous experience with GC1 (Puliga et al., 2017). We were aware that the tumor burden was different in the two models and hence WB using whole livers would have to be interpreted in that context. Decrease in p-Met-Y1234/1235, p-Stat3 and p-Erk1/2 was evident in the 5-week Rapamycin+GC1 group (Fig.7E). Rapamycin alone led to a decrease in p-Met-Y1234/1235 and p-Stat3 but no difference p-Erk1/2 (Fig.7E). Thus, combination of mTOR inhibition and GC1 more profoundly suppressed p-Met signaling, and may have contributed to an overall improved tumor response.
Short term treatment of Met-β-catenin mice with Rapamycin+GC1 affects tumor cell proliferation and survival through mTORC1 inhibition
To further address the basis of the therapeutic response observed after 5 weeks of treatment, we first determined the longitudinal impact of Rapamycin+GC1 on LW/BW in the Met-β-catenin mice. LW/BW ratio was assessed at the time of the initiation of treatment and thereafter at 2 weeks, 4 weeks and 5 weeks of treatment. A gradual increase in the tumor burden as reflected by increasing LW/BW was evident in controls from 5 and 7 weeks to 9 and 10 weeks, which remained static in the Rapamycin+GC1 group (Fig.8A). Next, we determined if the combination therapy elicited any biological response on tumor cell proliferation or cell survival at the earliest time point, i.e. at 2 weeks after treatment. Small clusters of tumor cells evident in both groups as seen by IHC for Myc-tag (Fig.8B), showed absence of PCNA-positive cells and increased number of TUNEL-positive cells in the treatment group versus controls (Fig.8C).
Figure 8. Two-week treatment of Met-β-catenin mice with Rapamycin+GC1 impacts mTORC1 and p-Erk signaling to reduce proliferation and increase cell death, to eventually affect HCC burden profoundly.
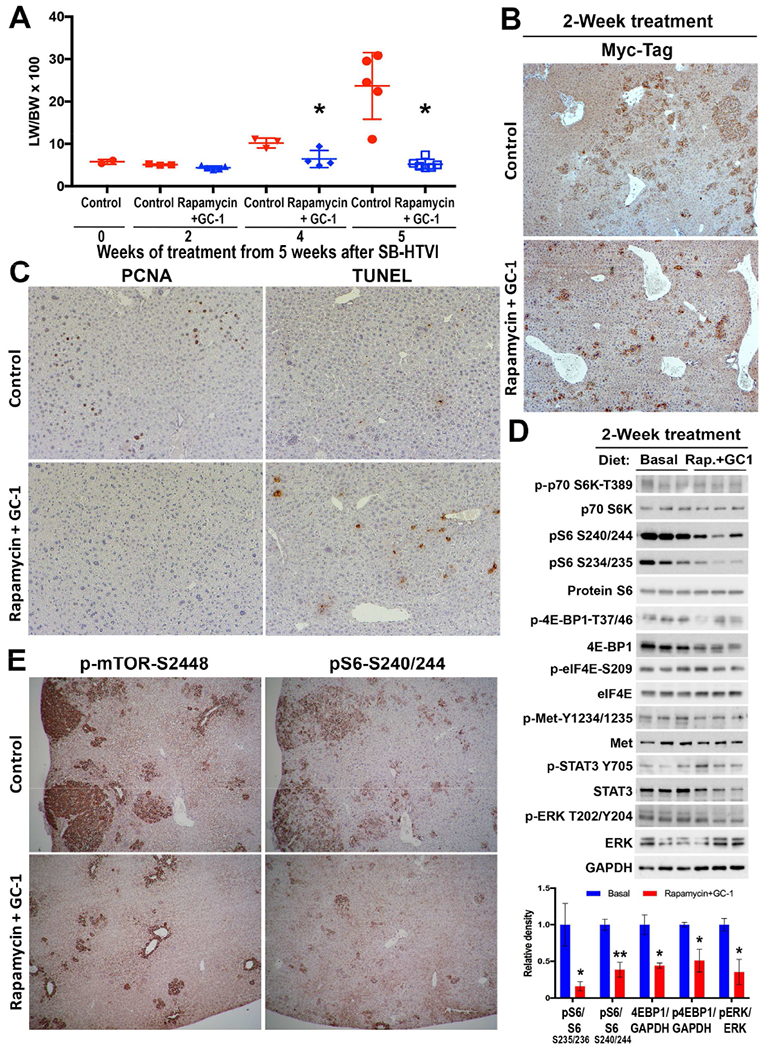
A. LW/BW was compared between controls and Rapamycin+GC1 group at 2, 4 and 5 weeks after intervention. Progressive increase in tumor burden was evident in controls but not in the combination treatment group. At 2 weeks of treatment, there was insignificant difference in LW/BW in the treatment group and controls (*p<0.05).
B. IHC for Myc-tag showed isolated cells or small clusters representing small tumor foci in the control and Rapamycin+GC1-treated Met-β-catenin mice at 2 weeks with marginally fewer and smaller foci seen in the treatment group (50×).
C. A notable decrease in the number of PCNA-positive tumor cells and an increase in the numbers of TUNEL-positive nuclei is evident in the Rapamycin+GC1 group as compared to basal diet fed controls at 2 weeks.
D. WB using liver lysates from Met-β-catenin mice on basal diet or Rapamycin+GC1 diet for 2 weeks show a notable decrease in p-S6-S235/236, p-S6-240/244, total 4EBP1 and p-4EBP1-T37/46 as well as p-Erk-T202/Y204 which was quantified and presented as bar graph (lower panel). WB for GAPDH depicts comparable protein loading.
E. Representative IHC shows a notable decrease in staining for p-mTOR-S2448 and pS6-S240/244 in 2-week Rapamycin+GC1 treatment versus controls.
Since the biological response to the drug treatment was visible at 2 weeks of treatment, and the small tumor foci were comparable in size and numbers between the two groups, we assessed downstream signaling at this stage in the two groups. Notable decreases in p-S6-S234-S235 and p-S6-S240-S244 were evident in the treatment group despite unchanged levels of p-p70S6K-T389 (Fig.8D,E). Total levels of p-4E-BP1-T37/46 and 4E-BP1 were modestly decreased while p-eIF4E-S209 or total eIF4E were not changed, after treatment (Fig.8D). Additionally, we saw modest decrease in p-Met-Y1234-1235 and in p-Erk-T202-Y204 (Fig.8D).
Discussion
Our current study identifies heretofore unrecognized zonal localization of p-mTOR-S2448 in normal livers in mice and humans. Only hepatocytes in zone-3 showed a basal presence of constitutively active mTORC1. This was a function of Wnt-β-catenin controlled constitutive expression of GS, which in turn results in highest intracellular glutamine levels in pericentral hepatocytes (Gebhardt and Coffer, 2013). Glutamine directly phosphorylates mTOR at Ser2448 in lysosomes (Jewell et al., 2015). Thus, it is likely that some of the roles of the Wnt-β-catenin pathway in zone-3 may be attributable to zonated mTORC1. It is likely that the Wnt-β-catenin-GS-glutamine-mTORC1 axis may be contributing to growth, metabolism, survival, protein biosynthesis, autophagy and transcription in zone-3 hepatocytes (Fig.S7). This axis may also be relevant to some of the pathophysiological events restricted to zone 3 cells such as accumulation of fat in hepatocytes seen in high-fat diet fed mice (Behari et al., 2014) and NASH patients (Chalasani et al., 2008); presence of basal high autophagy (Gebhardt and Coffer, 2013); increased glucose uptake by hepatocytes and increased glycolysis (Chafey et al., 2009); and lipogenesis (Gougelet et al., 2014). Such biological processes have been linked to mTOR activation independently (Sabatini, 2017; Saxton and Sabatini, 2017). Additionally, our observation provides an opportunity to explore novel links between processes not associated with mTOR such as bile acid synthesis and xenobiotic metabolism, which are classically evident in zone-3 hepatocytes.
CTNNB1 mutations in HCC are relatively common, affecting anywhere from 20-35% of all cases (Russell and Monga, 2018). CTNNB1 mutations in hepatocytes independent of their location within a liver lobule, can lead to β-catenin activation to regulate expression of key genes that encode for proteins regulation proliferation, survival, migration, cancer stem cell expansion, immune escape and angiogenesis. However, no specific mechanism by which β-catenin activation can contribute to hepatocarcinogenesis has been described. We identify a novel mechanism by which activating β-catenin mutations lead to overexpression of liver-specific Wnt target GLUL encoding for GS protein (Cieply et al., 2009; Zucman-Rossi et al., 2007) which leads to excessive glutamine levels and in turn mTORC1 activation to stimulate S6 family of ribosomal proteins involved in ribosomal biogenesis, and 4EBP1, which facilitates cap-dependent mRNA translation, to stimulate protein synthesis (Hay and Sonenberg, 2004). We show that β-catenin-mutated HCC are mTORC1-addicted owing to the GS-Glutamine-p-mTOR-S2448 axis and mTOR inhibition robs these tumors of their metabolic dependence, impeding ribosomal biogenesis (Pelletier et al., 2018) and protein synthesis (Chu et al., 2016), both critical in any tumor’s sustenance and expansion (Fig.S7).
We are not implying that only β-catenin-mutated HCCs are sensitive to mTOR inhibition and that non-β-catenin mutated HCCs will be resistant to Rapamycin-like agents. However, we believe that β-catenin-mutated HCC will be particularly sensitive to mTORC1 inhibition. In fact, there are well known mutations in TSC2 in a subset of HCC, which lead to mTORC1 activation and such HCC do respond to mTOR inhibition (Huynh et al., 2015). Likewise, mTOR activation may occur downstream of growth factor aberrations that are evident in subsets of HCC (Menon et al., 2012). In our own analysis, there were a subset of GS-negative HCC cases that were strongly p-mTOR-S2448-positive, and ranged from 4.4-7.7% between the two HCC cohorts.
Multiple studies have highlighted the critical role of mTOR activation in the establishment and progression of HCC (Villanueva et al., 2008). This has been corroborated in animal models where mTOR activation leads to the rapid formation of liver tumors (Huynh et al., 2009; Menon et al., 2012). These pre-clinical data provided a solid rationale for the Everolimus for Liver Cancer Evaluation (EVOLVE-1) trial, which was a randomized, double-blind, phase 3 study comparing Everolimus (an mTOR inhibitor) to placebo in HCC patients who had either progressed during or after treatment with Sorafenib, the first line therapy for unresectable HCC, or were intolerant to Sorafenib (Zhu et al., 2014). However, this multicenter trial in 546 patients showed no significant survival difference, or differences in time to progression between Everolimus- and placebo-treated patients although disease control rate was higher in the Everolimus (56.1%) compared to the placebo (45.1%) group. It is thought that lack of differences may have stemmed from recruiting “all-comers” without selecting patients enriched for mTOR-addicted tumors, since biomarkers are lacking (Llovet, 2014; Zhu et al., 2014). Indeed, Huynh et.al. reported loss of tuberous sclerosis complex 2 (TSC2) in a subset of HCC, which leads to increased mTOR signaling (Huynh et al., 2015). Using that rationale, subset analysis of HCC samples from the EVOLVE-1 trial found that 10.8% of all samples, which had low or undetectable TSC2, showed longer survival with Everolimus than placebo (Huynh et al., 2015).
CTNNB1 mutations in HCC usually co-occur with mutations in TERT (promoter), NFE2L2, MLL2, APOB, ARID2 and others (Schulze et al., 2015). And in almost all of these tumors whenever CTNNB1 is mutated, GS levels are upregulated, irrespective of co-occurring mutations (Schulze et al., 2015). Likewise, in major subsets of HCA and HB with CTNNB1 mutations, GS is upregulated (Tao et al., 2014; Zucman-Rossi et al., 2006). Our analysis of patient tumors also showed that most GS-positive tumors, irrespective of mutational spectra in the exon-3 of CTNNB1, were simultaneously positive for p-mTOR-S2448. Indeed, point-mutant versus Δ90-β-catenin in HCC when used in combination with c-Met showed similar β-catenin activation (Qiao et al., 2018). HCCs in TCGA database exhibiting mutations in CTNNB1 affecting S45 and S33 showed significant similarity in gene expression (Tao et al., 2016). We have also shown comparable activation of Wnt targets in HCC cases with exon-3 CTNNB1 mutations irrespective of the residue affected (Okabe et al., 2016). Another study showed some differences in the extent of the activation of the Wnt-β-catenin pathway due to differences in residues affected by mutations within the exon-3 of CTNNB1, however the overall activation of the pathway was comparable due to other concomitant events like gene duplication (Rebouissou et al., 2016). Thus, irrespective of mutant residue in exon-3 of CTNNB1, we observe a β-catenin-GS-Glutamine-mTORC1 axis in an array of hepatic tumors.
As a result of a consistent increase in mTORC1 activation in the GS-positive, CTNNB1-mutated HCC, as also seen in HCC in the Met-β-catenin mouse model that represents 11% of all human HCCs (Tao et al., 2016), these tumors showed increased susceptibility to Rapamycin and even more so to its combination with GC1. The reason why Rapamycin alone did not lead to a complete response could be multifactorial. The dose of Rapamycin may need further optimization. New second and third generation mTORC1 inhibitors with greater potency may be more efficacious (Xie et al., 2016). Lastly, since Met-β-catenin HCC model employs two oncogenic pathways, use of GC1 for c-Met inhibition likely complemented Rapamycin leading to a more profound response. GC1, like triiodothyronine, activates Wnt-β-catenin signaling to induce liver regeneration, but does not activate mutant-β-catenin (Puliga et al., 2017). However, it reduced p-Met-Y1234-1235 through an undetermined mechanism, but as a single agent, it only marginally reduced HCC burden in the Met-β-catenin model. In our current study, Rapamycin alone affected p-Met-Y1234-1235 and p-Stat3, but in combination with GC1, it notably reduced p-Erk1/2, which coincided with a greater decrease in tumor burden.
Identifying the crosstalk between the two major pathways conserved in evolution will likely have a major impact on understanding both physiological and pathologic processes. In the current study, we show mTOR addiction of the β-catenin-mutated HCCs, and paving a new way to target these subsets of HCCs since no anti-β-catenin inhibitors are currently available in the clinic. Future studies may unveil additional roles of the Wnt-LRP5/6-GS-Glutamine-mTORC1 axis in liver pathophysiology.
Limitations of Study
One limitation of our study is that it relied on retrospective analysis of patient tissues for demonstrating a notable correlation between CTNNB1 mutations, and immunostaining for GS and p-mTOR-S2448. The actual therapeutic relevance of this observation remains to be proven in the clinic. Even subset analysis retrospectively of patients that were enrolled previously in the EVOLVE-1 trial (Zhu et al., 2014), or SILVER trial, which tested effectiveness of Sirolimus (Rapamycin) as an immunosuppressive agent post liver transplantation for HCC (Geissler et al., 2016), may be useful. Both trials included all comers without any molecular stratification and it may be useful to address any benefit of these mTOR inhibitors in a subset of patients harboring CTNNB1 mutations. Lastly, since biopsies are not routine for the clinical management of HCC mostly due to lack of any actionable targets, it would be of high relevance to perform biopsies safely or use noninvasive modalities, such as circulating DNA analysis, to identify the CTNNB1-mutated subsets of HCC that may respond to mTOR inhibitors.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCES SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Satdarshan P. S. Monga, M.D. (smonga@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Hep3B cells (ATCC HB 8064: derived from 8 years old male juvenile) were cultured in the ATCC-formulated Dulbecco’s Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) in a humidified 5% CO2 incubator. One hundred units of penicillin and streptomycin were added to each cell culture media. An early passage Hep3B cell line, which was authenticated for morphology by optical observation was utilized. Absence of mycoplasma contamination was verified by the MycoAlert™ Mycoplasma detection kit (Lonza). For glutamine deprivation, cells were plated overnight in complete DMEM, briefly washed with phosphate-buffered saline (PBS) and then transferred into glutamine-free medium supplemented with 2% dialyzed FBS. Cells were either glutamine-starved or supplemented with 4mM Glutamine (ThermoFisher Scientific) for additional 24 hours before preparing whole cell lysates for protein analysis by western blot (WB).
Mice
Animals were maintained in accordance with national and international guidelines. Mice were group-housed (2–5 mice per cage) in standard mouse cages and maintained within temperatures of 18-23°C with 40-60% humidity under a 12h light/dark cycle with free access to water and standard mouse chow or specific diet for experimental purposes. All mice used in the study were were free of excluded rodent pathogens by the Division of Laboratory Animal Resources at the University of Pittsburgh and were in good health prior to the start of experiments. All transgenic and KO mouse lines were maintained on the immunocompetent C57BL/6 genetic background. For drug treatment experiments, 6-8 weeks old male immunocompetent FVB (Jackson Laboratory) mice were used for sleeping beauty transposon-transposase and hydrodynamic tail vein injection (SB-HTVI). Since HCC is a sexually dimorphic disease with preponderance in the males, only male mice were used for all tumor studies. Also, since SB-HTVI protocol works best in younger animals due to required hemodynamics for successful diversion of plasmid into the liver through hepatic vein, 6-8 week old mice were used for this protocol. Age and sex of mice used for genetic studies has been noted in appropriate sections.
For Alb-Cre/glutamine synthetase (GS)fl/fl mice, liver tissue was sampled in accordance with the German animal protection law and approved by local committees (Zentrale Einrichtung für Tierforschung und wissenschaftliche Tierschutzaufgaben, Heinrich-Heine-Universität Düsseldorf).
All experiments with Raptorfl/fl mice were conducted in accordance with the approved protocol by the Institutional Animal Care and Use Committee (IACUC) at UCSF. The other animal care and experiments were performed in accordance with the IACUC at the University of Pittsburgh.
Patients
Multiple human tumors were used in the study from multiple institutions, all following approval from respective Institutional Review Boards (IRB) which are listed appropriately in this section.
Fifteen hepatocellular adenomas (HCA) were used in the study and analyzed by WB, including 8 CTNNB1 mutated and 7 CTNNB1-non-mutated cases. Eleven were classical HCA without any suspicion of malignancy, 2 were borderline lesions between HCA and hepatocellular carcinoma (HCC) and 2 cases were HCC developed on HCA. All the cases were previously described (Rebouissou et al., 2016; Zucman-Rossi et al., 2006). The 5 non-tumor livers included in the analysis were taken from patients resected with primary liver tumors developed in the absence of cirrhosis. P-mTOR expression was evaluated by WB analyses as described methods details sections.
Additionally, fifty-five cases of hepatoblastoma (HB) from Children’s Hospital Pittsburgh were evaluated on 3 tissue microarrays (TMA) representing different areas of tumors with internal normal liver controls (Fig.S1). Detailed demographics and histological information for these cases is available in Table S1. The studies were approved under the IRB approval number PRO17090320. The TMA were stained using antibody against GS (Millipore) on the Ventana automated immunostainer (Ventana Medical Systems). The staining for p-mTOR-S2448 (Cell Signaling) was performed manually as described in methods details sections. Whole slide image capture of the tissue microarray was acquired using the Aperio XT slide scanner (Aperio Technologies). The staining was evaluated and scored by pediatric pathologist (S.R.). Staining was scored either as negative, 1+ (positive staining in up to 10% of section), 2+ (staining in 10-50% of section or 3+ (staining in greater than 50% of section). A staining of 2+ and 3+ was considered positive and no staining or 1+ staining was considered negative. All scores for each individual sample are included in Table S1.
A collection of 6 human HCC TMAs were next examined for GS (Ventana Medical Systems) and p-mTOR-S2448 (Cell Signaling) (Fig.S3). Study approval was obtained from the University of Pittsburgh (IRB number: PRO15060061). TMA were constructed from archival formalin-fixed paraffin-embedded tissue blocks from 256 HCC seen at the University of Pittsburgh Medical Center between 2009 and 2015. All tumor hematoxylin-and-eosin stained slides were reviewed, and representative areas were carefully selected for tissue microarray construction. Two, random 1.0 mm-sized cores were punched from each patient’s tumor and harvested into recipient blocks. The demographics and additional information and staining characteristics of these cases are included in Table S3. The staining was evaluated and scored by anatomic pathologist (A.S.) and scored as also described in the results section. Positive GS and p-mTOR-S2448 staining were defined as homogenous and intense staining occurring within most malignant hepatocytes within each core. All scores are provided for each individual sample in Table S3.
Lastly, a collection of 95 frozen and corresponding formalin-fixed, paraffin-embedded HCC specimens were used from the University of Greifswald (Greifswald, Germany). Tumors were divided in HCC with shorter survival/poorer outcome (HCCP; n = 45) and HCC with longer survival/better outcome (HCCB; n = 50), characterized by < 3 and > 3 years’ survival following partial liver resection, respectively. HCC specimens were collected at the Medical University of Greifswald (Greifswald, Germany). IRB approval was obtained at the Ethical Committee of the Medical University of Greifswald (Approval Number BB: 67/10). Informed consent was obtained from all individuals. Detailed demographics and additional patient and staining information is available in Table S2. All sections were stained for p-mTOR-S2448 (Cell Signaling) and GS (Abcam). Immunoreactivity for p-mTOR-S2448 and GS was estimated semi-quantitatively: upregulation of one and/or the other protein was defined when immunolabeling for the protein of interest was stronger in tumors when compared to corresponding surrounding non-neoplastic livers. All scores are provided for each individual section in Table S2.
METHODS DETAILS
Transient transfection (in vitro)
For transient knock down, Hep3B cells were transfected for 48 hours with 100 pmol CTNNB1 siRNA (Cell Signaling), 100 pmol mTOR siRNA (Cell Signaling), 100 pmol GLUL siRNA (Cell Signaling) or scrambled control siRNA (Cell Signaling) using the Lipofectamine RNAi Max (Invitrogen) according to the manufacturer’s protocol. After 24 hours, transfection media was removed, and wells were supplemented with DMEM or DMEM+ 4mM glutamine for 24hrs before harvesting for whole cell lysates for WB analysis.
Expression vectors (in vivo)
The constructs used for mouse injection, including pT3-EF5α-hMet-V5, pT3-EF5α-G12D-mutant-K-Ras, pT3-EF5α-YapS127A, pT3-EF1 ✔-Δ90-β-catenin, pCMV/sleeping beauty transposase (SB), pT3-EF5α-S45Y-β-catenin-Myc, pT3-EF5α-WT-β-catenin-Myc, pCMV-Cre have been described previously (Tao et al., 2014; Tao et al., 2016; Tao et al., 2017). Human NFE2L2 clone with G31A mutation, from pDONR223_NFE2L2_p.G31A vector (Addgene) was inserted into pT3-EF5α plasmid via the Gateway PCR cloning strategy (Invitrogen). We also generated T41A-mutant-β-catenin using Q5® Site-Directed Mutagenesis Kit (New England BioLabs) following manufacture’s instruction. We utilized pCI-neo beta catenin WT construct (Addgene) as a template with forward primer: 5’-CCACTgCCACAGCTCCTTCTCTGAGT-3’ and reverse primer: 5’-CACCAGAATGGATTCCAGAGTCCA-3’. Produced T41A-mutant-β-catenin was cloned into pT3-EF5α plasmid via the Gateway PCR cloning strategy (Invitrogen) along with an addition of a Myc tag. All the plasmids used for in vivo experiment were purified using the Endotoxin Free Maxi prep kit (Sigma-Aldrich).
Generation of conditional knockout mouse models
C57BL/6J mice were purchased from Jackson Laboratories for breeding. Generation of liver-specific β-catenin knockout (KO) mice (Tan et al., 2006), liver-specific Lrp5/6 double KO mice (Yang et al., 2014) and endothelial cells specific Wntless KO mice (Preziosi et al., 2018) have been described previously. Briefly, homozygous floxed mice for any gene were bred to Albumin-Cre (for liver-specific deletion) or Lyve1-Cre (for endothelial cell deletion) mice. Offspring carrying a floxed allele for a gene along with an allele of Cre were then bred back to the homozygous floxed mice for that gene. The mice generated this way for β-catenin are annotated as Alb-Cre/β-cateninfl/fl, for Lrp5/6, are labeled as Alb-Cre/Lrp5/6fl/fl, and for Wls, are labeled as Lyve1-Cre/Wlsfl/fl. Liver-specific GS deficient mice were generated as described recently (Qvartskhava et al., 2015). Briefly, loxP sites were inserted downstream of the 3′ end of exon 3 and between exon 1a and exon 1b of the GS gene, respectively (GenOway). Mice with floxed GS alleles (GSfl/fl) were crossed to Albumin-cre mice to eventually generate Alb-Cre/GSfl/fl mice. To generate Raptor conditional KO mice, 6-7-weeks old Raptof/f mice (Jackson Labatories) were used for study to address role of mTORC1 in the Met-β-catenin HCC model. Deletion of genes from hepatocytes or endothelial cells in respective KO mice was confirmed by PCR, WB, Immunohistochemistry (IHC) and immunofluorescence (IF) analysis as described in respective original studies describing the generation of these mice. At least 3 knockouts and 3 littermate age and sex matched controls for each genotype were used for basic characterization. All animals ranged from 8-12 weeks in age for analysis and were from either sex.
Animal models of hepatocellular cancer and hepatoblastoma
Six weeks old FVB male mice obtained from (Jackson Laboratories) were randomized into groups and subjected to the sleeping beauty transposon-transposase and hydrodynamic tail vein (SB-HTVI) protocol as described previously (Tao et al., 2014; Tao et al., 2016; Tao et al., 2017). Briefly, 20μg pT3-EF5α-hMet-V5 and pT3-EF5α-S45Y-β-catenin-Myc plasmids, or pT3-EF5α-G12D-mutant-K-Ras and pT3-EF5α-S45Y-β-catenin-Myc plasmids, 20μg pT3-EF5α-G31A-NFE2L2 and pT3-EF5α-T41A-β-catenin-Myc plasmids, or pT3-EF5α-YapS127A and pT3-EF5α-S45Y-β-catenin-Myc plasmids, or pT3-EF5α-hMet-V5 and pT3-EF5α-WT-β-catenin-Myc plasmids along with the transposase in a ratio of 25:1 were diluted in 2ml of normal saline (0.9% NaCl), filtered through 0.22 μm filter (Millipore), and hydrodynamically injected into the lateral tail vein of mice. To delete Raptor in the Met-β-catenin-transfected hepatocytes, Raptorf/f mice were injected with 20μg pT3-EF1α-c-Met and 20μg pT3-EF1α-Δ90-β-catenin and 40μg pCMV-Cre plasmids and transposase. All animals were sacrificed between 8-12 weeks of plasmids injections unless otherwise indicated.
Animal diets and treatment groups
In order to evaluate the therapeutic effect of rapamycin and GC-1 treatment on β-catenin-dependent HCC, we designed experiments and determined sample size and statistical method of computation based on our previous publications (Tao et al., 2014; Tao et al., 2016; Tao et al., 2017). Custom diets were prepared by Research Diets using AIN-93M mature rodent maintenance diet with of 14.2g Protein, 73.1g Carbohydrate and 4g Fat. For treatment groups, 19mg of Rapamycin or 19mg of Rapamycin + 4mg of GC1 added to 1kg of diet. Rapamycin was obtained from LC Laboratories. GC1 was purchased from Sigma-Aldrich. 6 weeks old FVB male mice (Jackson Laboratories) were subjected to SB-HTVI protocol using 5μg pT3-EF5α-hMet-V5 and 5μg pT3-EF5α-S45Y-β-catenin-Myc plasmids along with the transposase in a ratio of 25:1 diluted in 2.0ml of normal saline (0.9% NaCl), filtered through 0.22μm filter (Millipore), and injected hydrodynamically into the lateral tail vein of mice. Five weeks after injection, mice were randomly stratified into 3 groups and managed in a non-blinded manner. All mice were included in the study. One group was maintained on basal diet (n=5), another group on Rapamycin-supplemented diet (n=8), and another group on Rapamycin+GC1-supplemented diet (n=8). All animals were sacrificed 5 weeks after initiation of diet at 10-weeks post SB-HTVI. Analysis on all livers from each group from hereon, was performed in a blinded manner. For longitudinal assessment, the effect of Rapamycin + GC1 treatment on Met-β-catenin HCC model, additional mice that underwent the Met-β-catenin SB-HTVI protocol, were randomized into either basal diet group or Rapamycin+GC1 diet. Mice from both groups were then euthanized at 2 weeks after randomization (n=3/group) or 4 weeks (n=3 for basal diet; n=4 for Rapamycin + GC1). Livers from 2 weeks post treatment (7 weeks after SB-HTVI) were also assessed for molecular analysis by WB and IHC.
Glutamine Colorimetric Assay
To prepare the samples for glutamine colorimetric assay kit (Abcam), tissue samples were washed in 1× PBS and re-suspended in hydrolysis buffer on ice. Then, tissues were homogenized using a homogenizer with about 15 passes, then centrifuged at 4°C at 10,000g for 10 minutes. Deproteinization was performed on the supernatant, with the addition of ice cold 4M PCA to a final concentration of 1M. Samples were vortexed briefly and incubated on ice for 5 minutes, then centrifuged at 4°C at 13,000g for 2 minutes. An equal volume of 2M KOH was added to supernatant, vortexed and pH to 6.5-8. Samples were centrifuged at 4°C at 13,000g for 5 minutes. The supernatant from samples was transferred to newly, labeled tubes for glutamine assay. Briefly, 40μl of glutamine standard and diluted samples were added to 96-well plate. 2μl of hydrolysis mix was added to glutamine standards and sample wells and incubated for 30 minutes at 37°C. 50μl of glutamine reaction mix was added to wells and incubated for 60 minutes, protected from light. Absorbance was measured at OD-450nm on a microplate reader (Synergy HT, BioTek).
Immunohistochemistry
Mouse liver tissues were fixed for 48 hours in 10% neutralized formalin (Fisher Chemicals), transferred into 70% ethanol and then dehydrated and embedded in paraffin. For IHC, 4μm formalin-fixed sections were deparaffinized in graded xylene and ethanol and rinsed in PBS. To block endogenous peroxidase activity, the sections were incubated in 3% hydrogen peroxide (Fisher Chemicals). For antigen retrieval, samples were microwaved for 12 minutes in pH6 sodium citrate buffer (p-mTor S2448, GS, p-S6 S235/236, p-S6 S240/244) or Tris-EDTA buffer (Ki67), or were pressure cooked for 20 minutes in pH6 sodium citrate buffer (β-catenin). After cooling, samples were placed in 3% H2O2 for 10 minutes to quench endogenous peroxide activity. After washing with PBS, slides were blocked with Super Block (ScyTek Laboratories) for 10 minutes. Sections were incubated for overnight at 4°C with the primary antibodies. Sections were then incubated with species-specific biotinylated secondary antibodies (EMD Millipore) for 1 hour, at room temperature. Sections were incubated with Vectastain ABC Elite kit (Vector Laboratories) and signal was detected with DAB Peroxidase Substrate Kit (Vector Laboratories) followed by quenching in distilled water for five minutes. Slides were counterstained with hematoxylin (ThermoFisher Scientific), dehydrated to xylene (Fisher Chemicals) and coverslips applied with Cytoseal™ XYL (ThermoFisher Scientific). For H&E staining, samples were deparaffinized and stained with hematoxylin (ThermoFisher Scientific) and eosin (ThermoFisher Scientific), followed by dehydration to xylene and application of a coverslip. Terminal deoxynucleotidyl transferase mediated deoxyuridine triphosphate nick-end labeling (TUNEL) was performed as described in the protocol of the Peroxidase In Situ Apoptosis Detection Kit (EMD Millipore).
Immunofluorescence
Paraffin embedded liver sections (5μm) were dewaxed using xylene (Fisher Chemicals) and rehydrated by incubating the slices in ethanol (100, 95, 70 and 50% v/v, each 3×5 min) and washed in aqua bidest. Heat-induced epitope retrieval was performed for 20 minutes using a pressure cooker with 1 × pH6 citrate buffer. Sections were washed in PBS, blocked with PBS containing 5% fetal calf serum (FCS) for 1 hour at room temperature and incubated with primary antibodies in PBS containing 5% FCS overnight at 4°C. At the end of the incubation, liver samples were washed thrice and incubated with fluorochrome-conjugated secondary antibodies (goat anti-rabbit Cy3 and goat anti-mouse FITC) and Hoechst 34580 (Invitrogen) in PBS containing 5% FCS for 2h at room temperature. Liver sections were mounted using Fluoromount-G (SouthernBiotech) and pictures were acquired using the Cell Observer Z1 and Axiovision Software (Zeiss).
Protein extraction and Western Blot analysis
Frozen liver tissues were homogenized in ice-cold lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 10%glycerol) containing 1% Halt protease inhibitor cocktail (ThermoFisher Scientific). Lysates were subjected to brief sonication, centrifuged at 13,000g for 5 minutes to remove insoluble cell debris and the supernatant was recovered. Protein concentration was determined using Pierce bicinchoninic acid protein assay kit (ThermoFisher Scientific). Aliquots of 25-50μg of proteins were diluted and denatured by boiling in 2 × Laemmli sample buffer containing 2-mercaptoethanol (Bio-rad). Proteins (25-50μg per well) were separated in 4-15% or 4-20% gradient SDS-PAGE gels and transferred to PVDF membranes (Millipore). Membranes were blocked with 5% bovine serum albumin or non-fat milk (Lab Scientific), incubated overnight with primary antibodies, washed, and then incubated with HRP-linked species-specific secondary antibodies. Proteins were visualized by chemiluminescence and densitometry performed by ImageJ software or Image Lab software (Bio-Rad Laboratories) for eventual statistical analysis (Prism 7.0).
Ultrasound imaging
Ultrasound tumor imaging was performed using a Preclinical Vevo 3100 micro ultrasound imaging platform (Visualsonics). All scanning procedures were carried out by a trained radiology (H.Y.). Mice were anesthetized by isoflurane (3% for induction and 1.5% for maintenance) mixed with oxygen. Scans were performed at 1, 7, 14, 21, 28 and 35 days after start of diets. B-Mode or brightness mode imaging was used to acquire three-dimensional images of an area of interest and for identification of liver tumors. Images were quantified for the tumor volume using VevoLAB analysis software (Visualsonics).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
For all mouse experiments, sample size was pre-determined based on previous literature describing HTVI-mediated liver carcinogenesis (Tao et al., 2017). Accordingly, littermates were randomized into groups for HTVI and subsequent drug treatments (3 groups) and managed throughout the course of treatment in a non-blinded manner. All subsequent molecular, immunohistochemical and immunofluorescence analysis was performed in a blinded manner. All confidence intervals shown on the bar plots are presented as mean ± standard deviation (SD). Differences in mean values of liver volume and LW/BW ratio were analyzed by one-way ANOVA assuming normal Gaussian distribution with Geisser Greenhouse posttest correction. For differences in mean values of WB analysis, Student’s t test was used. For patient data, Kruskall-Wallis, Mann-Whitney test and Fisher’s exact test (2-sided) were utilized to assess statistical significance. P<0.05 was considered significant (*), p<0.01 was considered highly significant (**), p<0.005 was considered extremely significant (***) and so on. All statistical analysis on patient samples has been included in the results section and respective p values were included in the pertinent text and figure legends. All statistics were performed using GraphPad Prism 7.0 (GraphPad Software).
Supplementary Material
TABLE S3: Related to Figure 2. Demographics and additional information on the 256 HCC cases represented on the 6 TMA from the University of Pittsburgh Medical Center (Pittsburgh, PA, USA).
TABLE S2: Related to Figure 2. Demographics and additional information on 116 HCC samples procured from and assessed at the University of Greifswald (Greifswald, Germany).
HIGHLIGHTS.
mTORC1 activation is seen basally in pericentral hepatocytes due to Wnt/β-catenin
CTNNB1-mutated liver tumors are positive for GS and p-mTOR-S2448
CTNNB1-mutated hepatocellular cancers are addicted to mTORC1 for metabolism
Targeting β-catenin-GS-mTORC1 axis in liver tumors may enable precision medicine
Acknowledgements
This work was supported by NIH grants 1R01DK62277, 1R01DK100287, 1R01DK116993 and Endowed Chair for Experimental Pathology to S.P.M and by T32CA186873 (A.O.A.M.). Part of study was supported by R01CA204586 to S.P.M and X.C. Parts of the study was supported by Deutsche Forschungsgemeinschaft (DFG) through SFB 974.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Dr. Monga had grant funding and was a consultant for Abbvie and Dicerna but has no competing financial interests directly relevant to the current study. None of the other authors have any relevant competing interests to declare.
References:
- Altman BJ, Stine ZE, and Dang CV (2016). From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16, 749. [DOI] [PubMed] [Google Scholar]
- Behari J, Li H, Liu S, Stefanovic-Racic M, Alonso L, O’Donnell CP, Shiva S, Singamsetty S, Watanabe Y, Singh VP, et al. (2014). beta-catenin links hepatic metabolic zonation with lipid metabolism and diet-induced obesity in mice. Am J Pathol 184, 3284–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C, et al. (2006). Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell 10, 759–770. [DOI] [PubMed] [Google Scholar]
- Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, Kitajewski J, Kahn A, and Perret C (2002). New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 21, 8293–8301. [DOI] [PubMed] [Google Scholar]
- Chafey P, Finzi L, Boisgard R, Cauzac M, Clary G, Broussard C, Pegorier JP, Guillonneau F, Mayeux P, Camoin L, et al. (2009). Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 9, 3889–3900. [DOI] [PubMed] [Google Scholar]
- Chalasani N, Wilson L, Kleiner DE, Cummings OW, Brunt EM, Unalp A, and Network NCR (2008). Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J Hepatol 48, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Cargnello M, Topisirovic I, and Pelletier J (2016). Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol 26, 918–933. [DOI] [PubMed] [Google Scholar]
- Cieply B, Zeng G, Proverbs-Singh T, Geller DA, and Monga SP (2009). Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology 49, 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, and Nusse R (2012). Wnt/beta-catenin signaling and disease. Cell 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- Gebhardt R, and Coffer PJ (2013). Hepatic autophagy is differentially regulated in periportal and pericentral zones - a general mechanism relevant for other tissues? Cell Commun Signal 11, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler EK, Schnitzbauer AA, Zulke C, Lamby PE, Proneth A, Duvoux C, Burra P, Jauch KW, Rentsch M, Ganten TM, et al. (2016). Sirolimus Use in Liver Transplant Recipients With Hepatocellular Carcinoma: A Randomized, Multicenter, Open-Label Phase 3 Trial. Transplantation 100, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gougelet A, Torre C, Veber P, Sartor C, Bachelot L, Denechaud PD, Godard C, Moldes M, Burnol AF, Dubuquoy C, et al. (2014). T-cell factor 4 and beta-catenin chromatin occupancies pattern zonal liver metabolism in mice. Hepatology 59, 2344–2357. [DOI] [PubMed] [Google Scholar]
- Hay N, and Sonenberg N (2004). Upstream and downstream of mTOR. Genes Dev 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- Huynh H, Chow KH, Soo KC, Toh HC, Choo SP, Foo KF, Poon D, Ngo VC, and Tran E (2009). RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J Cell Mol Med 13, 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh H, Hao HX, Chan SL, Chen D, Ong R, Soo KC, Pochanard P, Yang D, Ruddy D, Liu M, et al. (2015). Loss of Tuberous Sclerosis Complex 2 (TSC2) Is Frequent in Hepatocellular Carcinoma and Predicts Response to mTORC1 Inhibitor Everolimus. Mol Cancer Ther 14, 1224–1235. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, and Guan KL (2015). Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw SH, Kuo I, and Eisenberg D (1995). Discovery of the ammonium substrate site on glutamine synthetase, a third cation binding site. Protein Sci 4, 2358–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM (2014). Liver cancer: time to evolve trial design after everolimus failure. Nat Rev Clin Oncol 11, 506–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeppen S, Koehle C, Buchmann A, and Schwarz M (2005). A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis 26, 239–248. [DOI] [PubMed] [Google Scholar]
- Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E, Bronson RT, Kwiatkowski DJ, and Manning BD (2012). Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal 5, ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monga SP (2015). beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 148, 1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Okabe H, Kinoshita H, Imai K, Nakagawa S, Higashi T, Arima K, Uchiyama H, Ikegami T, Harimoto N, Itoh S, et al. (2016). Diverse Basis of beta-Catenin Activation in Human Hepatocellular Carcinoma: Implications in Biology and Prognosis. PLoS One 11, e0152695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil MA, Lee SA, Macias E, Lam ET, Xu C, Jones KD, Ho C, Rodriguez-Puebla M, and Chen X (2009). Role of cyclin D1 as a mediator of c-Met- and beta-catenin-induced hepatocarcinogenesis. Cancer Res 69, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Thomas G, and Volarevic S (2018). Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer 18, 51–63. [DOI] [PubMed] [Google Scholar]
- Preziosi M, Okabe H, Poddar M, Singh S, and Monga SP (2018). Endothelial Wnts regulate β-catenin signaling in murine liver zonation and regeneration: A sequel to the Wnt-Wnt situation. Hepatology Communications 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puliga E, Min Q, Tao J, Zhang R, Pradhan-Sundd T, Poddar M, Singh S, Columbano A, Yu J, and Monga SP (2017). Thyroid Hormone Receptor-beta Agonist GC-1 Inhibits Met-beta-Catenin-Driven Hepatocellular Cancer. Am J Pathol 187, 2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Xu M, Tao J, Che L, Cigliano A, Monga SP, Calvisi DF, and Chen X (2018). Oncogenic potential of N-terminal deletion and S45Y mutant beta-catenin in promoting hepatocellular carcinoma development in mice. BMC Cancer 18, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvartskhava N, Lang PA, Gorg B, Pozdeev VI, Ortiz MP, Lang KS, Bidmon HJ, Lang E, Leibrock CB, Herebian D, et al. (2015). Hyperammonemia in gene-targeted mice lacking functional hepatic glutamine synthetase. Proc Natl Acad Sci U S A 112, 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouissou S, Franconi A, Calderaro J, Letouze E, Imbeaud S, Pilati C, Nault JC, Couchy G, Laurent A, Balabaud C, et al. (2016). Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology 64, 2047–2061. [DOI] [PubMed] [Google Scholar]
- Russell JO, and Monga SP (2018). Wnt/beta-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu Rev Pathol 13, 351–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM (2017). Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A 114, 11818–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Lan BY, Bedolli M, Feng S, and Hebrok M (2006). Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43, 817–825. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, and Sabatini DM (2010). mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104. [DOI] [PubMed] [Google Scholar]
- Steinhart Z, and Angers S (2018). Wnt signaling in development and tissue homeostasis. Development 145. [DOI] [PubMed] [Google Scholar]
- Tan X, Behari J, Cieply B, Michalopoulos GK, and Monga SP (2006). Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology 131, 1561–1572. [DOI] [PubMed] [Google Scholar]
- Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, Jiang L, Fan B, Terracciano L, Armeanu-Ebinger S, et al. (2014). Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 147, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Xu E, Zhao Y, Singh S, Li X, Couchy G, Chen X, Zucman-Rossi J, Chikina M, and Monga SP (2016). Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant beta-catenin. Hepatology 64, 1587–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Zhang R, Singh S, Poddar M, Xu E, Oertel M, Chen X, Ganesh S, Abrams M, and Monga SP (2017). Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 65, 1581–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, et al. (2008). Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 135, 1972–1983, 1983, e1971–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhao L, Fish M, Logan CY, and Nusse R (2015). Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature 524, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lu J, Edmunds LR, Kulkarni S, Dolezal J, Tao J, Ranganathan S, Jackson L, Fromherz M, Beer-Stolz D, et al. (2016). Coordinated Activities of Multiple Myc-dependent and Myc-independent Biosynthetic Pathways in Hepatoblastoma. J Biol Chem 291, 26241–26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Wang X, and Proud CG (2016). mTOR inhibitors in cancer therapy. F1000Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, Lang RA, Williams BO, and Monga SP (2014). beta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation! Hepatology 60, 964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, Poon RT, Blanc JF, Vogel A, Chen CL, et al. (2014). Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA 312, 57–67. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, Cunha AS, Bioulac-Sage P, and Perret C (2007). Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 26, 774–780. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, Bacq Y, Leteurtre E, Paradis V, Michalak S, et al. (2006). Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology 43, 515–524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S3: Related to Figure 2. Demographics and additional information on the 256 HCC cases represented on the 6 TMA from the University of Pittsburgh Medical Center (Pittsburgh, PA, USA).
TABLE S2: Related to Figure 2. Demographics and additional information on 116 HCC samples procured from and assessed at the University of Greifswald (Greifswald, Germany).