

Abstract
Crosstalk between metabolic and survival pathways is critical for cellular homeostasis, but the connectivity between these processes remains poorly defined. We used loss-of-function CRISPR/Cas9 knockout screening to identify metabolic genes capable of influencing cellular commitment to apoptosis, using sensitization to the BCL-2 inhibitor ABT-199 in BCL-2-dependent acute myeloid leukemia (AML) cell lines as a proxy for apoptotic disposition. This analysis revealed metabolic pathways that specifically cooperate with BCL-2 to sustain survival. In particular, our analysis singled out heme biosynthesis as an unappreciated apoptosis-modifying pathway. Although heme is broadly incorporated into the proteome, reduction of heme biosynthesis potentiates apoptosis through the loss of ETC activity, resulting in baseline depolarization of the mitochondrial membrane and an increased propensity to undergo apoptosis. Collectively, our findings chart the first apoptotic map of metabolism, motivating the design of metabolically-engaged combination chemotherapies and nominating heme biosynthesis as an apoptotic modulator in AML.
Graphical abstract
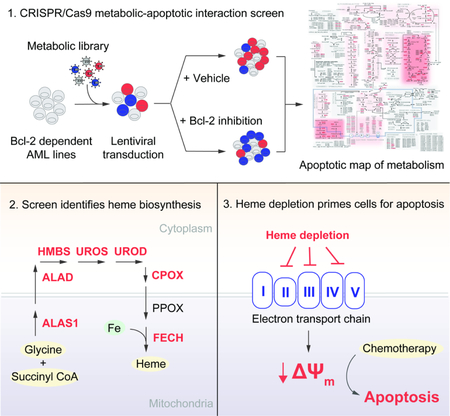
eTOC Blurb:
Using metabolically-focused CRISPR/Cas9 loss-of-function screens, Lin and Xie et al. map the metabolic-apoptotic interface in AML, revealing metabolic pathways that can be therapeutically targeted to augment chemotherapy. In particular, the authors identify heme biosynthesis as an actionable pathway whose prognostic importance is supported by clinical -omics data.
Introduction
To drive unbridled proliferation, cancer cells undergo extensive metabolic rewiring (Hanahan and Weinberg, 2011, DeBerardinis and Chandel, 2016). In theory, knowledge of the interactions between cancer metabolic pathways and survival mechanisms could be therapeutically exploited; in practice, the metabolic pathways most capable of promoting cell death remain largely unknown. One form of programmed death, intrinsic apoptosis, is principally regulated in the mitochondria, the primary metabolic organelle of the cell (Bhola and Letai, 2016, Chipuk et al., 2010). This colocalization makes the mitochondria a probable site for metabolic-apoptotic crosstalk, consistent with studies that demonstrate how rearrangement of mitochondrial networks affects both metabolism and intrinsic apoptosis (Martinou and Youle, 2011). Various metabolic pathways are already known to affect cellular apoptotic potential (Green et al., 2014). For instance, increased pentose phosphate pathway (PPP) flux indirectly resists caspase activation through NADPH-mediated redox inactivation of cytochrome c (Vaughn and Deshmukh, 2008). Permeabilization of the mitochondrial outer membrane (MOMP) by the apoptotic executor protein BAX has been causally linked to ceramide and sphingosine metabolism (von Haefen et al., 2002, Chipuk et al., 2012). Studies have also described how components of the apoptotic network can modulate metabolism (Andersen and Kornbluth, 2013, Vander Heiden and Thompson, 1999). Phosphorylation of pro-apoptotic proteins NOXA and BAD is known to promote glucose consumption through the PPP and glycolysis, respectively (Lowman et al., 2010, Danial et al., 2008), and the anti-apoptotic protein BCL-XL has been linked to oxidative phosphorylation through regulation of ATP synthase (Alavian et al., 2011, Chen et al., 2011).
Nevertheless, a full enumeration of the nodal interface between metabolic and apoptotic signaling has yet to be reported, although broad evidence for it can be found in studies that target metabolic processes to augment chemotherapy (Samudio et al., 2010, Pardee et al., 2018). This understanding would stand apart from existing knowledge of proliferative metabolic dependencies in cancer (Alvarez et al., 2017, Possemato et al., 2011) and could be used to design metabolically-driven combination therapies or identify cancers with metabolic alterations that predispose them to apoptosis. Motivated by this need, we used a CRISPR loss-of-function library targeting metabolic enzymes and transporters to screen a pair of BCL-2-dependent AML cell lines in the presence and absence of the BCL-2 inhibitor ABT-199. In this configuration, ABT-199 was deployed as a molecular probe for apoptosis. In screens treated with ABT-199, depletions within the compositional landscape of deleted genes, deconvoluted through deep sequencing and normalized to a vehicle-treated control screen, were interpreted as apoptotically reactive metabolic nodes. Using this approach, we produced the first apoptotic map of metabolism, a catalog of the metabolic pathways that are capable of modulating cellular commitment to apoptosis. We demonstrate how this knowledge can be used to uncover molecular mechanisms linking metabolism, apoptosis, and chemotherapeutic response and template the design of metabolically-engaged combination therapies.
Results
Designing and Validating a CRISPR/Cas9 Screening Library
We assembled a metabolically-focused CRISPR/Cas9 loss-of-function screening library comprised of 11,610 short guide RNA (sgRNA) constructs targeting 2322 metabolic enzymes and transporters (5 guides per gene) as well as 50 non-targeting control guides (Table S1). Among the genes are 150 control genes, selected for their essentiality or dispensability (Hart et al., 2014). Our library was cloned into an established lentiviral system (Shalem et al., 2014); all sgRNA sequences were retrieved from a published genome-wide library (Wang et al., 2014). Two AML cell lines, MOLM-13 and THP-1, were transduced with lentivirus, subjected to puromycin selection, treated with ABT-199 or DMSO (vehicle), and sampled weekly through 2 weeks of treatment (Figure 1A). The selected doses of ABT-199 corresponded to roughly 20–30% loss of cell viability in a three-day dose-response assay. We determined the composition of the sgRNA pools by deep sequencing. The three most depleted constructs per gene were averaged to produce a gene-level three score (TS) (Table S2). To validate our approach, we analyzed control genes by comparing the final and initial sgRNA pools from vehicle-treated screens, focusing on known-essential genes, known-non-essential genes, and non-targeting controls (Figure 1B, Figure S1A). Together, these findings affirm the basic functions of our CRISPR/Cas9 loss-of-function screening library.
Figure 1: Validation of CRISPR/Cas9-based screens for the identification of metabolic sensitizers to ABT-199-induced apoptosis.

(A) CRISPR/Cas9 loss-of-function screening strategy.
(B) Replicate-to-replicate comparison of gene-level essentiality phenotypes in MOLM-13. Essential controls are shown in red, non-essential controls in yellow, and non-targeting controls in blue.
(C) Gene-level representation of ABT-199 sensitization phenotypes from both cell lines, ranked by their log2-transformed three score (TS) (ABT-199-treated / DMSO-treated). Apoptotically reactive genes are denoted by the red box.
(D) Top-scoring genes (red), ranked by their log2(TS) values, displayed against each gene’s corresponding essentiality score (yellow).
(E) Results of pre-ranked GSEA performed with gene ontology gene sets, ordered by the negative log2 of gene set q-values.
Generation of a functional metabolic-apoptotic interaction map in AML cells
To understand which metabolic pathways influence apoptosis, we sought to identify which genes specifically depleted or enriched in ABT-199-treated populations relative to vehicle-treated populations. While the majority of screened genes did not meaningfully interact with ABT-199, a subset of 241 genes resided below the inflection point of the curve. This cutoff emphasizes approximately 10% of the screen and corresponds to a log2(TS) of −0.914 (Figure 1C). All genes scoring beyond this threshold yielded multiply corrected p-values less than 0.05 (Benjamini-Hochberg). Further study of this subset revealed little relationship between a given gene’s essentiality and its ability to potentiate cell death induced by ABT-199 (Pearson’s r = 0.1799) (Figure 1D), indicating that the dataset is distinct from existing studies that credential essential metabolic pathways in cancer.
To identify apoptotically reactive pathways, we performed pre-ranked Gene Set Enrichment Analysis (GSEA) on library genes, ordered by their capacity to sensitize both cell lines to ABT-199 (Figure 1E, Table S3) (Subramanian et al., 2005). Among the top-scoring pathways, GSEA highlighted oxidative phosphorylation, heme biosynthesis, and nucleoside biosynthesis (Figure S1B). Unfortunately, GSEA is limited by the scope of manually-curated gene sets. To better appreciate the connectivity of the reactive metabolic nodes identified in our screen, we overlaid our data atop a map of cellular metabolism (Figure 2). This allowed for the identification of scoring metabolic pathways, where greater than 20% of constituent genes scored individually. Notably, glycolysis and the PPP scored, as did folate processing and other pathways. Equally notable are complete metabolic pathways, including fatty acid β-oxidation, branched-chain amino acid catabolism, and steroid synthesis, which do not promote apoptosis when any of their components are knocked out. Finally, the uncoupling of genetic essentiality and apoptotic sensitization described above on a gene level is also evident on a pathway level, especially in pathways that score as apoptotic sensitizers (Figure S1C). This dataset represents the first comprehensive metabolic-apoptotic interaction map.
Figure 2: Metabolic-apoptotic interaction map.

Scoring library genes are highlighted in red; non-scoring library genes in black; pathway-relevant non-library genes in gray. Pathways with > 80% scoring genes are shaded dark red; pathways with 20 – 80% scoring genes are light red; pathways with < 20% scoring genes are not shaded.
Using the interaction map to predict apoptotically reactive metabolic paradigms
First, we used the interaction map to identify essential pathways whose ablation also specifically potentiated apoptosis. Two of the most reactive pathways, de novo purine and pyrimidine biosynthesis, are essential (Figure 3A,B). Antimetabolites targeting these pathways, such as 5-fluorouracil (5-FU), 6-mercaptopurine (6-MP), and gemcitabine, exhibit single-agent potency in many tumors. However, our data point beyond their roles as simple drug targets and speaks to their potential as therapy-augmenting targets, consistent with the role antimetabolites have long played in anchoring combination therapies (Peters et al., 2000, Smorenburg et al., 2001). We validated this prediction using dose-response assays that combined sublethal doses of antimetabolites with ABT-199 (Figure 3C, Figure S2A). Further, we phenocopied the effect of antimetabolite treatment on ABT-199 sensitivity using CRISPR-mediated knockouts of thymidylate synthetase and phosphoribosyl pyrophosphate, the respective targets of 5-FU and 6-MP (Figure 3D, Figure S2B). This suggests that the inhibition of de novo nucleotide synthesis is sufficient to prime AML cells to ABT-199-induced cell death.
Figure 3: Interaction map predicts apoptotic sensitization through targeting of nucleotide synthesis, TCA-associated nodes, and redox regulation.

Pathway genes that scored as sensitizers are highlighted in red.
(A, E, I) Gene-level representation of ABT-199-sensitization, ranked by log2(TS) values. Highlighted genes correspond to: (A) purine and pyrimidine biosynthesis, (E) TCA cycle, and (I) redox regulation.
(B, F, J) Annotated pathway schematics for: (B) purine/pyrimidine biosynthesis, (F) TCA cycle, and (J) redox regulation. Pharmacologic interrogations are shown in blue.
(C, G, K) Validation of pharmacologic sensitization of: (C) purine/pyrimidine biosynthesis, (G) TCA nodes, and (K) redox regulation to ABT-199-mediated cell death using 72hr growth inhibition (GI50) assays. Shown are relative 72hr GI50 values derived from ABT-199 dose-response curves in the presence and absence of background metabolic drugs, used at sublethal doses based on single-drug GI50 curves (Figure S2A).
(D, H, L) Fold change in 72hr GI50 values derived from ABT-199 dose-response curves for OCI-AML2 cells transduced with 2 sgRNAs targeting (D) TYMS or PPAT; (H) SLC25A1, GLS, OGDH, DLST, or DLD; and (L) GPX4, GCLC, HMGCR, or SEPHS1, relative to a non-targeting sgRNA.
*** p ≤ 0.001, **** p ≤ 0.0001 by student’s t-test; n=3. Data are mean ± SD.
Next, we observed that the TCA cycle, which did not score strongly as a whole, harbored reactive focalities: two subunits of the succinyl dehydrogenase complex scored, as did citrate synthase, aconitase, the mitochondrial citrate transporter CIC (SLC25A1), and the cytoplasmic citrate lyase (ACLY) (Figure 3E,F). Consistent with the notion that glutamine can anapleurotically replenish the TCA cycle (Altman et al., 2016), glutaminase (GLS) and three glutamine transporters (SLC1A5, SLC38A1, SLC38A2) scored (Figure 3E,F). These results frame a classic paradigm of metabolic rewiring in cancer: cells shunt carbon from glycolysis and glutaminolysis into citrate, which is transported by CIC and cleaved by ACLY to yield oxaloacetate and acetyl-CoA (Figure 3F), feeding de novo fatty acid biosynthesis and providing lipids for proliferating tumor cells (Menendez and Lupu, 2006, Rohrig and Schulze, 2016). Our screen data point to the concerted production of acetyl-CoA through the TCA cycle as reactive but stop short of implicating an anabolic role for acetyl-CoA. We validated the predicted cooperativities pharmacologically, combining ABT-199 with sublethal doses of the glutaminase inhibitor Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) and the CIC inhibitor 1,2,3-benzenetricarboxylic acid (BTA) (Figure 3G, Figure S2A), and genetically, using CRISPR knockout lines (Figure 3H, Figure S2B).
In some cases, the interaction map specified the apoptotic-reactivity of metabolic complexes within pathways. Dihydrolipoamide dehydrogenase (DLD), which encodes the E3 subunit of the pyruvate (PDH), a-ketoglutarate (aKGDH), and branched-chain amino acid dehydrogenase (BCKDH) complexes, scored as one of the most reactive genes in our screen. Between the PDH and BCKDH complexes, DLD was the only subunit that scored as reactive (Figure 3F). Meanwhile, OGDH and DLST, which encode the E1 and E2 subunits of the aKGDH complex, both scored, implying that this entire complex is reactive. CRISPR-mediated knockout of DLD, DLST, and OGDH sensitized cells to ABT-199 (Fig 3H, Figure S2B). These data speak to the ability of the screen to discriminate between related complexes.
The interaction map can also be used to synthesize disparate pathways. GPX4, a glutathione peroxidase that catalyzes the reduction of lipid peroxides, was one of the top sensitizers in our screen. Its catalytic activity requires a selenocysteine moiety, synthesized by selenocysteine synthase via selenophosphate synthetase (SEPHS1, SEPHS2). Selenocysteine incorporation entails isopentenylation of Sec-tRNA, which involves isopentenyl pyrophosphate, a mevalonate pathway intermediate (Fradejas et al., 2013). GPX4 also requires glutathione, synthesized by glutamate cysteine ligase (GCL) and glutathione synthetase (GSS). GSS and GCLC, which encodes the catalytic subunit of GCL, both scored. Sublethal inhibition of GSS using buthionine sulfoximine (BSO) and of HMG-CoA-reductase (HMGCR) by atorvastatin (ATS) was sufficient to sensitize AML cells to ABT-199-induced cell death (Figure 3K, Figure S2A). The latter point is consistent with a recent study demonstrating the ability of statins to potentiate the effect of ABT-199 (Lee et al., 2018). Both CRISPR-mediated genetic knockout and pharmacologic inhibition of each metabolic arm that feeds into GPX4, along with inhibition of GPX4 itself by RSL-3 or treatment with erastin, an inhibitor of the upstream cystine-glutamate antiporter system Xc-, enhanced ABT-199-induced cell death (Figure 3K,L, Figure S2A,B). The convergence of pro-apoptotic pathways upon GPX4 implicated ferroptosis, a lipid peroxide-driven form of oxidative cell death (Figure 3I, J) (Dixon et al., 2012). However, RSL-3 and erastin treatment potentiated ABT-199-induced cleavage of poly (ADP-ribose) polymerase (PARP) in a manner rescuable by the pan caspase inhibitor QVD (Figure S2C). We also observed that the potentiating effect of erastin on ABT-199 was rescuable by the antioxidant N-acetylcysteine (NAC) but not by the iron chelator deferoxamine (DFO) or by the inhibitors of lipid peroxidation ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1) (Figure S2D), while the potentiation of ABT-199 by RSL-3 was rescuable by treatment with each of NAC, DFO, Fer-1, and Lip-1, as expected in ferroptosis (Figure S2D). These data indicate that regulation of oxidative stress factors into cellular commitment to apoptosis but do not rule out a contribution of ferroptosis in the case of RSL-3 and its target GPX4.
Heme biosynthesis is downregulated in acute myeloid leukemia
Prior studies have established that AML cells often exhibit greater sensitivity to apoptosis-inducing therapies, including BCL-2 inhibitors, than other hematopoietic cells (Pan et al., 2014), but the underlying mechanisms are poorly understood. In order to assess whether downregulation of any of the pathways highlighted by the interaction map may contribute to this phenomenon, we analyzed published microarray data describing samples taken from healthy bone marrow or from patients with non-leukemic conditions (HBM), pre-leukemic myelodysplastic syndrome (MDS), and AML (Haferlach et al., 2010). Unsupervised clustering of metabolic pathway genes revealed that 1) the majority of genes were not differentially expressed between HBM, MDS, and AML samples, and 2) a subset of clustered genes were distinguished by their downregulation in AML relative to HBM / MDS (Figure 4A, denoted by red box). Notably, this cluster included genes encoding 6/8 enzymes in the heme biosynthetic pathway, also among the strongest scoring pathways in both GSEA and curated analyses (Figure 1E, Figure 2). Focusing on the heme biosynthetic genes, we observed relatively equivalent expression between HBM and MDS samples but significantly lower expression in AML samples, implying a relative reduction of heme biosynthesis in AML cells (Figure 4B). Global metabolomics performed on serum samples taken from a cohort of pediatric AML patients on diagnosis and upon remission also revealed that the latter contained higher levels of 5-aminolevulinic acid, the product of ALAS1, than measured at diagnosis (Figure 4C, Figure S3A).
Figure 4: Heme biosynthesis is downregulated in clinical AML.

(A) Heatmap depicting expression of metabolic pathway genes in samples from healthy bone marrow (HBM) n=74, myelodysplastic syndrome (MDS) n=206, or acute myeloid leukemia (AML) n=542. Inset: heme biosynthetic genes highlighted in red.
(B) Expression pattern of heme biosynthetic pathway genes in samples from HBM (n=74), MDS (n=206), or AML (n=542). **** p ≤ 0.0001 by Welch’s t-test.
(C) Comparison of 5-aminolevulinic acid levels from pediatric AML patients taken at diagnosis (n = 20) vs. upon remission (n = 7). ** p ≤ 0.01 by Welch’s t-test.
To define the relationship between heme biosynthetic gene expression and therapeutic response, we examined RNAseq data from 240 pediatric patients enrolled across three Children’s Oncology Group AML clinical trials. Gene expression analysis at diagnosis showed that patients who would later fail to achieve remission following two rounds of induction chemotherapy (nonresponders) expressed higher levels of heme biosynthetic genes compared to patients who were able to achieve remission (responders), consistent with the idea that AML cells that fail to downregulate heme biosynthesis may harbor intrinsic resistance to apoptosis-inducing chemotherapy (Figure S3B). An unbiased analysis of this dataset revealed that, across the same set of metabolic genes evaluated above (Figure 4A), heme biosynthetic genes were systematically upregulated in nonresponders (Figure S3C). We quantified the proportion of genes in each of the major metabolic pathways that exhibited significantly different expression in nonresponders vs. responders. Heme biosynthesis contained the highest proportion of significantly upregulated genes in patients that failed to achieve remission (Figure S3D). Together, these data support the possibility that heme homeostasis plays a role in dictating therapeutic response. The relative downregulation of heme biosynthesis we observe in AML vs. HBM, which coheres with the prevailing view that regulation of heme biosynthesis is coordinate with erythroid differentiation (Chiabrando et al., 2014), represents an explanation for the apoptotically-primed nature of AML cells and may partially account for the insensitivity of normal hematopoietic stem cells to BH3 mimetics. Conversely, AML cells that do not downregulate heme biosynthesis may be insufficiently primed for apoptosis, putting those patients at risk for failing pro-apoptotic induction chemotherapy.
Identifying a heme-centered pro-apoptotic metabolic network
Genes encoding 7/8 enzymes in the heme biosynthetic pathway scored in the screen (Figure 5A, B). To validate the proapoptotic effects of disrupting heme biosynthesis, we knocked out ALAS1, aminolevulinic acid dehydratase (ALAD), and ferrochelatase (FECH), encoding the first, second, and final steps of heme biosynthesis, respectively. In two cell lines, these genetic knockouts promoted cell death induced by ABT-199 (Figure 5C, Figure S2B). Next, we used succinylacetone (SA), an inhibitor of ALAD, to impede pathway flux. In a panel of AML lines, SA reduced cellular heme levels within 48 hours (Figure 5D, Figure S4A), and SA pretreatment potentiated apoptotic cell death induced by ABT-199 (Figure 5E). This effect was phenocopied with N-methyl protoporphyrin (NMP), a transition state analog of protoporphyrin IX (PPIX) that acts as an inhibitor of FECH (Figure S4B). The potentiation engendered by SA can be rescued by supplementing PPIX itself (Figure 5F). PPIX fails to rescue NMP, as both are substrates for FECH.
Figure 5: Modulation of intracellular heme potentiates ABT-199-induced apoptosis.

(A) Gene-level representation of ABT-199-sensitization, ranked by log2(TS) values. Heme biosynthesis genes and iron transporters are highlighted.
(B) Annotated pathway schematic of heme biosynthesis. Pharmacologic interrogations are shown in blue.
(C) Fold change in 72hr GI50 values, derived from ABT-199 dose-response curves for OCI-AML2 cells transduced with 2 sgRNAs targeting ALAS1, ALAD, or FECH, relative to a non-targeting sgRNA.
(D) Relative heme content after 48hr treatment with SA in six AML cell lines.
(E) Fold change in 72hr GI50 values derived from ABT-199 dose-response curves treated in the presence and absence of SA. Absolute GI50 values normalized to viability in SA alone.
(F) ABT-199 dose-response curves in OCI-AML2 cells pre-treated with background doses of 250µM SA or 250µM SA + 5µM PPIX for 48hr, then treated with an ABT-199 dilution series for 72h. Dose response curves normalized to the viability of SA alone or SA and PPIX alone.
(G) Immunoblots of cleaved PARP, cleaved caspase 3, and β-actin in cells treated for 48hr with SA (OCI-AML2: 100µM, MOLM-13: 750µM, THP-1: 2mM, MV4–11: 1mM, KG-1α: 1.5mM, HL-60: 2mM), 8hr treatment with ABT-199 (OCI-AML2: 50nM, MOLM-13: 30nM, THP-1: 200nM, MV4–11: 100nM, KG-1α: 25nM, HL-60: 50nM), or the combination.
(H) Percentage annexin V+ in cells treated with ABT-199 for 24hr (OCI-AML2: 75nM, MOLM-13: 25nM), SA for 72hr (OCI-AML2: 250µM, MOLM-13: 2mM), or the combination. Percentages of annexin V+ drug-treated cells normalized to signal from vehicle-treated cells.
(I) Immunoblots of Bax, Bak, and VDAC1 in OCI-AML2 cells treated with SA 100µM for 48hr, ABT-199 50nM for 8h, or the combination. Shown are mitochondrial fractions only; VDAC1 is shown as a mitochondrial loading control.
(J) ABT-199 dose-response curves in OCI-AML2 and MOLM-13 cells expressing shGFP or one of two shRNAs targeting Bax, treated in the presence and absence of SA (OCI-AML2: 300µM, MOLM-13: 600µM). Immunoblots of Bax.
(K) Quantification of OCI-AML2 cell populations cultured continuously in vehicle, SA 100µM, ABT-199 1.0µM, PPIX 1.0µM, or the stated combinations. Total cell number is an extrapolation from weekly growth rates.
(L) OCI-AML2 cells cultured continuously in vehicle, SA 100µM, cytarabine 100nM, PPIX 1.0µM, or the stated combinations.
(M) Percentage annexin V+ in primary AML patient cells treated with ABT-199 and/or SA for 24hr. n=3 for all samples except DP0285 (n=1).
** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 by student’s t-tests; n=3. Data are mean ± SD.
Across a panel of cell lines, SA treatment promoted ABT-199-induced cleavage of PARP and caspase 3 (Figure 5G). These results were corroborated by annexin positivity (Figure 5H). Furthermore, cells treated with both ABT-199 and SA revealed cooperative BAX accumulation and oligomerization in mitochondrial isolates, suggesting apoptosis driven by formation of BAX (but not BAK) pore complexes (Figure 5I). If SA potentiates ABT-199-induced cell death by modulating the apoptotic threshold, its effect should be rescuable by knockout of downstream apoptotic machinery and extrapolatable to additional drugs. Hairpin knockdown of BAX, which is preferentially bound by BCL-2 over BAK (Kale et al., 2018), neutralized the SA + ABT-199 combination (Figure 5J). To demonstrate SA’s broad pro-apoptotic potential as a combination chemotherapeutic partner, we continuously exposed AML cells to either ABT-199 or cytarabine, the backbone of AML induction therapy, both with and without SA. In these models, the addition of SA suppressed outgrowth through ten weeks, precluding the acquisition of resistance to either ABT-199 or cytarabine (Figure 5K,L). This phenotype was rescuable by PPIX.
Finally, we turned to primary AML patient samples as a model for clinical disease. We treated a panel of AML patient samples with ABT-199 +/− SA for 24 hours before quantifying the effect of SA on ABT-199-induced annexin positivity using flow cytometry. In each sample, we observed the inability of SA alone to drive apoptosis and the potentiating effect of SA on ABT-199-induced apoptosis (Figure 5M). These data suggest that the effect of SA treatment on ABT-199 sensitivity is conserved in AML patient samples, and likely represents an underestimate because this mechanism requires the depletion of both heme and heme-containing proteins (which takes 24–48 hours of SA exposure, while the viability of our patient samples was limited to 24–36 hours).
We also traced the pathways that produce the heme building blocks glycine, succinyl-CoA, and iron. Succinyl-CoA is produced by the reactive aKGDH complex; our screen points away from alternative sources, such as oxidation of odd chain fatty acids and branched chain amino acids. ALAS1 requires pyridoxal phosphate (PLP), a vitamin B6-derived cofactor produced by pyridoxal kinase (PDXK). PDXK scored and treatment of AML cells with 4-deoxypyridoxine hydrochloride (4-DPH), a vitamin B6 analog that inhibits PDXK, synergized with ABT-199 (Figure S4C). Iron, which is incorporated into PPIX by FECH to form heme b, is tightly regulated by the cell. DMT1 (SLC11A2) is the main entry point for nontransferrin-bound iron into the cell while mitoferrin 1 (SLC25A37) and mitoferrin 2 (SLC25A28) import ferrous iron into the mitochondrial matrix from the mitochondrial intermembrane space (Muckenthaler et al., 2017, Crielaard et al., 2017). All three iron importers scored (Figure 5A,B). In contrast, SLC40A1, which encodes the primary cellular iron exporter ferroportin, and HEPH, which encodes hephaestin, a ferroxidase that cooperates with ferroportin to mediate iron efflux, both scored as anti-apoptotic when knocked out (Figure 5A,B). Treatment of AML cells with the iron chelator deferoxamine (DFO) similarly potentiated the effect of ABT-199 (Figure S4C). Together, these results suggest that iron deficiency, especially in the mitochondria, where ferrous iron is incorporated into heme, moves the cell closer to apoptosis.
Inhibition of heme biosynthesis potentiates apoptosis through suppression of ETC function
The heme proteome serves varied functions centered about heme’s ability to act as an electron source, sink, or shuttle. To specify the pro-apoptotic consequence of inhibiting heme biosynthesis, we queried the UniProt database for heme-associating proteins, returning 106 unique genes, 83 in our library (Figure 6A). Each of the four top-scoring genes in this analysis (COX15, CYCS, COX10, SDHC) are associated with oxidative phosphorylation and the ETC; CYCS encodes cytochrome c, SDHC encodes the heme-incorporating subunit of succinate dehydrogenase (complex II), and COX15 and COX10 are required for the conversion of heme b to heme a, an obligate cofactor of complex IV. Thus, we hypothesized that heme biosynthesis exerts its apoptosis-sensitizing effects predominantly through modulation of the ETC.
Figure 6: Targeting heme biosynthesis primes the cell for apoptosis by disrupting electron transport chain integrity.

(A) 83 library genes that associate with heme, ranked by log2(TS) score.
(B) Schematic showing incorporation of heme species into electron transport chain complexes. Reactive genes are highlighted in red.
(C) Fold change in 72hr GI50 values derived from ABT-199 dose-response curves for OCI-AML2 cells transduced with 2 sgRNAs targeting COX10 or COX15, relative to a non-targeting sgRNA.
(D) Immunoblot of subunits of ETC complexes I – V in cells treated with SA or SA + PPIX 2.5µM for 48hr (OCI-AML2: 250µM, MOLM-13: 750µM).
(E) Basal OCR as measured by Seahorse XF Analyzer’s Mito Stress Test. Cells were treated with SA (OCI-AML2: 100µM, MOLM-13: 750µM) or SA and PPIX (OCI-AML2: 2.5µM, MOLM-13: 1.0µM).
(F) Enzymatic activity of complex IV in cells treated with SA (OCI-AML2: 250µM, MOLM-13: 750µM) or complex IV inhibitor sodium azide (OCI-AML2: 1mM, MOLM-13: 2mM) for 48hr. Enzymatic activity normalized to vehicle treatment.
(G) TMPD-driven oxygen consumption rate as measured by Seahorse XF Analyzer. Cells were treated for 48hr with SA (OCI-AML2: 250µM, MOLM-13: 750µM), resuspended in minimal media, and introduced to TMPD/ascorbate. Shown is the maximal OCR.
(H) Immunoblots of cleaved PARP, cleaved caspase 3, and β-actin in cells treated for 48h with SA (OCI-AML2: 100µM, MOLM-13: 750µM), 48h with sodium azide (OCI-AML2: 1mM, MOLM-13 2mM), 8h with ABT-199 (OCI-AML2: 50nM, MOLM-13: 30nM), or the stated combinations.
(I) JC-1 signal reported as ratios of red/green fluorescence normalized to vehicle, following 48hr treatment of cells with H2O (vehicle), SA (OCI-AML2: 250µM, MOLM-13: 750µM), or sodium azide (OCI-AML2: 500µM, MOLM-13: 2mM).
(J) JC-1 signal following 24hr treatment of primary AML patient cells with SA. All data reported are n =1.
(K) Immunoblots of cytochrome c, AIF, Smac, HtrA2, XIAP, cleaved PARP, cleaved caspase 3, and β-actin. OCI-AML2 cells were treated with DMSO, 100µM SA, 20nM ABT-199, or 100µM SA + 20nM ABT-199 for 24hr, collected, and fractionated. Cytoplasmic fractions are shown for drug-treatments. Cytoplasmic versus membrane/organellular distribution in untreated cells shown for reference.
(L) Model of heme-dependent apoptotic sensitization in AML cells. Heme depletion (right) drives baseline MMP depolarization through destabilization of heme-containing ETC complexes, sensitizing cells to MOMP and consequent release of pro-apoptotic factors.
* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 by student’s t-tests; n=3. Data are mean ± SD.
In the final step of heme synthesis, FECH catalyzes the formation of heme b, which is incorporated into complexes II and III of the ETC. Heme b is also subject to COX10-catalyzed farnesylation to form heme o, which is oxidized to heme a by ferredoxin (FDX1), ferredoxin reductase (FDXR), and COX15 (Moraes et al., 2004, Sheftel et al., 2010). Each of these enzymatic steps scored in our screen (Figure 6B) and were validated with genetic knockout (Figure 6C, Figure S2B). This pattern of genetic sensitization implies that focal loss of heme a biosynthesis is sufficient to sensitize to ABT-199, an explanation that is also consistent with sensitization driven by upstream depletion of heme b. In mammalian cells, the function of heme a as a prosthetic group is restricted to its known roles in cytochrome a and cytochrome a3 as part of complex IV. Beyond genes involved in the assembly of heme a, many of the complex IV assembly factors themselves scored highly in the screen, including SCO1, SCO2, COX10, COX11, COX15, and LRPPRC (Figure 2, Table S2).
To study the ability of heme depletion to affect the ETC, we treated cells with SA for 48 hours and monitored their ability to affect the integrity of heme-containing complexes II, III, and IV. We observed a relative preferential effect of heme depletion on complex IV versus complexes II and III by western blot (Figure 6D, Figure S5A), consistent with previous reports in astrocytoma cells (Atamna et al., 2002). This depletion could be rescued by administration of PPIX (Figure 6D). Treatment with NMP elicited the same effect but is not rescuable by PPIX (Figure S5B). Complexes I and V do not contain heme and were not depleted with SA or NMP treatment. To establish functional correlates, we measured cellular respiratory capacity. 48-hour SA treatment depressed basal cellular respiration in a manner partially rescuable by PPIX (Figure 6E). Additionally, the enzymatic activity of complex IV, along with complexes II and III, was attenuated in SA-treated cells (Figure 6F, Figure S5C). To further substantiate the loss of complex IV activity, we measured oxygen consumption driven by N,N,N’,N’-Tetramethyl-1,4-phenylendiamine (TMPD), an exogenous complex IV electron donor. SA-treated AML cells exhibited reduced complex IV-mediated oxygen consumption (Figure 6G).
To determine whether heme depletion’s pro-apoptotic effects are engendered through ETC disruption, in particular the two heme-containing, proton-pumping complexes III and IV, we treated AML cells with two and three-body combinations of ABT-199, SA, the complex III inhibitor antimycin A, and the complex IV inhibitor sodium azide. As expected, cells treated with either antimycin A and ABT-199, sodium azide and ABT-199, or SA and ABT-199 demonstrated synergistic induction of apoptosis by PARP or caspase 3 cleavage (Figure 6H, Figure S5D). Importantly, treatment with either antimycin A or sodium azide blocked further apoptotic sensitization by SA, evidenced through PARP and caspase 3 cleavage (Figure 6H, Figure S5D) and annexin V positivity (Figure S5E). The strength of our screening data (Figure 6B), taken with empiric data which demonstrates that complex IV is more sensitive to heme depletion (Figure S5A), initially led us to believe that complex IV may play a predominant role in the heme depletion phenotype. However, because individual inhibition of either complex is sufficient to fully prime the cell for ABT-199-induced apoptosis (likely a reflection of their successive arrangement), it is difficult to properly disentangle the relative contributions of targeting either of the two complexes relative to the full apoptotic potentiation phenotype. Regardless, these findings suggest that it is the disruption of heme-containing ETC complexes III and IV that account for the apoptosis-potentiating effect of heme depletion.
Heme depletion and resultant ETC dysfunction potentiates MOMP
To understand how ETC disruption by heme depletion promotes apoptosis, we first examined mitochondrial reactive oxygen species (ROS), which can be induced by ETC disruption (Murphy, 2009, Liu et al., 2002). However, treatment of AML cells with SA did not increase either cellular ROS or mitochondrial ROS (Figure S5F), and the addition of ROS sequestrants failed to reverse the observed sensitization between SA and ABT-199 (Figure S5G). This result has precedent, as disruption of COX activity was shown not to increase ROS production (Fukui et al., 2007). Moreover, heme acts as a cofactor in both ROS-producing and ROS-scavenging enzymes such as NADPH oxidase and catalase, respectively. An alternative explanation could concern a specific association between BCL-2 and a member(s) of the ETC (Chen and Pervaiz, 2010). However, this mechanism is unlikely to explain the observed effects, as SA was shown to similarly sensitize non-BCL-2-dependent cancers to their cognate BH3 mimetics. Sensitization was observed in a BCL-XL-dependent, JAK2-dependent model of myeloproliferative disease, a BCL-XL-dependent Burkitt’s lymphoma model, and a BCL-2/MCL-1-dependent murine B lymphocyte cell line (Figure S5H).
Another possibility may involve an imbalance in the pro-apoptotic vs. anti-apoptotic BCL-2 family members (Lin et al., 2016, Pan et al., 2014) Across a panel of BCL-2 family members in a panel of AML cell lines, we detected no significant differences in protein quantity between cells treated with vehicle or SA (Figure S6A). It is also possible that SA treatment may prime BAX itself. To test this, we probed SA-treated cells for a cryptic N-terminal epitope of BAX which is exposed upon pro-apoptotic activation; we observed no increased exposure of this epitope with SA treatment (Figure S6B). Separately, immunoprecipitation of BCL-2 revealed no increase in BCL-2-bound BAX in cells treated with SA, suggesting that the increased effect of ABT-199 is not simply due to increased liberation of BAX from BCL-2 (Figure S6C).
Instead, we considered the possibility that heme depletion-mediated disruption of complexes III and IV directly influences the mitochondrial membrane. Prior work has demonstrated the capacity for ETC dysregulation to affect mitochondrial membrane potential (MMP), thereby influencing the apoptotic threshold (Trotta et al., 2017). To test this, we treated cells with SA and measured their ability to aggregate JC-1 as a proxy for MMP. Cells treated with SA or subjected to genetic knockout of heme biosynthetic enzymes have a durably-depolarized MMP (Figure 6I, Figure S6D). This baseline depolarization may increase the dependency of the surviving cells on their anti-apoptotic BCL-2 family proteins, consistent with reports that (R)-2-HG-mediated complex IV inhibition sensitizes to BCL-2 inhibition. (Chan et al., 2015) To determine whether MMP dissipation is itself sufficient to sensitize to ABT-199, we treated a panel of cell lines with ABT-199 in the presence or absence of the mitochondrial uncouplers carbonyl cyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP) and carbonyl cyanide m-chlorophenyl hydrazine (CCCP). In each line, exposure to FCCP or CCCP engendered baseline MMP depolarization and elicited a more potent response from treatment with ABT-199 (Figure S6E,F). Finally, each of three primary AML samples tested exhibited a lowered MMP in response to SA treatment (Figure 6J), suggesting that this mechanism is conserved in primary AML samples.
These findings implied that SA-induced loss of MMP may be reflective of a broader loss of mitochondrial membrane integrity that could potentiate ABT-199-induced MOMP and cytochrome c release. BH3 profiling revealed that cells treated with SA were more strongly depolarized in response to equivalent exposures to peptides derived from the BH3-only molecules BIM and BID (Figure S6G). Because inhibition of BCL-2 by ABT-199 liberates pro-apoptotic BIM and BID (Pan et al., 2014), which then activate BAX and BAK, response to BIM and BID peptides in BH3 profiling is a proxy for treatment with ABT-199. Next, we demonstrated the effect of SA treatment on ABT-199-induced MMP depolarization by isolating mitochondria from cells treated with either SA or vehicle and subjecting those cells to direct treatment with either ABT-199 or vehicle. Mitochondria from cells pretreated with vehicle did not depolarize in response to low doses (10nM, 20nM) of ABT-199 (Figure S6H). In contrast, mitochondria from cells pretreated with SA exhibited dose-dependent depolarization in response to low doses of ABT-199 (Figure S6H). These data indicate that SA treatment lowers the threshold for mitochondrial depolarization, whether initiated through ABT-199 treatment or direct acquaintance with BIM/BID peptide.
Finally, we confirmed the potentiation of ABT-199-induced MOMP via SA by assaying for the translocation of mitochondrial proteins to the cytoplasm. In SA + ABT-199-treated cells, we observed a slight increase in drug-induced cytoplasmic cytochrome c relative to ABT-199 alone (Figure 6K), likely an underestimate given that cytochrome c is a hemeprotein and heme depletion decreased total cytochrome c (Figure S6I). We also considered the cytoplasmic release of other pro-apoptotic mitochondrial proteins: second mitochondria-derived activator of caspases (SMAC), apoptosis-inducing factor (AIF), and high-temperature requirement A2 (HtrA2). These factors were cooperatively released by the combination of SA and ABT-199 (Figure 6K,L). Each plays a distinct proapoptotic role: AIF drives chromatin condensation and subsequent DNA fragmentation; cytoplasmic SMAC binds inhibitor of apoptosis proteins (IAPs), stoichiometrically liberating caspases to drive apoptosis; cytoplasmic HtrA2 catalyzes enzymatic cleavage of IAPs. Consistent with the observed release of SMAC and HtrA2, we also observed cooperative cleavage of X-linked inhibitor of apoptosis protein (XIAP), caspase-3, and PARP in cells treated with both SA and ABT-199 (Figure 6K,L). Together, these data suggest that SA-driven ETC dysfunction leads to the loss of mitochondrial membrane integrity, potentiating ABT-199-induced MOMP and resultant apoptosis.
Discussion
The vast majority of altered metabolic pathways in cancer have not been clinically targeted. Part of the difficulty is that the metabolic requirements of normal, proliferating cells frequently resemble those of cancer cells (Vander Heiden, 2011), underscoring the value of cancer-specific dependencies that widen the therapeutic window. Several groups have identified dependencies resulting from the perturbation of key metabolic genes. For example, deletion of fumarate hydratase leads to a collateral dependence on heme oxygenase (Frezza et al., 2011). In glioblastoma, loss of enolase 1 can be exploited by targeting the compensatory isozyme encoded by enolase 2 (Muller et al., 2012). Neomorphic mutations in isocitrate dehydrogenase 1 and 2 (IDH1/2) are known to drive tumorigenesis in gliomas and AML (DeBerardinis and Chandel, 2016) and can be targeted by mutation-specific small molecules. Unfortunately, these strategies are highly contextual and difficult to extrapolate to other cancers.
An alternative approach leverages cancer metabolism for the purpose of priming the neoplasm to a second, apoptosis-inducing treatment (Zhao et al., 2013). Such a strategy requires a functional understanding of the metabolic-apoptotic interface. The systematic identification of apoptotically reactive metabolic nodes and pathways in AML presented here fills this need. Using our metabolic-apoptotic interaction map, we identified and experimentally validated several paradigms that exemplify crosstalk between metabolism and apoptosis. In particular, our interaction map nominated heme biosynthesis as an unappreciated metabolic vulnerability whose disruption can potentiate cancer cell death induced by pro-apoptotic chemotherapies. We tracked the pro-apoptotic consequence of heme depletion to the ETC and specified its reduction of the apoptotic threshold through the disassembly of heme-dependent ETC complexes. Open questions remain, namely the specific molecular mechanism by which ETC disruption potentiates ROS-independent MOMP. Nevertheless, leveraging suppressed heme biosynthesis to potentiate drug-induced apoptosis may prove to be a tractable approach for reasons that highlight broader themes.
First, gene-level and pathway-level apoptotic sensitization tends to be uncoupled from essentiality, especially in reactive pathways. Practically, this observation underscores the importance of screening in the proper context. Conceptually, this suggests that pathways which are more sensitizing than essential may offer a wider therapeutic window. For instance, AML cells tolerate SA treatment over months of culture, despite a reduced apoptotic threshold. Indeed, among major metabolic pathways, heme biosynthesis is selectively downregulated in AML cells; this downregulation is blunted in patients who fail to respond to induction chemotherapy. Coupled to the mechanistic work presented here, these observations suggest not only that baseline depletion of heme plays a role in priming AML cells to apoptosis, potentially undergirding their chemosensitivity, but also that this pro-apoptotic advantage can be amplified through additional suppression of heme biosynthesis. Second, meaningful metabolic-apoptotic interactions tend to form clusters which can be mapped to identify network interactions. By tracing the pathways that source heme’s originating substrates (succinyl-CoA, glycine, iron) and utilize its downstream products (heme b, heme a), we were able to functionally diagram heme metabolism, bolstering our confidence in heme’s status as an apoptotic sensitizer. Third, a holistic approach to the study of metabolism permits an unbiased assessment of the factors that causally influence a given pathway. For instance, although many studies have described perturbation of the ETC as a method of potentiating apoptosis (Trotta et al., 2017, Kruspig et al., 2016), our study distinguishes heme biosynthesis as the metabolic pathway most directly and potently amenable to intervention.
Finally, by contextualizing cancer metabolism within the study of apoptosis, these findings recast our understanding of cancer metabolism in a therapeutic light. Some of the proposed metabolic adjuvants involve a mere repurposing of established chemotherapies (antimetabolites), while others push emerging (ferroptosis/oxidative stress) or underdeveloped (heme) metabolic paradigms toward therapy. In principle, the entire interaction map could be mined for actionable nodes or pathways. Information from the map may also be adaptable for higher-order applications. The burgeoning use of metabolomics to study cancer, coupled with emerging analyses that associate metabolic changes with tumor initiation or progression (Reznik et al., 2018), raises the possibility that personalized metabolic data could be harnessed to inform cancer treatment. Presently, this type of directed application suffers from a lack of organizing principles. Our work in AML offers at least one such opportunity.
The optimal treatment of AML in elderly patients, who are frequently unable to tolerate intensive chemotherapy, remains an open question. Do you administer high dose cytotoxic chemotherapy, which affords the best chance of remission but is frequently intolerable, or hypomethylating agents, which are better tolerated but often offer little more than palliation (Ferrara and Schiffer, 2013, Dohner et al., 2015)? Here, characterization of heme pathway activity could serve as a molecular biomarker, distinguishing those whose neoplasms would most likely respond to cytotoxic, apoptosis-inducing chemotherapy from those who would be more likely to suffer without clinical benefit. Eventually, one might envision a scenario where a clinical metabolite panel, representing apoptotically reactive pathways, could be used to optimize the selection of patients to be treated with BH3 mimetics or other apoptosis-inducing therapies.
In sum, the findings presented here outline a network of functional relationships between metabolism and apoptosis, the first such interaction map reported in the literature. This understanding is valuable, in part because it motivates the design of metabolically-informed chemotherapeutic strategies, but also because widespread appreciation of the principles defined here have the potential to reshape the way we perceive the interconnectivity and attendant co-dysregulation of metabolism and apoptosis in cancer.
Limitations of study
The screening approaches detailed here are limited by the scope of the CRISPR library employed. While our library was comprised of metabolic enzymes and transporters to enhance potential druggability and translatability, subsequent iterations would be improved by including nonenzymatic metabolic targets. Separately, our library produces single-gene knockouts, rendering it prone to false-negatives caused by isoform/paralog-mediated compensation. This may account for the nonreactivity of all five hexokinase isoforms in our map, despite that the hexokinase inhibitor 2-deoxyglucose is known to be a chemosensitizer; this shortcoming could be addressed using multiple-knockout CRISPR systems. Finally, although ABT-199 and its target, BCL-2, have been well studied in the context of apoptosis, we are unable to exclude contributions from non-apoptotic modes of cell death such as necrosis, pyroptosis, ferroptosis, necroptosis, and others.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-β-Actin (clone 13E5) antibody | Cell Signaling Technology | Cat# 4970, RRID:AB_2223172 |
| Rabbit monoclonal anti-AIF (clone D39D2) antibody | Cell Signaling Technology | Cat# 5318, RRID:AB_10634755 |
| Rabbit monoclonal anti-Smac/Diablo (D20A5) antibody | Cell Signaling Technology | Cat# 9745, RRID:AB_11220423 |
| Rabbit monoclonal anti-HtrA2/Omi (clone D20A5) antibody | Cell Signaling Technology | Cat# 9745, RRID:AB_11220423 |
| Rabbit monoclonal anti-Bax (D2E11) antibody | Cell Signaling Technology | Cat# 5023, RRID:AB_10557411 |
| Rabbit monoclonal anti-Bak (clone D4E4) antibody | Cell Signaling Technology | Cat# 12105, RRID:AB_2716685 |
| Mouse monoclonal anti-cleaved-PARP (Asp214) (clone 19F4) antibody | Cell Signaling Technology | Cat# 9546, RRID:AB_2160593 |
| Mouse monoclonal anti-cleaved-caspase 3 (Asp175) (clone 5A1E) antibody | Cell Signaling Technology | Cat# 9664, RRID:AB_2070042 |
| Rabbit polyclonal anti-Na,K-ATPase antibody | Cell Signaling Technology | Cat# 3010, RRID:AB_2060983 |
| Rabbit monoclonal anti-cytochrome c (clone D18C7) antibody | Cell Signaling Technology | Cat# 11940, RRID:AB_2637071 |
| Rabbit monoclonal anti-X-IAP (clone 3B6) antibody | Cell Signaling Technology | Cat# 2045, RRID:AB_2214866 |
| Rabbit monoclonal anti-VDAC1 (clone EPR10852(B)) antibody | Abcam | Cat# ab154856, RRID:AB_2687466 |
| Total OXPHOS Human WB Antibody Cocktail: 1. Mouse monoclonal anti-Complex I subunit NDUFB8 antibody (ab110242) 2. Mouse monoclonal anti-Complex II subunit 30kDa antibody (ab14714) 3. Mouse monoclonal anti-Complex III subunit Core 2 antibody (ab14745) 4. Mouse monoclonal anti-Complex IV subunit II antibody (ab110258) 5. Mouse monoclonal anti-ATP synthase subunit alpha antibody (ab14748) |
Abcam | Cat#ab110411: 1. Cat# ab110242, RRID:AB_10859122; 2. Cat# ab14714, RRID:AB_301432; 3. Cat# ab14745, RRID:AB_2213640; |
| Rabbit polyclonal anti-Bax (N-terminus) | EMD Millipore | Cat# 06–499, RRID:AB_310143 |
| Rabbit monoclonal anti-Bcl-2 (clone D55G8) antibody | Cell Signaling Technology | Cat# 4223, RRID:AB_1903909 |
| Rabbit monoclonal anti-Bcl-xL (clone 54H6) antibody | Cell Signaling Technology | Cat# 2764, RRID:AB_2228008 |
| Rabbit monoclonal anti-Mcl-1 (clone S-19) antibody | Santa Cruz Biotechnology | Cat# sc-819, RRID:AB_2144105 |
| Rabbit monoclonal anti-Bim (clone C34C5) antibody | Cell Signaling Technology | Cat# 2933, RRID:AB_1030947 |
| Rabbit polyclonal anti-Bid antibody | Cell Signaling Technology | Cat# 2002, RRID:AB_10692485 |
| Rabbit monoclonal anti-total Bad (clone D24A9) antibody | Cell Signaling Technology | Cat# 9239, RRID:AB_2062127 |
| Rabbit monoclonal anti-phospho-Bad (S112) (clone 40A9) antibody | Cell Signaling Technology | Cat# 5284, RRID:AB_560884 |
| Rabbit monoclonal anti-phospho-Bad (S136) (clone D25H8) antibody | Cell Signaling Technology | Cat# 4366, RRID:AB_10547878 |
| Rabbit monoclonal anti-PUMA (clone D30C10) antibody | Cell Signaling Technology | Cat# 12450 |
| Rabbit monoclonal anti-NOXA (clone D8L7U) antibody | Cell Signaling Technology | Cat# 14766 |
| Bacterial and Virus Strains | ||
| lentiCRISPR v2 | Sanjana et al. Nat. Methods 2014 | https://www.addgene.org/52961/ |
| Envelop VSV-G expression plasmid | T. Reya | https://www.addgene.org/14888/ |
| PsPAX2 packaging vector | D. Trono | https://www.addgene.org/12260/ |
| E. cloni 10G ELITE electrocompetent cells | Lucigen | 60052–3 |
| Biological Samples | ||
| Human AML bone marrow, peripheral blood samples | Duke Biorepository | IRB #: Pro00006268 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Venetoclax (ABT-199) | Selleck Chemicals | S8048; CAS: 1257044–40-8 |
| 4,6-Dioxoheptanoic acid (Succinyl acetone) | Sigma-Aldrich | D1415; CAS: 51568– 18-4 |
| Protoporphyrin IX | Sigma-Aldrich | P8293; CAS: 553– 12-8 |
| N-methyl protoporphyrin IX | Santa Cruz Biotechnology | CAS: 79236–56-9 |
| Mercaptopurine (6-MP) | Selleck Chemicals | S1305; CAS: 50–44- 2 |
| Fluorouracil (5-FU) | Selleck Chemicals | S1209; CAS: 51–21- 8 |
| Gemcitabine | ApexBio | A8437; CAS: 95058– 81-4 |
| Erastin | Selleck Chemicals | S7242; CAS: 571203–78-6 |
| RSL-3 | Selleck Chemicals | S8155; CAS: 1219810–16-8 |
| Ferrostatin-1 | Sigma-Aldrich | SML0853, CAS: 347174–05-4 |
| Liproxstatin-1 | Sigma-Aldrich | SML1414; CAS: 950455–15-9 |
| DL-Buthionine-sulfoximine | Sigma-Aldrich | 19176; CAS: 5072– 26-4 |
| Atorvastatin | Selleck Chemicals | S2077; CAS: 134523–03-8 |
| 1,2,3-Benzenetricarboxylic acid | Sigma-Aldrich | B4201; CAS: 732304–21-1 |
| BPTES | Sigma-Aldrich | SML0601; CAS: 314045–39-1 |
| Sodium azide | Mallinckrodt | 1953–57 |
| Antimycin A from Streptomyces sp. | Sigma-Aldrich | A8674; CAS: 1397–94-0 |
| Rotenone | Cayman Chemicals | 13995; CAS: 83–79-4 |
| Deferoxamine | Sigma-Aldrich | D9533; CAS: 138–14-7 |
| 4-Deoxypyridoxine hydrochloride | Santa Cruz Biotechnology | D0501; CAS: 148–51-6 |
| Cytarabine | ApexBio | A8405; CAS: 147–94-4 |
| Doxorubicin | ApexBio | A1832; CAS: 25316–40-9 |
| Oxalic acid | Sigma-Aldrich | 241172; CAS: 144–62-7 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma-Aldrich | C2920; CAS: 370–86-5 |
| Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) | Sigma-Aldrich | C2759; CAS: 555–60-2 |
| Bim | New England Peptide | MRPEIWIAQELRRI GDEFNA |
| Bid | New England Peptide | EDIIRNIARHLAQVG DSMDR |
| QVD-Oph hydrate | Sigma-Aldrich | SML0063 |
| Oligomycin | Sigma-Aldrich | 75351; CAS: 579– 13-5 |
| N,N,N’,N’-Tetramethyl-1,4-phenylendiamine (TMPD) | Sigma-Aldrich | T7394; CAS: 100– 22-1 |
| Rotenone | Sigma-Aldrich | R8875; CAS: 83–79- 4 |
| Ascorbate | Sigma-Aldrich | A7631; CAS 134–03- 2 |
| Critical Commercial Assays | ||
| Cell Fractionation Kit | Cell Signaling Technology | #9038 |
| JC-1 Mitochondrial Membrane Potential Assay Kit | Cayman Chemical | 10009172 |
| Seahorse XF96 V3 PS Cell Culture Microplates | Agilent | 101085–004 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | 103015–100 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7570 |
| Complex IV Human Enzyme Activity Microplate Assay | Abcam | Cat# 109909 |
| Mitochondria Isolation Kit for Cultured Cells | Abcam | Cat# 110170 |
| MitoTox Complex II + III OXPHOS Activity Assay Kit | Abcam | Cat# 109905 |
| APC Annexin V | BD Biosciences | Cat# 550474 |
| 7-AAD Cell Viability Solution | BD Via-Probe | Cat# 555815 |
| H2DCFDA | Thermo Fisher Scientific | D399 |
| MitoSOX Red Mitochondrial Superoxide Indicator | Thermo Fisher Scientific | M36008 |
| Experimental Models: Cell Lines | ||
| Human: OCI-AML2 line | A. Letai | RRID: CVCL_1619 |
| Human: MOLM-13 line | ATCC | RRID: CVCL_2119 |
| Human: THP-1 line | ATCC | RRID: CVCL_0006 |
| Human: MV4–11 | ATCC | RRID: CVCL_0064 |
| Human: KG-1a | ATCC | RRID: CVCL_1824 |
| Human: HL-60 | ATCC | RRID: CVCL_0002 |
| Human: HEL | ATCC | RRID: CVCL_2481 |
| Human: BJAB | S. Dave | RRID: CVCL_5711 |
| Human: OCI-LY1 | S. Dave | RRID: CVCL_1879 |
| Human: SU-DHL-4 | S. Dave | RRID: CVCL_0539 |
| Human: 293T | D. Sabatini | RRID: CVCL_0063 |
| Oligonucleotides | ||
| BAX shRNA; TRCN0000033470 | TRC Library | GCCCACCAGCTCTGAGCAGAT |
| BAX shRNA; TRCN0000033471 | TRC Library | GCCGGAACTGATCAGAACCAT |
| GFP shRNA; TRCN0000072181 | TRC Library | ACAACAGCCACAACGTCTATA |
| ArrayF | IDT | TAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCG |
| ArrayR | IDT | ACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
| Recombinant DNA | ||
| Metabolically-focused sgRNA library | This paper | Table S1 |
| Software and Algorithms | ||
| GraphPad Prism software | GraphPad Software | https://www.graphpad.com/ |
| R | R Core Team, 2015 | https://www.r-project.org/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kris Wood (kris.wood@duke.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines:
All cell lines were cultured at 37 °C in 5% CO 2 and grown in RPMI-1640 with 10% fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies). The OCI-AML2 cell line was generously gifted by Anthony Letai (Dana Farber Cancer Institute). BJAB, OCI-LY1, and SU-DHL-4 cell lines were gifted by Sandeep Dave (Duke University). MOLM-13, THP-1, MV4–11, KG-1a, HL-60, and HEL cell lines were purchased from Duke University Cell Culture Facility (CCF) or American Type Culture Collection (ATCC). All cell lines were authenticated using short tandem repeat (STR) profiling.
Patient samples / primary cell culture:
Primary AML patient cells were obtained through informed consent from Duke University Medical Center according to protocols approved by the Duke Health Institutional Review Board. Samples were Ficoll purified and the mononuclear fraction was extracted, viably frozen in 90% FBS + 10% dimethyl sulfoxide (DMSO), and stored in liquid nitrogen until subsequent use. Frozen mononuclear cells were thawed, resuspended and washed in RPMI, and treated with ABT-199 and/or SA for the appropriate time. Cells were then stained with annexin V and analyzed using flow cytometry (detailed above) or assessed for their mitochondrial membrane potential (detailed above). It should be noted that this approach was necessarily limited by the availability of samples and the difficulty of culturing primary samples in vitro; using the samples available to us, upwards of 40–50% of control-treated primary AML cells exhibited spontaneous death within 24–36 hours, limiting the number of experimental replicates and the diversity of assays we could perform.
METHOD DETAILS
Pooled CRISPR Screening
THP-1 or MOLM-13 cells were seeded into six-well plates at a density of 3E6 cells per well and transduced at an MOI of 0.2. A total of 144E6 cells per cell line were transduced in 48 wells. 24 hours after viral transduction, cells were replated into puromycin-containing media. A sample was collected at 48 hours of puromycin exposure to confirm library coverage in the transduced population. Transduced cells were expanded in puromycin for a total of 10 days prior to drug introduction, at which point the transduced cell population was split into vehicle (DMSO) and ABT-199 treatment conditions and maintained for up to three weeks. All conditions were performed in replicate. Cells were counted, replated, and drug replenished every 3–4 days. At any given point during the screen, each replicate was represented by a minimum of 12E6 cells, sufficient to provide 1000x coverage of the library (~1000 cells per unique sgRNA). Samples of 25E6 cells were collected upon screen initiation, termination, and at weekly intervals. Following completion of the screens, DNA was extracted (DNeasy Blood & Tissue Kit, QIAGEN) and prepared for sequencing as previously described (Shalem et al., 2014).
Screen analysis
Deep sequencing was performed on an Illumina Nextseq platform (75 bp, single-ended) to identify differences in library composition. All sequencing was performed by Hudson Alpha Institute for Biotechnology. Barcoded reads were matched and binned into guide-level counts. Determinations of genetic essentiality and drug sensitization/resistance were made by evaluating differential guide compositions between the initial population and subsequent drug-treated and vehicle-treated cells populations. Briefly, the fractional representation (FR) for a guide within a sample was normalized to the sum of all guides attributed to that sample. A direct comparison between two samples entailed the quotient of the respective FRs, which we term the depletion metric (DM). The five guide-level DMs for each gene were then collapsed to gene-level scores by taking the average. Genes represented by fewer than 5 guides per condition were excluded from analysis. In our 2322-gene library, 7 genes (representing 0.3% of the total library) were excluded. Genetic essentiality was calculated by considering the depletion/enrichment of the vehicle-treated population over time (DMSOfinal / initial). Drug sensitization/resistance was calculated by considering the depletion/enrichment of the drug-treated population relative to the vehicle-treated population (Drugfinal / DMSOfinal). All depletion/enrichment effects are reported as log2 ratios. All described manipulations were performed in R (https://www.r-project.org/). Because many metabolic genes are known to be essential to cellular viability (Hart et al., 2015), determining the effect of cell-essential genetic loss on apoptosis is difficult. To this point, a subset of essential metabolic genes will lose representation in our screen before the 10-day puromycin selection period is over; our screen does not capture the effect of these genes, which represent a trivial fraction of our library, on apoptosis. The remaining cell-essential genes are captured by the screen. Since our analysis normalizes the effect of gene knockout + drug treatment to gene knockout alone, the interpretation of these genes does not require additional correction and is unchanged except that they necessarily suffer from reduced resolution.
Cloning of constructs:
CRISPR constructs were cloned following previously published methods (Shalem et al., 2014) using previously characterized sgRNAs (Wang et al., 2014). sgRNA inserts were synthesized by CustomArray of the form:
GGAAAGGACGAAACACCGxrefxrefxrefXXGTTTTAGAGCTAGAAATAGCAAGTTAAA ATAAGGC
“X” denotes unique 20mer sgRNA sequence (see “20-mer sequences” below).
The oligo pool was diluted 1:100 in water and amplified using NEB Phusion Hotstart Flex enzyme master mix and the primers ArrayF and Array R (see Key Resources Table above).
PCR protocol: 98 °C/30 s, 18 × [98 °C/10 s, 63 °C/10 s, 72 °C/15 s], 72 °C/3 min
Inserts were cleaned with Axygen PCR clean-up beads (1.8×; Fisher Scientific) and resuspended in molecular biology grade water. lentiCRISPRv2 (Addgene ID 52961) was digested with BsmBI (Thermo Fisher) for 2 h at 37 °C. The ~13 kB band was gel-extracted after size-selection on a 1% agarose gel. Using 100 ng of cut lentiCRISPRv2 and 40 ng of sgRNA oligos, a 20 µL Gibson assembly reaction was performed (30 min, 50 °C). Following Gibson assembly, 1 µL of the reaction was transformed into electrocompetent Lucigen cells, spread on LB-ampicillin plates and incubated overnight. Single colonies were picked and grown overnight in liquid culture at 37 °C. Plasmid extraction was performed using a Plasmid miniprep kit (Qiagen). DNA was used to make lentivirus as described below.
shRNA glycerol stocks were obtained from the Duke Functional Genomics Core Facility. Glycerol stocks were streaked out on LB/Amp plates overnight. Subsequently, colonies were picked and grown overnight in liquid culture at 37 °C. Plasmid extraction was performed using a Plasmi d miniprep kit (Qiagen). DNA was used to make lentivirus as described below. The identity, TRC number, and target sequences of the hairpins are listed in the Key Resources Table above.
Lentivirus production and transduction:
HEK 293 T cells were grown in 10 cm to ~50% confluence. Per-plate transfection was performed using Fugene6 (Promega), 6.2 µg of psPAX2, 0.620 µg pVSVg, 6.25 µg of CRISPR plasmid. After 30 min of incubation at room temperature, the mixture was added to the cells and incubated overnight. The next day harvest media was added (DMEM 30% FBS). After two consecutive 24hr collections, the harvested virus was passed through a 0.45 µm filter. Transductions were performed by plating 1 million cells in 1.4mL RPMI media into six-well dishes, then adding 0.6mL virus and 2 µg polybrene. The cells were then centrifuged at 2500 RPM for 1h at 37°C and incubated overnight at 37°C. The following day, the transduced cells were replated in fresh media into 10cm plates. After 24hrs culture, cells were selected with puromycin (2ug/mL).
GI50 assay
Cells were seeded in 96-well plates at 10,000 cells per well and treated with a 10-fold serial dilution of indicated drug. For instance, treatment with ABT-199 yielded final drug concentrations of 20, 2, 0.2, 0.02, 0.002, 0.0002, 0.00002, and 0.000002µM. A final well was treated with only vehicle (DMSO or water). CellTiter-Glo luminescent viability assay (Promega) was used to measure cell viability 72 hours after addition of drug. Each treatment condition was represented by three individual experiments. Relative viability was calculated by normalizing raw luminescence counts to vehicle-treated wells. For experiments involving two drugs, a second background drug was kept at a constant concentration across all wells except for the vehicle control. GI50 values were calculated by fitting each individual experiment to a 4-parameter logistic curve using GraphPad/Prism 6 software and selecting the dose at which cell viability equals 50% of DMSO-treated viability. Absolute GI50 values normalized to viability in background drug alone to permit observation of supra-additive effects, which are read out as fold changes in ABT-199 GI50 value of less than 1.0. GI50 assays were first performed to obtain the dose-response curves for each metabolic drug singly. Doses used in background with ABT-199 were chosen based on those curves.
Background drug doses were: 5-FU (OCI-AML2: 2µM, MOLM-13: 1.25µM), 6-MP (OCI-AML2: 0.4µM, MOLM-13: 1µM), gemcitabine (OCI-AML2: 4nM, MOLM-13: 4nM), BTA (OCI-AML2: 2.5mM, MOLM-13: 3mM), BPTES (OCI-AML2: 5µM, MOLM-13: 5µM), erastin (OCI-AML2: 4µM, MOLM-13: 0.3µM), RSL-3 (OCI-AML2: 90nM, MOLM-13: 40nM), BSO (OCI-AML2: 50µM, MOLM-13: 50µM), ATS (OCI-AML2: 1.5µM, MOLM-13: 1µM), and SA (OCI-AML2: 100µM, MOLM-13: 750µM, THP-1: 2mM, MV-411: 1mM, KG-1α: 1.5mM, HL-60: 2mM).
Immunoblotting:
Immunoblotting was performed as previously described (Lin et al., 2016). Briefly, cells were resuspended in lysis buffer, incubated on ice for 15 min, then clarified at 13,000 RPM, 4 °C, for 10 min. Protein was quantified using the Bradford method and lysates were made with NuPage Sample Buffer (4×). Membranes were probed with primary antibodies recognizing β-Actin (CST#4970) at a 1:2000 dilution; AIF (CST #5318), Smac (CST #15108), HtrA2/Omi (CST #9745), Hsp60 (AB #45134), Bax (CST #5023), Bak (CST #12105), cleaved-PARP (CST #9546), Na,K-ATPase (CST #3010), and VDAC1 (AB #154856) at a 1:1000 dilution; and cytochrome c (CST #11940), c-caspase 3 (CST #9664), X-IAP (CST #2045), and Total OXPHOS Human WB Antibody Cocktail (ab110411) at a 1:500 dilution. HRP-linked anti-Rabbit (CST #7074) and anti-Mouse (CST #7076) secondary antibodies were applied at a 1:5000 dilution.
For cell fractionation experiments, the Cell Fractionation Kit (CST #9038) was used according to the manufacturer’s instructions. The Bradford method was additionally used to normalize protein concentrations of all samples in the cytoplasmic and membrane-bound fractions.
Heme assay:
Relative intracellular heme levels were assessed in cells treated with SA (OCI-AML2: 100µM, MOLM-13: 750µM, THP-1: 2mM, MV4–11: 1mM, KG-1α: 1.5mM, HL-60: 2mM) for 48h, using a published protocol (Sassa, 1976). 3E6 cells per sample were collected, washed with PBS, resuspended in 500uL of 20mM oxalic acid (Sigma Aldrich), and stored for 16–24hrs at 4 °C. The next day, 500uL of 2M oxalic acid wa s added. The sample was split evenly in two: half of each sample was boiled at 96 °C for 30 min and half was stored in a closed box at RT for 30 min. After centrifuging for 2 min at 16,000g, 200uL of each sample were transferred to a 96-well black plate. Fluorescence intensity was measured at excitation 400nm and emission 620 nm.
Annexin V staining:
Annexin V staining was performed to determine the percentage of cells undergoing apoptosis. 250,000 cells were plated in six-well plates and pre-treated with the indicated amount of SA, azide, or vehicle (H2O) for 48hr, with ABT-199 or vehicle (DMSO) for 8hr, or the combination. Upon collection, cells were washed twice with PBS, resuspended in 100uL 1X Annexin V binding buffer (BD Biosciences) containing 5uL Annexin V stain conjugated to APC (allophycocyanin) (BD Biosciences) and 5uL 7-AAD (BD Biosciences). Phosphatidylserine externalization was measured using APC-conjugated Annexin V and 7-AAD was used as a viability probe. Following a 15min incubation at RT, the samples were analyzed using flow cytometer BD FACSCanto II. Gating strategy was defined using untreated/unstained cells. Analysis of flow cytometry data was performed with FlowJo v10.
For six patient samples, cells were plated and treated for 24hr with ABT-199 and SA (DP0285: ABT-199 200nM, SA 100uM; DP0454: ABT-199 200nM, SA 1mM; DP0451: ABT-199 200nM, SA 1mM; DP0452: ABT-199 200nM, SA 1mM; DP0415: ABT-199 200nM, SA 1mM; DP0436: ABT-199 400nM, SA 1mM).
ROS detection:
ROS production was detected using the probes H2DCF-DA (Thermo Fisher) and MitoSOX Red (Thermo Fisher). Cells were treated with SA for 24h, and 3 million per sample were collected, washed with PBS, and incubated in 2.5 µM DCFDA or 5 µM MitoSOX Red at 37°C for 10 minutes. Cells were washed, resuspended in fresh PBS, and analyzed using flow cytometer BD FACSCanto II.
Time-to-progression assay:
A time-to-progression assay was performed as previously described (Lin et al., 2016). Cells were plated into 10 cm plates at 1E6 cells per plate. Seven days later, the cells counted using a Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter). All cells were centrifuged, counted, and up to 1E6 cells re-plated into a 10 cm plate with fresh treatment. This procedure was repeated weekly for 6 weeks. Weekly growth rates (µ) were calculated from the number of cells plated the previous week (N0) and the number counted the current week (N) according to the formula ln N = ln N0 + µ*t; where t is elapsed time or 168 hr. These growth rates were then used to project the total virtual cell number.
Membrane potential assay:
Collapse of mitochondrial membrane potential was measured using the JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman Chemical), according to manufacturer instructions. Cells were treated with SA, azide, or vehicle for 48h. At 500,000 cells per sample, samples were incubated in 1X JC-1 stain in RPMI media for 20 minutes for 37°C. JC-1 signal was measured and reported as ratios of red (excitation: 535nm, emission: 590nm) over green (excitation: 535nm, emission: 590nm) fluorescence, normalized to vehicle. Primary AML patient cells were treated with SA for 24hr (DP0285: SA 100uM; DP0454: SA 1mM; DP0451: SA 1mM).
Complex IV enzymatic activity:
Enzymatic activity of Complex IV, or cytochrome c oxidase, was measured in cells using the Complex IV Human Enzyme Activity Microplate Assay (Abcam), according to manufacturer instructions. Cells were treated with SA, azide, or vehicle for 48h, before collection, counting, and isolation of 4 million cells per sample. A Tecan Infinite M1000 plate reader was used to measure decreases in absorbance at OD 550nm. Enzymatic rate was calculated as the average slope over 2hr.
Complex II + III enzymatic activity:
Cells were treated with SA, antimycin A, or vehicle for 48h, before collection of 25 million cells per condition. Mitochondria were isolated using the Mitochondria Isolation Kit for Cultured Cells (Abcam), according to manufacturer instructions. Combined enzymatic activities of Complex II + III in isolated mitochondria samples were measured using the MitoTox Complex II + III OXPHOS Activity Assay Kit (Abcam), according to manufacturer instructions. A Bradford assay was additionally performed to normalize complex IV protein concentrations before plating. A Tecan Infinite M1000 plate reader was used to measure increases in absorbance at OD 550nm. Enzymatic rate was calculated as the average slope over 2hr.
BH3 Profiling
BH3 profiling assays were performed as previously described (Sarosiek et al., 2013). Briefly, OCI-AML2 cells were seeded in a six-plate at a density of 2E6 cells/well and treated with SA or vehicle control for 48hr prior to BH3 profiling. Following pretreatment, 3E6 cells were collected, washed with PBS, and resuspended in Newmeyer buffer before digitonin permeabilization, exposure to BH3 peptides, and kinetic monitoring of mitochondrial transmembrane potential loss using JC-1. Fluorescence measurements were taken at 590nM at 30C every 5 minutes for 3 hours. In the experiment shown in Figure S6G, cells were treated with BIM and BID at a final concentration of 100µM. The experiment shown in Figure S6H was performed analogously except that mitochondrial isolates were used instead of pelleted cells and ABT-199 at a final concentration of 10nM and 20nM was used instead of BIM and BID.
Seahorse measurements:
Basal oxygen consumption rate was measured using a Mito Stress test Kit and XF96 Extracellular Flux Analyzer (Seahorse Bioscience), according to manufacturer’s instructions. Cells were resuspended in XF Assay Medium (Seahorse Bioscience) supplemented with 10mM glucose, 1mM pyruvate, and 2mM glutamine. 200,000 cells/well were plated in the XF96 plates pre-coated with Cell-Tak (Corning) and centrifuged to immobilize cells on the bottom of the wells. The plate was incubated in a non-CO2 incubator at 37°C for 2hr to equilibrate. OCR measu rements, taken every 6min, were collected at baseline and after the sequential addition of oligomycin 1uM (final concentration), FCCP 0.5uM, and rotenone 0.75uM + antimycin A 1.5uM.
TMPD-driven oxygen consumption rate was measured using the XF96 Extracellular Flux Analyzer (Seahorse Bioscience). Cells were resuspended in XF Assay Medium (Seahorse Bioscience), not supplemented. 200,000 cells/well were plated in the XF96 plates pre-coated with Cell-Tak (Corning) and centrifuged to immobilize cells on the bottom of the wells. The plate was incubated in a non-CO2 incubator at 37°C for 2hr to equilibrate. OCR measu rements, taken every 6min, were collected at baseline and after the sequential addition of TMPD 0.5mM + ascorbate 10mM + antimycin A 2uM + ADP 1mM, oligomycin 2uM, and azide 20mM.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in R or in Prism 7.0 (GraphPad). All results are shown as mean ± SD. Unless noted otherwise, p values were determined using unpaired, two-tailed Student’s t tests. P-values < 0.05 were considered significant. For Figure 5a, b, the two-tailed unpaired Student’s t-test with Welch’s correction was used owing to differing sample sizes.
Analysis of microarray data
Quantile-normalized data was downloaded from Gene Expression Omnibus Accession No. GSE13159. Probe intensities per gene were averaged, median-centered, and clustered using complete linkage with Euclidean distance. All analyses and visualization these data were performed in R (https://www.r-project.org/).
Analysis of RNAseq data
Normalized, on-diagnosis RNAseq data from Children’s Oncology Group trials CCG-2961, AAML03P1, and AAML0531 were downloaded from the TARGET Data Matrix (https://ocg.cancer.gov/programs/target/data-matrix ). All analyses and visualization of these data were performed in R (https://www.r-project.org/ ).
DATA AND SOFTWARE AVAILABILITY
The microarray data analyzed in this paper (Haferlach et al., 2010) can be found on GEO: GSE13159.
RNAseq data from Children’s Oncology Group trials CCG-2961, AAML03P1, and AAML0531 was downloaded from the TARGET Data Matrix.
Metabolomics data is from a study conducted under the Children’s Oncology Group-AAML1031 Clinical Trial (NCT01371981). Data was provided by Jatinder Lamba and can be downloaded from the Metabolomics Workbench, study “ST000270.”
Supplementary Material
Highlights:
CRISPR/Cas9 loss-of-function screening produces the first apoptotic map of metabolism
Analysis of the map nominates heme biosynthesis as a modifier of apoptotic proclivity
Heme depletion primes cells for apoptosis by disrupting the electron transport chain
Acknowledgements:
We thank the members of the K.C.W. laboratory as well as Christopher Newgard, Nancy Andrews, and Jason Locasale for providing feedback. This work was supported by Duke University School of Medicine start-up funds and support from the Duke Cancer Institute (K.C.W.), NIH awards (R01CA207083 to K.C.W., F30CA206348 to K.H.L., F32CA206234 to R.S.S.), the Duke Medical Scientist Training Program (T32 GM007171 to K.H.L.), and the Duke Undergraduate Research Office (to A.X. and J.C.R.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation or the NIH. We dedicate this work to the memory of our friend, Kimberly Brigati Wang, and her courageous fight against AML.
T.S. Pardee has speaking relationships with Abbvie, Agios, Celgene, Novartis, Amgen, Pharmacyclics, receives commercial research support from Jazz Pharmaceuticals, Karyopharm, Rafael Pharmaceuticals, and is on the advisory board of BioPharma. T.S. Pardee is also employed by Rafael Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
The remaining authors have no competing interests to declare.
References
- ALAVIAN KN, LI H, COLLIS L, BONANNI L, ZENG L, SACCHETTI S, LAZROVE E, NABILI P, FLAHERTY B, GRAHAM M, CHEN Y, MESSERLI SM, MARIGGIO MA, RAHNER C, MCNAY E, SHORE GC, SMITH PJ, HARDWICK JM & JONAS EA 2011. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol, 13, 1224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALTMAN BJ, STINE ZE & DANG CV 2016. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer, 16, 619–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALVAREZ SW, SVIDERSKIY VO, TERZI EM, PAPAGIANNAKOPOULOS T, MOREIRA AL, ADAMS S, SABATINI DM, BIRSOY K & POSSEMATO R 2017. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature, 551, 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDERSEN JL & KORNBLUTH S 2013. The tangled circuitry of metabolism and apoptosis. Mol Cell, 49, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATAMNA H, KILLILEA DW, KILLILEA AN & AMES BN 2002. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc Natl Acad Sci U S A, 99, 14807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHOLA PD & LETAI A 2016. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell, 61, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN SM, THOMAS D, CORCES-ZIMMERMAN MR, XAVY S, RASTOGI S, HONG WJ, ZHAO F, MEDEIROS BC, TYVOLL DA & MAJETI R 2015. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med, 21, 178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN YB, AON MA, HSU YT, SOANE L, TENG X, MCCAFFERY JM, CHENG WC, QI B, LI H, ALAVIAN KN, DAYHOFF-BRANNIGAN M, ZOU S, PINEDA FJ, O’ROURKE B, KO YH, PEDERSEN PL, KACZMAREK LK, JONAS EA & HARDWICK JM 2011. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol, 195, 263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN ZX & PERVAIZ S 2010. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ, 17, 408–20. [DOI] [PubMed] [Google Scholar]
- CHIABRANDO D, MERCURIO S & TOLOSANO E 2014. Heme and erythropoieis: more than a structural role. Haematologica, 99, 973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIPUK JE, MCSTAY GP, BHARTI A, KUWANA T, CLARKE CJ, SISKIND LJ, OBEID LM & GREEN DR 2012. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell, 148, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIPUK JE, MOLDOVEANU T, LLAMBI F, PARSONS MJ & GREEN DR 2010. The BCL-2 family reunion. Mol Cell, 37, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRIELAARD BJ, LAMMERS T & RIVELLA S 2017. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov, 16, 400–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANIAL NN, WALENSKY LD, ZHANG CY, CHOI CS, FISHER JK, MOLINA AJ, DATTA SR, PITTER KL, BIRD GH, WIKSTROM JD, DEENEY JT, ROBERTSON K, MORASH J, KULKARNI A, NESCHEN S, KIM S, GREENBERG ME, CORKEY BE, SHIRIHAI OS, SHULMAN GI, LOWELL BB & KORSMEYER SJ 2008. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med, 14, 144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEBERARDINIS RJ & CHANDEL NS 2016. Fundamentals of cancer metabolism. Sci Adv, 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIXON SJ, LEMBERG KM, LAMPRECHT MR, SKOUTA R, ZAITSEV EM, GLEASON CE, PATEL DN, BAUER AJ, CANTLEY AM, YANG WS, MORRISON B 3RD & STOCKWELL BR 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 149, 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOHNER H, WEISDORF DJ & BLOOMFIELD CD 2015. Acute Myeloid Leukemia. N Engl J Med, 373, 1136–52. [DOI] [PubMed] [Google Scholar]
- FERRARA F & SCHIFFER CA 2013. Acute myeloid leukaemia in adults. Lancet, 381, 484–95. [DOI] [PubMed] [Google Scholar]
- FRADEJAS N, CARLSON BA, RIJNTJES E, BECKER NP, TOBE R & SCHWEIZER U 2013. Mammalian Trit1 is a tRNA([Ser]Sec)-isopentenyl transferase required for full selenoprotein expression. Biochem J, 450, 427–32. [DOI] [PubMed] [Google Scholar]
- FREZZA C, ZHENG L, FOLGER O, RAJAGOPALAN KN, MACKENZIE ED, JERBY L, MICARONI M, CHANETON B, ADAM J, HEDLEY A, KALNA G, TOMLINSON IP, POLLARD PJ, WATSON DG, DEBERARDINIS RJ, SHLOMI T, RUPPIN E & GOTTLIEB E 2011. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature, 477, 225–8. [DOI] [PubMed] [Google Scholar]
- FUKUI H, DIAZ F, GARCIA S & MORAES CT 2007. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A, 104, 14163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREEN DR, GALLUZZI L & KROEMER G 2014. Cell biology. Metabolic control of cell death. Science, 345, 1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAFERLACH T, KOHLMANN A, WIECZOREK L, BASSO G, KRONNIE GT, BENE MC, DE VOS J, HERNANDEZ JM, HOFMANN WK, MILLS KI, GILKES A, CHIARETTI S, SHURTLEFF SA, KIPPS TJ, RASSENTI LZ, YEOH AE, PAPENHAUSEN PR, LIU WM, WILLIAMS PM & FOA R 2010. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol, 28, 2529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANAHAN D & WEINBERG RA 2011. Hallmarks of cancer: the next generation. Cell, 144, 646–74. [DOI] [PubMed] [Google Scholar]
- HART T, BROWN KR, SIRCOULOMB F, ROTTAPEL R & MOFFAT J 2014. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol Syst Biol, 10, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HART T, CHANDRASHEKHAR M, AREGGER M, STEINHART Z, BROWN KR, MACLEOD G, MIS M, ZIMMERMANN M, FRADET-TURCOTTE A, SUN S, MERO P, DIRKS P, SIDHU S, ROTH FP, RISSLAND OS, DUROCHER D, ANGERS S & MOFFAT J 2015. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell, 163, 1515–26. [DOI] [PubMed] [Google Scholar]
- KALE J, OSTERLUND EJ & ANDREWS DW 2018. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ, 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRUSPIG B, VALTER K, SKENDER B, ZHIVOTOVSKY B & GOGVADZE V 2016. Targeting succinate:ubiquinone reductase potentiates the efficacy of anticancer therapy. Biochim Biophys Acta, 1863, 2065–71. [DOI] [PubMed] [Google Scholar]
- LEE JS, ROBERTS A, JUAREZ D, VO TT, BHATT S, HERZOG LO, MALLYA S, BELLIN RJ, AGARWAL SK, SALEM AH, XU T, JIA J, LI L, HANNA JR, DAVIDS MS, FLEISCHMAN AG, O’BRIEN S, LAM LT, LEVERSON JD, LETAI A, SCHATZ JH & FRUMAN DA 2018. Statins enhance efficacy of venetoclax in blood cancers. Sci Transl Med, 10. [DOI] [PMC free article] [PubMed]
- LIN KH, WINTER PS, XIE A, ROTH C, MARTZ CA, STEIN EM, ANDERSON GR, TINGLEY JP & WOOD KC 2016. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci Rep, 6, 27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Y, FISKUM G & SCHUBERT D 2002. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem, 80, 780–7. [DOI] [PubMed] [Google Scholar]
- LOWMAN XH, MCDONNELL MA, KOSLOSKE A, ODUMADE OA, JENNESS C, KARIM CB, JEMMERSON R & KELEKAR A 2010. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol Cell, 40, 823–33. [DOI] [PubMed] [Google Scholar]
- MARTINOU JC & YOULE RJ 2011. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell, 21, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENENDEZ JA & LUPU R 2006. Oncogenic properties of the endogenous fatty acid metabolism: molecular pathology of fatty acid synthase in cancer cells. Curr Opin Clin Nutr Metab Care, 9, 346–57. [DOI] [PubMed] [Google Scholar]
- MORAES CT, DIAZ F & BARRIENTOS A 2004. Defects in the biosynthesis of mitochondrial heme c and heme a in yeast and mammals. Biochim Biophys Acta, 1659, 153–9. [DOI] [PubMed] [Google Scholar]
- MUCKENTHALER MU, RIVELLA S, HENTZE MW & GALY B 2017. A Red Carpet for Iron Metabolism. Cell, 168, 344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER FL, COLLA S, AQUILANTI E, MANZO VE, GENOVESE G, LEE J, EISENSON D, NARURKAR R, DENG P, NEZI L, LEE MA, HU B, HU J, SAHIN E, ONG D, FLETCHER-SANANIKONE E, HO D, KWONG L, BRENNAN C, WANG YA, CHIN L & DEPINHO RA 2012. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature, 488, 337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY MP 2009. How mitochondria produce reactive oxygen species. Biochem J, 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAN R, HOGDAL LJ, BENITO JM, BUCCI D, HAN L, BORTHAKUR G, CORTES J, DEANGELO DJ, DEBOSE L, MU H, DOHNER H, GAIDZIK VI, GALINSKY I, GOLFMAN LS, HAFERLACH T, HARUTYUNYAN KG, HU J, LEVERSON JD, MARCUCCI G, MUSCHEN M, NEWMAN R, PARK E, RUVOLO PP, RUVOLO V, RYAN J, SCHINDELA S, ZWEIDLER-MCKAY P, STONE RM, KANTARJIAN H, ANDREEFF M, KONOPLEVA M & LETAI AG 2014. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov, 4, 362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARDEE TS, ANDERSON RG, PLADNA KM, ISOM S, GHIRALDELI LP, MILLER LD, CHOU JW, JIN G, ZHANG W, ELLIS LR, BERENZON D, HOWARD DS, HURD DD, MANUEL M, DRALLE S, LYERLY S & POWELL BL 2018. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res [DOI] [PMC free article] [PubMed]
- PETERS GJ, VAN DER WILT CL, VAN MOORSEL CJ, KROEP JR, BERGMAN AM & ACKLAND SP 2000. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther, 87, 227–53. [DOI] [PubMed] [Google Scholar]
- POSSEMATO R, MARKS KM, SHAUL YD, PACOLD ME, KIM D, BIRSOY K, SETHUMADHAVAN S, WOO HK, JANG HG, JHA AK, CHEN WW, BARRETT FG, STRANSKY N, TSUN ZY, COWLEY GS, BARRETINA J, KALAANY NY, HSU PP, OTTINA K, CHAN AM, YUAN B, GARRAWAY LA, ROOT DE, MINO-KENUDSON M, BRACHTEL EF, DRIGGERS EM & SABATINI DM 2011. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature, 476, 346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REZNIK E, LUNA A, AKSOY BA, LIU EM, LA K, OSTROVNAYA I, CREIGHTON CJ, HAKIMI AA & SANDER C 2018. A Landscape of Metabolic Variation across Tumor Types. Cell Syst, 6, 301–313 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROHRIG F & SCHULZE A 2016. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer, 16, 732–749. [DOI] [PubMed] [Google Scholar]
- SAMUDIO I, HARMANCEY R, FIEGL M, KANTARJIAN H, KONOPLEVA M, KORCHIN B, KALUARACHCHI K, BORNMANN W, DUVVURI S, TAEGTMEYER H & ANDREEFF M 2010. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest, 120, 142–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAROSIEK KA, CHI X, BACHMAN JA, SIMS JJ, MONTERO J, PATEL L, FLANAGAN A, ANDREWS DW, SORGER P & LETAI A 2013. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell, 51, 751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SASSA S 1976. Sequential induction of heme pathway enzymes during erythroid differentiation of mouse Friend leukemia virus-infected cells. J Exp Med, 143, 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHALEM O, SANJANA NE, HARTENIAN E, SHI X, SCOTT DA, MIKKELSON T, HECKL D, EBERT BL, ROOT DE, DOENCH JG & ZHANG F 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEFTEL AD, STEHLING O, PIERIK AJ, ELSASSER HP, MUHLENHOFF U, WEBERT H, HOBLER A, HANNEMANN F, BERNHARDT R & LILL R 2010. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A, 107, 11775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMORENBURG CH, SPARREBOOM A, BONTENBAL M & VERWEIJ J 2001. Combination chemotherapy of the taxanes and antimetabolites: its use and limitations. Eur J Cancer, 37, 2310–23. [DOI] [PubMed] [Google Scholar]
- SUBRAMANIAN A, TAMAYO P, MOOTHA VK, MUKHERJEE S, EBERT BL, GILLETTE MA, PAULOVICH A, POMEROY SL, GOLUB TR, LANDER ES & MESIROV JP 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A, 102, 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TROTTA AP, GELLES JD, SERASINGHE MN, LOI P, ARBISER JL & CHIPUK JE 2017. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J Biol Chem, 292, 11727–11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANDER HEIDEN MG 2011. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov, 10, 671–84. [DOI] [PubMed] [Google Scholar]
- VANDER HEIDEN MG & THOMPSON CB 1999. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol, 1, E209–16. [DOI] [PubMed] [Google Scholar]
- VAUGHN AE & DESHMUKH M 2008. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol, 10, 1477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VON HAEFEN C, WIEDER T, GILLISSEN B, STARCK L, GRAUPNER V, DORKEN B & DANIEL PT 2002. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene, 21, 4009–19. [DOI] [PubMed] [Google Scholar]
- WANG T, WEI JJ, SABATINI DM & LANDER ES 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science, 343, 80–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Y, BUTLER EB & TAN M 2013. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis, 4, e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data analyzed in this paper (Haferlach et al., 2010) can be found on GEO: GSE13159.
RNAseq data from Children’s Oncology Group trials CCG-2961, AAML03P1, and AAML0531 was downloaded from the TARGET Data Matrix.
Metabolomics data is from a study conducted under the Children’s Oncology Group-AAML1031 Clinical Trial (NCT01371981). Data was provided by Jatinder Lamba and can be downloaded from the Metabolomics Workbench, study “ST000270.”