

Abstract
Genes encoding the small GTPases of the Ras superfamily are among the most frequently mutated or dysregulated in human cancer. No systematic studies, however, have yet been conducted for assessing the implications of small GTPases in the metastatic transformation of colorectal cancer (CRC). By utilizing a recently established high-throughput multiple-reaction monitoring (MRM)-based workflow together with stable isotope labeling by amino acids in cell culture (SILAC), we investigated comprehensively the relative expression of the small GTPase proteome in a pair of matched primary/metastatic CRC cell lines (SW480/SW620). Among the 83 quantified small GTPases, 25 exhibited at least a 1.5-fold difference in protein expression in SW480 and SW620 cells. In particular, SAR1B protein was found to be substantially down-regulated in SW620 relative to SW480 cells. In addition, bioinformatic analyses revealed that diminished SAR1B mRNA expression is significantly associated with higher CRC stages and unfavorable patient prognosis, in support of a potential role of SAR1B in suppressing CRC metastasis. In addition, diminished SAR1B expression could stimulate epithelial–mesenchymal transition (EMT), thereby promoting motility and in vitro metastasis of SW480 cells. In summary, we profiled systematically, by employing an MRM-based targeted proteomic method, the differential expression of small GTPase proteins in a matched pair of primary/metastatic CRC cell lines. Our results revealed the potential roles of SAR1B in suppressing CRC metastasis and in the prognosis of CRC patients.
Graphical Abstract
INTRODUCTION
Colorectal cancer (CRC) is a major cause of cancer-associated deaths worldwide, and it ranks third in terms of incidence but second in terms of mortality.1,2 According to the stage definition by the American Joint Committee on Cancer (AJCC), five-year survival rates ranged from 93.2% for stage I to 8.1% for stage IV CRC patients.3 Approximately 40–50% of all patients with CRC will ultimately develop into metastatic disease either at the time of diagnosis or develop distant relapses after therapy, of which the median overall survival is less than two years.4 Therefore, a better understanding about the metastatic transformation of CRC cells could lead to more precise therapeutic strategies that improve patient outcomes.
The Ras superfamily of small GTPases (20–35 kDa) function as molecular switches that play important roles in carcinogenesis and tumor progression by regulating intra-cellular trafficking, cell signaling and malignant secretion.5,6 Among them, KRAS, one of the most frequently mutated oncogenes that drive colorectal tumorigenesis, is mutated in 40% of sporadic CRC.7 In addition to mutations, aberrant regulation of small GTPase expression has been shown to have a role in CRC progression. For instance, overexpression of RAB3C promoted colorectal tumor progression in vitro and in vivo.8 Moreover, miR-27b and miR-204-5p suppressed proliferation and invasion of CRC cells by down-regulating RAB3D and RAB22A, respectively,9,10 and diminished expression of RAB25 was found to be associated with reduced metastatic potential of CRC cells in vivo.11 Therefore, we reason that a systematic investigation about small GTPases associated with metastatic transformation of CRC cells may lead to novel molecular targets for the prognosis and therapeutic intervention of CRC. In this study, we applied a recently established high-throughput, targeted proteomic approach to robustly quantify the differential expression of small GTPases in the SW480/SW620 matched primary/metastatic cells derived from the same patient.12 We also examined, by using bioinformatic analyses, whether the aberrant expression of these small GTPases were dysregulated in clinical samples and showed prognostic values. Finally, we identified SAR1B as a potential metastasis suppressor and a prognostic biomarker that could modulate the migration and invasion of CRC cells in vitro by regulating epithelial–mesenchymal transition (EMT).
MATERIALS AND METHODS
Cell Culture.
The CRC cell lines used in this study were SW480 (ATCC# CCL-228) and SW620 (ATCC# CCL-227), which were established from a primary human colon adenocarcinoma and a lymph node metastasis of the same patient, respectively. Both cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen-Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen-Gibco, Carlsbad, CA) and penicillin/ streptomycin (PS, 100 IU/mL) in a humidified atmosphere with 5% CO2 at 37 °C, and the culture medium was changed in every 2 to 3 days as necessary.
For stable isotope labeling by amino acids in cell culture (SILAC) experiments, “heavy” and “light” SILAC DMEM media were freshly prepared by adding 0.146 g/L [13C6, 15N2]-l-lysine (Lys-8) and 0.84 g/L [13C6]-l-arginine (Arg-6) (Cambridge Isotopes Inc.) or the corresponding unlabeled lysine (Lys-0) and arginine (Arg-0) to the DMEM medium depleted of l-lysine and l-arginine (Thermo Scientific Pierce). The SILAC media were further supplemented with 10% dialyzed FBS (Corning) and PS (100 IU/mL). SW480 and SW620 cells were cultured in the heavy-DMEM medium for at least 10 days or six cell doublings to ensure complete heavy-isotope incorporation.
Sample Preparation and LC-MS/MS Analyses.
To assess the differential expression of small GTPases in SW480 and SW620 cells, we conducted SILAC-based quantitative proteomic experiments with forward and reverse labeling strategies. Briefly, we quantified the lysates of light-labeled SW480 cells and heavy-labeled SW620 cells by Bradford assay (Bio-Rad) and combined them at a 1:1 ratio (by mass) in the forward labeling experiments (Figure 1A). The reverse labeling experiment was conducted in the opposite way. The mixed cell lysate (100 μg in total) was mixed with 4× Laemmli sample buffer (Bio-Rad) in the presence of 10% (v/v) 2-mercaptoethanol as a reducing agent, heated for 5 min at 95 °C, loaded onto a nonreducing, homemade 10% SDS-PAGE gel, and separated by electrophoresis at 120 V and at room temperature for 40 min. To assist the identification of molecular weight region for small GTPase proteins, 5 μL protein molecular weight standard solution (Precision Plus Protein Kaleidoscope Standards, Bio-Rad) was loaded into a separate lane of the same gel. The gel band corresponding to the molecular weight range of 15–37 kDa (with an approximate surface area of 6 × 5 mm) was excised and further cut into 1 mm3-volume cubes. The proteins in the gel slices were then reduced with 20 mM dithiothreitol, alkylated with 55 mM iodoacetamide, and digested in-gel with trypsin at an enzyme/protein ratio of 1:100 in 50 mM ammonium bicarbonate (pH 7.8) at 37 °C overnight. After tryptic digestion, the peptide mixtures were desalted and subjected to LC-MRM analyses on a TSQ Vantage triple-quadruple mass spectrometer, which was equipped with a nanoelectrospray ionization source and coupled with an EASY-nLC II HPLC system (Thermo Fisher Scientific, San Jose, CA). Approximately 4% of the aforementioned tryptic peptide mixture was automatically loaded onto a 4 cm trapping column (150 μm i.d.) packed with ReproSil-Pur 120 C18-AQ resin (5 μm in particle size and 120 Å in pore size, Dr. Maisch GmbH HPLC) at 3 μL/min. The trapping column was connected to a 20 cm fused silica analytical column (75 μm i.d.) packed with ReproSil-Pur 120 C18-AQ resin (3 μm in particle size and 120 Å in pore size, Dr. Maisch GmbH HPLC). A 157 min linear gradient of 2–35% acetonitrile in 0.1% formic acid was employed for peptide separation, and the flow rate was 230 nL/min. The spray voltage was set at 1.8 kV. Ions were isolated in both Q1 and Q3 using 0.7 fwhm resolution, where the cycle time was set at 5 s. The optimal collisional energy for each targeted peptide was calculated using a linear equation specific to the TSQ Vantage instrument and the precursor mass-to-charge ratio (m/z), according to the default setting in Skyline.13
Figure 1.

MRM-based targeted quantitative profiling of small GTPases associated with CRC metastasis. (A) A schematic diagram depicting the targeted quantitative proteomic workflow, relying on forward SILAC labeling, in-gel fractionation, and scheduled LC-MRM analysis. (B) Correlation between the Log2-transformed SILAC ratios (Log2R) obtained from one forward and one reverse SILAC labeling experiments with a relatively high correlation coefficient (R2 = 0.9280). (C) Venn diagrams displaying the overlap between quantified small GTPases in the triplicate SILAC experiments obtained from MRM and DDA analysis, respectively, and the comparison between the performances of the two methods. (D) A bar chart showing significantly up- and down-regulated (>1.5-fold) small GTPases quantified from three LC-MRM experiments.
To enable high-throughput quantitative analysis, we applied a previously developed scheduled LC-MRM method, where the mass spectrometer was programmed to acquire the MS/MS of the precursor ions for a limited number of peptides in each 6 min retention time (RT) window.12 The MRM data for all targeted peptides were manually inspected to ensure correct peak picking. In this regard, the dot-product (dotp)14 value has to exceed 0.80. In addition, the iRT value represents an intrinsic property (i.e., hydrophobicity) of a peptide;15 hence, a substantial deviation of measured RT from that projected from the linear plot of RT over iRT signals a false-positive detection. The relative levels of small GTPase peptides were calculated directly from the ratios of sum of peak areas for all MRM transitions for the light over heavy form of the peptides, where no normalization was imposed. In this context, it is worth noting that the ratios for the total peak areas for the light- over heavy-labeled peptides in the two forward SILAC and one reverse SILAC labeling experiments were 1.11, 0.93, and 1.09, respectively, suggesting that the 1:1 mixing based on Bradford assay is reasonably accurate. The ratios obtained from different peptides from the same small GTPase were then averaged to yield the protein ratio.
Data Availability.
The processed MRM data are provided in Table S1. The Skyline MRM library (including the transition list for quantification and the iRT file) for human small GTPase proteome and the raw files obtained from the LC-MRM analyses of small GTPases for the SW480/SW620 cell lines were deposited into PeptideAtlas with the identifier number of PASS01326 (http://www.peptideatlas.org/PASS/PASS01326).
Transwell Migration and Invasion Assay.
Transwell chambers (Corning Inc., Corning, NY, U.S.A.) were rehydrated at 37 °C, 1 × 105 cells were then added to the top chamber in serum-free medium, and the bottom chamber was filled with medium containing 10% fetal bovine serum. The invasion assay was conducted under the same conditions except that the transwell membranes were precoated with Matrigel (Corning, NY). The cells were cultured at 37 °C for 48 h in a humidified incubator containing 5% CO2. To quantify migrated or invaded cells, the cells from the top side of the membrane were gently removed using a cotton-tipped swab, and invading cells attached to the bottom side of the membrane were fixed with 70% ethanol and stained with 0.5% crystal violet. Cell numbers from five representative fields were counted for each insert.
RESULTS
Targeted Quantitative Profiling of Differential Expression of Small GTPases during Metastatic Transformation of CRC Cells.
The SW480/SW620 pair of isogenic, primary/metastatic CRC cell lines constitute a valuable in vitro cellular model for studying CRC metastasis. The SW620 cell line was isolated from the lymph node metastatic site of the same patient as its nonmetastatic counterpart (i.e., SW480).16 This pair of cell lines have been widely explored with various comparative shotgun and targeted proteomics approaches at the whole proteome and secretome levels to discover potential biomarkers and therapeutic targets for CRC.17–22
To systematically investigate the differential protein expression of small GTPases in the paired SW480/SW620 cells, we employed a previously established targeted proteomic workflow, which involves SILAC, SDS-PAGE fractionation, and scheduled MRM analysis (Figure 1A).12 In this vein, we incorporated, into the MRM library, only those tryptic peptides that could be uniquely assigned to specific protein forms of small GTPases proteins. SW480/SW620 cells are known to harbor the commonly observed KRAS G12 V mutations. The mutation-containing peptide sequence, however, is also present in NRAS and HRAS. Hence, this peptide was excluded in our MRM analysis, and our current MRM workflow only includes unmutated small GTPase peptides. To obtain reliable quantification results, we carried out SILAC experiments in triplicate, with two sets of forward labeling (Figure 1A) and one set of reverse labeling. In this vein, the throughput for the method is high, where the entire library of small GTPase tryptic peptides could be monitored in two LC-MRM runs with retention time scheduling by using the iRT algorithm.15 We also examined the run-to-run reproducibility of the MRM-based quantification. As illustrated in Figure 1B, the Log2-transformed SILAC ratios for all of the quantified small GTPases obtained from forward and reverse SILAC labeling experiments exhibited an excellent linear fit (R2 = 0.9280).
MRM analyses facilitated reproducible quantification of 83 small GTPases in each of the three SILAC labeling experiments (Table S1 and Figure S1). By contrast, analysis of the same samples by LC-MS/MS in the data-dependent acquisition (DDA) mode only led to the identification of 37 and 35 small GTPases in the two forward SILAC samples (F1 and F2, respectively) and 31 small GTPases in the reverse SILAC sample (R; Figure 1C). Moreover, MRM exhibited better sensitivity than the shotgun proteomic approach, as reflected by the substantially increased coverage of the small GTPase proteome in two one-dimensional LC-MRM runs (Figure 1C).
We also analyzed the previously published shotgun or targeted proteomic data acquired from the SW480/SW620 paired cell lines. Figure S2 shows the Venn diagrams representing small GTPases quantified in this study as compared to those reported by three independent proteomic studies.18,20,21 Thus, scheduled LC-MRM analysis, together with SDS-PAGE fractionation, which substantially reduces complexity of background proteome, provided an increased depth in coverage of the small GTPase proteome. Moreover, the enhanced reproducibility and sensitivity of the MRM approach facilitated reliable high-throughput quantification of small GTPases with a relatively small amount of protein mixture, where 100 μg was employed for gel fractionation and only 4% of the ensuing tryptic digestion mixture was injected for LC-MRM analysis. It is of note that the method reported by Cai et al.,21 which utilizes the acyl-phosphate GTP affinity probe, enabled proteome-wide enrichment of GTP-binding proteins, not solely restricted to small GTPases.
The above results demonstrated that the scheduled LC-MRM approach facilitates highly sensitive and reproducible quantitative profiling of small GTPases in CRC cell lines with an elevated throughput and depth of coverage.
Validation of Differential Protein Expression by Western Blot Analyses.
Among the 83 quantified small GTPases, 7 and 18 proteins were significantly up- and down-regulated (with >1.5-fold change), respectively, in the metastatic (SW620) relative to the primary (SW480) CRC cells (Figure 1D). In this vein, we employed 1.5-fold change as a cutoff based on the mean relative standard deviation (RSD; 6.7%, Table S1) of the measurement results of all small GTPases. Notably, several Rho small GTPases exhibited significantly altered expression, including RHOB, RHOF, and RHOG. Figure S3A displays the selected-ion chromatograms (SICs) for these small GTPases. Down-regulation of RHOB and up-regulation of RHOG in SW620 relative to SW480 cells were previously reported in two independent studies.21,23 Importantly, RHOB is a well-recognized tumor suppressor for CRC and clear-cell renal cell carcinoma (ccRCC).23–25 Since the protein abundance of RHOB was down-regulated by ~2-fold in SW620 over SW480 cells, our results validated a known suppressor for CRC metastasis.
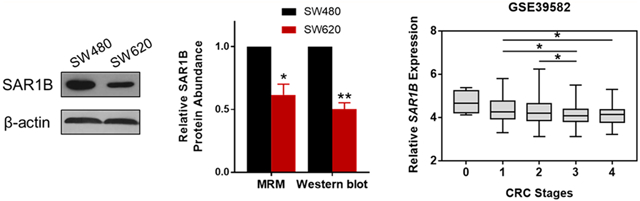
Among the small GTPases with altered protein expression, RAB6B displayed a pronouncedly decreased expression (by ~10-fold) in SW620 relative to SW480 cells (Figure 1D). Owing to the high degree of sequence homology among the three RAB6 isoforms (RAB6A/B/C), common antibody-based approach including Western blot may be susceptible to cross-reactivity and hence could be difficult to distinguish the different protein isoforms. By selecting proteotypic peptides which have distinct sequences for different protein isoforms, our MRM-based approach successfully revealed the substantial down-regulation of RAB6B, but not RAB6A/C, in SW620 relative to SW480 cells (Figure S3B). Meanwhile, Western blot analyses validated the MRM data for RAB6A and pan-RAB6 proteins (RAB6A/B/C) but failed to distinguish RAB6A/C from RAB6B (Figure S3B). In addition, we verified the robustness and accuracy of the MRM-based quantitation for other GTPases. As shown in Figures 2 and S4, we confirmed the up-regulation of SAR1A and RAB27A, and down-regulation of SAR1B and ARF4 in the metastatic SW620 cells relative to the primary SW480 cells. Taken together, the MRM method constitutes a high-throughput and accurate approach for quantifying small GTPases.
Figure 2.

Validation of differential expression of SAR1A and SAR1B in SW480/SW620 cells. Selected-ion chromatograms (SICs) for the quantification of tryptic peptides: (A) TDAISEEK from SAR1A and (B) EMFGLYGQTTGK from SAR1B, in one forward and one reverse SILAC labeling experiments. Western blot validation of the differentially expressed SAR1A (A) and SAR1B (B) proteins in SW480/SW620 cells, and quantitative comparison of protein ratios obtained from LC-MRM and Western blot analyses (n = 3). The p values were calculated by using a paired two-tailed Student’s t test (*, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01). The data represent the mean and standard deviation of results obtained from three parallel experiments.
Potential Roles of SAR1A and SAR1B in CRC Progression.
We sought to examine, among the differentially expressed small GTPase proteins, which can potentially promote or suppress CRC progression. Therefore, we assessed the gene expression data from patients in The Cancer Genome Atlas Colon Adenocarcinoma (TCGA-COAD) data set, and we found that there were significantly lower mRNA expressions of ARF4, RAB6B, RHOF, and SAR1B genes in CRC tissues than in normal tissues (n = 50; p < 0.05, paired Student’s t-test) (Figures 3A and S5).
Figure 3.

Down-regulation of SAR1B in CRC confers worse patient prognosis and associates with higher disease stages. Comparison of SAR1B expression levels in paired CRC tissues (CRC) with adjacent nontumor tissues (normal) in (A) TCGA-COAD (n = 50) and (B) GSE21510 (n = 44) cohorts. The p values were calculated by using a paired two-tailed Student’s t test (****, p < 0.0001). Kaplan–Meier survival analysis of CRC patients stratified by the median SAR1B mRNA expression in (C) GSE17536 (n = 232) and (D) GSE39582 (n = 585) cohorts. The log-rank (Mantel–Cox) test was used to calculate the p values. (E) Correlation of SAR1B mRNA expression with different CRC stages in the GSE39582 (n = 585) cohort. The p values were calculated by using an unpaired two-tailed Student’s t test (*, 0.01 ≤ p < 0.05).
We also interrogated other CRC patient data sets accessible from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database, including GSE14333 (n = 290, Ludwig Institute for Cancer Research), GSE17536 (n = 232, Vanderbilt University), GSE21510 (n = 274, Juntendo University), and GSE39582 (n = 585, Ligue Nationale contre le Cancer).26–29 We again observed significantly lower mRNA expressions of ARF4, RAB6B, RHOF, and SAR1B in CRC tissues than in normal tissues (Figures 3B and S5). However, higher mRNA expression of ARF4, RAB6B, and RHOF was significantly associated with poorer CRC patient survival, which is contradictory to their plausible roles in suppressing metastasis (Figures S5C, S5F, and S5I). The discrepancy between the proteomic data and the bioinformatic data may arise from high degree of patient-to-patient variability. In addition, bioinformatic analysis was performed on the basis of mRNA expression levels; thus, transcript isoforms resulting from alternative splicing and post-translational modifications of proteins can further complicate the comparison between mRNA and protein levels.30 In this vein, a moderate correlation reflected by the Pearson’s correlation coefficients in the range of 0.39–0.79 from cell-line data and 0.6 from tissue-derived data were observed between mRNA levels and protein abundance.31
We further investigated the prognostic relevance of SAR1A and SAR1B in CRC patient cohorts. We observed that SAR1A expression is consistently up-regulated in human CRC tissues compared to the adjacent normal tissues in both TCGA-COAD and GSE21510 cohorts (Figures S6A,B). Kaplan–Meier survival analyses showed that CRC patients with higher SAR1A expression presented a higher risk of death [hazard ratio (HR) = 2.353, 95% confidence interval (CI) = 1.349 to 4.106, Log-rank p = 0.0026], and those with higher SAR1B expression displayed a more favorable outcome (HR = 0.5915, 95% CI = 0.3723 to 0.9398, Log-rank p = 0.0262; Figures 3C,D and S6C,D). These observations are in keeping with the quantitative proteomic data showing the down-regulated SAR1B and up-regulated SAR1A in SW620 over SW480 cells. Furthermore, lower SAR1B mRNA expression is significantly correlated with more advanced pathological stages of CRC (Figure 3E).
Therefore, the observation of a progressive diminution of SAR1B mRNA expression with increasing CRC stage and the significant association between SAR1B down-regulation and worse patient survival in several large cohorts (290, 232, 274, and 585 patients for the GSE14333, GSE17536, GSE21510, and GSE39582 data sets, respectively) further substantiate the relevance of SAR1B in CRC.
Collectively, these findings substantiated SAR1B as a potential suppressor and a prognostic biomarker for CRC progression, and SAR1A and SAR1B may assume distinct roles in disease development. Nonetheless, further investigation and functional characterizations of SAR1B as a diagnostic biomarker in patient biopsies or serum samples are warranted.
SAR1B Knockdown Led to Elevated in Vitro Migration and Invasion of SW480 Cells by Modulating Epithelial–Mesenchymal Transition (EMT).
To explore the potential roles of SAR1B in CRC metastasis, we next examined how the migratory and invasive abilities of CRC cells are modulated by genetic depletion of SAR1B. We found that shRNA-mediated knockdown of SAR1B in SW480 cells elicited marked elevations in both the migratory and invasive abilities, as manifested by results from both transwell migration/invasion assay (Figure 4A,B) and wound-healing assay (Figure 4C,D). These observations are in accordance with the acquisition of a mesenchymal phenotype.
Figure 4.

Depletion of SAR1B modulates the migratory and invasive capacities of SW480 cells. (A) Representative images and (B) quantification results for migration/invasion assay showing the effects of stable SAR1B knockdown (shSAR1B) on the in vitro migratory and invasive abilities of SW480 cells compared to the control (shScramble). (C) Representative images and (D) quantification results for wound healing assay showing the effects of stable SAR1B knockdown (shSAR1B) on the wound healing abilities of SW480 cells compared to the control (shScramble). (E) RT-qPCR expression analyses of EMT marker genes in SW480 cells with stable shScramble or shSAR1B knockdown. Gene expression was normalized to the relative expression of GAPDH. “n.d.”, not detectable. (F) Cell proliferation of shScramble/shSAR1B SW480 cells. The p values were calculated by using a paired two-tailed Student’s t test (*, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01). The data represent the mean and standard deviation of results obtained from three parallel experiments.
We next performed quantitative reverse transcription PCR (RT-qPCR) experiments to assess the expression of EMT markers in SW480 cells upon shRNA-mediated stable knockdown of SAR1B. The results indeed demonstrated that knockdown of SAR1B led to a significant reduction in the levels of epithelial marker E-cadherin (CDH1) and increased mRNA expression of the mesenchymal marker vimentin (VIM), indicating a shift to a more mesenchymal phenotype (Figure 4E). We, however, did not observe any significant changes in mRNA expression for other mesenchymal markers, including SNAI1/2 and ZEB1/2. In addition, the mRNA level of CDH2 gene was not detectable in SW480 or SW620 cells. Likewise, loss of SAR1B did not alter the proliferation rate of SW480 cells (Figure 4F). Together, SAR1B may contribute to suppression of CRC motility and metastasis partly through modulating EMT, but not by regulating proliferation.
RAB31 Promotes Proliferation, Migration, and Invasion of SW480 Cells in Vitro.
RAB31 was previously shown to play a role in breast cancer proliferation and acquired tamoxifen resistance.32,33 Our targeted proteomic data revealed a more than 4-fold down-regulation of RAB31 in the metastatic SW620 cells, which was validated by Western blot analysis (Figures S7A,B). However, there were no significant differences in RAB31 expression between CRC and normal tissues in the TCGA-COAD data set (Figure S7C). Higher RAB31 mRNA expression is significantly correlated with more advanced pathological stages of CRC (Figure S7D). Moreover, Kaplan–Meier survival analyses of the GSE14333 and GSE17536 data sets revealed that higher RAB31 mRNA levels are correlated with worse disease-free survival (DFS; Figures S7E,F), which argues against the hypothesis that RAB31 may serve as a suppressor for CRC metastasis.
We further investigated whether knock-down of RAB31 affects the behavior of SW480 cells by assessing cell proliferation and transwell migration/invasion. The siRNA-mediated knockdown of RAB31 in SW480 cells was confirmed by Western blot analysis (Figure S8A). We observed significantly diminished cell proliferation, in vitro migration and invasion of SW480 cells after genetic depletion of RAB31 (Figures S8B,C), which is accompanied by a diminished level of mesenchymal marker Slug (Figure S8A). Although RAB31 is substantially down-regulated in SW620 cells, it appeared to be a metastasis driver instead of a suppressor based on the cellular experiments. Therefore, these results underscored that some of the dysregulated small GTPases may be simply accompanied by CRC metastasis and they may not modulate metastatic transformation of CRC. This observation emphasizes the importance of validating the findings made from proteomic experiments with bioinformatic analysis and cell-based assay.
DISCUSSION
In this study, we employed our recently developed MRM-based targeted proteomic method for high-throughput, reproducible and in-depth quantification of small GTPases in the matched primary/metastatic CRC cell lines (i.e., SW480/ SW620). This approach, involving the use of metabolic labeling with SILAC and SDS-PAGE fractionation, is a readily accessible, facile, and inexpensive strategy. The method facilitated the quantification of 83 quantified small GTPases. Among them we found that SAR1A and SAR1B proteins were significantly elevated and diminished, respectively, in SW620 over SW480 cells. Furthermore, lower levels of SAR1B mRNA expression was significantly associated with poorer survival in CRC patients and with elevated disease stages, suggesting the potential role of SAR1B in suppressing CRC progression. Our results also indicated that loss of SAR1B conferred elevated in vitro migratory and invasive abilities of SW480 cells, accompanied by reduced E-cadherin and elevated vimentin expression. Metastasis consists of multiple steps of cellular transformation, during which the acquisition of enhanced cell motility, migratory capability and invasiveness is essential to the early stage of metastatic dissemination.34 Hence, we reason that diminished SAR1B promotes motility and in vitro metastasis of SW480 cells partly through modulating EMT maker gene expression.
The two SAR1 proteins, SAR1A and SAR1B, are ubiquitously expressed in many types of cancer. Aberrant expression or mutations in the SAR1A and SAR1B genes were shown to play an important role in regulating cholesterol biosynthesis.35,36 SAR1B promotes secretion of lipoproteins apoB48 and apoB100, whereas overexpression of SAR1A displays an opposite effect, preferentially blocking the secretion of the lipid-laden particles.36 Furthermore, a reduction in total serum cholesterol could be a signal of occult CRC.37 We, therefore, speculate that the crucial role of SAR1B in lipid biosynthesis and secretion may contribute to CRC progression. Interestingly, a previous study showed that the expressions of SAR1B and SAR1A were significantly decreased and increased, respectively, in the intestinal biopsies of patients with Anderson’s disease as compared to healthy individuals.38 Hence, SAR1A and SAR1B may display functionally redundant, yet distinct roles in regulating vesicular trafficking and cargo transport. Although we demonstrated that SAR1A is up-regulated in the metastatic SW620 cells and displayed prognostic values in large patient cohorts, the detailed molecular mechanisms underlying the role of SAR1A in CRC progression warrant further investigation.
In conclusion, we described the use of a previously established MRM-based targeted proteomic method for high-throughput quantitative measurement of small GTPases associated with CRC metastasis by utilizing paired SW480/SW620 cell lines. Among the 25 small GTPases that exhibited differential expression with at least a 1.5-fold change in metastatic SW620 relative to primary SW480 cells, we found that SAR1B was significantly down-regulated during metastatic transformation and the SAR1B mRNA expression was significantly correlated with disease stages and prognosis in CRC patient cohorts. We also reported that SAR1B depletion in SW480 cells promoted cell motility and transwell invasion, partly through modulation of EMT. Collectively, these results suggested that SAR1B may serve as a prognostic biomarker and a potential suppressor for CRC metastasis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA210072 to Y. W.), and M. H. was supported in part by an NRSA Institutional Training Grant (T32 ES018827).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.9b00938.
Detailed experimental conditions, LC-MRM traces and Western blot results, and results from bioinformatic analysis of public available data(PDF)
Excel table for the LC-MRM quantification data (XLSX)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL; Miller KD; Fedewa SA; Ahnen DJ; Meester RGS; Barzi A; Jemal A Ca-Cancer J. Clin. 2017, 67, 177–193. [DOI] [PubMed] [Google Scholar]
- (2).Bray F; Ferlay J; Soerjomataram I; Siegel RL; Torre LA; Jemal A Ca-Cancer J. Clin 2018, 68, 394–424. [DOI] [PubMed] [Google Scholar]
- (3).O’Connell JB; Maggard MA; Ko CY J. Natl. Cancer I 2004, 96, 1420–1425. [DOI] [PubMed] [Google Scholar]
- (4).Cartwright TH Clin. Colorectal Cancer 2012, 11, 155–66. [DOI] [PubMed] [Google Scholar]
- (5).Colicelli J Sci. Signaling 2004, 2004, RE13. [Google Scholar]
- (6).Porther N; Barbieri MA Small GTPases 2015, 6, 135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Armaghany T; Wilson JD; Chu Q; Mills G Gastrointest. Cancer Res 2012, 5, 19–27. [PMC free article] [PubMed] [Google Scholar]
- (8).Su CY; Lim C; Jan YH; Chang YC; Chen CL; Hsiao M Faseb J. 2014, 28. [Google Scholar]
- (9).Luo Y; Yu SY; Chen JJ; Qin J; Qiu YE; Zhong M; Chen M Oncotarget 2018, 9, 3830–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yin Y; Zhang B; Wang W; Fei B; Quan C; Zhang J; Song M; Bian Z; Wang Q; Ni S; Hu Y; Mao Y; Zhou L; Wang Y; Yu J; Du X; Hua D; Huang Z Clin. Cancer Res 2014, 20, 6187–99. [DOI] [PubMed] [Google Scholar]
- (11).Goldenring JR; Nam KT Br. J. Cancer 2011, 104, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang M; Qi TYF; Li L; Zhang G; Wang YS Cancer Res. 2018, 78, 5431–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kawahara R; Bollinger JG; Rivera C; Ribeiro AC; Brandao TB; Paes Leme AF; MacCoss MJ Proteomics 2016, 16, 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Proteomics 2012, 12, 1111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Leibovitz A; Stinson JC; McCombs WB; McCoy CE; Mazur KC; Mabry ND Cancer Res. 1976, 36, 4562–9. [PubMed] [Google Scholar]
- (17).Xue H; Lu B; Zhang J; Wu M; Huang Q; Wu Q; Sheng H; Wu D; Hu J; Lai MJ Proteome Res. 2010, 9, 545–55. [DOI] [PubMed] [Google Scholar]
- (18).Ghosh D; Yu H; Tan XF; Lim TK; Zubaidah RM; Tan HT; Chung MC; Lin QJ Proteome Res. 2011, 10, 4373–87. [DOI] [PubMed] [Google Scholar]
- (19).Lei YL; Huang K; Gao C; Lau QC; Pan H; Xie K; Li JY; Liu R; Zhang T; Xie N; Nai HS; Wu H; Dong Q; Zhao X; Nice EC; Huang CH; Wei YQ Mol. Cell. Proteomics 2011, 10, M110.005397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lee JG; McKinney KQ; Pavlopoulos AJ; Park JH; Hwang SJ Proteomics 2015, 113, 326–36. [DOI] [PubMed] [Google Scholar]
- (21).Cai R; Huang M; Wang YS Anal. Chem. 2018, 90, 14339–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Torres S; Garcia-Palmero I; Marin-Vicente C; Bartolome RA; Calvino E; Fernandez-Acenero MJ; Casal JI J. Proteome Res. 2018, 17, 252–264. [DOI] [PubMed] [Google Scholar]
- (23).Arsic N; Ho-Pun-Cheung A; Evelyne C; Assenat E; Jarlier M; Anguille C; Colard M; Pezet M; Roux P; Gadea G PLoS One 2017, 12, No. e0172125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu M; Tang Q; Qiu M; Lang N; Li M; Zheng Y; Bi F FEBS Lett. 2011, 585, 2998–3005. [DOI] [PubMed] [Google Scholar]
- (25).Chen WH; Niu SX; Ma X; Zhang P; Gao Y; Fan Y; Pang HG; Gong HJ; Shen DL; Gu LY; Zhang Y; Zhang X PLoS One 2016, 11, No. e0157599. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (26).Jorissen RN; Gibbs P; Christie M; Prakash S; Lipton L; Desai J; Kerr D; Aaltonen LA; Arango D; Kruhoffer M; Orntoft TF; Andersen CL; Gruidl M; Kamath VP; Eschrich S; Yeatman TJ; Sieber OM Clin. Cancer Res. 2009, 15, 7642–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Freeman TJ; Smith JJ; Chen X; Washington MK; Roland JT; Means AL; Eschrich SA; Yeatman TJ; Deane NG; Beauchamp RD Gastroenterology 2012, 142, 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tsukamoto S; Ishikawa T; Iida S; Ishiguro M; Mogushi K; Mizushima H; Uetake H; Tanaka H; Sugihara K Clin. Cancer Res 2011, 17, 2444–50. [DOI] [PubMed] [Google Scholar]
- (29).Marisa L; de Reynies A; Duval A; Selves J; Gaub MP; Vescovo L; Etienne-Grimaldi MC; Schiappa R; Guenot D; Ayadi M; Kirzin S; Chazal M; Flejou JF; Benchimol D; Berger A; Lagarde A; Pencreach E; Piard F; Elias D; Parc Y; Olschwang S; Milano G; Laurent-Puig P; Boige V PLoS Med. 2013, 10, No. e1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Liu Y; Beyer A; Aebersold R Cell 2016, 165, 535–50. [DOI] [PubMed] [Google Scholar]
- (31).Edfors F; Danielsson F; Hallstrom BM; Kall L; Lundberg E; Ponten F; Forsstrom B; Uhlen M Mol. Syst. Biol 2016, 12, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Grismayer B; Solch S; Seubert B; Kirchner T; Schafer S; Baretton G; Schmitt M; Luther T; Kruger A; Kotzsch M; Magdolen V Mol. Cancer 2012, 11, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Huang M; Wang Y Anal. Chem 2018, 90, 14551–14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Justus CR; Leffler N; Ruiz-Echevarria M; Yang LV J. Visualized Exp 2014, 51046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jones B; Jones EL; Bonney SA; Patel HN; Mensenkamp AR; Eichenbaum-Voline S; Rudling M; Myrdal U; Annesi G; Naik S; Meadows N; Quattrone A; Islam SA; Naoumova RP; Angelin B; Infante R; Levy E; Roy CC; Freemont PS; Scott J; Shoulders CC Nat. Genet 2003, 34, 29–31. [DOI] [PubMed] [Google Scholar]
- (36).Fryer LGD; Jones B; Duncan EJ; Hutchison CE; Ozkan T; Williams PA; Alder O; Nieuwdorp M; Townley AK; Mensenkamp AR; Stephens DJ; Dallinga-Thie GM; Shoulders CC J. Biol. Chem 2014, 289, 4244–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mamtani R; Lewis JD; Scott FI; Ahmad T; Goldberg DS; Datta J; Yang YX; Boursi B PLoS Med. 2016, 13, No. e1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Georges A; Bonneau J; Bonnefont-Rousselot D; Champigneulle J; Rabes JP; Abifadel M; Aparicio T; Guenedet JC; Bruckert E; Boileau C; Morali A; Varret M; Aggerbeck LP; Samson-Bouma ME Orphanet J. Rare Dis 2011, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The processed MRM data are provided in Table S1. The Skyline MRM library (including the transition list for quantification and the iRT file) for human small GTPase proteome and the raw files obtained from the LC-MRM analyses of small GTPases for the SW480/SW620 cell lines were deposited into PeptideAtlas with the identifier number of PASS01326 (http://www.peptideatlas.org/PASS/PASS01326).