

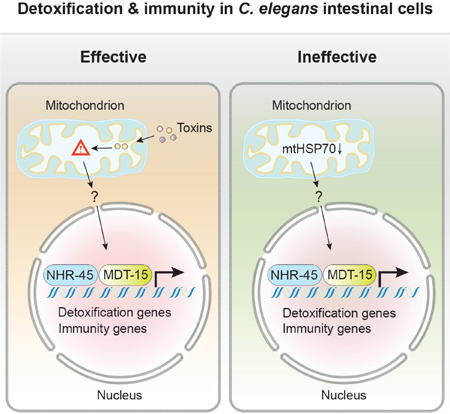
Summary
In Caenorhabditis elegans, mitochondrial dysfunction caused by mutation or toxins activates programs of detoxification and immune response. A genetic screen for mutations that constitutively induce C. elegans mitochondrial defense revealed reduction of function mutations in the mitochondrial chaperone hsp-6/mtHSP70 and gain of function mutations in the Mediator component mdt-15/MED15. The activation of detoxification and immune responses is transcriptionally mediated by mdt-15/MED15 and nuclear hormone receptor nhr-45. Mitochondrial dysfunction triggers redistribution of intestinal mitochondria that requires the mitochondrial Rho GTPase miro-1 and its adaptor trak-1/TRAK1 but not nhr-45-regulated responses. Disabling the mdt-15/nhr-45 pathway renders animals more susceptible to a mitochondrial toxin or pathogenic Pseudomonas aeruginosa, but paradoxically improves health and extends lifespan in animals with mitochondrial dysfunction caused by a mutation. Thus, some of the health deficits in mitochondrial disorders may be caused by the ineffective activation of detoxification and immune responses, which may be inhibited to improve health.
Graphical Abstract
eTOC blurb
Mitochondrial dysfunction activates a xenobiotic response, but the physiological consequences are unknown. Mao et al. show that MDT-15 and NHR-45 regulate a mitochondrial dysfunction-induced xenobiotic response, and analyze its physiological role in C. elegans detoxification, immunity and lifespan.
Introduction
Mitochondria are organelles with a bacterial origin that perform diverse functions in eukaryotic cells (Nunnari and Suomalainen, 2012). Thousands of eukaryotic nuclear genes encode proteins with ancient bacterial ancestry that are transported to the mitochondrion and coordinate with just a few dozen genes that remain in the mitochondrial genome to assemble, replicate, and regulate the mitochondrion. In Caenorhabditis elegans, mitochondrial dysfunction caused by mutation, gene inactivation, or toxins triggers detoxification and immune responses, perhaps because bacterial pathogens take advantage of the microbial origin of the mitochondrion to evolve toxins and virulence factors from their weaponry of bacterial conflict. A key mitochondrial function is the electron transport chain that is composed of dozens of proteins from both the nuclear and mitochondrial genomes and transduces redox energy of metabolism to a mitochondrial proton gradient which powers the synthesis of ATP by ATP synthase. A broad range of age-related human diseases, including neurodegeneration, cardiac disease, diabetes and cancer, are caused by decreased activity of the respiratory chain (Vafai and Mootha, 2012). Mutations in the genes encoding subunits of mitochondrial respiratory chain elicit devastating early onset diseases, such as Leigh syndrome, and are largely untreatable. However, alleviating the pathology of mitochondrial dysfunction to relieve some symptoms can be achievable if the response pathways can be modulated.
The mitochondrion plays a key role in the innate immune system, contributing ATP and reactive oxygen species (ROS), and serving as a signaling hub for antiviral and antibacterial immunity (West et al., 2011). Disruption of the mitochondrial respiratory chain in Caenorhabditis elegans or mice activates immune responses (Kim et al., 2016; Liu et al., 2014; Pellegrino et al., 2014; West et al., 2015). Surveillance of mitochondrial integrity is part of a broader surveillance of core cellular machineries, including mitochondria, ribosomes, proteasomes, etc, that couples to detoxification and immune responses (Govindan et al., 2015; Lehrbach and Ruvkun, 2016; Liu et al., 2014; Melo and Ruvkun, 2012). In these surveillance systems, even disruptions of the cellular pathways by mutation or RNAi inactivation are interpreted by C. elegans response pathways as a pathogen attack, so that drug detoxification and immune responses are mounted for the C. elegans genetic dysfunction misperceived as a pathogen attack. However, how mitochondrial dysfunction signals to the activation of immune and detoxification responses, and the effect of this response on animal physiology and health is unknown.
In this study, we constructed a xenobiotic reporter gene that responds specifically to mitochondrial dysfunction, and performed a genetic screen to identify genes that encode proteins that surveil mitochondrial dysfunction and couple to induce xenobiotic responses. We found that a hypomorphic mutation of mitochondrial chaperone hsp-6/mtHSP70 activates this xenobiotic response through MDT-15/NHR-45 transcriptional signaling. Uncoupling this pathway by removing mdt-15 or nhr-45 disables these detoxification and immune responses, and makes the animal more sensitive to the mitochondrial inhibitor antimycin or the pathogen bacteria Pseudomonas aeruginosa. However, decoupling this xenobiotic response from mitochondrial surveillance improves the health and extends life span, suggesting the pathologic effects of mitochondrial mutations are partially caused by the ineffective induction of detoxification and immune response genes. Moreover, the intestine of C. elegans displays unique mitochondrial relocalization upon mitochondrial dysfunction. And we show that this relocalization of mitochondria occurs in a MIRO-1/TRAK-1-dependent and NHR-45-independent manner.
Results
A xenobiotic reporter that is tightly coupled to mitochondrial dysfunction
Gene expression analysis of C. elegans with a mitochondrial defect caused by RNAi inactivation of the mitochondrial AAA protease spg-7/AFG3L2 revealed a strong induction of genes involved in detoxification and immunity (Figure S1A) (Nargund et al., 2012). For example, the C. elegans cytochrome P450 cyp-14A4 is induced about 100-fold by mitochondrial dysfunction (Figure S1A). This C. elegans cytochrome gene is most closely related to the mammalian CYP2C2 clade which shows signs of positive selection in mammals, favoring a role in detoxification (Thomas, 2007). Cytochrome P450s are heme proteins that use molecular oxygen to hydroxylate small molecule substrates as steps in the detoxification of toxins (Guengerich, 2008; Thomas, 2007). This induction of C. elegans detoxification and immunity genes by a genetic defect in the mitochondria is consistent with a model that mitochondrial dysfunction in nature is often caused by toxins from pathogenic bacteria, rather than by mutations, and is thus interpreted by eukaryotic surveillance pathways as likely to be toxin-induced (Figure S1B). Consistent with the view that mitochondrial mutations and toxins produce the same detoxification response, a cyp-14A4 promoter GFP fusion gene is strongly induced by treatment with a low dose antimycin, a Streptomyces mitochondrial toxin that inhibits complex III (Figure 1A and S1C). Higher doses of antimycin cause developmental delay or larval arrest in addition to induction of the cyp-14A4p::gfp fusion gene (Figure 1A). Activation of cyp-14A4p::gfp is also observed after RNAi inactivation of the genes that encode mitochondrial chaperones hsp-6/DnaK/mtHSP70, hsp-60/GroEL/mtHSP60 (Yoneda et al., 2004), or protease spg-7/AFG3L2 (Figure 1B, S1D and S1E).
Figure 1. A particular cytochrome p450 gene is tightly coupled to mitochondrial dysfunction.

(A) Synchronized L1 larvae carrying a cyp-14A4p::gfp transgene were grown in M9 with E. coli OP50 and indicated concentration of antimycin at 20°C for th ree days. A red rectangle indicates larval delay or arrest. Scale bar, 500 μm.
(B) Synchronized L1 larvae of cyp-14A4p::gfp animals feeding on control RNAi or RNAi of hsp-6, hsp-60 or spg-7 at 20°C for three days. Scale bar, 100 μm.
(C) L4 larvae carrying cyp-14A4p::gfp of indicated genetic background were photographed. Scale bar, 100 μm.
(D) Synchronized L1 larvae of wild-type and hsp-6(mg585) animals were grown at 20°C or 25°C for two days. Scale bar, 2 mm.hsp-6: mitochondrial DnaK/Hsp70; hsp-60: mitochondrial GroEL/Hsp60; spg-7: mitochondrial AAA protease, AFG3L2.
The human pathogenic bacteria P. aeruginosa secretes pyoverdin, an iron-binding siderophore that sequesters iron and damages C. elegans mitochondria when wild type C. elegans is grown on P. aeruginosa (Kim et al., 2002; Kirienko et al., 2015). C. elegans cultured with P. aeruginosa as a food source also induces cyp-14A4p::gfp (Figure S1F and S1G). Hydrogen peroxide which damages mitochondria, also activates cyp-14A4p::gfp (Figure S1H and S1I); heat shock stress slightly reduced cyp-14A4p::gfp but tunicamycin which inhibits endoplasmic reticulum function does not activate cyp-14A4p::gfp (Figure S1H–S1K).
To establish the specificity of cyp-14A4p::gfp induction for mitochondrial dysfunction, we inactivated a range of essential genes and tested for cyp-14A4p::gfp activation. Inactivation by RNAi of 379 essential genes encoding C. elegans core components, ranging from ribosomes to vacuolar proton pumps, activates xenobiotic responses and bacterial avoidance behavior, both countermeasures to a perceived bacterial attack (Melo and Ruvkun, 2012) (Table S1). When animals were challenged by each of these 379 RNAi gene inactivations representing a wide range of cellular assaults, cyp-14A4p::gfp was induced almost exclusively by the inactivation of nuclear genes that encode mitochondrial proteins (Figure S1L–S1O and Table S1); RNAi inactivation of genes required for other core cellular processes, such as microtubule, actin cytoskeleton, proteasomes, ribosomes, and ER, did not induce cyp-14A4p::gfp (Figure S1N). Thus the induction of cyp-14A4 gene is tightly coupled to mitochondrial dysfunction. cyp-14A4 is one of nearly 100 C. elegans cytochrome p450 genes; distinct suites of cytochrome p450 as well as distinct sets of other classes of detoxification genes such as UDP-glycosyltransferase genes and ABC transporter genes are coupled to other cellular dysfunctions, such as the ribosome, the cytoskeleton, the proteasome, etc. (Govindan et al., 2015; Lehrbach and Ruvkun, 2016; Liu et al., 2014; Melo and Ruvkun, 2012). The expression of cyp-14A4p::gfp is predominantly in the intestine that is likely to be the first line of defense against toxins and virulence factors from the complex bacterial diet consumed by C. elegans (Melo and Ruvkun, 2012; Samuel et al., 2016). The intestine is the major site of C. elegans detoxification; in mammals, cells in the gut-derived liver are specialized for detoxification and are a major site of cytochrome p450 expression. C. elegans has no liver, but those detoxification functions reside in the 32 intestinal cells which serve both gut and liver functions (McGhee, 2007).
hsp-6 hypomorphic mutant activates xenobiotic response
To discern the molecular pathways that couple the mitochondrial dysfunction to the induction of xenobiotic responses, an ethyl methanesulfonate (EMS) mutagenesis genetic screen of a parent strain with no mitochondrial defects carrying the cyp-14A4p::gfp fusion gene was performed. This screen of 20,000 F2 generation progeny after the EMS mutagenesis yielded 8 mutants that express cyp-14A4p::gfp (Figure S1P). After checking for the induction of cyp-14A4p::gfp continuing in progeny, deep sequencing of mutant genomes and genetic mapping of candidate molecular lesions (Bigelow et al., 2009; Sarin et al., 2008) identified mutations in 7 genes, including a Pro386Ser (CCA to TCA) missense mutation in the gene hsp-6(mg583) allele (Figure S1Q and Table S1). C. elegans hsp-6, an orthologue of human mtHSP70 (Figure S1S), encodes a mitochondrial chaperone that is essential for mitochondrial protein import and proteostasis (Heschl and Baillie, 1989; Kim et al., 2016; Kimura et al., 2007). C. elegans HSP-6, as well as human mtHSP70, is most closely related to bacterial or Archaea DnaK (Figure S1S), consistent with its migration along with thousands of other bacterial genes to the animal nuclear genome after the capture of the bacterial genome more than a billion years ago to become the mitochondria. The mg583 mutation is a conserved Pro386 to Ser substitution (Figure S1S). RNAi inactivation of hsp-6 gene or a probable null deletion allele hsp-6(tm515) (Figure S1R) causes a larval lethal phenotype; the viable hsp-6(mg583) allele is likely a reduction-of-function mutation. CRISPR-Cas9 was used to generate an independent allele, hsp-6(mg585), with the same missense mutation Pro386Ser (Figure S1Q) (Arribere et al., 2014; Ward, 2015). The expression of cyp-14A4p::gfp was activated in both hsp-6(mg583) and hsp-6(mg585) mutants (Figure 1C, S1T and S1U). hsp-6(mg585) mutant animals grow slowly and are less fertile than wild type (Figure S1V–S1X). Mitochondrial mutations in C. elegans trigger an aversive behavioral response, as animals misinterpret the dysfunctions caused by their own genetic dowry as a bacterial pathogen attack and modulate their behavior to attempt to escape the benign E. coli bacterial lawn they misperceive as pathogenic (Melo and Ruvkun, 2012). hsp-6(mg585) mutant animals show this aversive behavior (Figure 1D).
mRNA sequencing of hsp-6(mg585) revealed thousands of genes that are two-fold or more upregulated or downregulated (Table S1). Comparison of the hsp-6(mg585) mRNA-seq data and microarray data of spg-7 RNAi (Nargund et al., 2012) or microarray of hsp-6 RNAi (Kim et al., 2016) showed a high consistency and revealed that characteristic mitochondrial stress responses are activated in hsp-6(mg585) mutant (Figure S1Y–S1Z).
If the gene expression responses to mitochondrial dysfunction are key homeostatic responses to this dysfunction, mutants that disrupt these responses might render weak mitochondrial mutations such as hsp-6(mg585) more severe. We previously identified 42 gene inactivations that disrupt up-regulation of hsp-6 in response to mitochondrial dysfunction (Liu et al., 2014). We tested whether any of these gene inactivations enhance the phenotype of hsp-6(mg585). Eight of the 42 gene inactivations that are viable in wild type caused larval arrest in combination with the hsp-6(mg585) allele (Table S1). These gene inactivations may interfere the homeostatic mitochondrial responses to hsp-6(mg585), rendering the mutation more detrimental to mitochondrial function.
MDT-15 couples mitochondrial surveillance to xenobiotic response
Our genetic screen for mutations that activate expression of the cyp-14A4p::gfp detoxification fusion gene also identified a dominant mutant on Chromosome III. (Figure S1P, S2A and Table S1). Genetic mapping and genome sequencing of this gain-of-function mutant generated a list of 6 candidate mutations in the genetic interval from −1.44 to 1.14 on Chromosome III. One of the candidate mutations is a Pro117Ser (CCA to TCA) substitution in the mdt-15 gene. Amazingly, this P117S mutation is exactly the same proline residue that is mutated in a gain-of-function mdt-15 allele that emerged from an independent genetic screen: the mdt-15(et14) gain-of-function allele (Pro117Leu (CCA to CTA) substitution) (Figure S2A) was identified in a genetic screen for suppression of glucose and cold sensitivity of mutations in the adiponectin receptor homologue, paqr-2 (Svensk et al., 2013). Due to the close linkage (0.3 mu) of mdt-15 (genetic position: Chr III, −1.44) and paqr-2 (Chr III, −1.77), we did not try to separate mdt-15(et14) from paqr-2(tm3410). Instead we generated two independent mdt-15 alleles: mg584 with a Pro117Ser (CCA to AGT) mutation and mg640 with a Pro117Leu (CCA to TTG) mutation by CRISPR-Cas9 (Figure S2B). Both of mg584 and mg640 in a clean non-mutagenized genetic background activated the expression of the cyp-14A4p::gfp (Figure 2A, S2C and S2D), validating that these mdt-15 mutations cause the activation of cyp-14A4.
Figure 2. mdt-15 is essential for mitochondrial surveillance.

(A and B) L4 larvae carrying cyp-14A4p::gfp of indicated genetic background were photographed. Scale bars, 100 μm.
(C) L4 larvae of wild-type and mdt-15(tm2182lf) carrying cyp-14A4p::gfp were treated with antimycin for one day, and then photographed. Scale bar, 100 μm.
(D) Synchronized L1 larvae of wild-type and mdt-15(tm2182lf) animals were grown in M9 with E. coli OP50 and indicated concentration of antimycin at 20°C for three days. A red rectangle indicates larval delay or arrest. Scale bar, 500 μm.
C. elegans mdt-15 is annotated to encode a Mediator subunit homologous to human MED15 (Yang et al., 2006), but that homology cannot be detected via simple human BLASTP analysis using a C. elegans query. Mediator is a multisubunit transcriptional activation complex in eukaryotes. Orthologues of C. elegans MDT-15 can be easily detected by BLASTP to parasitic nematodes such as Ancylostoma ceylanicum over the entire length of the protein (Figure S2J). Human MED15 or C. elegans MDT-15 are transcriptional coactivators for fatty acid metabolism genes (Lee et al., 2015; Taubert et al., 2006; Yang et al., 2006).
Consistent with the mdt-15 gain-of-function mutations activating detoxification, mdt-15 gene activity is required for the induction of the detoxifying CYPs after the detection of mitochondrial dysfunction: the induction of cyp-14A4p::gfp in the hsp-6(mg585) mutant or by antimycin treatment is abolished in a loss-of-function mdt-15(tm2182) deletion mutant (Figure 2B, 2C, S2B and S2E–S2G). And mdt-15(tm2182lf) animals are more sensitive to antimycin than wild type, indicating that the mdt-15 detoxification responses are salient (Figure 2D).
mRNA-Seq analysis of RNA from the gain-of-function mdt-15(mg584gf) mutant showed the majority of the up-regulated or down-regulated mRNAs in mdt-15(mg584gf) were the same as those mRNAs up- or down-regulated in the hsp-6(mg585) mutant (Figure S2H and Table S2). Genes involved in detoxification and immunity responses, but not mitochondrial quality control are induced by mdt-15(mg584gf) (Figure S2I).
NHR-45 associates with MDT-15 to promote xenobiotic response
As a transcriptional coactivator MDT-15 is expected to work with transcription factors to promote gene expression. The two known MDT-15 interacting partners are sterol regulatory element binding protein SBP-1 and nuclear hormone receptor NHR-49 (Taubert et al., 2006; Yang et al., 2006); however, RNAi of either sbp-1 or nhr-49 does not compromise the activation of cyp-14A4p::gfp in the hsp-6(mg585) mutant background (Figure S3A and S3B), suggesting that MDT-15 might associate with other transcriptional factors for xenobiotic responses. To genetically identify the genes that are required for these mitochondrial detoxification responses, we conducted a second genetic screen after EMS mutagenesis for mutations that suppress the induction of the cyp-14A4p::gfp in hsp-6(mg585) animals. From 10,000 mutagenized F2 generation animals that were screened, 12 mutants that decrease the expression of cyp-14A4p::gfp in hsp-6(mg585) were isolated (Figure S3C and Table S3). One of the mutants carries a Cys39Phe (TGC to TTC) missense mutation disrupting one of the conserved Cys ligands in the Zn finger DNA-binding domain of nhr-45 (Figure S3D). nhr-45 encodes one of 284 C. elegans nuclear hormone receptors (NHR), and is most closely related to human hepatocyte nuclear factor 4 (HNF4). To confirm that the lesion in nhr-45 is the causative mutation, we generated an independent null allele of nhr-45(mg641) with a “stop” codon by CRISPR-Cas9 (Figure S3D). The loss-of-function nhr-45(mg641) mutant strongly disrupts the activation of cyp-14A4p::gfp in hsp-6(mg585) mutant or by antimycin (Figure 3A, 3B, S3E and S3F). Induction of cyp-14A4p::gfp in the mdt-15(mg584gf) mutant is also dependent on nhr-45 (Figure 3C and S3G). Comparison of mRNA-Seq analysis of hsp-6(mg585);nhr-45(mg641) mutant and hsp-6(mg585) mutant suggests that genes involved in detoxification and immunity, but not mitochondrial quality control, are regulated by nhr-45 (Figure. S3H and Table S3).
Figure 3. Regulation of xenobiotic responses by MDT-15/NHR-45.

(A and C) L4 larvae carrying cyp-14A4p::gfp of indicated genetic background were photographed. Scale bars, 100 μm.
(B) L4 larvae of wild-type and nhr-45(mg641) animals carrying cyp-14A4p::gfp were treated with antimycin for one day, and then photographed. Scale bar, 100 μm.
(D) Animals expressing an integrated array mgIs76[nhr-45p::nhr-45::mScarlet] were grown at 20°C until adulthood. Antimycin treatment was performed with L4 larvae. Scale bar, 40 μm.
(E) Synchronized L1 larvae of wild-type and nhr-45(mg641) animals were grown in M9 with E. coli OP50 and indicated concentration of antimycin at 20°C fo r three days. Red rectangle indicates larval delay or arrest. Scale bar, 500 μm.
(F) Survival analysis of wild-type, mdt-15(tm2182lf), and nhr-45(mg641) on P. aeruginosa PA14 strain.
mScarlet (Bindels et al., 2017) tagged NHR-45 protein expressed from its own promoter was detected in intestinal nuclei with or without antimycin (Figure 3D), suggesting that the intestine is a center of mitochondrial surveillance and detoxification response. This is consistent with a bacterial source of many mitochondrial assaults. The nhr-45-mediated transcriptional cascade is an essential response to mitochondrial dysfunction, because nhr-45(mg641) mutant animals are more sensitive to antimycin (Figure 3E). Moreover, compared to the wild-type animals, mdt-15(tm2182lf) and nhr-45(mg641) mutants died more rapidly when grown on P. aeruginosa PA14 strain (Figure 3F and S3I). Thus MDT-15/NHR-45-mediated detoxification and immune responses are salient to defense from a bacterial pathogen that attacks the mitochondrion.
The mitochondrial unfolded protein response (UPRmt), which is activated by mitochondrial dysfunction, promotes the restoration and recovery of mitochondrial function (Lin and Haynes, 2016). RNAi of atfs-1, one of the key regulators of UPRmt (Nargund et al., 2012), but not mdt-15 or nhr-45, suppresses the induction of hsp-6p::gfp, the benchmark reporter for UPRmt, in the hsp-6(mg585) mutant (Figure S3J and S3K). Other than UPRmt, disruption of mitochondrial proteostasis by RNAi of hsp-6 induces a mitochondrial-to-cytosolic stress response (MCSR) and elevates the capability of cytosolic protein folding (Kim et al., 2016). The expression of hsp-16.2, the marker gene of MCSR, was increased 15-fold in hsp-6(mg585) mutant, consistent with a previous report (Kim et al., 2016) (Figure S3L). The induction of hsp-16.2 was abolished by RNAi of dve-1 and hsf-1, two key regulators of MCSR, but not nhr-45 (Figure S3L). Moreover, we asked if factors regulating UPRmt and MCSR are involved in the MDT-15/NHR-45-mediated xenobiotic response. RNAi of atfs-1, but not lin-65 (Tian et al., 2016), dve-1, pod-2 or fasn-1 (Kim et al., 2016), blocked the activation of cyp-14A4p::gfp in the hsp-6(mg585) mutant (Figure S3M–S3P); RNAi of hsf-1 further increased cyp-14A4p::gfp by 57% (Figure S3M and S3N3). Therefore, MDT-15/NHR-45 mediates a transcriptional program for detoxification and immunity that is distinct from UPRmt and MCSR.
The xenobiotic response program is detrimental to the health of the hsp-6(mg585) mutant
While the early detection of a bacterial toxin attack on the mitochondrion and the detoxification of that attack offers selective advantages, if the cause of the mitochondrial dysfunction is a mutation, the induction of detoxification response genes would be futile and could compromise health and immunity. For example, the use of iron in the heme protein cytochrome p450 and the use of valuable oxygen in the p450 hydroxylation of target xenobiotic molecules could actually render a mitochondrial mutant more unhealthy. To test this model, we assayed the effects of detoxification responses during mitochondrial stress caused by hsp-6(mg585) by monitoring the speed of development from egg to L4 larva. The rate of development of hsp-6(mg585) is slower than wild type while the nhr-45(mg641) or mdt-15(mg584gf) single mutants develop at almost the same rate as wild-type (Figure 4A and S4A). Dramatically, hsp-6(mg585);nhr-45(mg641) animals, in which the detoxification response is attenuated, showed markedly improved developmental rate compared to hsp-6(mg585) (Figure 4A). Conversely, the hsp-6(mg585);mdt-15(mg584gf) double mutant, in which the detoxification response is further activated by the mdt-15 gain-of-function allele, dramatically exacerbated the developmental delay of hsp-6(mg585) (Figure S4A).
Figure 4. Detrimental effects of xenobiotic responses on hsp-6(mg585) animals.

(A) Egg laying was performed to adult animals of indicated genetic background at 20°C for 1h. After removal of the mothers, eggs were grown at 20°C for 56h or 72h. Worms that reach L4 larval stage were counted. *** p<0.001; ns, not significant.
(B) Quantification of movements in day 10 adults of indicated genetic background. **** p<0.0001. (C-E) Lifespan analysis of animals with indicated genetic background. **** p<0.0001; ns, not significant.
Furthermore, we monitored the locomotion of each strain at the L4 larval stage, day 1 adults and day 10 adults. When the animals were young (L4 larvae or day 1 adults), nhr-45(mg641) mutants moved as fast as wild-type (Figure S4B–S4D), and the speed of the hsp-6(mg585) mutant was about 48% of that of wild-type at L4 larvae and about 30% at day one adults (Figure S4B–S4D). Compared to the hsp-6(mg585) mutant, the hsp-6(mg585);nhr-45(mg641) mutant showed a 25% increase in speed at the L4 larval stage and 20% increase at day 1 adult (Figure S6B–S6D). When the animals were old (day 10 adults), nhr-45(mg641) mutants had a 170% increase of movement speed compared to wild-type, and hsp-6(mg585);nhr-45(mg641) mutants had a 130% increase compared to the hsp-6(mg585) mutant (Figure 4B and S4D). These observations support the hypothesis that activation of detoxification is a burden to the already malfunctioning mitochondria in mitochondrial mutant animals for which detoxification is futile. Movement of nhr-45(mg641) mutants is significantly improved compared to wild-type only at day 10 adults; this may be caused by the decreased activity of mitochondrial function during aging of wild type animals that in turn activates a burdensome detoxification program if nhr-45 is operational.
Non-null mutations or gene inactivations of non-essential subunits of C. elegans mitochondrial genes extend lifespan (Dillin et al., 2002; Lee et al., 2003; Yanos et al., 2012). To explore the relationship between mitochondrial surveillance and lifespan, we monitored the lifespan in hsp-6(mg585) and nhr-45(mg641) mutants. At 20°C, the median survival of hsp-6(mg585) (20 days) is 25% longer than wild type (16 days) (Figure 4C–4E and S4H). Although the single nhr-45(mg641) mutant does not affect lifespan (16 days), the hsp-6(mg585);nhr-45(mg641) double mutant has a 20% longer lifespan (24 days) than hsp-6(mg585), indicating that disruption of the detoxification response in the hsp-6(mg585) mutant is beneficial.
Mitochondrial dysfunction triggers mitochondrial relocalization in intestinal cells
To test whether NHR-45 regulates mitochondrial function or ATP production. We first evaluated the mtDNA copy number in the various mutant strains. In the hsp-6(mg585) mutant, the mtDNA copy numbers were 46% increased compared to wild-type. The loss-of-function of nhr-45 has no effect on mtDNA copy number in either wild-type or the hsp-6(mg585) mutant (Figure 5A). We monitored the ATP levels of animals with mitochondria challenged by P. aeruginosa infection or the hsp-6(mg585) mutation. When wild-type animals were cultured on P. aeruginosa, the ATP level showed 26% decrease (Figure S5A). The ATP level in hsp-6(mg585) mutant was 42% decreased compared to wild-type, and the ATP level in the hsp-6(mg585);nhr-45(mg641) mutant was 14% increased compared to hsp-6(mg585) mutants (Figure S5B). These data further confirmed that the nhr-45-mediated response does not lead to increased mitochondrial biogenesis or function.
Figure 5. Mitochondrial dysfunction triggers mitochondrial relocalization in intestine.

(A) Single L4 larva of indicated genetic background was picked and qPCR was performed toward mitochondrial DNA with genomic DNA as the control. *** p<0.001; ns, not significant.
(B and C) Day one adults of indicated genetic background animals with mgTi33[vha-6p::tomm-20(1–54)::mScarlet] transgene were photographed. Antimycin treatment was performed at L4 larvae. Scale bars, 10 μm.
Bacterial attacks on mitochondria are likely to begin where C. elegans first encounters bacteria, in the intestine. And because free radical production by eukaryotes is a major axis of antibacterial immunity, the mitochondria of the intestine are a possible front line of attack and defense. To better understand the mitochondrial morphology or localization in intestinal cells, we monitored mitochondria in the intestine of wild type treated with a natural mitochondrial toxin or in various mitochondrial mutants. To label mitochondria in the intestine, the N-terminal region of TOMM-20 was fused with mScarlet under the control of an intestine-specific promoter (vha-6p) (Bindels et al., 2017; Ichishita et al., 2008). In wild-type animals, a tubular pattern of mitochondria are broadly distributed in the 32 intestinal cells; after antimycin treatment, a dramatic redistribution of mitochondria to a narrow region adjacent to the plasma membrane facing the intestinal lumen is triggered (Figure 5B and S5C). Time course treatment of antimycin indicates that 88% of animals had relocalized the mitochondrial pattern to this luminal distribution within 4 h (Figure S5D). The same polarized distribution pattern of mitochondria was observed after antimycin treatment as analysed by a ges-1 promoter driven GFP-fused with a mitochondrial leader sequence (Figure S5E and S5F). C. elegans genetic lesions that disrupt mitochondrial function induced a similar redistribution of intestinal mitochondria, for example, in the hsp-6(mg585) mutant, as well as mutations in mitochondrial respiratory chain components such as clk-1/COQ7, isp-1/Rieske, and nduf-7/NDUFS7 (Figure 5B, S5C, S5G and S5H). And this is not likely due to the elevation of mitochondrial biogenesis, as the fluorescence of the mitochondrial protein fusion transgene did not change in these mutants (Figure S5I–S5K).
Antimycin treatment also caused the polarized mitochondrial distribution in the nhr-45(mg641) mutant; thus the intestinal mitochondrial movement is not dependent on NHR-45-regulated transcription (Figure S5L and S5M). In D. melanogaster, mitochondrial transport in neurons is mediated by a highly conserved mitochondrial Rho GTPase dMiro/Miro and its binding partner kinesin-associated mitochondrial adaptor Milton/TRAK1 (Hirokawa et al., 2010). In C. elegans, MIRO-1/Miro facilitates mitochondrial translocation into injured axons to promote axon regeneration (Han et al., 2016). In animals carrying null alleles of miro-1(tm1966) or trak-1(tm1572) (Figure S5N and S5O), the intestinal mitochondria showed an abnormal and clustered structure, and no longer redistributed toward the lumen of the intestine after antimycin treatment (Fig. 5C, S5P and S5Q), demonstrating that mitochondrial redistribution requires MIRO-1 and TRAK-1.
Dissection of detoxification response and fat metabolism in the mdt-15(mg584gf) mutant.
The nhr-45(mg641) and mdt-15(tm2182lf) mutants are both hypersensitive to antimycin (Figure 2D and 3E). However, nhr-45(mg641) and mdt-15(tm2182lf) had distinct interactions with hsp-6(mg585). Compared to hsp-6(mg585), the hsp-6(mg585);nhr-45(mg641) double mutant showed improved larval development (Figure 4A); in contrast, hsp-6(mg585);mdt-15(tm2182lf) is larval lethal or sterile (data not shown). Consistent with this, RNAi of mdt-15 in hsp-6(mg585) animals is also larval lethal (Figure S4I). Thus, in the hsp-6(mg585) genetic background, both mdt-15 gain-of-function and loss-of-function are detrimental, which made us suspect that pathways in addition to xenobiotic responses are affected in mdt-15 mutants.
One of the well-characterized transcriptional programs mediated by MDT-15 is the fatty acid biosynthesis pathway. MDT-15 collaborates with sterol regulatory element binding protein SBP-1 to promote expression of fatty acid metabolism enzymes, including fat-5 and fat-7 (Yang et al., 2006). From RNAseq analysis, fat-5 and fat-7 are upregulated in the mdt-15(mg584gf) mutant, but downregulated in hsp-6(mg585) mutant (Figure S2I). Indeed, a fat-7p::FAT-7::GFP reporter was decreased 68% in hsp-6(mg585) (Figure S4J and S4K) and the mRNA level of fat-5 was decreased 64% in hsp-6(mg585) (Figure S4L). The induction of fat-7p::FAT-7::GFP in mdt-15(mg584gf) is dependent on sbp-1 but not nhr-45 (Figure S4M and S4N). Gas chromatography analysis showed that the fatty acid composition, especially unsaturated fatty acids, is abnormal in hsp-6(mg585) (Figure S4O). RNAi of sbp-1 in hsp-6(mg585) animals causes larval lethality, reminiscent of mdt-15 inhibition in hsp-6(mg585) (Figure S4I). SBP-1 regulates fatty acid metabolism to prolong animal life span (Han et al., 2017; Yang et al., 2006). The median survival of mdt-15(mg584gf) (18 days) is longer than wild-type (16 days) (Figure S4E and S4H); however, hsp-6(mg585);mdt-15(mg584gf) mutants (19 days) had a similar life span to hsp-6(mg585) (20 days) (Figure S4F and S4H). This may be due to simultaneous activation of fatty acid biosynthesis and detoxification. We disrupted the detoxification response pathway by introducing nhr-45(mg641) into mdt-15(mg584gf), and robust extension of life span was now observed, 21 days compared to 18 days (Figure S4G and S4H). Finally, we disrupted the fatty acid pathway by sbp-1 RNAi in the mdt-15(mg584gf) strain: RNAi of sbp-1 renders the lifespan of mdt-15(mg584gf) even shorter than wild type (Figure S4P–S4S). This genetic dissection of mdt-15 function in fatty acid metabolism and mitochondrial surveillance indicates that MDT-15 functions by associating with different transcriptional factors to mediate various downstream functions.
Discussion
We have shown that the assessment of impaired mitochondrial function caused by a toxin or a mutation triggers a xenobiotic response through activation of MDT-15 and NHR-45. This response is key to survival of a mitochondrial toxin: animals carrying mutations in these xenobiotic response pathway genes cannot survive antimycin doses that wild type can survive and die rapidly on a pathogen that targets the mitochondrion (Figure 2D, 3E and 3F). Conversely, we have shown that the mdt-15(mg584gf) mutant causes constitutive activation of detoxification responses, even though no mitochondrial insult is detected by the surveillance system (Figure 2A), and an mdt-15 loss-of-function allele interrupts mitochondrial surveillance (Figure 2B and 2C). Thus, MDT-15 and NHR-45 serve as a key coupling transcription factors from the upstream mitochondrial surveillance system to coordinate detoxification responses. Increase or attenuation of mitochondrial response can be achieved by manipulating MDT-15 activity. However, these ineffective detoxification and immune responses to a mitochondrial mutation are actually deleterious; a mutation in nhr-45 that disrupts these burdensome detoxification responses actually renders the animals healthier as measured by growth rate, locomotion or lifespan (Figure 4A–4E). The hsp-6(mg585) mutation prolongs lifespan, however, these animals are very sick throughout their lives. In contrast, hsp-6(mg585);nhr-45(mg641) mutant animals that do not activate an ineffective detoxification response not only live longer, but also have improved health status, such as better movement and a faster rate of growth (Figure 4A and 4B). Thus the activation of detoxification in a mitochondrial mutant is actually harmful to health. It is remarkable that nhr-45(mg641) does not affect the development or locomotion when the animals are young (Figure 4A, S4B and S4C), however, dramatically suppresses the decline of movement during aging (Figure 4B). We suspect that without any mitochondrial mutation, toxin or pathogen, mitochondria are not challenged during development or young adults; however, the xenobiotic response is induced during aging as the mitochondrial electron transport chain collapses and mitochondrial function declines. Therefore, even though nhr-45(mg641) does not affect the life span (Figure 4D), it benefits the health of aged animals (Figure 4B).
This system of detoxification and immunity is a product of billions of years of evolution to protect animals from invading pathogens, which are the most frequent cause of mitochondrial dysfunction. But this system has not evolved to protect animals from mutations. When their core cellular components are disrupted by mutation, eukaryotes may misinterpret the dysfunction as caused by toxins and activate xenobiotic responses. These detoxifying enzymes may cause unnecessary reallocation of nutrients such as iron and oxygen or affect normal metabolism since many xenobiotic enzymes have internal targets as well. It is possible that some of the health consequences of mitochondrial disease, for example, may be due to the ineffective activation of detoxification and immune responses. Inhibition of these detoxification and immunity outputs may improve some disease symptoms without the need to actually improve mitochondrial function.
There were more factors identified from our two rounds of genetic screens. First, among the five other mutations that induced cyp-14A4p::gfp expression were two probable null alleles in the pumilio factor puf-9 (Table S1), which is an orthologue of S. cerevisae Puf3, known to regulate mRNA abundance of mitochondrial ETC complex IV assembly (Olivas and Parker, 2000). Second, the ligand for NHR-45 could, for example, be a bacterial toxin, or a C. elegans small molecule signal of mitochondrial dysfunction. Interestingly, another of the 7 alleles that induce cyp-14A4p::gfp expression was in dhs-28, an orthologue of mammalian 17-beta-hydroxysteroid dehydrogenase (Table S1). Finally, among the other mutations identified in our genetic screen for mutations that suppress the induction of the cyp-14A4p::gfp in hsp-6(mg585) were four independent probable loss-of-function alleles of ipmk-1, an inositol polyphosphate multikinase (Table S3). In yeast, inositol polyphosphate multikinase has been implicated in transcriptional regulation (Odom et al., 2000) and thus may serve as a transcriptional cofactor like mdt-15.
Mitochondrial dysfunction induced by toxin or mutations also triggers a dramatic movement of mitochondria to the lumen of C. elegans intestinal cells. We monitored mitochondrial behavior in the intestine rather than in the more traditionally observed muscles because the anti-mitochondrial toxins and virulence factors produced by bacteria may enter the animal across the single membrane that separates the lumen of the gut from the cytoplasm of these large cells. In a similar manner, a C. elegans defense response in the intestine is a cell membrane away from the diverse types of bacteria that C. elegans consumes or avoids in the wild (Samuel et al., 2016). We suspected that the redistribution of mitochondria to the apical surface of intestine may represent a defensive fortification; however miro-1(tm1966) is even more resistant to P. aeruginosa and trak-1(tm1572) showed similar survival to wild-type (Figure S5R). Thus this relocalization pattern of intestinal mitochondria is unlikely related to immune response, which is consistent with our observation that nhr-45, which mediates xenobiotic response, does not affect this relocalization.
Limitations of study
Nuclear hormone receptors (NHR) function as transcription factors that regulate suites of downstream genes. The activity of NHRs is often modulated by small molecule “ligands”, for example, steroid and thyroid hormones or other types of molecules. Our results show that C. elegans NHR-45 mediates xenobiotic responses when mitochondria are disrupted. However, the “ligand” of NHR-45 and how aberrant mitochondria transmit this “ligand” to NHR-45 nuclear gene targets is unknown. NHR-45::mScarlet is constitutively localized to intestinal nuclei independent of mitochondrial status (Figure 3D). An attractive hypothesis that is not yet addressed in this study is that mitochondrial dysfunction causes increased abundance of a small molecule, perhaps derived from the mitochondrion, which then activate NHR-45 transcriptional activity in the nucleus. Our future study will focus on identifying the “ligand” of NHR-45, which will uncover the mysterious bridge that links mitochondrial dysfunction to xenobiotic response gene activation.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gary Ruvkun (ruvkun@molbio.mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C. elegans strains N2 wild-type, XA7702 (mdt-15(tm2182) III), DMS303 (nIs590[fat-7p::fat-7::GFP + lin-15(+)]) and SJ4143 (zcIs17[ges-1p::gfp(mit)]) were obtained from the Caenorhabditis Genetic Center (CGC), and FX01966 (miro-1(tm1966) IV) and FX01572 (trak-1(tm1572) I) were obtained from National BioResource Project (NBRP/Mitani lab). For cyp-14A4p::gfp or NHR-45::mScarlet transgenic animals (cyp-14A4p::gfp::cyp-14A4 3’UTR) or (nhr-45p::nhr-45::mScarlet::nhr-45 3’UTR) plasmid was injected into N2. The extrachromosomal arrays were integrated onto C. elegans genome by EMS. For vha-6p::tomm-20(1–54)::mScarlet transgenic animals, (pCFJ910::vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR) plasmid was injected into N2 following the miniMOS protocol (Frokjaer-Jensen et al., 2014). For CRISPR of hsp-6(mg585), mdt-15(mg584), mdt-15(mg640) and nhr-45(mg641) mutants, we chose dpy-10(cn64) as the co-CRISPR marker (Arribere et al., 2014) and pJW1285 (Addgene) to express both of guide-RNA (gRNA) and Cas9 enzyme (Ward, 2015).
Microbe Strains
E. coli OP50 and P. aeruginosa PA14 were cultured overnight in LB at 37°C. 100 μl of liquid culture was seeded on plates. E. coli HT115 was used to perform RNAi and was cultured overnight at 37°C in LB containing 100 μg/ml ampicillin.
METHOD DETAILS
Drug treatment
Antimycin A (Sigma, A8674) was dissolved in ethanol at 25 mg/ml. For larval development assay in liquid, synchronized L1 worms were grown in M9 with amply E. coli OP50 with indicated concentration of antimycin. For antimycin treatment on plates, synchronized L1 worms were grown with either E. coli OP50 or HT115 (DE3) with designated RNAi clones at 20°C for 2 days, and 500 μl of M9 with 6.25 μg/ml antimycin was added onto each plates. After 24 hours, fluorescence was monitored.
Microscopy
The fluorescent signals of transgenic animals were photographed by Zeiss AX10 Zoom.V16 microscope. Intestinal mitochondrial examination with (vha-6p::tomm-20(1–54)::mScarlet) or (ges-1p::gfp(mit)), and subcellular localization of NHR-45::mScarlet were carried out on the Leica TCS SP8 confocal microscope. Photographs were analyzed by Fiji-ImageJ.
mRNA-seq analysis
Around 200 L4 worms were hand picked and frozen by liquid nitrogen. Total RNA was isolated by TRIzol extraction (Thermo Fisher #15596026). mRNA was isolated by Poly(A) magnetic beads (New England BioLabs #E7490), and libraries was constructed by NEBNext Ultra RNA Library Prep Kit for Illumina (New England BioLabs #E7530S) and sequenced on an Illumina HiSeq2500 instrument, results in approximately 16 million reads per sample on average. STAR aligner (Dobin et al., 2013) was used to map sequencing reads to transcripts in C. elegans ce10 reference genome. Read counts for individual transcripts were produced with HTSeq-count (Anders et al., 2015), followed by the estimation of expression values and detection of differentially expressed transcripts using EdgeR (Robinson et al., 2010). Differentially expressed genes were defined by at least 2-fold change with FDR less than 0.001.
Western blot
Worms were collected from liquid or washed off plates. Worm lysates were generated by TissueLyser with steel beads (Qiagen #69989), and re-suspended with NuPAGE™ LDS sample buffer (Thermo Fisher #NP0007) and heated at 70 °C for 10 minutes. Samples were loaded onto NuPAGE™ 4–12% Bis-Tris Protein Gels (Thermo Fisher #NP0323BOX) and run with NuPAGE™ MES SDS running buffer (Thermo Fisher #NP0002). After semi-dry transfer, PVDF membrane (Millipore #IPVH00010) was blocked with 5% nonfat milk, and probed with designated primary and secondary antibodies, and developed with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher #34096) and visualized by Amersham Hyperfilm (GE Healthcare #28906845).
Fatty acid analysis
~40000 L4 stage animals were washed off the plates with water, rinsed five times and the pellet was frozen at −80°C. Fatty acid methyl esters (FAME) we re prepared as described (Miquel and Browse, 1992). In brief, 1 ml of 2.5% (v/v) H2SO4 in methanol was added to the pellets and samples were heated at 80°C for 1 h and cooled to room temperature foll owing by addition of 1.5 ml of 0.9% NaCl. FAME were extracted into organic phase with 1 ml of hexane after shaking and spinning down at low speed. GC of FAME was performed on a HP6890N (Agilent) apparatus equipped with a DB-23 column (30 m × 250 μm × 0.25 μm). Each experiment was repeated at least three times.
Lifespan analysis
Animals were synchronized by egg-laying and grown till L4 stage as day 0. Adults were separated from their progenies by manually transferring to new plates. Survival was examined on a daily basis.
Determination of ATP level
50 L4 larvae were picked from mix population grown at 20°C and frozen in liquid nitrogen. Worm lysis was performed with 90°C for 15 min. The ATP concent ration was measured by “ATP Determination Kit, Thermo Fisher A22066”, and was normalized by the protein concentration that was measured by “Pierce™ BCA Protein Assay Kit, Thermo Fisher 23225”.
Determination of mtDNA copy number
8 L4 larva were individually picked from mix population grown at 20°C into 10 μl of worm lysis buffer (50 mM KCl, 2.5 mM MgCl2 10 mM Tris pH 8.3, 0.5% NP40, 0.5% Tween 20, 0.01% Gelatine and 100 μg/ml protease K). Worm lysis is performed on PCR machinery at 60°C for 1 h and 95°C for 15 min. qPCR was performed toward mtDNA with genomic DNA (act-1) as control by iQ™ SYBR® Green Supermix (BIORAD, #1708880).
RT-qPCR
Around 50 L4 larvae were picked from mixed population grown at 20°C and frozen by liquid nitrogen. Total RNA was isolated by TRIzol extraction (Thermo Fisher #15596026). The cDNA was generated by ProtoScript® II First Strand cDNA Synthesis Kit (New England BioLabs, #E6560L). qPCR was performed toward interested genes with act-1 as control by iQ™ SYBR® Green Supermix (BIO-RAD, #1708880).
Monitoring of locomotion
~50 L4 larvel were picked and grown at 20°C for 10 days. On day 0 (L4 larvae), day 1 (day 1 adults) and day 10 (day 10 adults), worms were picked onto bacteria free NGM plates and photographed directly by Zeiss AX10 Zoom.V16 microscope. The movement of worms was recorded by continuing picturing every 0.5 seconds for 30 seconds in total. The speed of movement was analyzed and calculated by AxioVision (Zeiss).
QUANTIFICATION AND STATISTICAL ANALYSIS
The fluorescent intensities of GFP reporters were quantified by Fiji-ImageJ and student’s t test was performed by GraphPad Prism. The Log-rank (Mantel-Cox) test was used to compare survival curves for lifespan analyses.
Supplementary Material
Table S1. Mitochondrial dysfunction activates xenobiotic responses. Related to Figure 1.
Table S2. mdt-15 regulates xenobiotic responses. Related to Figure 2.
Table S3. nhr-45 mediates xenobiotic responses. Related to Figure 3.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-GFP | Fisher Scientific | NC9777966 |
| anti-actin | Abcam | ab3280 |
| Bacterial and Virus Strains | ||
| E. coli: Strain OP50 | CGC | N/A |
| E. coli: Strain HT115 | CGC | N/A |
| P. aeruginosa: Strain PA14 | F. M. Ausubel lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| antimycin A | Sigma | A8674 |
| NuPAGE™ LDS Sample Buffer (4X) | Thermo Fisher | NP0007 |
| NuPAGE™ 4–12% Bis-Tris Protein Gels | Thermo Fisher | NP0323BOX |
| NuPAGE™ MES SDS running buffer | Thermo Fisher | NP0002 |
| Immobilon-P PVDF Membrane | Millipore | IPVH00010 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher | 34096 |
| Amersham Hyperfilm MP | GE Healthcare | 28906845 |
| TRIzol | Thermo Fisher | 15596026 |
| Critical Commercial Assays | ||
| ATP determination kit | Thermo Fisher | A22066 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher | 23225 |
| NEBNext Ultra RNA Library Prep Kit | New England BioLabs | E7530S |
| iQ™ SYBR® Green Supermix | Bio-Rad | 1708880 |
| ProtoScript® II First Strand cDNA Synthesis Kit | New England BioLabs | E6560L |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Bristol (N2) wild-type | CGC | N2 |
| C. elegans: GR2250 (mgIs73[cyp-14A4p::gfp::cyp-14A4 3’UTR + myo-2p::mcherry] V] | This study | N/A |
| C. elegans: GR2248 (hsp-6(mg583) V) | This study | N/A |
| C. elegans: GR2249 (hsp-6(mg585) V) | This study | N/A |
| C. elegans: GR2252 (hsp-6(mg585); mgIs73[cyp-14A4p::gfp::cyp-14A4 3’UTR + myo-2p::mcherry] V) | This study | N/A |
| C. elegans: GR2247 (mdt-15(mg584) III) | This study | N/A |
| C. elegans: GR2251 (mdt-15(mg640) III) | This study | N/A |
| C. elegans: GR3275 (mdt-15(mg584) III; mgIs73[cyp-14A4p::gfp::cyp-14A4 3’UTR + myo-2p::mcherry] V) | This study | N/A |
| C. elegans: XA7702 (mdt-15(tm2182) III) | CGC | WormBase: XA7702 |
| C. elegans: GR2263 (nhr-45(mg641) X) | This study | N/A |
| C. elegans: GR2264 (mdt-15(mg584) III; nhr-45(mg641) X) | This study | N/A |
| C. elegans: GR2265 (hsp-6(mg585) V; nhr-45(mg641) X) | This study | N/A |
| C. elegans: GR2266 (mgIs76[nhr-45p::nhr-45::mScarlet::nhr-45 3’UTR + myo-2p::gfp]) | This study | N/A |
| C. elegans: GR3276 (hsp-6(mg585); zcIs13[hsp-6p::gfp] V) | This study | N/A |
| C. elegans: DMS303 (nIs590[fat-7p::fat-7::GFP + lin-15(+)]) | CGC | WormBase: DMS303 |
| C. elegans: GR3277 (hsp-6(mg585) V; nIs590[fat-7p::fat-7::GFP + lin-15(+)]) | This study | N/A |
| C. elegans: GR3278 (mdt-15(mg584) III; nIs590[fat-7p::fat-7::GFP + lin-15(+)]) | This study | N/A |
| C. elegans: GR2267 (mgTi33[vha-6p::tomm-20(1−54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3279 (hsp-6(mg585) V; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: SJ4143 (zcIs17[ges-1p::gfp(mit)]) | CGC | WormBase: SJ4143 |
| C. elegans: FX01966 (miro-1(tm1966) IV) | NBRP/Mitani lab | WormBase: WBVar00250926 |
| C. elegans: FX01572 (trak-1(tm1572) I) | NBRP/Mitani lab | WormBase: WBVar00250558 |
| C. elegans: GR3280 (miro-1(tm1966) IV; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3281 (trak-1(tm1572) I; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3282 (clk-1(qm30) III; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3283 (nduf-7(et19) I; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3284 (isp-1(qm150) IV; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3285 (hsp-6(mg585) V; nhr-45(mg641) X; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| C. elegans: GR3286 (nhr-45(mg641) X; mgTi33[vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR]) | This study | N/A |
| Oligonucleotides | ||
| guide-RNA reverse: caagacatctcgcaataggagg | This study | N/A |
| hsp-6(mg585) guide-RNA Forward: tctgggttgacagcctttgagtttaagagctatgctggaaacag | This study | N/A |
| hsp-6(mg585) donor oligo: gacggcctcgtctgggttgacagcctttgaactaacttttccgaagattt cttgaacagt | This study | N/A |
| mdt-15(mg584) or (mg640) guide-RNA forward: tttcttgcctgagctgatgtgtttaagagctatgctggaaacag | This study | N/A |
| mdt-15(mg584) donor oligo: acaggtggatttcttgcctgagctgatgtactttgtggatctggaggca caggagctcga | This study | N/A |
| mdt-15(mg640) donor oligo: acaggtggatttcttgcctgagctgatgtcaattgtggatctggaggca caggagctcga | This study | N/A |
| nhr-45(mg641) guide-RNA Forward: aaagtgctgaccgtgtgcatgtttaagagctatgctggaaacag | This study | N/A |
| nhr-45(mg641) donor oligo: ggataccaaagtgctgaccgtgtgcatcatgatcaggtctcacacac cagacacgacatG | This study | N/A |
| Recombinant DNA | ||
| cyp-14A4p::gfp::cyp-14A4 3’UTR | This study | N/A |
| nhr-45p::nhr-45::mScarlet::nhr-45 3’UTR | This study | N/A |
| vha-6p::tomm-20(1–54)::mScarlet::nhr-45 3’UTR | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism 6 | GraphPad | N/A |
| Fiji-ImageJ | Fiji | N/A |
| AxioVision | Zeiss | N/A |
Highlights.
Mitochondrial dysfunction activates a xenobiotic response.
MDT-15 and NHR-45 coordinate the xenobiotic response.
The xenobiotic response is detrimental to mitochondrial mutant animals.
Mitochondrial dysfunction triggers mitochondrial relocalization in the intestine.
Acknowledgements
We thank Caenorhabditis Genetics Center and National BioRescource Project (Tokyo, Japan) for providing strains. K.M. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2213–15). This work is supported by a grant from the National Institute of Health awarded to G.R. (NIH AG16636).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References:
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow H, Doitsidou M, Sarin S, and Hobert O (2009). MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods 6, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14, 53–56. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, and Kenyon C (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, LaBella M, Pozniakovsky A, Moerman DG, and Jorgensen EM (2014). Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat Methods 11, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan JA, Jayamani E, Zhang X, Breen P, Larkins-Ford J, Mylonakis E, and Ruvkun G (2015). Lipid signalling couples translational surveillance to systemic detoxification in Caenorhabditis elegans. Nature cell biology 17, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP (2008). Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21, 70–83. [DOI] [PubMed] [Google Scholar]
- Han S, Schroeder EA, Silva-Garcia CG, Hebestreit K, Mair WB, and Brunet A (2017). Monounsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 544, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Baig HS, and Hammarlund M (2016). Mitochondria Localize to Injured Axons to Support Regeneration. Neuron 92, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heschl MF, and Baillie DL (1989). Characterization of the hsp70 multigene family of Caenorhabditis elegans. DNA 8, 233–243. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, and Tanaka Y (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. [DOI] [PubMed] [Google Scholar]
- Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, and Oka T (2008). An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem 143, 449–454. [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, et al. (2002). A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626. [DOI] [PubMed] [Google Scholar]
- Kim HE, Grant AR, Simic MS, Kohnz RA, Nomura DK, Durieux J, Riera CE, Sanchez M, Kapernick E, Wolff S, et al. (2016). Lipid Biosynthesis Coordinates a Mitochondrial-to-Cytosolic Stress Response. Cell 166, 1539–1552 e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Tanaka N, Nakamura N, Takano S, and Ohkuma S (2007). Knockdown of mitochondrial heat shock protein 70 promotes progeria-like phenotypes in caenorhabditis elegans. The Journal of biological chemistry 282, 5910–5918. [DOI] [PubMed] [Google Scholar]
- Kirienko NV, Ausubel FM, and Ruvkun G (2015). Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America 112, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Jeong DE, Son HG, Yamaoka Y, Kim H, Seo K, Khan AA, Roh TY, Moon DW, Lee Y, et al. (2015). SREBP and MDT-15 protect C. elegans from glucose-induced accelerated aging by preventing accumulation of saturated fat. Genes Dev 29, 2490–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, and Ruvkun G (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Lehrbach NJ, and Ruvkun G (2016). Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, and Haynes CM (2016). Metabolism and the UPR(mt). Molecular cell 61, 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel BS, Breen PC, and Ruvkun G (2014). Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508, 406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD (2007). The C. elegans intestine. WormBook, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, and Ruvkun G (2012). Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel M, and Browse J (1992). Arabidopsis mutants deficient in polyunsaturated fatty acid synthesis. Biochemical and genetic characterization of a plant oleoyl-phosphatidylcholine desaturase. The Journal of biological chemistry 267, 1502–1509. [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, and Haynes CM (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, and Suomalainen A (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom AR, Stahlberg A, Wente SR, and York JD (2000). A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science 287, 2026–2029. [DOI] [PubMed] [Google Scholar]
- Olivas W, and Parker R (2000). The Puf3 protein is a transcript-specific regulator of mRNA degradation in yeast. The EMBO journal 19, 6602–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, and Haynes CM (2014). Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516, 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel BS, Rowedder H, Braendle C, Felix MA, and Ruvkun G (2016). Caenorhabditis elegans responses to bacteria from its natural habitats. Proceedings of the National Academy of Sciences of the United States of America 113, E3941–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O’Meara MM, Pe’er I, and Hobert O (2008). Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods 5, 865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensk E, Stahlman M, Andersson CH, Johansson M, Boren J, and Pilon M (2013). PAQR-2 regulates fatty acid desaturation during cold adaptation in C. elegans. PLoS Genet 9, e1003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert S, Van Gilst MR, Hansen M, and Yamamoto KR (2006). A Mediator subunit, MDT-15, integrates regulation of fatty acid metabolism by NHR-49-dependent and -independent pathways in C. elegans. Genes Dev 20, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH (2007). Rapid birth-death evolution specific to xenobiotic cytochrome P450 genes in vertebrates. PLoS Genet 3, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, and Dillin A (2016). Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell 165, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, and Mootha VK (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. [DOI] [PubMed] [Google Scholar]
- Ward JD (2015). Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 199, 363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, and Ghosh S (2011). Mitochondria in innate immune responses. Nature reviews. Immunology 11, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Vought BW, Satterlee JS, Walker AK, Jim Sun ZY, Watts JL, DeBeaumont R, Saito RM, Hyberts SG, Yang S, et al. (2006). An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442, 700–704. [DOI] [PubMed] [Google Scholar]
- Yanos ME, Bennett CF, and Kaeberlein M (2012). Genome-Wide RNAi Longevity Screens in Caenorhabditis elegans. Curr Genomics 13, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, and Ron D (2004). Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of cell science 117, 4055–4066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Mitochondrial dysfunction activates xenobiotic responses. Related to Figure 1.
Table S2. mdt-15 regulates xenobiotic responses. Related to Figure 2.
Table S3. nhr-45 mediates xenobiotic responses. Related to Figure 3.