

SUMMARY
Bacterial microcompartments (BMCs) are composed of an enzymatic core encapsulated by a selectively permeable protein shell that enhances catalytic efficiency. Many pathogenic bacteria derive competitive advantages from their BMC-based catabolism, implicating BMCs as drug targets. BMC shells are of interest for bioengineering due to their diverse and selective permeability properties and because they self-assemble. A complete understanding of shell composition and organization is a prerequisite for biotechnological applications. Here, we report the cryo-EM structure of a BMC shell at 3.0 Å resolution, using an image processing strategy that allowed us to determine the previously uncharacterized structural details of the interactions formed by the BMC-TS and BMC-TD shell subunits in the context of the assembled shell. We found unexpected structural plasticity among these interactions, resulting in distinct shell populations assembled from varying numbers of the BMC-TS and BMC-TD subunits. We discuss the implications of these findings on shell assembly and function.
Keywords: Bacterial microcompartments, microbiology, metabolosome, modular assembly, bioengineering, structural biology, permeability, cryo-electron microscopy
Graphical Abstract
eTOC summary:
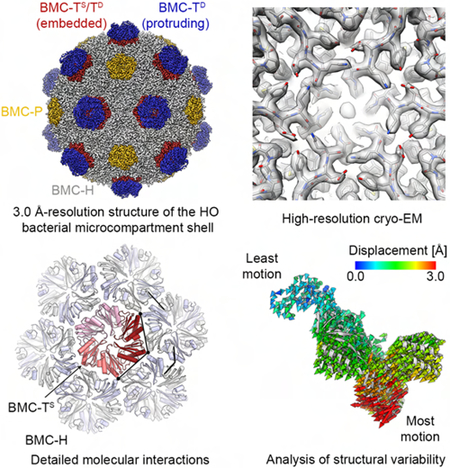
Greber et al. use high-resolution cryo-electron microscopy to reveal the structural plasticity in a bacterial microcompartment shell and the complete blueprint for its assembly.
INTRODUCTION
Bacterial microcompartments (BMCs) are composed of a proteinaceous shell and encapsulated enzymes, thereby enabling the sequestration of biochemical reaction pathways from the cytosol, preventing loss of metabolites to side-reactions, or exposure of the cell to large amounts of toxic intermediates (Kerfeld et al., 2018). BMCs are found across the bacterial kingdom where they serve as organelles for CO2 fixation in cyanobacteria and some chemoautotrophs, or the catabolism of a variety of organic compounds in heterotrophic bacteria (Axen et al., 2014; Erbilgin et al., 2014; Havemann and Bobik, 2003; Heldt et al., 2009; Herring et al., 2018; Penrod and Roth, 2006; Zarzycki et al., 2015). Importantly, BMCs have been implicated in virulence of some bacteria, including Salmonella, by providing a competitive growth advantage to the pathogen over other bacteria (Heithoff et al., 1999; Jakobson and Tullman-Ercek, 2016; Kaval and Garsin, 2018; Thiennimitr et al., 2011).
While there is a functional diversity of BMCs due to the variety of enzymes they encapsulate, all BMCs are bounded by a selectively permeable protein shell composed of up to four distinct types of proteins, BMC-H, BMC-TS, BMC-TD and BMCP, which collectively make up the icosahedral shell (Fig. 1A, B). BMC-P proteins contain the pfam03319 domain and assemble into pentamers that serve as vertices (Sutter et al., 2017; Tanaka et al., 2008). The facets of the shells are composed of BMC-H, BMC-TS and BMC-TD proteins. BMC-H proteins are ubiquitous among all BMCs and comprise a single pfam00936 domain (Fig. 1A, B). Six BMC-H monomers form a cyclically symmetric hexagon with a convex and concave side, and are typically perforated by a constitutively open central pore (Kerfeld et al., 2005). All BMC-T-type subunits are composed of a tandem fusion of two pfam00936 domains (Fig. 1A, B). BMC-TS assembles into trimers that assume pseudo-hexameric symmetry, thereby recapitulating the shape of the BMC-H hexamer. BMC-TD proteins are also composed of a tandem fusion on pfam00936 domains, but with secondary structure elements that are circularly permuted relative to BMC-H and BMC-TS (Fig. 1B). Moreover, BMC-TD trimers dimerize across their concave faces (Fig. 1A), forming an inner chamber. The pores in the BMC-TD trimers are larger than those typically observed in other shell proteins (Fig. 1A) and are gated by conformational changes of an absolutely conserved arginine residue. The stacking and gating of BMC-TD trimers has been suggested to be critical for conducting of large metabolites across BMC shells through an air-lock mechanism (Cai et al., 2013; Klein et al., 2009; Larsson et al., 2017). Based on crystal structures of individual shell proteins, it is known that pore gating of BMC-TD can be influenced by the presence of small-molecule ligands (Larsson et al., 2017).
Fig. 1. Component structures of the HO BMC shell.

(A) Cartoon representation of structures of the BMC shell proteins found in the HO shell, viewed from the side (top row) and from the top onto the concave side (bottom row; view point indicated by a blue arrow in the side view). One protein chain each is colored according to secondary structure (helix: red, sheet: yellow, first and last secondary structure elements colored darker) and marked N- and C-termini.
(B) Secondary structure diagrams of the shell proteins depicted in a. BMC-H consists of a single pfam00936 domain. HO-BMC-TS1 is a member of the BMC-TS-type that comprises a fusion of two pfam00936 domains. HO-BMC-TD2 and HO-BMC-TD3 are BMC-TD-type shell proteins that are a fusion of a permuted pfam00936 domain where β4 and α3 are located at the N-terminus. BMC-P contains the all-β-sheet pfam03319 domain.
Recombinantly expressed 6.5 MDa shells derived from the Haliangium ochraceum (HO) BMC, a microcompartment of unknown physiological function, are composed of five types of shell proteins: one BMC-P, one BMC-H, one BMC-TS (BMC-TS1), and two BMC-TD paralogs (BMC-TD2 and BMC-TD3). Based on the enzymes encoded in proximity to the shell proteins of the HO BMC, it is a type of metabolosome, catabolic BMCs known to sequester aldehyde intermediates (Axen et al., 2014; Bobik et al., 2015; Kerfeld et al., 2018; Kerfeld and Erbilgin, 2015). The HO BMC shell serves as a prototypical metabolosome shell, containing all three of the known facet building blocks, reflecting the need to allow several types of metabolites to cross metabolosome shells. Notably, this modular architecture, using grossly structurally equivalent proteins (hexamers and pseudohexameric trimers) with distinctly different permeability properties, distinguishes BMCs from other proteinaceous shell-based systems such as bacterial encapsulin nanocompartments (Sutter et al., 2008) with only one shell protein component. Moreover, even in the larger virus structures there is typically only one type of protein occupying icosahedrally equivalent positions (Prasad and Schmid, 2012). The relative complexity of BMC shells and their ability to incorporate building blocks that allow passage of different small molecules reflects one evolutionary solution to the key functional requirement of selective permeability for several types of small molecules simultaneously, which is further elaborated by the incorporation of stacked and gated BMC-TD proteins, which form distinctive protuberances from the shell surface.
The X-ray crystal structure of the HO BMC shell (Sutter et al., 2017) revealed the overall shell architecture with a triangulation number of pseudo T = 9 (Sutter et al., 2017). The BMC-H hexamer constitutes the base unit of the shell facet as it interacts with all other building block types, including other BMC-H hexamers. In contrast, the BMC-P pentamer, the BMC-TS trimer, and the BMC-TD dimer of trimers interact with BMC-H building blocks exclusively. Due to the averaging inherent to the method of X-ray diffraction from crystals, the differences arising from the presence of the three different proteins BMC-TS1 and BMC-TD2/TD3 on the icosahedral threefold axes could not be resolved in the crystal structure. How the pattern of selective and specific interactions, which precludes interactions between the BMC-T paralogs, arises to form an organized and stable 6.5 MDa shell is a fundamental question of self-assembly.
Here, we present the cryo-EM structure of the HO BMC shell at 3.0 Å resolution. A detailed symmetry-free analysis of the cryo-EM dataset allowed us to determine the specific interactions of the BMC-H hexamers with the trimeric BMC-TS subunit (BMC-TS1) and the BMC-TD stacked trimers in the context of the intact shell. Furthermore, we were able to distinguish and characterize structural variations of the shell, thereby obtaining evidence that the interactions governing BMC shell assembly exhibit a marked degree of plasticity, which may enable the formation of different BMC sizes and shapes in their native hosts (Havemann and Bobik, 2003; Iancu et al., 2007; Schmid et al., 2006). We discuss possible implications of our findings for shell assembly, dynamics, and permeability.
RESULTS
Cryo-EM structure determination
To determine the cryo-EM structure of the HO BMC shell, we collected electron micrographs of frozen-hydrated shell particles using a FEI Titan transmission electron microscope equipped with a direct electron detector camera, enabling correction of beam-induced specimen motion and accounting for radiation damage by dose weighing (Zheng et al., 2017). Three-dimensional maximum-likelihood-based image classification (Scheres, 2012) (Fig. 2) identified one highly populated 3D class exhibiting high-resolution features, of which we determined the icosahedrally symmetrized structure at an overall resolution of 3.0 Å by employing per-particle defocus refinement implemented in FREALIGN (Grigorieff, 2007; Grigorieff, 2016) (Figs. 2, 3). The resulting cryo-EM map shows excellent side chain density (Fig. 2D) for most protein subunits, including the protruding trimers of the BMC-TD (BMC-TD2/TD3) subunits (Fig. 3B), which could be interpreted only in terms of poly-alanines in the X-ray crystal structure of this BMC shell (Sutter et al., 2017). We built, refined, and validated an atomic model for all BMC-H and BMC-P subunits and the protruding trimers of the BMC-TD proteins according to our cryo-EM map (Fig. 2B, Table S1). In this cryo-EM map, the shell-embedded trimer of the BMC-TD subunits exhibited fragmented density due to the averaging of all three BMC-T paralogs and was interpreted by a model in which the side chains were truncated at the Cβ position. To resolve the structural heterogeneity of the shell-embedded BMCTS and BMC-TD in order to enable a complete interpretation of the interactions governing HO BMC shell assembly, we applied a focused 3D classification strategy based on symmetry expansion of the cryo-EM dataset followed by 3D refinement without imposing symmetry (see STAR Methods for details). This strategy enabled us to successfully distinguish BMC-TS from BMC-TD (Figs. 4, 5), detect different conformations within the BMC-TD double-stacked trimers that are linked to structural variations of the shell (Figs. 6–8), and build and refine atomic coordinate models for these cryo-EM reconstructions at 3.3–3.4 Å resolution (Fig. S1, Table S1).
Fig. 2. Cryo-EM structure determination.

(A) Data processing workflow for high-resolution reconstruction of the structure of the HO shell. See text and STAR Methods for details.
(B) Resolution estimation for the cryo-EM map. Black: Fourier Shell Correlation (FSC) curve between two cryo-EM half-maps obtained by frequency-limited refinement (see Methods), indicating a resolution of 3.0 Å according to the FSC = 0.143 criterion (Rosenthal and Henderson, 2003; Scheres and Chen, 2012). Red: FSC curve between the cryo-EM map and refined coordinates, indicating good correlation to 3.1 Å according to the FSC = 0.5 criterion (Rosenthal and Henderson, 2003). These criteria (FSC = 0.143 for half-map FSCs and FSC = 0.5 for model vs. map FSCs) are applied throughout this work.
(C) Local resolution estimate for the cryo-EM reconstruction of the HO shell. Most of the shell is resolved at 3 Å or better, while the protruding BMC-T modules are still resolved at better than 3.5 Å resolution.
(D) View of the cryo-EM density with the fitted atomic coordinate model.
Fig. 3. Cryo-EM reconstruction of the HO BMC shell.

(A) High-resolution cryo-EM reconstruction of the HO BMC shell.
(B) Cryo-EM density for the protruding BMC-TD trimer. Central arginine residue R70 gating the BMC-TD2 pore is labeled. See also Table S1.
Fig. 4. Data processing for determination of BMC-T structures in the context of the assembled shell.

The entire BMC shell dataset was initially classified into 10 classes with icosahedral symmetry imposed. All 10 classes obtained in this initial classification step are shown at identical thresholds, with the exception of class 1, which is shown at slightly reduced threshold because it exhibited markedly lower density levels, likely due to classification of broken or otherwise aberrant particles or false positive particle picks into this class. Selected classes (6 and 10) were refined and symmetry expanded. Subsequently, one BMC-T position (indicated by a dotted line) was classified using a mask. The resulting sub-datasets were reconstructed, showing clearly the difference between the single-layer BMC-TS1 and the double-layer BMCTD2/TD3 at the classified position, and again subclassified to obtain more homogeneous datasets. The final subclassified datasets were again reconstructed, and the final volumes were masked around the classified BMC-T position and sharpened using the post-processing function in RELION for visualization and structure interpretation.
Fig. 5. Structure and interactions of BMC-TS in the HO BMC shell.

(A) Interfaces between the HO BMC-TS1 trimer (red hues) and the BMC-H hexamers (alternating grey/light blue) viewed from the outside of the shell. The pseudohexameric BMC-TS trimer forms two types of interfaces with BMC-H subunits (indicated). Straight/bent lines indicate the planar and bent hexamer interface; the positions of one neighboring BMC-P and BMC-T are indicated.
(B, C) Detailed views of the two interfaces (B, interface 1; C, interface 2). Dashed lines indicate hydrogen bonds and salt bridges. Residues discussed in the text are labeled. See also Figure S1.
Fig. 6. Global structural variations between HO BMC shell subpopulations.

(A) Slices of the 3D maps of the main BMC shell population (left) and the compacted BMC shell population (right).
(B) Views of the densities of the main particle population (blue) and the compacted shell (light grey).
(C) Difference density resulting from the subtraction of the cryo-EM density of the compacted shell from the density of the main shell population is shown in light green, superposed onto the reconstruction of full map of the compacted state (grey). The difference is smallest at the BMC-P positions.
(D) Side view of the asymmetric unit of the main shell population colored and labeled by subunit type. The corresponding model from the compacted shell is shown in lighter colors.
(E) Same view as D, with color-coded displacement vectors indicating the differences of Cα positions between the compacted and main shell conformations. BMC-P is least displaced while the shell-embedded ring at the BMC-T position is most displaced. See also Figure S1, Table S1.
Fig. 8. Structure, conformations, and interactions of BMC-TD in the HO BMC shell.

(A) Closed conformation of the shell-embedded BMC-TD layer (dark red) in the main population of shell particles (viewed from the inside of the shell).
(B) Intermediate conformation of the inner BMC-TD ring in the main population of shell particles. Note that density for the gating arginines is still present, while the overall structure of the inner BMC-TD subunits has already undergone conformational changes (yellow state in C, D).
(C) Comparison of BMC-TD with closed inner pore (protruding trimer blue, embedded trimer brown) with BMC-TD with intermediate conformation of the inner BMC-TD ring (yellow).
(D) Detailed view of boxed region in C with additional superposition of the structure of ligand bound CcmP (green) in fully open conformation (PDB ID 5LT5) (Larsson et al., 2017).
(E) Structures of BMC-TD with closed pore and the intermediate conformation were superposed based on surrounding BMC-H, and Cα-Cα distances were plotted as color coded arrows.
(F) Same as D, but viewed from the side along the pore axis (two BMC-TD subunits are hidden to reveal the view into the channel).
(G) Weak density (purple) above the closed pore in the outer BMC-TD layer (blue) may correspond to the C-terminus of BMC-TD2. The unsharpened final map was used to generate this figure because the weak additional density is fragmented in the sharpened map.
(H) Comparison of overall conformation of BMC-TD trimers from the BMC main and compacted shell populations (intermediate and open states, respectively).
(I) Open conformation of the pore in the population of compacted shell particles (smaller apparent diameter; cyan state in H). Note the absence of density for gating arginine residues in the shell-embedded layer (dark red) and the presence of clear density for these residues in the protruding trimer in the background (blue). See also Figures S1, S4–S6, Table S1.
Specific interactions of BMC-TS in the assembled BMC shell
To address more subtle global structural variations (see below) that could hamper the detailed analysis of individual subunits, we computationally sorted our cryo-EM dataset into 10 classes while applying icosahedral symmetry. Subsequently, using the focused classification strategy outlined above (Fig. 4, see STAR Methods for details), we individually resolved the BMC-TD and BMC-TS building blocks that occupy the three-fold axes of the icosahedral shell, allowing us to determine the structure of BMC-TS (BMC-TS1, gene ID Hoch-5812) in the HO shell (Figs. 5, S1). This shell-embedded structure aligns very well to the crystal structure of the isolated protein (PDB ID 5DIH) (Aussignargues et al., 2016), with a RMSD of 0.5 Å over 535 aligned Cα atoms. Our structure shows that the same BMC-H residues that are involved in inter-hexamer contacts also form the interface with BMC-TS (Sutter et al., 2017). Because BMC-TS is a fusion of two pfam00936 domains, two different BMC-TS-BMC-H interfaces exist (Fig. 5A). At each of these interfaces, BMC-H interacts with one BMC-TS domain through an interaction between absolutely conserved lysine residues at the center of the mostly flat interface (Fig. 5B, C); this lysine was previously observed to mediate the hexamer-hexamer contacts in BMC-H (Sutter et al., 2017). In contrast to these conserved interactions, other contacts differ between the two types of interfaces formed by BMC-TS. In one interface, involving pfam00936 domains of two separate BMC-TS chains (Fig. 5A, B), positively charged residues (R194 on BMC-TS1 and R78 on BMC-H) form hydrogen bonds to backbone carbonyl oxygens and one salt bridge (BMC-H R78 to BMC-TS1 E198) across the interface (Fig. 5B). In the other interface, involving the N- and C-terminal pfam00936 domains of a single BMC-TS (Fig. 5C), the side chain of R78 on BMC-H predominantly assumes a different conformation that precludes hydrogen bonding with BMC-TS, although there is weak density indicating that a subpopulation might be within hydrogen bonding distance of BMC-TS1 Q100, which occupies the position equivalent to E198 of the C-terminal pfam00936 domain of BMC-TS1. The contacts at the corner where BMC-TS meets two BMC-H subunits are formed by conserved small residues (A68 and A169 on BMC-TS1), a property that is typical for all BMC shell proteins because a bulky residue in this position would very likely prevent shell formation.
Structural homology of BMC-TS across diverse BMC shells
Phylogenetic analysis of 319 BMC-TS proteins with significant sequence homology to HO-BMC-TS1 (for details see STAR Methods, redundancy corrected with a 90% identity cut-off) shows distinct clades that are generally consistent with BMC type (Fig. S2A; type names according to (Axen et al., 2014)). HO BMC-TS1 falls into a clade with SPU (putatively Sugar Phosphate Utilizing) BMCs, which may hint at the enzymatic reactions catalyzed inside the functionally uncharacterized HO shell. The primary structure of the HO BMC-TS protein is only distantly related to other structurally characterized BMC-TS proteins, and an initial Cα root-mean-square deviation (RMSD) when aligning the HO BMC-TS with the BMC-TS from the propanediol utilization (PDU) BMC (PDB code 3PAC, (Pang et al., 2011)) is high (3.6 Å; Fig. S2B). However, the poor overall alignment arises from slightly different orientations between the two component pfam00936 domains of the two BMC-TS homologs. Indeed, when the HO BMC-TS and the PDU BMC-TS are split into their constituent structural domains, the individual domains align very well, with Cα RMSDs of < 1 Å (Fig. S2C). This demonstrates the strong structural homology despite a moderate 36% identity and 55% similarity between the amino acid sequences of the PDU and HO BMC-TS proteins. Despite HO BMC-TS1 being in a separate clade from the BMC-TS of the well characterized PDU metabolosome, the high structural conservation of the domains indicate that it is a valuable model for the interactions with BMC-H and its integration into BMC shells in general.
Variations of the overall shell structure
The finely sampled 3D classification of the HO shell dataset into 10 classes (Fig. S4) resulted in 3 classes in which the HO BMC shell appeared to exhibit a smaller diameter, of which we selected the class with the smallest apparent diameter for further analysis and comparisons (Fig. 6A–C). Interestingly, this class also contained higher density, albeit weak, inside the shell compared to the solvent surrounding the shell, unlike other classes (Fig. 6A). This diffuse density might arise either from protein molecules encapsulated by chance during recombinant expression and assembly of the HO BMC shell in E. coli, or alternatively from an electron-dense solute accumulated inside these shells. This difference in encapsulated density between the two classes correlates with structural variations in the shell subunits. Comparison of the refined structure of this variant class to the structure of the largest class (used for BMC-TS structure determination) shows that these differences are smallest at the BMC-P positions (Fig. 6C–E), while the BMC-H and BMC-T building blocks bulge inward in this seemingly compacted shell conformation (Video S1). This initial assessment is confirmed by more detailed analysis of the displacement of individual Cα positions between classes (Fig. 6D, E).
We next sought to determine the origin of these structural variations, and whether these represent a dynamic conformational equilibrium or static structural differences. Our rationale was that if these classes represent interconverting conformational states, they should be identical except for their conformational difference; any compositional differences would argue for the observed differences to be static. We therefore classified one BMC-T position in several shell populations (classes 4, 5, 6, 9, and 10 in Fig. 4) into BMC-TD and BMC-TS classes (Fig. 7A, B) to obtain the relative frequencies of BMC-TS and BMC-TD trimers in shells in these classes. This analysis revealed that all classes contain approximately equal proportions of BMC-TS and BMC-TD subunits (Fig. 7B), with slight differences. However, it is important to note that these proportions apply only to the particle class as a whole, and individual shells will have a range of BMC-TS:BMC-TD ratios (shown for the main population in Fig. 7C). Given the structural differences observed in the presence of similar BMC-TS:BMC-TD ratios, and the much larger variation between shells within one class than across classes, we hypothesized that there might be a difference in the relative arrangement of the BMC-TD and BMC-TS subunits in the shell.
Fig. 7. Classification of four adjacent BMC-T positions and analysis of the distribution of BMC-TS or BMC-TD in the HO BMC shell.

(A) Left: Region of density showing the arrangement of classified BMC-T positions (masks used for classification shown in light green and light blue). Classification using the blue mask resulted in reconstructions sorted for the central BMC-T trimer (shown in the center). Right: Difference density obtained by subtracting the reconstruction classified for a central BMC-TD from the reconstruction classified for a central BMC-TS (the two reconstructions shown in the center).
(B) Average percentages of BMC-TS and BMC-TD in selected HO shell 3D classes.
(C) Histogram of number of BMC-TS subunits in HO shells (main population, class 6). The number of BMC-TS counts that are not divisible by 3 indicates that the error rate of the classification at the single BMC-T subunit level is on the order of 0.25 % (given approx. 16 % of shells with one classification error and 60 BMC-T monomer positions per shell).
(D) Visualization of difference density resulting from subtraction of BMC-TS maps from BMC-TD maps for the compacted class 10 (only positive difference density shown), revealing a different pattern for this class (compare to panel A, above). See also Figures S7, S8, Table S1.
We therefore computed a fully asymmetric 3D reconstruction of the HO BMC shell at 4.4 Å resolution from 11,100 particle images (Fig. S3). If BMC-TS and BMC-TD proteins were arranged in a fixed pattern across the shell, this pattern would likely be resolved in the asymmetric reconstruction. In contrast, for a fully random distribution of BMC-TS vs. BMC-TD, we anticipated observing uniform density levels for all BMC-T positions on the shell. However, the resulting cryo-EM map shows variable density for the protruding BMC-TD trimers, indicating that the distribution of BMC-TD and BMC-TS on the shell is neither completely random nor adhering to a fully fixed pattern. The observed variable density for the outer BMC-TD trimer is best explained by local patterns, which may partially drive image alignment during cryo-EM map refinement, but cannot be fully resolved because the global pattern varies from shell to shell. Quite strikingly, in the asymmetric reconstruction of the main shell population, weak and strong density for protruding BMC-TD trimers is observed at adjacent BMC-T positions (Fig. S3A, B). To investigate whether this pattern is a general feature of the HO shell, we calculated refined cryo-EM maps for the BMC-TD and BMC-TS subclasses for each of the shell classes for which we had previously determined the BMC-TD:BMC-TS ratio and then computed difference densities between the BMC-TD and BMC-TS subclasses. For most classes, including the main population (Fig. 7A) this analysis confirmed our observation derived from the asymmetric reconstruction, with a preference for unequal types of BMC-T subunits located next to each other (Fig. 7A, S3A, B). However, we found that the distribution bias was reversed in the compacted shell population (Fig. 7D). This indicates that the structural variation between these shell populations is most likely explained by different patterning of their BMC-T subunits, arising stochastically during shell assembly, which might influence the position or conformation of intervening BMC-H subunits. Therefore, in spite of the structural and compositional similarities between the main shell population and this compacted class of shells, these structures are probably not conformational variants that are able to interconvert, but structural variants of the HO BMC shell that arise from differing distributions of the BMC-TS and BMC-TD subunits across the shell.
BMC-TD2 and BMC-TD3 are functionally distinct but structurally interchangeable
Next, we addressed the structure of the BMC-TD proteins in the shell, using the same approach as for resolving BMC-TS (see Fig. 4 and STAR Methods). The HO shell contains two members of the BMC-TD protein family (Fig. 1A, B), BMC-TD2 (Hoch-5816) and BMC-TD3 (Hoch-3341). BMC-TD proteins were first identified in α- and β-carboxysomes (CsoS1D and CcmP, respectively). Although only a few loci encoding the functionally well characterized PDU and EUT BMCs contain a gene encoding a BMC-TD (Fig. S4A), recent bioinformatic surveys (Axen et al., 2014) and structural studies (proteins from the Rhodococcus and Mycobacterium microcompartment (RMM) (Mallette and Kimber, 2017)) indicate that BMCs containing these distinctive “stacked” shell building blocks with their internal chambers and gated pores may be more widespread than previously known.
The two BMC-TD paralogs found in the HO shell are members of distinct clades (Fig. S4A). BMC-TD3 clusters with proteins associated with the SPU (potentially Sugar-Phosphate Utilizing) BMC while BMC-TD2 does not group with a particular BMC type; its closest relatives are the α-cyanobacterial CsoS1D proteins (Fig. S4A). Crystal structures of the isolated proteins show that BMC-TD2 and BMC-TD3 are very similar, with an RMSD of 0.42 Å over 181 aligned Cα atoms. Because of this, we were unable to classify BMC-TD2 and BMC-TD3 into distinct particle populations (Fig. 4), but can assume that our cryo-EM map represents both variants with sufficient accuracy for the purpose of our analysis. We verified that the HO shells contain both BMC-TD2 and BMC-TD3 (Sutter et al., 2017); consistent with the presence of both BMC-TD paralogs, we observe intermediate size side chain densities at positions of amino acid sequence differences between the two proteins (e.g. BMC-TD2 residues F10 or W39, Fig. S4B). Nevertheless, the overall quality of the density for the composite BMC-TD2/D3 is excellent, highlighting the structural homology between BMC-TD2 and BMC-TD3 and suggesting that different members of the BMC-TD class fulfill the same structural role.
BMC-TD2 and BMC-TD3 are, however, only 57% identical (74% similarity) in primary structure. This raises the question whether the sequence differences between these two essentially structurally identical building blocks are important for their function. While most differences between BMC-TD2 and BMC-TD3 involve surface-exposed residues that are not involved in inter-subunit interactions, several significant substitutions (red in Fig. S4B) face their interior cavities. A sequence analysis of the close homologs of BMC-TD3 (clade indicated in Fig. S4A) shows that the interior residues E166 and R127 are highly conserved among BMC-TD3 homologs (Fig. S5A), but are not found in BMC-TD2 (Fig. S4B). Those residues are located in a region on the interior cavity close to the interface between the two layers of BMC-TD3 (Fig. S5B). The electron density in the crystal structure of the isolated BMC-TD3 protein (Sutter et al., 2017) indicates the presence of a glycerol molecule as well as further additional density near those residues (Fig. S5C), suggesting that this area binds small molecules that could potentially regulate the opening/closing of the pore. There are conserved interior cavity residues for BMCTD2 as well, but these are broadly conserved among BMC-TD clades (Fig. S6A, B and (Sutter et al., 2017)).
If those regions are involved in regulation of shell permeability by binding small molecules involved in the BMC reaction as suggested previously (Cai et al., 2013; Larsson et al., 2017), this could indicate that BMC-TD2 and BMC-TD3 have a preference for different small molecule ligands. In agreement with this hypothesis, BMC-TD3 is encoded distal to the main BMC operon (which contains all the other shell proteins and two additional BMC-Ps which are not part of our structure) and could therefore represent a separately regulated component of the shell that conducts a metabolite facultatively. Interestingly, this genomic organization is also found in Deltaprotebacteria bacterium CG2_30_63_29, which harbors the closest homologs to both BMC-TD2 and BMC-TD3 (gene IDs AUK47_22055 and AUK_11260, respectively), which is consistent with the hypothesis that BMC-TD3 was a later evolutionary addition to functionally expand the permeability properties of the shell.
Structure and conformational differences of shell-embedded BMC-TD
As detailed above, BMC-TD proteins occur in a wide range of BMCs and are thought to be important modulators of shell permeability. Due to the high structural similarity between BMC-TD2 and BMC-TD3, as observed in the isolated crystal structures (Sutter et al., 2017), we could not further sub-classify the composite BMC-TD2/3 class into separate BMC-TD2 and BMC-TD3 classes, and the two proteins are assumed to be structurally equivalent for our analysis.
The asymmetric 3D classification revealed two distinct conformations for the shell-embedded trimer of BMC-TD in the main shell population (Figs. 8A, B, S1, Video S1). The first conformation exhibits a closed pore at the inside of the shell (Fig. 8A), with three clearly resolved gating arginine residues forming salt bridges to glutamate residues of the neighboring subunit (Fig. 8A; also shown for the protruding BMC-TD trimer in Fig. 3B), as observed previously (Sutter et al., 2017). The second conformation (Fig. 8B) exhibits an outward displacement of secondary structure elements and loops surrounding the pore (Fig. 8B–D). A more detailed analysis shows that the BMC-TD assembly performs a slight rigid-body-like motion that widens the shell-embedded BMC-TD ring overall, and positions residues near the inner arginine gate towards the shell interior and away from the pore axis (Fig. 8E, F). Nevertheless, density for the gating arginines is still present, albeit weaker than for the fully closed state (Fig. 8B). The outer trimer of BMC-TD, protruding from the shell surface, shows the gating arginines in the closed conformation in all of our reconstructions. Weak density that is seen on the outside of the gate residues could not be definitively interpreted but may correspond to the C-terminus of BMC-TD2 (Fig. 8G), given its location in the immediate vicinity of A205, the most C-terminal built BMC-TD2 residue. A comparison of the HO composite BMC-TD with the structures of carboxysomal CcmP (Larsson et al., 2017) or CsoS1D (Klein et al., 2009) in their open states shows that the widened BMC-TD conformation in our structure corresponds to an intermediate state between the fully closed and fully open states observed in the structures of the isolated BMC-TD proteins (Fig. 8D).
We also analyzed the BMC-TD subunits found in the compacted shell subpopulation. Interestingly, even though the overall conformation of the shell-embedded BMC-TD ring resembled the partially widened BMC-TD ring in the main shell population, there was no density for the gating arginine residues of the shell-embedded BMC-TD trimer in these shells (Fig. 8H, I). We could not detect a fully closed state even using focused 3D classification after partial signal subtraction (not shown), a cryo-EM method designed to detect conformational changes of small substructures in the context of larger assemblies (Bai et al., 2015). The absence of density for the gating arginine side chains in this reconstruction (Fig. 8I) indicates that these side chains are likely flexible, and that they might be able to allow passage of small molecules between the shell lumen and the cavity between the two BMC-TD rings in this conformation. The presence of an open inner pore and a closed outer pore (Fig. 8G) is consistent with the previously proposed airlock-like mechanism for small-molecule shuttling across BMC shells (Cai et al., 2013; Klein et al., 2009; Larsson et al., 2017).
The observation of these conformational differences of the BMC-TD dimers of trimers in the context of the assembled shell may reflect inherent conformational flexibility of these proteins stemming from their ability to selectively open and close to enable shuttling of reactants into or out of the shell (Cai et al., 2013; Klein et al., 2009; Larsson et al., 2017). However, while selective gating appears to depend on the presence of small molecule ligands, the prevalence of an open or near-open conformation in a distinct population of shells in our preparation might be related to the overall conformation of the shell, possibly to accommodate the slightly different positions of the surrounding BMC-H subunits (Video S1). The conformational flexibility of BMC-TD trimers may therefore not only serve ligand-controlled small molecule conductivity, but also play a role in accommodating variations in shell assembly, unlike for the BMC-TS variant, which appear to be more rigid, as we could not resolve different conformational states in their case (not shown).
BMC-TS and BMC-TD may contribute to HO shell stability and architectural plasticity
Biochemical experiments show that deletion of BMC-TS1 in a recombinant expression system reduces HO BMC shell yield approximately 10-fold (Lassila et al., 2014), consistent with the idea that BMC-TS1 contributes significantly to the stability of the shell. This is in agreement with the binding energies for the interfaces formed by all BMC-T variants within the shell, which, derived from our structures, reveal clearly higher binding energy for BMC-TS1 compared to BMC-TD2 (Table 1). Accordingly, individual deletions of BMC-TD2 or BMC-TD3 have a far less severe impact on shell yield (Lassila et al., 2014). Nevertheless, BMC-TD building blocks are efficiently incorporated into BMC shells, suggesting that even though a single BMC-TS1 interaction with the surrounding BMC-H hexamers is more stable than one occupied by BMC-TD2/TD3, both BMC-TS and BMC-TD support assembly of stable shells. This is agreement with the high variability in the BMC-TD:BMC-TS ratio in our HO BMC shell preparation (Fig. 7C), suggesting that assembly of HO BMC shells is extraordinarily robust to a range of BMC-T compositions.
Table 1. Calculation of binding energies and buried surface area.
Interface statistics between of BMC-TD2 (two different states) and BMC-TS1 interaction with the surrounding hexamers in the main shell population, calculated using PISA (Krissinel and Henrick, 2007).
| ΔGint (kcal/mol) |
Buried area (Å2) |
|
|---|---|---|
| BMC-TD2 (closed) | −17.5 | 6044 |
| BMC-TD2 (intermediate] | −18.2 | 6682 |
| BMC-TS1 | −33.3 | 7198 |
Why, then, do the structural variations observed in the global shell classes correlate with different distribution patterns of BMC-T variants? The sites of incorporation of BMC-T type building blocks are separated by intervening BMC-Hs, and there are no direct interactions between one BMC-T trimer (or dimer of trimers) and its most proximal BMC-T neighbors. However, the positions of the BMC-TS and BMC-TD trimers within the shell layer differ slightly, with the inner layer of BMC-TD being embedded more deeply in the shell than the single BMC-TS ring (Fig. 9A). Therefore, one possible source of the variations in shell geometry we observed in the compacted class of shells, with largest differences near BMC-T subunits and smallest differences at BMC-P positions (Fig. 6), is a slight tilt of the BMC-H hexamers that occupy the space between neighboring BMC-T modules, depending on the BMC-T variant that they are contacting. To investigate this hypothesis further, we performed a 3D classification of the symmetry-expanded dataset of shell particles (see above). We sequentially classified four neighboring BMC-T positions (blue and green masks in Fig. 7A; Figs. S7, S8) to obtain cryo-EM maps with defined BMC-TS or BMC-TD occupancies at these positions. Comparison of these cryo-EM density maps (Fig. 9B) suggest that indeed, BMC-H subunits next to BMC-TD proteins might be tilted outwards slightly compared to BMC-H subunits next to BMC-TS (Video S2). Even though the differences are subtle, these observations provide a possible structural mechanism for the origin of the slight variations in shell geometry that we observe in our cryo-EM data.
Fig. 9. Analysis of BMC-TS and BMC-TD interactions with intervening BMC-H.

(A) Comparison of the positions of BMC-TS and BMC-TD as they are embedded in the shell. BMC-TD sits slightly lower in the surrounding shell formed by BMC-H, which probably leads to small changes of interaction geometry.
(B) View of two reconstructions after sorting for four neighboring BMC-T positions. Left: Central BMC-TS surrounded by three BMC-TD building blocks; right: Central BMC-TD surrounded by three BMC-TS building blocks. See also Figures S7, S8.
DISCUSSION
Here we present a complete structural analysis of the HO BMC shell system, which consists of one BMC-H and one BMC-P type protein as well as two BMC-TD and one BMC-TS. Accordingly, our model contains representatives of all of the major classes of shell proteins, enabling us to define molecularly the complete set of specific non-covalent interactions that underlie the construction of this 6.5 MDa icosahedral shell. Specifically, our structure provides the first molecular description of the interactions between BMC-H and BMC-TS in an assembled shell. BMC-H proteins are a component of all BMC shells identified to date, and BMC-TS proteins are nearly as ubiquitous, though usually present in smaller numbers (Axen et al., 2014). Due to the high structural conservation of BMC-TS subunits, even in the presence of only moderate sequence conservation (Fig. S2), our structure can serve as a blueprint for the BMC-TS:BMC-H interaction, and indeed the interactions of all major types of shell proteins, across a wide range of functionally diverse BMC shells.
Furthermore, our cryo-EM reconstructions reveal that the HO BMC shell exhibits both global and local structural variability. We observed three different states of the shell-embedded trimer of BMC-TD subunits in the intact shell (Fig. 8). Notably, the one that most resembles previously observed open states of BMC-TD homologs was found in a particle population with a slightly different geometry of the overall shell (Fig. 6). Even though this finding is based on an ensemble of cryo-EM particle images and icosahedral averaging, which may result in averaging of slightly different conformations, these observations suggest that the flexibility of the BMCTD subunits that we observe may aid in accommodating slight variation in the overall shell geometry that are induced by the distribution of the BMC-TD and BMCTS variants across the shell. Even though one of these pore states resembles an open state, and the ring of gating arginines at the inside of the shell is not resolved in the density of this compacted shell population, actual gating and solute exchange may require the binding of a small molecule cofactor, as observed in structures of the related CcmP protein in isolation (Larsson et al., 2017). This idea is compatible with the observation that the gating arginines in the outer BMC-TD ring are closed in our reconstructions, and with the possible containment of solute in the relatively compact shell population, as suggested by the elevated density levels inside the shell (Fig. 6A). However, we note that we cannot exclude the possibility that the source of the elevated density inside the compacted shells is an encapsulated macromolecule or possibly a small molecule that might not be able to pass through even a conducting BMC-TD channel. Nevertheless, our observations are in good agreement with an airlock-like mechanism of BMC-TD gating (Cai et al., 2013; Klein et al., 2009; Larsson et al., 2017), thereby expanding earlier observations from isolated structures to a fully assembled BMC shell.
Binding of such small molecule ligands may control the gating of individual BMC-TD pores locally (Cai et al., 2013; Klein et al., 2009; Larsson et al., 2017; Mallette and Kimber, 2017), and largely independently of the composition of the surrounding shell. The presence of two BMC-TD paralogs in different locations of the H. ochraceum, genome and the differences in the residues facing their interior cavity (Figs. S4–S6) suggest a preference for different regulatory molecules, a different substrate, or ability to respond to different physiological conditions, in spite of minimal structural difference between them. Our finding that the proportion of BMC-TD subunits in HO shells can vary considerably (Fig. 7C) suggests that the incorporation of BMC-T subunits may be largely stochastic, possibly dependent on the available concentration of free subunits in the cell. Encoding one of the BMC-TD proteins away from the main BMC operon (Lassila et al., 2014) may allow independent regulation of its expression, and therefore dynamic adaptation of shell composition to the requirements of the cellular metabolism. BMC-TD proteins may therefore represent a modular way to enhance the metabolic versatility of BMCs that have BMC-TD in a different genomic location, including HO-type metabolosomes and carboxysomes (Fig. S4). It is worth noting that given the tighter interaction of BMC-TS with the surrounding BMC-H subunits in the shell compared to BMC-TD (Table 1), the presence of substantial numbers of BMC-TD in the HO BMC shells cannot be fully explained by considering local thermodynamics alone. This suggests that other effects, possibly irreversible incorporation of BMC-T subunits into growing shells or local geometric distortions that make accommodation of one BMCT variant preferred over the other, might play a role. Additionally, based on the extensive interactions between shell proteins observed in the full shell structure, it is likely that subunits cannot be exchanged once the shell is fully formed and changes of shell protein composition will only apply to newly formed shells.
The observation of correlated changes between overall shell geometry (Fig. 6) and the conformation of shell-embedded small-molecule conducting pore proteins (Fig. 8) raises the intriguing hypothesis that under some circumstances, the overall shell conformation might be dynamically coupled to the conformation of the pore proteins, giving rise to cooperative pore opening or closing throughout the shell. However, our current data do not provide direct evidence for such a mechanism because the shell classes that show such conformational differences also differ in the arrangement of BMC-TD and BMC-TS subunits, indicating that what we observe in our dataset are static, rather than dynamic, structural differences between shell classes. It is worth noting that the majority of shell subpopulations analyzed exhibits a bias towards incorporation of unequal types of BMC-T subunits in neighboring positions (Fig. 7A), indicating that, possibly due to better compatibility of the conformation of the intervening BMC-H subunits, this arrangement of BMC-T subunits may be slightly favored overall.
The modularity of BMC shells and their ability to incorporate unequal subunits in symmetry-related positions contrasts with other shell systems such as the bacterial T = 1 encapsulin shell (Sutter et al., 2008) or viral and bacteriophage capsids, which employ building blocks that are evolutionarily unrelated to the BMC fold. These properties may reflect a functional adaptation to the requirements of a metabolic compartment evolved to perform multi-step reaction pathways with high efficiency. The shell protein pores are analogous to membrane protein transporters or channels of lipid-bound compartments, which are crucial to the reactions that are performed inside the compartment. Similar to how a variety of transporters and channels are required for every lipid membrane-based compartment, a metabolosome shell needs to allow the selective passage of different substrates, products, and enzyme cofactors, while retaining full integration with the requirements of cellular metabolism. Our high-resolution cryo-EM structure, containing representatives of all of the major BMC shell protein subunits, reveals the structural details of all the interactions in the HO-shell, providing a blueprint for assembly of diverse shells. These findings provide structural insight into the diverse BMC shells found in nature that assemble from homologous subunits, and facilitate efforts to engineer its properties for biotechnological applications.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cheryl A. Kerfeld (ckerfeld@lbl.gov).
METHOD DETAILS
Cryo-EM specimen preparation and data acquisition
Cryo-EM specimens were prepared as described (Sutter et al., 2017). Cryo-EM data for high-resolution structure determination were acquired in a semi-automated fashion using LEGINON (Suloway et al., 2005) on a Titan low-base cryo-electron microscope (FEI Company) using a side-entry holder (Gatan) and a K2 Summit direct electron detector (Gatan). Dose-fractionated movies were acquired using a total dose of 25 electrons/Å2 fractionated into 30 frames, a total exposure time of 4.5 sec, defocus values of −1.0 to −3.0 μm, and a magnification of 48,543 x, resulting in a pixel size of 1.03 Å on the object scale.
Initial cryo-EM data processing and analysis
A total of 982 dose-fractionated movies were collected. The movie frames were aligned and dose-weighed using MOTIONCOR2 (Zheng et al., 2017) and defocus was estimated using CTFFIND4 (Rohou and Grigorieff, 2015) from within RELION (Scheres, 2012). Initially, roughly 1,000 particles were picked manually from 42 micrographs and subjected to reference-free 2D classification in RELION (Scheres, 2012) to generate templates for automated particle selection. After inspection of micrograph quality (rejection of micrographs showing poor power spectra or large amounts of contamination), roughly 31,800 particles were selected automatically in RELION 1.4 (Scheres, 2015) from 928 retained micrographs using the templates generated in the previous step. Further processing was performed in RELION (Scheres, 2012) and is detailed in Fig. 2A. All particle images were initially subjected to 3D refinement to obtain initial angular assignments, applying icosahedral symmetry and using as an initial reference the low-resolution reconstruction of the HO BMC shell (EMD-8747) (Sutter et al., 2017). The resulting particle alignment parameters were used for 3D classification without image realignment, yielding one poor class exhibiting very low density values, and three classes exhibiting good high-resolution features. Because these classes exhibited slight differences in shell diameter, only one class was selected and refined, using two fully independent particle half-sets (gold standard refinement (Rosenthal and Henderson, 2003; Scheres and Chen, 2012)), yielding a reconstruction of the HO shell at 3.2 Å resolution. The resolution of this reconstruction was subsequently improved to 3.0 Å (Fig. 2B) by employing per-particle defocus refinement and Ewald sphere correction in FREALIGN (Grigorieff, 2007; Grigorieff, 2016; Wolf et al., 2006). The main contribution to this improvement came from per-particle defocus refinement, in agreement with theoretical considerations indicating that Ewald sphere curvature would become severely limiting only at higher resolutions for a 440 Å-diameter particle (DeRosier, 2000; Wolf et al., 2006). Overfitting of high-resolution information was prevented by using a frequency-limited refinement, excluding data beyond 3.6 Å resolution (Grigorieff, 2016). The resulting map shows excellent resolution (Fig. 2C), as determined using the local resolution function in RELION 2 (Kimanius et al., 2016; Scheres, 2012), with clear side chain density that in many cases allows the identification of rotamers (Fig. 2D).
Asymmetric reconstruction and classification for BMC-T variants
Resolving the structures of the BMC-TS (BMC-TS1) and BMC-TD (BMC-TD2/D3) subunits in the context of the intact shell required computational sorting of the cryo-EM particle images into homogeneous subsets for at least one BMC-T position in the shell. The presence of three different BMC-T variants at the icosahedral 3-fold axes breaks the symmetry and would lead to averaged density if icosahedral symmetry were applied to the whole particle. In an initial attempt of asymmetric analysis (not shown), we refined 27,800 particles retained after an initial 2D classification without imposing symmetry to calculate a 3D reconstruction at 4.1 Å resolution. Based on this reconstruction, classification to distinguish BMC-TS from BMC-TD was attempted using localized reconstruction (Ilca et al., 2015). However, even though 3D classification without alignment was possible from the resulting sub-particles, particle alignment and refinement failed when using this approach, possibly because the remaining signal for a single BMC-T trimer after signal-subtraction was too weak to drive accurate alignment. Therefore, we had to employ a different strategy for asymmetric analysis that exploits the signal of the entire shell particle to guide high-resolution particle image alignment.
While computing a fully asymmetric reconstruction of the HO BMC shell is possible from relatively small numbers of particles (Fig. S3) and would in principle allow classification of individual BMC-T positions, higher-resolution reconstructions can be obtained by symmetry expanding the dataset before classification, followed by symmetry-free 3D reconstruction. The symmetry expansion procedure implemented in RELION 2 (Kimanius et al., 2016) creates 60 copies of each particle image and assigns all icosahedral symmetry-related orientations to the copies, thereby placing every BMC-TS/TD monomer of every physical particle in the dataset at each BMC-T position in the resulting 3D reconstruction. 3D classification, restricted to one BMC-T position by computational masking, subsequently allows classification of all BMC-T positions in the entire dataset into either BMC-TS or BMCTD (Fig. 4).
More specifically, we initially classified the icosahedrally symmetric reconstruction from all particle images (see above) into 10 classes (Fig. 4). As noted above, the resulting classes differed in shell diameter. We therefore selected two classes, first, the class exhibiting the best high-resolution features and containing the most particles, and second the class with the smallest apparent shell diameter. After map refinement of these two classes, we symmetry-expanded the particle dataset by transforming the alignment parameters of each particle into all 60 symmetry-related orientations (symmetry operators for I2 icosahedral symmetry) using the corresponding command in RELION 2 (Kimanius et al., 2016). The symmetry expansion procedure places every BMC-T trimer of each (physical) shell particle at every symmetry-related position of the shell reconstruction. From this symmetry-expanded alignment parameter files, the particle images could be classified (with a mask) or refined (without a mask, allowing the signal of the entire particle to drive the alignment). Using a mask to limit classification to a single BMC-T trimer, this procedure allowed for 3D classification of different BMC-T variants at this one position (indicated in Fig. 4; the remaining BMC-T positions will remain averages).
As detailed in Figs. 4, 7, and S8, trimeric BMC-TS (36 % of the dataset) and double-stacked BMC-TD (64 % of the dataset) were separated in a first round of classification of the main particle population, using alignment-free classification focused on a single BMC-T position by applying a mask. The BMC-TS data subset was subsequently refined (using limited angular searches to prevent randomization of the classified BMC-T position, but without masking) to 3.4 Å resolution (Fig. S1A, B). The BMC-TD dataset was processed similarly. The class containing most of the particle images was refined and subsequently reclassified using masked alignment-free 3D classification. This step separated two conformations of the shell-embedded trimer of the BMC-TD assembly, with intermediate and closed pore states, respectively. The final data subsets were refined to 3.3 Å resolution (Fig. S1A, C). Both the BMC-TS and BMC-TD maps show excellent side chain density, as expected at a resolution approaching 3 Å.
The particle population giving rise to a reconstruction of the compacted shell was classified similarly. After initial refinement of the class (to 3.3 Å using the same procedure as described for the refinement of the main population, see above), local masked classification was performed using 4 classes, of which one showed excellent density for BMC-TD. This class was subsequently refined to 3.6 Å resolution and shows a intermediate state of the BMC-TD structure surrounding the pore, similarly to one of the subclasses obtained from classification of the main particle population. Because we could not detect a particle population with a closed pore in the compacted shell dataset, we additionally attempted focused 3D classification after subtraction of residual background signal (Bai et al., 2015). However, the resulting focused 3D classification did not detect a subpopulation of particles with closed inner pore either.
Classification of four neighboring BMC-T positions
The classification of four neighboring BMC-T positions was performed similarly as described above for structure determination of the BMC-T paralogs. However, during the classification process for structure determination described above, homogeneity of the resulting classes was paramount, and more classes than strictly necessary for separation of BMC-T variants were used (resulting e.g. in detection of conformational states or elimination of poor quality particles). The classification performed here aimed to assign all BMC-T trimers to either BMC-TS or BMC-TD. Consequently, the number of classes used was chosen to match exactly the number of combinations of BMC-TS vs. BMC-TD expected to avoid losing particles into poorly defined and not interpretable classes that would lead to inaccuracy of the numbers derived from the in silico experiment.
The classification was performed in three steps (Fig. S8) because simultaneous classification of all four positions (or also three positions) at once sometimes failed to detect rare classes as more highly populated classes started dominating the classification. In a first step, the central position (position 1; Fig. S8) was classified into BMC-TS and BMC-TD classes (2 classes used; Fig. 7A). In the second step, both of these classes were sub-classified into four classes to analyze two neighboring BMCT positions (positions 2 and 3). In the third step, all of these classes (2 × 4) were sub-classified again into two classes (position 4). Overall, this resulted in 24=16 classes that represent all combinations of BMC-TS and BMC-TD at four positions (Fig. S8).
Atomic model building and refinement
Atomic models were built and fully refined for one asymmetric unit of the icosahedral reconstructions of the main and compacted BMC shell populations and for the BMC-T-containing sub-volumes obtained from focused classification and refinement. In the icosahedrally symmetrized reconstructions, the shell-embedded BMC-T ring was interpreted using a model of BMC-TD2 with side chains truncated at the Cβ position due to fragmented density. Model building was performed in COOT (Emsley et al., 2010), based on the X-ray crystal structure of the BMC shell (Sutter et al., 2017) for BMC-H and the X-ray crystal structures of isolated BMC-TS1 and BMCTD2 (Aussignargues et al., 2016; Sutter et al., 2017). The resulting atomic models were refined using the real space refinement program in PHENIX (Adams et al., 2010). Model vs. map FSC curves were computed using the MTRIAGE program in PHENIX (Adams et al., 2010). Protein structures were visualized with UCSF Chimera (Pettersen et al., 2004) and Pymol (The PyMOL Molecular Graphics System, Version 1.7 Schrödinger, LLC).
Sequence analysis.
BMC-TS and BMC-TD homolog protein sequences were gathered using BLAST against the full Uniprot dataset and HMMs (Eddy, 2008) generated using established members of the BMC-TS (e.g. HO-BMC-TS1, PduT) and BMC-TD (e.g. HO-BMC-TD2/3, CcmP)-type were used to identify BMC proteins of the same subtype (e-value scores to the subtype HMM <1e-20, calculated by HMMSCAN (Eddy, 2008)). Sequences were corrected for redundancy using a 90% identity cutoff using BLASTCLUST, aligned using ClustalX (Larkin et al., 2007) with default parameters, and phylogenetic trees were generated using phyml with default parameters. Conservation was mapped onto the structure using HOMOLMAPPER (Rockwell and Lagarias, 2007).
QUANTIFICATION AND STATISTICAL ANLYSIS
Quantifications of BMC-T type (Fig. 7B, C) used assignments derived from maximum-likelihood 3D classification algorithms implemented in RELION (Scheres, 2012).
DATA AND SOFTWARE AVAILABILITY
Data resources
The cryo-EM maps and refined coordinate models reported here were deposited in the Electron Microscopy Data Bank and Protein Data Bank with accession codes given in Table S1.
Software availability
This study did not generate new software. All software used in this study has been previously published and is publicly available. The script used for rendering of Cα displacement vectors was based on the methodology and script described on the PYMOL website (https://pymolwiki.org/index.php/Modevectors).
Supplementary Material
Video S1, related to Figures 5, 6, 8. Global and local structural differences in the HO BMC shell.
Part 1: Global structural differences between the main and compacted shell states.
Part 2: Correlated global and local structural differences of a BMC-TD module and neighboring BMC-H subunits (closed and open pore states in main and compacted shell states, respectively). Note the change of overall geometry and of the inner BMC-TD ring.
Part 3: Local structural variability of a BMC-TD module in the main shell population (closed and intermediate states).
Video S2, related to Fig. 9. BMC-H tilting depending on identity of neighboring BMC-T. Morph between two maps classified for four BMC-T positions (maps as shown in Fig. 9B), focused on two BMC-T positions (central BMC-T and one corner of classified region) and colored by radius from red to blue. The intervening BMC-H building block shows different tilts depending on the identity oft he neighboring BMC-T module.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Preparation of HO shell | (Sutter et al., 2017) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nonidet P40 substitute | Sigma-Aldrich | Cat# 11332473001 |
| Deposited Data | ||
| Coordinates HO shell main population | this paper | PDB ID 6MZX |
| Map HO shell main population | this paper | EMD-9309 |
| Coordinates HO shell compacted | this paper | PDB ID 6MZY |
| Map HO shell compacted | this paper | EMD-9310 |
| Coordinates for subvolume with BMC-TS | this paper | PDB ID 6N06 |
| Map for subvolume with BMC-TS | this paper | EMD-9311 |
| Coordinates BMC-TD (main population, closed) | this paper | PDB ID 6MZU |
| Map BMC-TD (main population, closed) | this paper | EMD-9307 |
| Coordinates BMC-TD (main population, widened) | this paper | PDB ID 6MZV |
| Map BMC-TD (main population, widened) | this paper | EMD-9308 |
| Coordinates BMC-TD (compacted population, open) | this paper | PDB ID 6N07 |
| Map BMC-TD (compacted population, open) | this paper | EMD-9312 |
| Coordinates BMC-T four positions (TDTSTSTS) | this paper | PDB ID 6N0F |
| Map BMC-T four positions (TDTSTSTS) | this paper | EMD-9314 |
| Coordinates BMC-T four positions (TDTDTDTD) | this paper | PDB ID 6N09 |
| Map BMC-T four positions (TDTDTDTD) | this paper | EMD-9313 |
| Coordinates BMC-T four positions (TSTDTDTD) | this paper | PDB ID 6N0G |
| Map BMC-T four positions (TSTDTDTD) | this paper | EMD-9315 |
| Map HO shell C1 refinement | this paper | EMD-9296 |
| Coordinates HO shell X-ray structure | (Sutter et al., 2017) | PDB ID 5V74 |
| Map HO shell low-resolution cryo-EM (initial reference) | (Sutter et al., 2017) | EMD-8747 |
| Coordinates of ligand-bound CcmP, fully open | (Larsson et al., 2017) | PDB ID 5LT5 |
| Coordinates of BMC-TS from Citrobacter freundii | (Pang et al., 2011) | PDB ID 3PAC |
| Coordinates of BMC-TS from Haliangium ochraceum | (Aussignargues et al., 2016) | PDB ID 5DIH |
| Software and Algorithms | ||
| MOTIONCOR2 | (Zheng et al., 2017) | N/A |
| CTFFIND 4 | (Rohou and Grigorieff, 2015) | N/A |
| LEGINON | (Suloway et al., 2005) | N/A |
| RELION 1.4 | (Scheres, 2012) | N/A |
| RELION 2.0 | (Kimanius et al., 2016) | N/A |
| FREALIGN 9 | (Grigorieff, 2007; Grigorieff, 2016) | N/A |
| PHENIX 1.14 | (Adams et al., 2010) | N/A |
| COOT 0.8.1 | (Emsley et al., 2010) | N/A |
| HMMSCAN | (Eddy, 2008) | N/A |
| CLUSTAL X | (Larkin et al., 2007) | N/A |
| HOMOLMAPPER | (Rockwell and Lagarias, 2007) | N/A |
| UCSF CHIMERA 1.11 | (Pettersen et al., 2004) | N/A |
| PYMOL 1.7 | Schrödinger, LLC | N/A |
| Other | ||
| C-flat 1.2/1.3 holey carbon films on Cu 400 mesh grid | Protochips, Morrisville, USA | Cat# CF-1.2/1.3–4C |
Highlights.
3.0 Å resolution cryo-EM structure of the H. ochraceum microcompartment shell
Asymmetric data processing strategy resolves complete blueprint for shell assembly
Structure of shell-embedded BMC-TS shell subunit reveals its specific interactions
Insights into structural and compositional variability of the HO BMC shell
Acknowledgements
We thank Eva Nogales for access to the electron microscopy facility at the University of California at Berkeley, Patricia Grob and Daniel Toso for support with electron microscopy, Abhiram Chintangal for computing support, Kristin Parent for critical reading of the manuscript and helpful suggestions, and Jamie Cate for sharing of PYMOL scripts. This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (NIAID) grant 5 R01 AI114975–05 and the U.S. Department of Energy, Basic Energy Sciences, Contract DE-FG02–91ER20021. We acknowledge the use of the resources of the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science user facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02–05CH11231. B.J.G. was supported by fellowships from the Swiss National Science Foundation (projects P300PA_160983, P300PA_174355).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no competing interests.
Data and materials availability
The cryo-EM maps and refined coordinate models reported here were deposited in the Electron Microscopy Data Bank and Protein Data Bank with the accession codes given in Table S1.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aussignargues C, Pandelia M-E, Sutter M, Plegaria JS, Zarzycki J, Turmo A, Huang J, Ducat DC, Hegg EL, Gibney BR, et al. (2016). Structure and Function of a Bacterial Microcompartment Shell Protein Engineered to Bind a [4Fe-4S] Cluster. J Am Chem Soc 138, 5262–5270. [DOI] [PubMed] [Google Scholar]
- Axen SD, Erbilgin O, and Kerfeld CA (2014). A taxonomy of bacterial microcompartment loci constructed by a novel scoring method. PLoS Comput Biol 10, e1003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X-C, Rajendra E, Yang G, Shi Y, and Scheres SH (2015). Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife 4, e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobik TA, Lehman BP, and Yeates TO (2015). Bacterial microcompartments: widespread prokaryotic organelles for isolation and optimization of metabolic pathways. Mol Microbiol 98, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F, Sutter M, Cameron JC, Stanley DN, Kinney JN, and Kerfeld CA (2013). The structure of CcmP, a tandem bacterial microcompartment domain protein from the β-carboxysome, forms a subcompartment within a microcompartment. J Biol Chem 288, 16055–16063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier DJ (2000). Correction of high-resolution data for curvature of the Ewald sphere. Ultramicroscopy 81, 83–98. [DOI] [PubMed] [Google Scholar]
- Eddy SR (2008). A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput Biol 4, e1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbilgin O, McDonald KL, and Kerfeld CA (2014). Characterization of a planctomycetal organelle: a novel bacterial microcompartment for the aerobic degradation of plant saccharides. Appl Environ Microb 80, 2193–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N (2007). FREALIGN: high-resolution refinement of single particle structures. J Struc Biol 157, 117–125. [DOI] [PubMed] [Google Scholar]
- Grigorieff N (2016). Frealign: An Exploratory Tool for Single-Particle Cryo-EM. Meth Enzymol 579, 191–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havemann GD, and Bobik TA (2003). Protein content of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol 185, 5086–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heithoff DM, Conner CP, Hentschel U, Govantes F, Hanna PC, and Mahan MJ (1999). Coordinate intracellular expression of Salmonella genes induced during infection. J Bacteriol 181, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt D, Frank S, Seyedarabi A, Ladikis D, Parsons JB, Warren MJ, and Pickersgill RW (2009). Structure of a trimeric bacterial microcompartment shell protein, EtuB, associated with ethanol utilization in Clostridium kluyveri. Biochem J 423, 199–207. [DOI] [PubMed] [Google Scholar]
- Herring TI, Harris TN, Chowdhury C, Mohanty SK, and Bobik TA (2018). A Bacterial Microcompartment Is Used for Choline Fermentation by Escherichia coli 536. J Bacteriol 200, e00764–00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu CV, Ding HJ, Morris DM, Dias DP, Gonzales AD, Martino A, and Jensen GJ (2007). The structure of isolated Synechococcus strain WH8102 carboxysomes as revealed by electron cryotomography. J Mol Biol 372, 764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilca SL, Kotecha A, Sun X, Poranen MM, Stuart DI, and Huiskonen JT (2015). Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat Commun 6, 8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobson CM, and Tullman-Ercek D (2016). Dumpster Diving in the Gut: Bacterial Microcompartments as Part of a Host-Associated Lifestyle. PLoS Pathog 12, e1005558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaval KG, and Garsin DA (2018). Ethanolamine Utilization in Bacteria. MBio 9, e00066–00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfeld CA, Aussignargues C, Zarzycki J, Cai F, and Sutter M (2018). Bacterial microcompartments. Nat Rev Microbiol 16, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfeld CA, and Erbilgin O (2015). Bacterial microcompartments and the modular construction of microbial metabolism. Trends Microbiol 23, 22–34. [DOI] [PubMed] [Google Scholar]
- Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, and Yeates TO (2005). Protein structures forming the shell of primitive bacterial organelles. Science 309, 936–938. [DOI] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, e18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MG, Zwart P, Bagby SC, Cai F, Chisholm SW, Heinhorst S, Cannon GC, and Kerfeld CA (2009). Identification and structural analysis of a novel carboxysome shell protein with implications for metabolite transport. J Mol Biol 392, 319–333. [DOI] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Larsson AM, Hasse D, Valegård K, and Andersson I (2017). Crystal structures of β-carboxysome shell protein CcmP: ligand binding correlates with the closed or open central pore. J Exp Bot 68, 3857–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassila JK, Bernstein SL, Kinney JN, Axen SD, and Kerfeld CA (2014). Assembly of robust bacterial microcompartment shells using building blocks from an organelle of unknown function. J Mol Biol 426, 2217–2228. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, and Adams PD (2017). Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallette E, and Kimber MS (2017). A Complete Structural Inventory of the Mycobacterial Microcompartment Shell Proteins Constrains Models of Global Architecture and Transport. J Biol Chem 292, 1197–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang A, Warren MJ, and Pickersgill RW (2011). Structure of PduT, a trimeric bacterial microcompartment protein with a 4Fe-4S cluster-binding site. Acta Crystallogr D Biol Crystallogr 67, 91–96. [DOI] [PubMed] [Google Scholar]
- Penrod JT, and Roth JR (2006). Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol 188, 2865–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Prasad BVV, and Schmid MF (2012). Principles of virus structural organization. Adv Exp Med Biol 726, 17–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell NC, and Lagarias JC (2007). Flexible mapping of homology onto structure with homolmapper. BMC Bioinform 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, and Henderson R (2003). Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333, 721–745. [DOI] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2015). Semi-automated selection of cryo-EM particles in RELION-1.3. J Struct Biol 189, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, and Chen S (2012). Prevention of overfitting in cryo-EM structure determination. Nat Meth 9, 853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MF, Paredes AM, Khant HA, Soyer F, Aldrich HC, Chiu W, and Shively JM (2006). Structure of Halothiobacillus neapolitanus carboxysomes by cryo-electron tomography. J Mol Biol 364, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Sutter M, Boehringer D, Gutmann S, Guenther S, Prangishvili D, Loessner MJ, Stetter KO, Weber-Ban E, and Ban N (2008). Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat Struct Mol Biol 15, 939–947. [DOI] [PubMed] [Google Scholar]
- Sutter M, Greber B, Aussignargues C, and Kerfeld CA (2017). Assembly principles and structure of a 6.5-MDa bacterial microcompartment shell. Science 356, 1293–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Kerfeld CA, Sawaya MR, Cai F, Heinhorst S, Cannon GC, and Yeates TO (2008). Atomic-level models of the bacterial carboxysome shell. Science 319, 1083–1086. [DOI] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, and Bäumler AJ (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci USA 108, 17480–17485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M, Derosier DJ, and Grigorieff N (2006). Ewald sphere correction for single-particle electron microscopy. Ultramicroscopy 106, 376–382. [DOI] [PubMed] [Google Scholar]
- Zarzycki J, Erbilgin O, and Kerfeld CA (2015). Bioinformatic characterization of glycyl radical enzyme-associated bacterial microcompartments. Appl Environ Microb 81, 8315–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Meth 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1, related to Figures 5, 6, 8. Global and local structural differences in the HO BMC shell.
Part 1: Global structural differences between the main and compacted shell states.
Part 2: Correlated global and local structural differences of a BMC-TD module and neighboring BMC-H subunits (closed and open pore states in main and compacted shell states, respectively). Note the change of overall geometry and of the inner BMC-TD ring.
Part 3: Local structural variability of a BMC-TD module in the main shell population (closed and intermediate states).
Video S2, related to Fig. 9. BMC-H tilting depending on identity of neighboring BMC-T. Morph between two maps classified for four BMC-T positions (maps as shown in Fig. 9B), focused on two BMC-T positions (central BMC-T and one corner of classified region) and colored by radius from red to blue. The intervening BMC-H building block shows different tilts depending on the identity oft he neighboring BMC-T module.
Data Availability Statement
Data resources
The cryo-EM maps and refined coordinate models reported here were deposited in the Electron Microscopy Data Bank and Protein Data Bank with accession codes given in Table S1.
Software availability
This study did not generate new software. All software used in this study has been previously published and is publicly available. The script used for rendering of Cα displacement vectors was based on the methodology and script described on the PYMOL website (https://pymolwiki.org/index.php/Modevectors).