

Abstract
Advances over the past 25 years have revealed much about how the structural properties of membranes and associated proteins are linked to the thermodynamics and kinetics of membrane protein (MP) folding. At the same time biochemical progress has outlined how cellular proteostasis networks mediate MP folding and manage misfolding in the cell. When combined with results from genomic sequencing, these studies have established paradigms for how MP folding and misfolding are linked to the molecular etiologies of a variety of diseases. This emerging framework has paved the way for the development of a new class of small molecule “pharmacological chaperones” that bind to and stabilize misfolded MP variants, some of which are now in clinical use. In this review, we comprehensively outline current perspectives on the folding and misfolding of integral MPs as well as the mechanisms of cellular MP quality control. Based on these perspectives, we highlight new opportunities for innovations that bridge our molecular understanding of the energetics of MP folding with the nuanced complexity of biological systems. Given the many linkages between MP misfolding and human disease, we also examine some of the exciting opportunities to leverage these advances to address emerging challenges in the development of therapeutics and precision medicine.
1. Introduction: Linking Biophysical Studies with Membrane Protein Folding in the Cell
I am waylaid by Beauty...
Oh, savage Beauty, suffer me to pass!
From “Assault”, by Edna St. Vincent Millay (1921)
By definition, integral membrane proteins (MPs) are components of lipid bilayers and cannot be extracted into solution without first dissolving the bilayer. Given that these molecules are embedded in the bilayer, their conformational state is influenced by an array of weak, competing interactions between the protein and other components of the bilayer. Such complex solvation shapes the conformational energetics of MPs in a manner that is distinct from water-soluble proteins. This divergence is highlighted by the existence of a specialized ensemble of quality control proteins that are specifically devoted to facilitating MP folding and to managing their misfolding in the cell. These quality control proteins constitute an important component of the web of molecular chaperones and other proteins that stabilize the cellular proteome: the so-called “proteostasis network”. Efforts to rationalize how the conformational equilibria associated with the folding and misfolding of MPs influence their interactions with quality control machinery and the proteostasis network are still in their infancy but are relevant to understanding and treating many diseases.
Our understanding of the structure and conformational energetics of MPs lags behind that of water-soluble proteins. The first experimentally determined structure of a MP was generated in 1985 (the photosynthetic reaction center), which was nearly 30 years after completion of the first high resolution structure of a water-soluble protein (myoglobin, 1958).1,2 In that same time frame the first studies demonstrating the reversible folding of polytopic membrane proteins were reported.3−6 The first quantitative studies of the folding energetics of polytopic MPs were not published until the mid-1990s,7,8 long after Anfinsen’s classic early 1960s work demonstrating the reversibility of ribonuclease unfolding.9,10 The first detailed studies of the structure–stability relationships for a MP were published in 1992.11−13 Nevertheless, after a slow start, physicochemical studies of MP folding have rapidly advanced in recent years, revealing many mechanistic insights into the folding of purified MPs in model membranes. With reverence, we refer the reader to some of the excellent previous reviews of MP folding involving isolated proteins, most of which include coverage of topics that we do not endeavor to rereview in this work, in particular the structural basis for MP stability.14−31
An emerging frontier in studies of MP folding is integration of studies of MP folding using purified proteins with studies of the folding of MPs in the context of living cells. There has been a wealth of parallel progress in recent years in these disciplines, which now have much to offer each other. Additional impetus for bridge-building is provided by recent advances in human genomics, which have highlighted numerous relationships between defects in MP folding and human disease that may be addressable using emerging chemical tools. Here, we endeavor to review results from studies of purified MPs that are of particular importance for understanding how MPs fold in the context of living cells. We also examine recent progress in the myriad of studies devoted to identifying the key molecular players for managing MP folding and misfolding in vivo. In particular, we focus on the chaperones and other proteins that comprise the folding quality control system of the endoplasmic reticulum (ER), which serves as the primary site of MP assembly in eukaryotic cells. Finally, we examine how MP misfolding under physiological conditions contributes to numerous diseases and examine emerging chemical biology and medicinal chemistry approaches that directly address defects in MP folding to treat these diseases. It is hoped that this review will stimulate cross-talk between traditionally disparate areas of study, resulting in synergy that results in true “bench to bedside” progress that both illuminates the detailed chemical basis for key life processes and also is of great benefit to humankind.
2. Intrinsic Differences between Membrane Proteins and Water-Soluble Proteins
2.1. The Membrane Environment and the Native Structures of Membrane Proteins
Natively folded MPs adopt conformational states that are partly, or in some cases nearly completely, embedded within the membrane. With a handful of important exceptions (such as the cyclooxygenases and caveolins),32−34 the vast majority of mammalian MPs have at least one segment that spans the bilayer. These transmembrane (TM) segments typically consist of an α helix with a hydrophobic stretch of 18–28 amino acids flanked by polar residues.35−37 β Barrel MPs composed of antiparallel transmembrane beta sheets are found in mitochondrial and prokaryotic outer membranes. For both classes of MPs, polar side chains near the edge of TM domains interact with lipid head groups and water molecules in a manner that helps stabilize their native topological orientation in the membrane.38,39 Energetic barriers associated with the translocation of these and other polar groups across membrane is likely to prohibit many topological rearrangements.40 With some notable exceptions (see section 3.7), it seems likely that few helical MPs are capable of efficient spontaneous insertion across the bilayer. This energetic constraint restricts the number of topological orientations that are kinetically accessible to integral MPs.41 In most cases, the native topology (or something close to it) must be established cotranslationally with the assistance of the Sec translocon protein complex (see section 4.1) or a related membrane-integrative system such as Tim/Tom mitochondrial membrane translocases. The Sec translocon effectively circumvents the insertion barrier by providing hydrophobic segments access to the membrane core through a lateral opening within its transbilayer pore.42 This represents one of enabling strategies developed by nature to allow polypeptides to fold to a functional state within lipid bilayers on biologically relevant time scales.
After MP translation, the physicochemical properties of the lipid bilayer enforce constraints on the conformational equilibria of integral MPs. For instance, the hydrophobic nature of the membrane core imposes a steep energetic penalty associated with the solvation of unpaired hydrogen bond donors and acceptors.14,15 This essentially forces the backbone of TM segments to adopt a regular secondary structure within the bilayer so that the hydrogen bonding potential of the backbone amide protons and carbonyl oxygens are satisfied by intramolecular interactions. This requirement is satisfied both for MPs with α-helical transmembrane segments and those that form transmembrane beta barrels. As a result of these energetic constraints, the central portions of naturally evolved transmembrane segments are enriched in the hydrophobic amino acids, and polar residues are typically rare within the membrane core (Figure 1). This sequence bias is particularly pronounced on the lipid-exposed surface of the TM domain.43,44 Nevertheless, in light of the presence of backbone carbonyl oxygens, amide hydrogens, and the occasional polar side chain, TM segments are actually quite rich in hydrogen bonding groups. This abundance of polar groups provides ample opportunities to form intramolecular hydrogen bonds in a manner that provides some structural plasticity.45,46 Furthermore, though the membrane core is hydrophobic, the dynamics and imperfect packing of lipid acyl chains allows water to penetrate the bilayer to a surprisingly high degree.47,48 Bound water molecules are often observed in the TM domains of MP crystal structures.49 Biophysical experiments such as pulse radiolysis and FT-IR spectroscopy have also provided confirmation that water molecules are often associated with TM domains under native-like conditions.50 The presence of water within the bilayer helps explain a number of anomalous structural features that have been observed within the TM domains of some MPs. For example, aquaporins and some other membrane proteins have “re-entrant” strands of residues that extend into one face of the membrane and return to the same face without spanning the bilayer. The residues in these extended membrane-buried segments appear to have unsatisfied backbone hydrogen bonding potentials and possibly interact with water molecules.51−53 There are also TM helices in polytopic membrane proteins that are surprisingly polar.54,55 Access of water to the membrane may also help to explain the observation that the kinetic barriers associated with rearrangement of membrane-buried hydrogen bonds in flexible MPs can be surprisingly low.46 Even without buried water, TM helices are often kinked due to proline residues and/or native tertiary contacts.46,56,57 Some MPs also feature helices bearing a pi bulge or helical break.52,58 Certain MPs also feature sizable gaps (fenestrations) within their TM domains connecting the membrane phase to polar cavities within the protein core.59−61 Together, these observations suggest the surprising conformational diversity of MPs may arise partly from the appreciable hydration of proteins within membranes. As the pace of MP structure determination continues to accelerate (http://blanco.biomol.uci.edu/mpstruc/), we likely will continue to find even more exciting twists and turns that underlie their biochemical functions.
Figure 1.
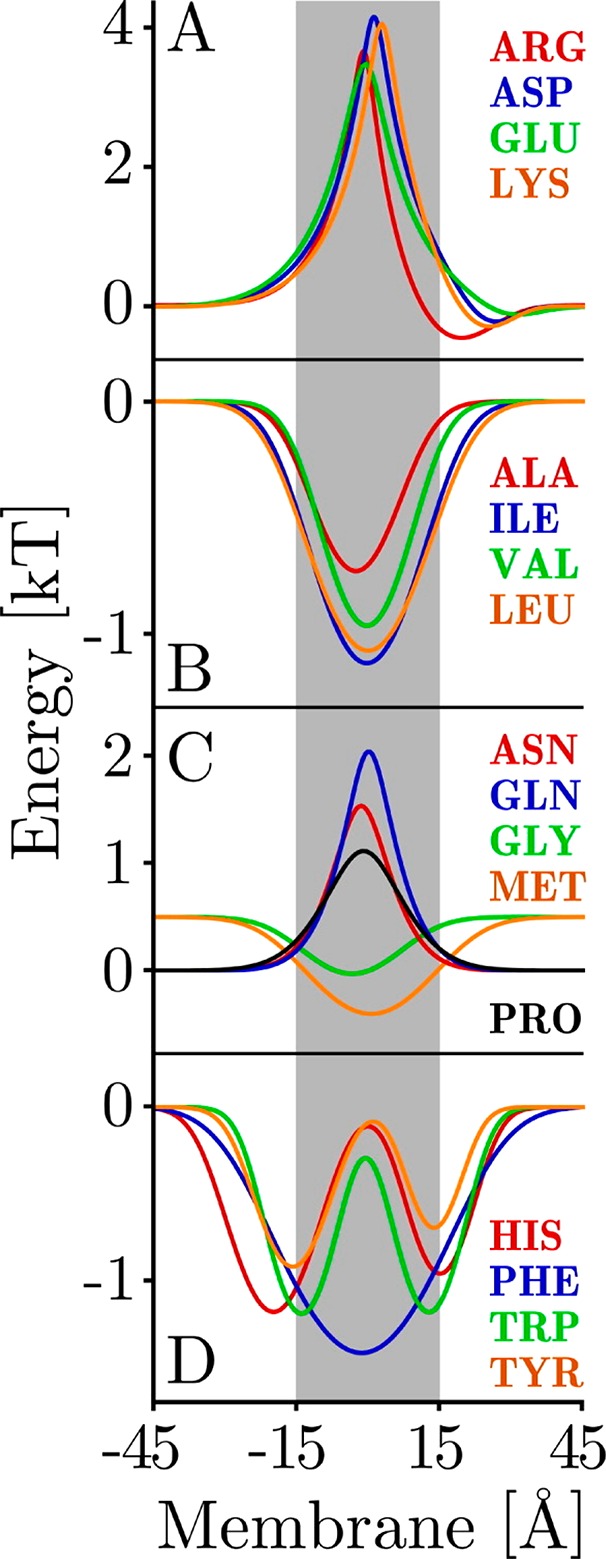
Depth-dependent statistical distributions of amino acids within transmembrane domains. Experimental depth dependent compositional biases within transmembrane domains were used to train a probabilistic potential energy function (A) charged, (B) hydrophobic, (C) polar, and (D) aromatic amino acid. Lower energies correspond to higher probabilities of finding the corresponding residue at a given depth. An X-coordinate of 0 Å corresponds to the center of the membrane normal. Reprinted with permission from ref (44). Copyright 2005 John Wiley and Sons.
MPs are often sensitive to the physical properties of their bilayer solvent. Most lipids are roughly cylindrical in shape and are arranged with their long axes orthogonal to the plane of the membrane, which allows them to neatly pack into a two-dimensional sheets. However, in part due to the abundance of unsaturated fatty acids in mesophilic organisms, there is typically a gradient of lateral pressure extending from the bilayer interface into the highly dynamic bilayer core, where the fluidity can approach that of liquid hydrocarbons.62 The lateral pressure exerted by lipid acyl chains has a direct impact on the conformational equilibria of MPs.63−66 Moreover, lateral pressure can be tuned by membrane curvature, which is another factor that influences the conformational energetics of MPs.67 Highly curved membranes are sometimes enriched in lipid packing defects, which can lower the energetic barriers associated with the insertion of proteins across the membrane.68,69 Membrane curvature is, of course, critical for many biological processes.70,71 Surface binding proteins such as caveolins, proteins containing amphipathic helices, and those containing BAR domains can dynamically control membrane curvature. This manipulation of curvature is required for many cellular processes such as vesicle budding, transport, and fusion.72−74 Lateral pressure may also influence the orientation of TM domains. In some cases, TM helices are tilted relative to the bilayer normal, which may occur as a result of a mismatch between the length of the TM domain and the thickness of the membrane (hydrophobic mismatch, Figure 2).75,76 In other cases, it appears that the span of TM segments is asymmetrically adjustable.77 Thus, thickness represents an additional membrane property that may tune the conformational states of integral MPs (Figure 2).
Figure 2.
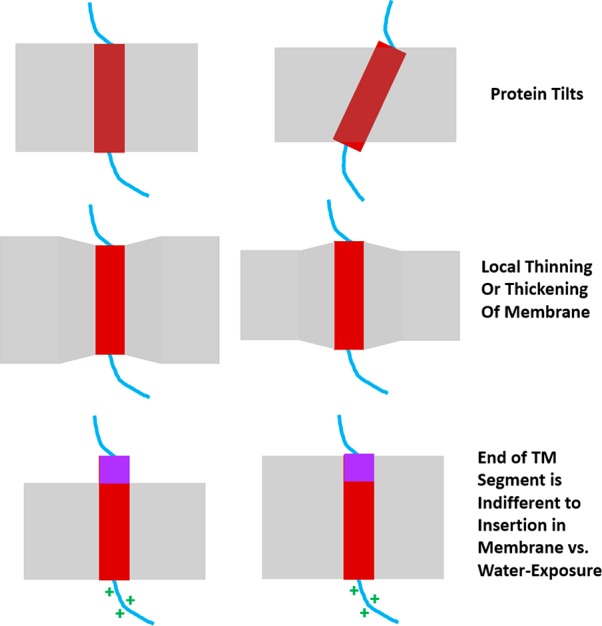
Adaptation of transmembrane domains to variations in bilayer thickness. Cartoons depict ways TM domains adapt to changes in bilayer thickness. A thinning of the membrane may cause TM domains to tilt with respect to the membrane (top). Alternatively, the bilayer may be locally distorted in order to facilitate the solvation of a lengthy TM domain (middle). Thickening of the bilayer may also result in the extension of TM helices out of the bilayer when sites located at the end of the helix have similar preferences for aqueous exposure or membrane burial (bottom).
Native membranes have protein-to-lipid mass ratios (P/L) ranging from 0.25 to 5.78 MPs occupy 20% of the surface area of red blood cells,79 which have a P/L of 1.3. Under nonphysiological conditions in which the concentration of MPs within the bilayer is typically much lower, membrane thickness is largely determined by lipid composition. Saturated acyl chains produce thicker bilayers, whereas unsaturated chains dynamically splay outward in a manner that allows the two leaflets to pack more closely together. In contrast, the rigid, flat surface of cholesterol stabilizes extended conformational states of adjacent phospholipid acyl chains in a manner that typically increases membrane thickness and lipid conformational order.80 There is also evidence to suggest that, in the context of protein-rich environments of cellular membranes, the properties of the MPs themselves influence membrane thickness, which varies from organelle to organelle.81 In some cases, proteins satisfy the energetic strain associated with hydrophobic mismatch of transmembrane segments by altering the packing of their annular lipids in order to change the thickness of the local bilayer (hydrophobic matching, see Figure 2).82 Other MPs may form oligomers to reduce the solvent-accessible surface area of mismatched TM domains.83,84 In many cases, the formation of transmembrane oligomers helps to optimize van der Waals interactions through interhelical packing85,86 and can reduce unfavorable clashes between polar side chains and acyl chains within the membrane core.87 It has been suggested that 60% of all single span plasma membrane proteins form homodimers.88 Together, the physical constraints imposed by the bilayer significantly restrict the conformational space that is accessible to integral MPs. Indeed, Bowie and co-workers have argued that the total number of possible folds that are accessible to MPs is limited relative to soluble proteins.89
A distinctive trait of plasma, endosome, and lysosome membranes relative to those of the nucleus, mitochondria, endoplasmic reticulum, and Golgi is the presence of higher concentrations of both cholesterol and sphingolipids in the former (Figure 3).7890−92 Cholesterol has an ordering effect on the chains of neighboring lipids even when the bilayer remains in the disordered phase.93,94 One of the ways that sphingolipids differ from glycerolipids is that the sphingosine backbone includes a potential hydrogen bond-donating moiety that may lead to enhanced lipid–lipid and interfacial water–lipid interactions.95,96 Whether the presence of high levels of cholesterol and sphingolipids in membranes alter the energetics of membrane protein folding and stability is largely unexplored, but seems likely. In this regard, an interesting preliminary observations is that high levels of cholesterol in the membrane tend to promote alignment of otherwise tilted transmembrane helices with the bilayer normal.97 It has also been reported that plasma MPs tend to have longer TM segments than resident MPs of the Golgi and endoplasmic reticulum.37
Figure 3.
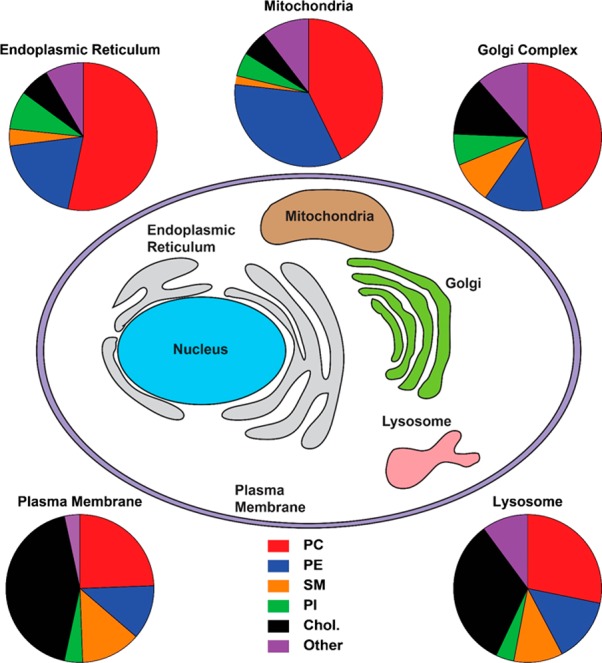
Lipid compositions of membranes from various organelles in mammalian cells. Lipid compositions of organelle membranes from rat liver cells (derived from multiple sources) are shown. Abbreviations: PC: phosphatidylcholine, PE: phosphatidylethanolamine, SM: sphingomyelin, PI: phosphatidylinositol, Chol: cholesterol. The data on lipid composition is from ref (92).
The original fluid mosaic model98 continues to serve as a point of reference for our understanding of the organization and dynamics of biological membranes. Nevertheless, the simplistic assumptions of this model have been subject to a variety of clarifications, revisions, and updates over the years.99−101 The controversy associated with “lipid rafts” is of particular note with respect to MP folding.
It has long been appreciated that bilayers are capable of forming a liquid-ordered phase (Lo) at certain temperatures and lipid compositions (especially those rich in cholesterol and sphingolipids). Lipids within Lo phase membranes exhibit conformational, axial rotational, and diffusional order that is somewhere between those within dynamic liquid-disordered (Ld, also referred to as the fluid, liquid crystalline, or Lα phase) and highly ordered gel phase membranes.102−104 Various cholesterol and sphingolipid-rich membranes such as those of myelin, caveolae, and the apical surfaces of some epithelial cells may sometimes have characteristics akin to Lo phase membranes.105−108 Like synthetic Lo membranes, these natural membranes are also resistant to detergent solubilization.109−111 These considerations have contributed to the hypothesis that the plasma membranes of higher organisms contain phase-separated “lipid rafts” that exhibit Lo-like phase behavior.,102112−115
Macroscopic Ld and Lo phases are capable of coexisting within a single lipid vesicle in a manner that can be visualized by confocal fluorescence microscopy.116,117 However, until very recently118 there was little evidence to suggest that the intact membranes of living cells were capable of undergoing robust phase separation. For this reason, the very existence of lipid rafts and their potential biological relevance have proven controversial.119,120
The existence of lipid rafts is typically debated in the context of eukaryotic plasma membranes. Direct observation of coexisting phases in the plasma membranes of living cells has proven extremely challenging. However, macroscopic phase separation does occur upon lowering the temperature (to well below physiological temperature) in giant plasma membrane-derived vesicles” (GPMVs) that have been blebbed from eukaryotic plasma membranes.113,117,120 Many different membrane proteins, especially those that are palmitoylated, appear to preferentially partition into the Lo phase under these conditions.121−124 Others are enriched at the interface of the Lo and Ld phases.125−128 However, how these observations pertain to the behavior of intact bilayers under physiological conditions remains the subject of active investigation
There is considerable evidence to suggest that small (<50 nm) ordered domains are capable of transiently forming in the context of otherwise disordered plasma membranes. Over the past 15 years, a classic series of studies from the Veatch, Keller, and Baird laboratories have offered a satisfying explanation for the dynamic coexistence of less-ordered and more-ordered phases within plasma membranes.129−132 This explanation is based on appreciation of the fact that the phase diagrams of multicomponent systems (in this case for membrane bilayers) sometimes have compositions for which there is a critical temperature, Tc.133 Critical points, which are a general feature of multidimensional phase diagrams, occur at the limit of the two-phase region in which the Lo and Ld phases coexist. At a critical point, the compositions and populations of both phases are equal and lipids randomly fluctuate between phases in a manner mediated by thermal energy (kBT). Below Tc (at fixed membrane composition) the membrane demixes into two stable macroscopic phases. Above Tc, the phases appear to mix into a single phase. Nevertheless, ensembles of lipids are still capable of transiently sampling ordered phases above Tc through “critical fluctuations.” As the temperature is increased above Tc, the size of these fluctuations (their “correlation length”) decreases. The shapes of these transient ordered domains are irregular as a result of the reduced line tension between phases. Notably, the physical basis for this framework can be modeled reasonably well using a two-dimensional Ising model, which provides a mechanistic framework that can be used to rationalize membrane organization.134
Remarkably, studies of GPMVs from mammalian plasma membranes exhibit critical behavior at reduced temperatures, with Tc values on the order of 10–20° below 37 °C.129,131,132 This phenomenon has been documented in GPMVs derived from multiple cell types through a series of painstaking and rigorous results.129−132 These collective observations suggest that small portions (<50 nm) of the plasma membrane are likely to transiently sample ordered phases at physiological temperature. These phases likely have only slight differences in composition and order relative to the comingled disordered phase. This framework offers a very satisfying resolution to the “lipid raft” controversy by providing a generalized physical explanation for the extensive number of nuanced biochemical and biophysical observations suggesting biological membranes do not behave as ideal liquid phase assemblies. Plasma membranes do contain more-highly disordered domains in coexistence with more fluid microdomains. However, not only are these domains transient—mere fluctuations!—but the differences in the lipid compositions and order between the less and more highly ordered domains are likely to be quite modest.135 Indeed, imaging mass spectrometry studies have documented that cholesterol is distributed uniformly across intact mammalian plasma membranes,136,137 though sphingomyelin appears to form small clusters.138,139
The biological implications of critical behavior within biological membranes is just beginning to be explored. For example, critical behavior provides a quantitative framework that accounts for how certain receptors undergo spatial clustering and oligomerization.134 It is interesting to ponder how the dynamic jostling of MPs between percolating more- and less-ordered phases in the plasma membrane might alter MP stability and the energetics of oligomerization. This is clearly an avenue for future exploration. At the same time, we note that appreciation of critical behavior in biological membrane will ultimately need to be melded with what is understood about of how cytoskeletal attachment points, lipid asymmetry, and MP crowding alter membrane-based phenomena, including MP folding and stability.
2.2. Tolerance versus Adaptation to Varying Membrane Environment
Longstanding interest in how lipids interact with MPs has heightened in recent years as advances in both computational and experimental structural biology (especially mass spectrometry) have provided new details on the nature of these interactions.140,141 Moreover, biochemical and biophysical studies have revealed some of the ways by which MP function is regulated by lipids that act as allosteric ligands.142−146 For instance, phosphatidylinositol-4,5-diphosphate (PIP2) and polyunsaturated fatty acids (PUFAs) regulate the function of many different MPs through specific binding interactions.147,148 The functions of MPs can also be tuned by variations in bulk lipid composition.149,150
Stoichiometric complexes between certain proteins and lipids have been found to promote the stability and organization of native MP complexes.151−155 The formation of correct membrane topology and folding of certain proteins sometimes requires specific protein–lipid interactions.156−159 MP folding and stability also depends on both bulk membrane composition and the physical properties of the bilayer.63,160−164 Disruption of lipid-MP interactions is likely responsible for some diseases.165
In addition to adapting to the chemical and physical properties of the membrane environment, native MP conformations must, to some extent, also have evolved to tolerate variations in membrane lipid composition.166,167 Eukaryotic MPs must remain folded and functional in the face of fairly dramatic changes in lipid composition that occur as proteins are shuttled from the ER to the Golgi and beyond. Each of these organelles has its own distinctive lipid composition (Figure 3),91,92 which likely plays a role in the tuning of the structure and function of resident MPs. It has been empirically shown that the subset of MPs that reside within these organelles exhibit distinct distributions of TM domain lengths, which implies these proteins may have been tailored to fold and function within distinct membranes.37 Nevertheless, there are several lines of evidence to suggest their native structures typically persist across divergent membranes. For instance, the lipids of archaebacteria have exotic structures compared to eubacteria and eukaryotes,168 yet the native structures MPs from archaea appear to be similar to those of the homologous proteins from eubacteria and eukaryotes (Figure 4).166,169 Furthermore, certain lipid biosynthetic pathways have been completely knocked out in E. coli to dramatically alter membrane lipid composition and yet result in only limited influence on cellular viability.170,171 This implies that most of the proteins that reside within these reformatted membranes retain function. Indeed, it has been shown that there is no single type of phospholipid that E. coli cannot survive without.171,172 Given this apparent tolerance, it is perhaps unsurprising that a great many MPs retain their native fold within the artificial environment of detergent micelles, which have very different physical chemical properties than bona fide membranes. Indeed, some MPs continue to fold and function even when solubilized by amphipathic polymers, which shield the hydrophobic portions of the molecule from water.31,173,174
Figure 4.
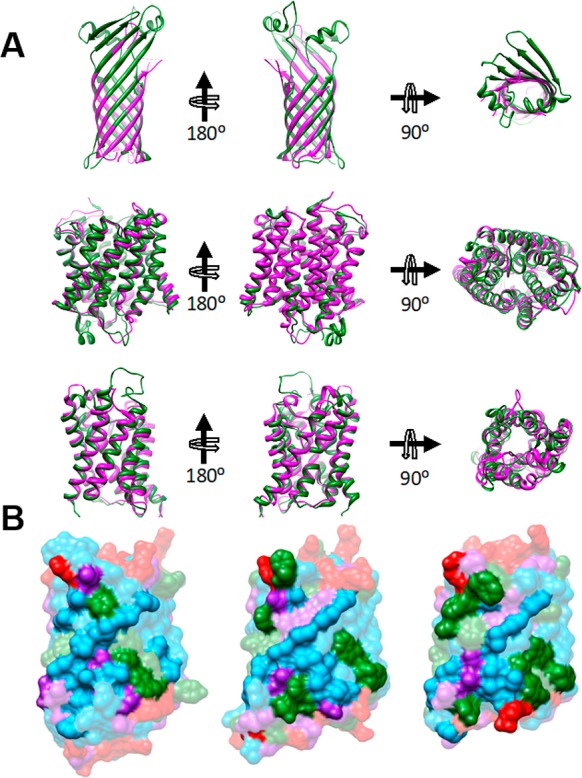
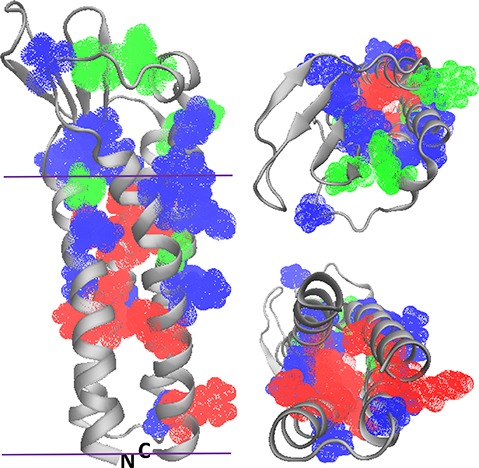
Comparison of MP structures from the disparate domains of life. (A) Superpositions of structures of thermophilic archaeal MPs on those of mesophilic counterparts reveal high similarity. Superposition of porins TtoA from Thermus thermophilus (green, PDB ID: 3DZM) and OmpA from E. coli (magenta, PDB ID: 1QJP). (Middle panel) Superposition of ammonium transporters Amt-1 from the archaeal hyperthermophile A. fulgidus (green, PDB ID: 2B2H) and AmtB from E. coli (magenta, PDB ID: 1U77). (Bottom panel) Superposition of aquaporin from A. fulgidus (green, PDB ID: 3NE2) with AqpZ from E. coli (magenta, PDB ID: 1RC2). View onto the membrane surface is from the periplasm/extracellular space. (B) The lipid-contact faces of aquaporins from three domains of life exhibit common features. Shown are aquaporins from a hyperthermophilic archaebacterium, A. fulgidus (PDB ID: 3NE2, left), E. coli (PDB ID: 1RC2, center), and O. airies (sheep) (PDB ID: 3M9I, right). Residues are colored as follows; red: polar residues; blue: large hydrophobic; green: aromatic/His; purple: small (Gly, Ala, Ser, Cys). Figure used with permission from ref (172). Copyright 2015 ACS.
All together, the growing body of data regarding how MPs interact with bilayers and specific lipids indicates that MPs have evolved in concert with the membranes in which they reside to satisfy the imperative of being able to robustly fold and function even in membranes of varying compositions, while in many cases also being appropriately regulated by specific lipid binding and varying membrane properties. When considering this dichotomy from the perspective of MP folding, it should not be surprising that there are examples where specific lipid interactions are required for folding.156−159
3. Kinetics and Thermodynamics of Membrane Protein Folding
3.1. Conformational Stability and the Physiologically Relevant Unfolded States of Integral Membrane Proteins
Proteins sample a continuum of conformational states regardless of whether they reside in water or in a lipid bilayer. The relative abundance of molecules that adopt a given conformational state is primarily dictated by the kinetic and/or thermodynamic barriers that separate this state from competing conformations.175 The magnitude of these energetic barriers is largely determined by the primary structure of the protein, how the molecule is solvated, and the abundance of cofactors that bind and stabilize certain conformations. Only a small subset of compactly folded, energetically accessible conformational states are capable of mediating protein function. Thus, the fraction of protein that is functional may largely depend on the free energy difference between the native ensemble and the lowest energy nonfunctional conformational state(s) populated under native conditions, which we will refer to hereafter as the physiological unfolded state (Figure 5). Physiological unfolded states are likely akin to transient partially unfolded states of water-soluble proteins that lack ordered structure in one or more subdomains.176,177 However, far less is known about the physiological unfolded states of helical integral MPs, or how they exchange with the native state. This uncertainty constitutes a central caveat to ongoing discussions of MP folding and stability. Nevertheless, some educated guesses about the properties of the physiological unfolded states of α-helical MPs can be made in light of the physicochemical properties of these proteins and of the membrane itself. First, given their sheer hydrophobicity, polytopic α-helical MPs are likely to remain confined within the membrane throughout most of their lifespan, regardless of their conformational state. Given the low dielectric constant within this environment,14,40 hydrophobic TM segments are likely to retain their helical secondary structure, even under conditions in which the native tertiary structure is lost. Thus, the physiological unfolded states likely constitute bundles of weakly interacting helices within the membrane; an ensemble of structures akin to the first stage of the classic 2-stage model for MP folding originally suggested by Popot and Engelman.14
Figure 5.
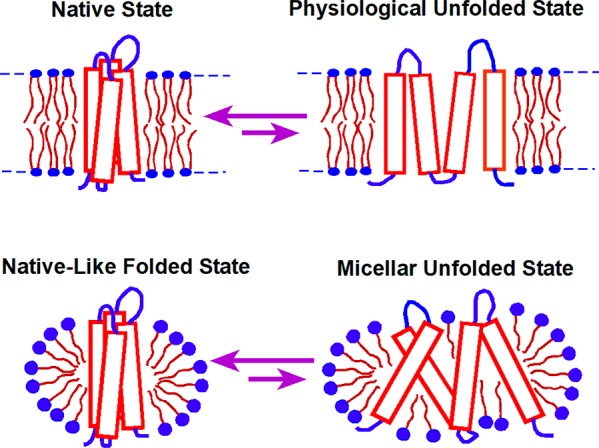
Folding equilibrium for a MP in lipid bilayers versus in detergent micelles. The native conformation is often the most favorable conformational state for MPs in both bilayers and micelles. However, the structural properties of the accessible denatured states, the energies between folded and unfolded states, and the magnitude of the energetic barriers that separate them from the native state may often differ in bilayers and in micelles.
Though technical barriers have largely prevented characterization of physiologically relevant unfolded states in their native environment, a handful of studies have provided clues about the properties of non-native states that are energetically accessible in vitro.3,178−182 We focus on two recent examples. Solution nuclear magnetic resonance (NMR) studies of peripheral myelin protein 22 (PMP22) in micelles have revealed that the folded form of this protein is in equilibrium with a conformational state in which the N-terminal TM segment is fully dissociated from the other three TM segments, the latter of which interact in a molten globule-like manner (Figure 6).182 Furthermore, this conformational state is promoted by the pathogenic L16P mutation within its first TM domain. In a second example, NMR studies of the KCNQ1 channel voltage sensor domain have also revealed that mutations known to promote cellular mistrafficking of the full length channel also perturb how the TM helices interact within LMPG micelles.183 None of the 47 mutations examined in that study caused the protein to dissociate from the micelle or to transition to a random coil state. However, the NMR spectra of mistrafficked KCNQ1 variants exhibited peak broadening that is consistent with molten globular structure, a well-known folding intermediate state for many water-soluble proteins (see section 5.4.1).183,184 The extent to which these conformational transitions in detergent micelles relate to physiological unfolded states is not yet well-established. Nevertheless, the observed effects of these destabilizing mutations in vitro are intriguing. In the following, we summarize current knowledge regarding the conformational equilibria of integral MPs, with how folding/unfolding transitions relate to the molecular basis of MP misfolding and disease.
Figure 6.
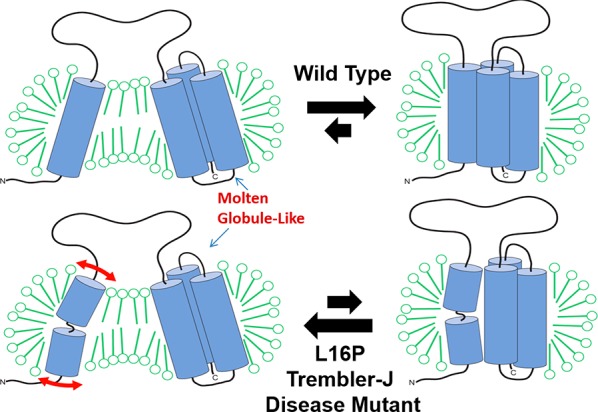
Folding equilibrium for WT and L16P mutant PMP22. This is determined using NMR and other methods under conditions where PMP22 is solubilized in tetradecylphosphocholine micelles at 25 °C. The L16P disease mutation site is located in the first TM helix with the proline substitution resulting in the flexible hinge illustrated in the lower left panel. We suggest that the “partially unfolded state” depicted on the left may actually be similar to the true physiological unfolded state. Further unfolding is restrained by the short loops connect TM2 to TM3 and TM3 to TM4. Reprinted with permission from ref (182). Copyright 2011 Cell Press.
3.2. Native Membrane Protein Structures Are Thermodynamically Stable
Christian Anfinsen’s landmark investigations into the folding of RNase A established that the native conformations of water-soluble proteins tend to reside within free energy minima.9,10 Like all conformational transitions, it stands to reason that MP folding should also serve to reduce the free energy of the system. However, MPs must navigate their conformational energy landscapes within a much more complex solvent, which is likely to alter the kinetic barriers to folding. For this reason, early investigations of MP folding were mindful of the possibility that the functional structures of integral MPs could be kinetically accessible yet thermodynamically unstable. Pioneering investigations of the α-helical MP bacteriorhodopsin (bR) demonstrated the native fold could be regenerated from the SDS-denatured state upon addition of cholate and/or soy bean lipids.3 This key observation echoed Anfinsen’s finding that the primary structure contains all of the information needed for the protein to achieve its functional structure under native-like conditions. This conclusion was also supported by subsequent observations that natively folded bR could be regenerated from denatured proteolytic bR fragments.6 Since these investigations there have been numerous studies of MP folding in mixed micelles, bicelles, and synthetic lipid bilayers185,186 showing that a wide array of β-barrel and α-helical MPs are capable of reclaiming their native structures regardless of whether the proteins are first denatured using organic solvent,187 chaotropes,164,188−192 anionic detergents,8,46,193−195 steric trapping,196,197 or mechanical force.198−200 Thus, there is now ample evidence to suggest the native conformations of integral MPs are thermodynamically stable relative to an array of non-native denatured states including the “physiological unfolded state” described in section 3.1 and Figure 5. It is also clear that some denatured integral MPs can find their way back to the native conformation on experimentally accessible time scales. Nevertheless, even when a MP reaches its thermodynamically favored functional conformation, it is not always clear that such folded states are at equilibrium with physiological unfolded states, as is discussed further below.
Experimental investigations of water-soluble proteins most often utilize concentrated urea or guanidinium to induce global unfolding. Although certain proteins retain residual structure under these conditions,201 the denatured ensemble of most water-soluble proteins is dominated by random coil structure. This lack of well-defined intramolecular interactions provides a useful reference state in investigations of the contributions of intramolecular interactions to conformational stability. However, many α-helical MPs cannot generally be unfolded in this way,202 and even those that exhibit sensitivity to urea typically retain helical secondary structure within a diverse array of commonly employed membranes and membrane mimetics (Figure 7).164,190−192 In contrast, β-barrel MPs globally unfold and partition into the aqueous phase in concentrated urea solutions,188 a transition that can be rendered fully reversible. Under controlled conditions,189 dilution of protein/denaturant solutions in the presence of lipid vesicles results in the spontaneous transfer of denatured β-barrel proteins into the bilayer in a manner that is reversibly coupled to folding. The energetics of these folding transitions are highly sensitive to the experimental conditions (especially the properties of the lipid bilayer) and vary considerably among members of the β-barrel family.23,24,186,188,203 Nevertheless, in many cases β-barrel folding appears to be extremely favorable under ambient conditions. Indeed, the strong driving force associated with this reaction appears to provide some of the energy needed for the sorting of proteins destined for the outer membrane in the periplasm.204 Ongoing studies of the mechanisms by which molecular chaperones assist in the folding and assembly of β-barrels offer the potential to provide additional insights into the energetics of bacterial proteostasis systems.205,206 Beyond the biological relevance of these measurements, the nature of this transition has also provided unique opportunities to evaluate the transfer free energies of amino acid side chains from the aqueous phase into the lipid bilayer.189,207−209 Together these special properties of β-barrels have made them an extremely useful system for investigating MP insertion and folding.
Figure 7.
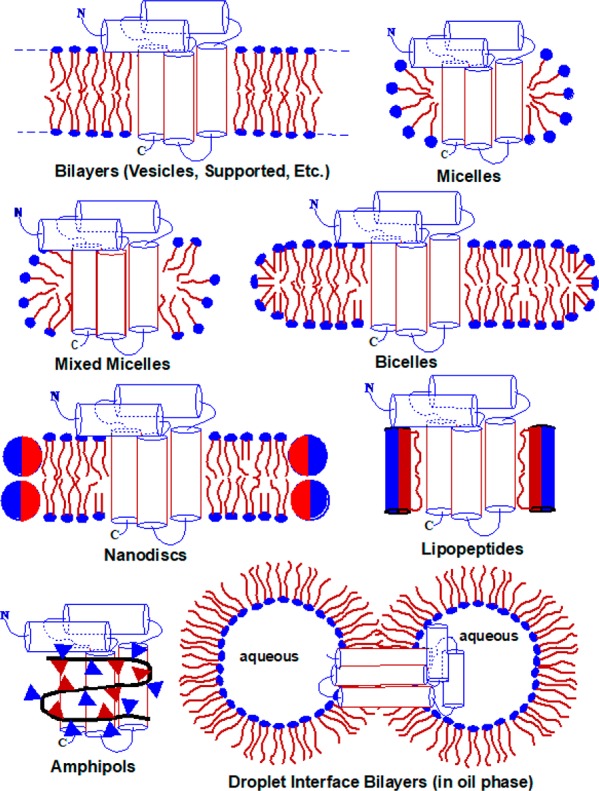
Classes of model membrane used in studies of purified MPs. Not illustrated here are “lipodisqs”, which resemble nanodiscs except that a synthetic amphipathic polymer is used to stabilize the edge of the bilayered disc instead of an amphipathic protein.
In contrast to β-barrels, α-helical MPs typically cannot be driven from membranes into the aqueous phase without excessive aggregation (with exceptions described in section 3.7). In the cell, most α-helical MPs are cotranslationally inserted into the membrane, obviating the need for them to transiently reside in the aqueous phase. Furthermore, the removal of helical proteins from the membrane under physiological conditions requires hydrolysis of hundreds of ATP molecules210 and is typically coupled to proteolysis or to the formation of ordered aggregates known as aggresomes.211 Thus, α-helical MPs are unlikely to sample fully hydrated states under physiological conditions. Accordingly, it is likely that the physiological folding trajectories within the membrane primarily involve transitions between non-native helical intermediate states. Experimental efforts to assess the conformational stability of these proteins have most often employed anionic detergents (typically SDS) to induce denaturation in the context of mixed micelle solvents.3,5,8 Helical MPs typically retain secondary structure but usually lose their native tertiary structure in the SDS-denatured state.8,212,213 When the concentration of denaturant is expressed in terms of detergent mole fraction,8 equilibrium unfolding transitions typically exhibit the markings of a cooperative two-state unfolding reaction.8,194,195,214−216 In the context of these conformational transitions, mutagenesis studies have revealed that, despite vast differences in solvation, the stabilization afforded by native hydrogen bonds and packing interactions are on par with those of water-soluble proteins.46,203,217 The interpretation of these measurements is certainly complicated by the presence of residual structure in the denatured state. The loss of native contacts upon denaturation is likely coupled to the formation of a spectrum of weaker non-native interactions in the denatured ensemble. Nevertheless, this likely parallels the physiological reaction coordinate in which the native conformation must compete with a spectrum of alternative arrangements of weakly interacting helices in the physiological unfolded state. Overall, these observations suggest that the effective strength of native interactions are likely to be considerably lower than would expected based on the dielectric constant within the bilayer.14,40 In addition to the presence of modest levels of water in the membrane core, the protein itself offers hydrogen bonding groups that are capable of competing with native interactions.45 A better understanding of the structural properties of the physiological unfolded state within biological membranes is needed in order to clarify the true stability of the native fold relative to physiological unfolded states.
3.3. Native Membrane Protein Structures Can Be Kinetically Stable
Cells are nonequilibrium systems, and kinetic control of chemical reactions is a mechanistic pillar of biomolecular regulation. With regard to protein folding and assembly in the cell, many chaperone-assisted folding and degradation networks appear to be under kinetic control.218−221 Thus, in some cases slow unfolding (kinetic stability) may be an essential property of long-lived MPs. Pioneering investigations into the relaxation kinetics of bR in mixed micelles as a function of the mole fraction of the denaturing detergent SDS (XSDS) was interpreted to suggest the half-life of bR unfolding is on the order of thousands of years in lipid bicelles.213 Though the unfolding of bR is undoubtedly slow, this estimation required a lengthy extrapolation from a condition in which the protein resides within an SDS-rich mixed micelle to a condition in which the protein resides within a DMPC-rich, SDS-free bicelle. Linear extrapolations of rate constants and ΔG values across wide ranges of detergent mole fractions have since proven unreliable.196,197 Despite this caveat, a number of subsequent observations have suggested that at least some MPs may unfold slowly. Bowie and co-workers showed that the half-life for dissociation of the subunits of diacylglycerol kinase in beta-octylglucoside micelles (considered mildly destabilizing relative to more ideal model membranes) is on the order of 2 weeks.222 A recent investigation of the E. coli intramembrane protease GlpG revealed that unfolding requires weeks in lipid bicelles as monitored by steric trapping,210 a timespan in reasonable agreement with its extrapolated rate constants for SDS-mediated denaturation.194 Indeed, the application of magnetic tweezers to GlpG also revealed the native state resides within a steep energy well.223 It remains unclear whether high kinetic stability is a common property of integral MPs or whether this is a special trait of these particular proteins, each of which also exhibits considerable thermodynamic stability.8,194,196,197 DsbB exhibits modest thermodynamic stability and folds and unfolds relatively rapidly.224,225 Nevertheless, the metastable human protein PMP22, which is only marginally stable in DPC micelles, requires hours for relaxation under this condition.195 The notion that some helical MPs fold into thermodynamically preferred native states that are then effectively kinetically trapped potentially has wide-ranging implications for MP folding in the cell. Considerations regarding the kinetics of conformational exchange may also be a relevant factor in efforts to develop small molecules that correct the folding and stability of disease-linked MPs, as is discussed further below.
Could kinetic entrapment of the native state of some MPs be biochemically tunable? Interestingly, it has been demonstrated that the binding of retinal to the apoform of bR appears to decrease its rate of unfolding by over 10 orders of magnitude.226 The physical basis for this effect suggests the binding of small molecules may potentially play a general role in the tuning of the kinetic and/or thermodynamic stability of integral MPs.227−232 Because small molecules tend to selectively bind to natively folded proteins, binding should universally decrease the rate of unfolding and increase thermodynamic stability in a manner related to the binding affinity and ligand concentration, provided ligand dissociation occurs prior to formation of the transition state for unfolding (Figure 8). Indeed, many G protein-coupled receptors (GPCRs) are known to bind agonists and antagonists with nanomolar to picomolar affinity, resulting in increased protein stability.233,234 Specific interactions of lipids with proteins also frequently appears to enhance the stability of MP oligomers.151 Thus, it is quite plausible that MPs rely on specific lipid and/or physiological small molecule interactions to tune their relaxation kinetics in a manner that alters their cellular trafficking and turnover. In many cases, the extent to which drug binding influences the unfolding kinetics of MPs may also be relevant to their mechanism of drug action, as is discussed further below (see section 6.2). It is also noteworthy that, in some cases, metabolite binding to certain MPs appear to destabilize the folded state, leading to regulated degradation.228,235,236 Additional investigations into the linkage between ligand binding, kinetic stability, and the cellular proteostasis of integral MPs are needed.
Figure 8.
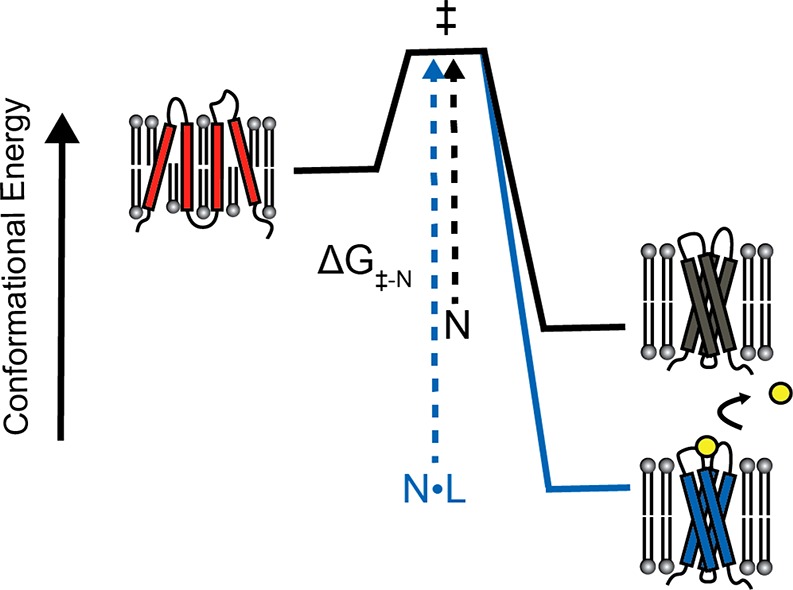
Effect of ligand binding on the kinetic stability of integral membrane proteins. If the native binding pocket is disrupted prior to the rate limiting step for unfolding, then excess ligand will selectively stabilize the native conformation (N) relative to the transition state for unfolding (‡) and the denatured state (red). In this case, the rate of unfolding and the fraction of unfolded protein at equilibrium will be decreased in the presence of ligand.
3.4. Membrane Protein Folding Kinetics
Anfinsen’s formative experiments provided a framework for understanding how proteins select their functional structures from a vast sea of competing non-native conformations. Nevertheless, Leventhal subsequently noted that this thermodynamic perspective did not provide an obvious explanation for the fact that proteins navigate this immense conformational space within remarkably short time scales.237 Considerable efforts spanning decades sought to elucidate the nature of the kinetic intermediates involved in the rapid folding of water-soluble proteins. These investigations revealed considerable heterogeneity in the pathways by which proteins achieve their native secondary, tertiary, and quaternary structures. Many proteins appear to fold through a discrete set of structurally defined intermediates,238,239 while others appear to fold through an array of parallel pathways.240 Despite this mechanistic heterogeneity, the fact that certain structural intermediates seem to form more readily than others suggests that proteins solve this kinetic dilemma through a biased search: the formation of early intermediate structures (or a folding core) dramatically reduces the accessible search space for subsequent transitions. Though considerably less is known about MP folding kinetics, it is clear that even the physiological unfolded state of MPs tends to retain secondary structure within the bilayer in a manner that should severely constrict their conformational search. Accordingly, the topological and secondary structural constraints imposed by the bilayer may vastly simplify the MP folding problem.241 Nevertheless, technical challenges have plagued efforts both to measure the rate of protein folding within the membrane and to elucidate the factors that influence the kinetics of this process. It is therefore unclear whether the kinetic mechanisms that govern MP folding reactions parallel those of soluble proteins.
Pioneering investigations into MP folding kinetics focused on the folding of SDS-denatured bR in bicelles (see Figure 7). Spectroscopic investigations of this process revealed that, much like the folding of soluble proteins, the folding of bR occurs through a series of transient structural intermediates.7 Observations of this process using multiple spectroscopic tools suggested early intermediates involve the formation of tertiary contacts while the binding of the retinal ligand and the extension of the native helices occur later.7,242,243 Nevertheless, these investigations were insufficient to reveal the nature of the structural transitions that limit the rate of folding. Efforts to probe the structural properties of the transition state for BR folding under conditions in which folding occurs through a single phase were initially probed using phi-value analysis.244 Phi-value analysis is applied to proteins in the form of a mutagenic approach to identify native tertiary contacts that are formed within the transition state.245 However, interpretation of these kinetic measurements was complicated by the fact that variations in the concentrations of lipids and detergents obscured the influence of mutations on the rate of bR folding under these conditions.246 Under more controlled conditions, a subsequent analysis of an array of bR mutants distributed throughout its three-dimensional structure failed to identify any native tertiary contacts that appreciably limit the rate of folding.247 The apparent absence of a folding core is interesting considering that the kinetics of water-soluble protein folding is typically rate-limited by the formation of sequence-distant tertiary contacts.248 Based on these results, it was proposed that bR folding is rate-limited by a topological search, in which preformed TM helices sample an array of interhelical contacts.247 This interpretation was supported by the recent finding that bR folding can be accelerated by simply reducing the size of the bicelle, which is likely to reduce the degrees of freedom in the denatured ensemble.249 Thus, it appears the energetic barrier to bR folding is rooted in conformational entropy, at least in bicelles. These investigations paint a picture of the conformational energy landscape of bR that resembles the hypothetical champagne glass-shaped landscape originally described by Dill and Chan (Figure 9);175 the protein must explore a variety of near-isoenergetic orientations of preformed helices before eventually stumbling upon the native topology. Nevertheless, given the artificial nature of the micellar/bicellar solvent used for these studies, it is uncertain whether these findings can be fully extrapolated to the mechanism of bR folding within natural membranes, much less to other polytopic MPs.
Figure 9.
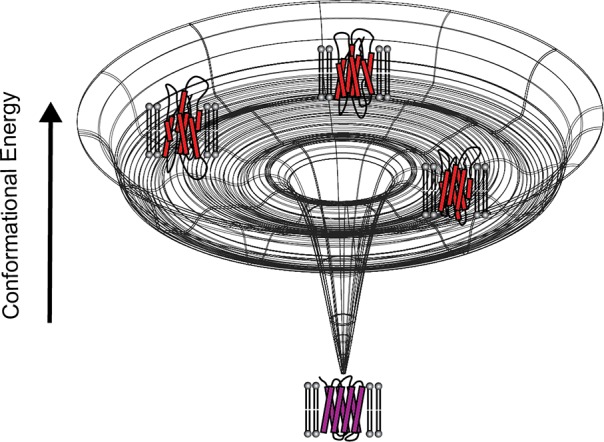
Hypothetical morphology of the conformational energy landscape for bR in DMPC/CHAPSO/SDS bicelles. This cartoon depicts a hypothetical energy landscape that describe the conformational energetics of bR in bicelles. The upper rim of the conformational energy landscape represents the random coil state, which is unlikely to be sampled within membranous environments. Instead, the TM segments are likely to persist in an ensemble of helical bundles within the denatured state, which is represented by the secondary basin of the energy landscape. To find the native conformation, helical TM segments must explore various topological configuration until the native topology is achieved and folding can proceed downhill. It is emphasized that, for many membrane proteins, the folding funnel may be much more complicated than as proposed here for the well-characterized case of BR.
Phi-value analysis has been employed to evaluate the nature of the transition state of two other α-helical MPs to date. An analysis of the kinetic effects of 12 alanine mutants enabled phi-value analysis of the E. coli disulfide bond reducing protein B (DsbB),250 an α-helical MP that folds by way of a single observable kinetic intermediate in mixed micelles. The results of this analysis revealed that two residues near the edge of a TM domain appear to be involved in the rate limiting step for the formation of the intermediate and that native contacts appear to propagate from this region within the intermediate state.250 Similarly, an exhaustive kinetic analysis of 69 GlpG variants also identified two residues near the cytosolic edges of two neighboring N-terminal helices that appear to form native contacts in the transition state.194 In contrast with the findings for bR folding, the folding of these two proteins appears to be rate-limited by the formation of native contacts near the edges of specific TM domains. Though these results are potentially suggestive of mechanistic differences between bR, DsbB, and GlpG, caution must be exercised when comparing these studies. It is possible that differences in the mixed micelle/bicelle components may alter the denatured state ensemble in a way that fundamentally distorts their folding trajectories. Nevertheless, these findings highlight the potential for mechanistic diversity in the folding kinetics of helical MPs. Moving forward, it may be particularly interesting to consider how folding pathways may vary for helical MPs containing stable soluble domains, which often appear to have evolved from soluble proteins.251 Can the rapid folding of a soluble domain seed folding within the membrane, or vice versa? Additional studies are needed to explore how folding pathways may be navigated under such circumstances.
Unlike α-helical MPs, investigations of the folding kinetics of β-barrels are less muddled by the potential influence of residual structure in the denatured state (although see181). Furthermore, investigations of the folding kinetics of β-barrels can be carried out using true lipid bilayers, which is a distinct advantage relative to kinetic investigations of α-helical MPs. Nevertheless, the kinetic mechanism(s) that modulate the folding of β-barrels in vitro still appear to be exquisitely complex. Initial studies of outer membrane protein (OMP) folding kinetics from aqueous solution to the folded form in lipid vesicles were conducted by Jahnig and co-workers69,205,206,252,253 and subsequently continued in an extensive study by Kleinschmidt and Tamm.186,254−257 Many of the kinetic constraints of these reactions have been characterized, including the general magnitude of the activation energies associated with rate-limiting transitions. However, the interpretation of β-barrel folding kinetics is complicated by the fact that structural transitions coincide with the transfer of the protein from the aqueous phase to the membrane interface and eventually from the interface to the membrane core. Indeed, the rate limiting transitions for β-barrel folding are sensitive to the lipid-to-protein ratio, to the composition of the lipid head groups and chain lengths, and to the lateral pressure of the bilayer.258 Nevertheless, phi-value analysis of the β-barrel protein PagP revealed that the rate-limiting step for folding likely involves the formation of numerous native-like interactions between side chains that are coupled with the transfer of the protein from the interface to the membrane core.259 This transition state presumably also involves protein–lipid contacts that distort the bilayer, as the rate of OMP folding is accelerated by conditions that introduce lipid packing defects.260 A recent comprehensive investigation of the folding kinetics of OmpA revealed that the folding of this protein occurs with no fewer than five intermediate states, some of which are off-pathway, even under the most optimal conditions.261 Given the vast array of intermediates that accumulate in vitro along with the fact that the folding of purified β-barrels is rate-limited by its transfer into the bilayer under certain experimental conditions, it is perhaps unsurprising that outer MP biogenesis in cells relies on the activity of BamA,262 a chaperone that catalyzes the insertion of β-barrels into the membrane.263 The reader is also referred to elegant studies of the interactions of unfolded OmpA with the periplasmic chaperone Skp, which helps the nascent porin reach the other membrane without aggregating or prematurely forming tertiary structure.206,264
3.5. Helical Membrane Protein Folding within Lipid Bilayers
Kinetic and thermodynamic investigations of folding and unfolding in micelles, bicelles, and synthetic membranes have yielded considerable insights into the conformational energetics of integral MPs. However, given the drawbacks of these artificial solvents, there is still much to be learned about the conformational equilibria of MPs in their native environments. This is especially true for α-helical MPs, for which biophysical studies in lipid bilayers are few and far between. Nevertheless, several recent breakthroughs have paved the way for the next generation of folding studies.198 For instance, a recent report from the Booth lab demonstrated that E. coli LeuT, a structural homologue of neurotransmitter sodium symporters, can be reversibly unfolded by urea in the context of a variety of synthetic liposomes, provided that submicellar concentrations of β-octylglucoside are included to facilitate the equilibration of urea across the bilayer.164 Under these conditions, urea induces a partial loss of secondary structure and a complete loss of function,164 as would be anticipated for a physiological unfolded state. Unlike the OMPs, urea does not lead to dissociation of LeuT from the bilayer to the aqueous phase. Instead, the results show that the structural properties of its denatured state and the free energy of unfolding can be tuned by lipid head groups and by the lateral pressure within the bilayer.164 The native conformation is modestly favored (2.5–3.8 kcal/mol) over the corresponding denatured ensembles in vesicles of varying composition.164 This suggests that differences in the bilayer can tune the properties of the unfolded state to reshape the relevant features of the conformational energy landscape, a key consideration for eukaryotic MPs that must traffic through a range of different membranes within the secretory pathway.172 The Booth lab has demonstrated that several transporter proteins are susceptible to denaturation by urea,164,190,191 which may open the door for comparative studies on a range of other transporters using this approach. Thus, the unique properties of these proteins may provide an opportunity to explore a range of hypotheses regarding the nature of α-helical MP folding within the bilayer.
An additional class of next-generation experiments have sought to do away with chemical denaturants entirely. The development of steric trapping approaches, which couple the energetics of biotin–streptavidin binding to the occlusion of intra or intermolecular contacts,265 has been employed to probe the partial unfolding of helical MPs and the dissociation of helical oligomers within micelles, bicelles, and membranes (see more detailed reviews in refs (21 and 266)). This approach offers several key advantages, including the fact that the denatured or dissociated state remains embedded within the membrane environment. Initial applications to measure the strength of glycophorin A dimers demonstrated the power of this technique, as dimerization was found to be stronger in synthetic membranes than in micelles.267 Strikingly, subsequent investigation found that natural lipid compositions significantly weaken this dimerization.268
Application of steric trapping to the unfolding of GlpG, which contains six TM domains, has provided evidence that this protein unfolds through a series of subglobal unfolding transitions.197 These results were recently echoed by single molecule studies in which GlpG was mechanically unfolded within a lipid bicelle.269 This denaturant-free approach probes the dissociation of TM helices within lipid bicelles as the N- and C-termini are pulled apart laterally using magnetic tweezers (Figure 10). A recent application of this approach to the ClC chloride transporter revealed that the intact subdomains of this protein are capable of separating prior to force-mediated subglobal unfolding within the bilayer.199 Interestingly, each of these magnetic tweezer studies has provided compelling evidence that both the native and partially unfolded forms of these proteins are kinetically stable, a clear indication that excursions between the native and partially unfolded forms occur on a time scale of minutes to hours. The apparent spectrum of partially unfolded forms that are accessible by these techniques as well as the marginal free energy differences that separate them is reminiscent of the transient partially unfolded forms of soluble proteins that are observable by hydrogen/deuterium exchange.176 Indeed, it has long been postulated that TM helices and/or helical bundles may behave as domain-like structural units.14,40,270 Thus, this interpretation of the conformational energy landscape seems quite plausible in light of recent observations.
Figure 10.

Single-molecule forced unfolding of a MP, GlpG. (A) Schematic of the single-molecule magnetic tweezers experiment for studying the unfolding and refolding of GlpG in a bicelle. The protein termini have been conjugated with DNA, with the end of one DNA molecule being surface anchored and the other end being attached to a bead that can be pulled away from the surface to force unfolding of the protein in the plane of the bicelle bilayer. (B) Representative force–extension curves for repeated GlpG unfolding and refolding transitions. (C) The energy landscape for folding/unfolding of GlpG in bicelles, where kf0 and ku0 are the kinetic rates for folding and unfolding at zero force, ΔG is the unfolding free energy, and ΔGu† and ΔGf† are the kinetic energy barriers for unfolding and folding, respectively. Reprinted with adaptations from ref (269). Copyright 2015 Springer Nature.
Next-generation approaches to study the conformational transitions within lipid bicelles and synthetic liposomes will play a critical role in ongoing efforts to rationalize the conformational energy landscapes of integral MPs. However, additional steps will be needed to bridge the current gap between MP biophysics and the gritty reality of biological membranes. In this regard, advances in quantitative microscopy have provided new insights into how MPs move and interact within eukaryotic plasma membranes. Recent theoretical and methodological advances from the Hristova lab have yielded a quantitative fluorescence resonance energy transfer (FRET) approach for the determination of equilibrium constants for MP dimerization within the plasma membranes of live cells.199,271,272 Emerging applications of this technique have revealed that, in contrast with established views, several receptor tyrosine kinases including fibroblast growth factor (FGF) and vascular endothelial growth factor 2 (VEGFR-2) form dimers and autophosphorylate in the absence of activating ligands.273 Recent advances in fluorescence cross-correlation spectroscopy have also provided an additional route to measure equilibrium dissociation constants, as well as the lateral diffusion coefficients for monomers and oligomers within the plasma membrane of live mammalian cells.274,275 Advanced applications of super-resolution microscopy have also provided a fascinating glimpse into how critical fluctuations of membrane phases132 drive the sorting and activation of B cell receptors within the plasma membrane.134 These and other emerging advances in microscopy and single particle tracking show great promise for future efforts to understand how MPs exist within their native cellular environment.
3.6. Misfolding of Purified Membrane Proteins
Misfolding is very often the unwanted companion of scientists seeking to reconstitute purified MPs into model membranes. Misfolding of nascent MPs also routinely occurs under physiological conditions, which is part of the reason that cells have an elaborate system for detecting, correcting, and sometimes degrading misfolded MPs (see section 4). MP misfolding in the cell often results in a pathogenic loss of MP function or in the formation of toxic aggregates. Nevertheless, despite considerable biomedical relevance, there have been relatively few structural studies of MP misfolding in vitro. The most extensively developed of these studies involved E. coli diacylglycerol kinase (DAGK),276 a 122 residue homotrimer in which each subunit contains three TM helices.277,278
Pioneering studies in the Bowie lab quantified the thermodynamic stability of DAGK in mixed micelles.8 Wild type (WT) DAGK exhibits considerable thermodynamic stability under these conditions. Moreover, the Bowie lab found that DAGK seems to be structurally and catalytically tolerant of mutations.279,280 This paved the way to a long-term study by the Sanders lab of a library of 120 single-cysteine DAGK mutants generated starting with a catalytically native-like quadruple mutant form of DAGK in which both native Cys residues were mutated to Ala (C46A, C13A) and that also contained W117R and S118T mutations. It was soon discovered that, unlike the WT protein, many variants within this single-Cys library are highly prone to misfolding in vitro.281 These variants therefore afforded an opportunity to systematically probe MP misfolding.
Exploration of DAGK misfolding benefited from two additional properties of this small yet complex membrane enzyme.192 First, in the presence of concentrated urea or guanidinium, it is possible to solubilize DAGK in the absence of any detergent or lipid. In concentrated urea under acidic conditions, DAGK retains some secondary structure but loses its quaternary and tertiary structure. However, at low pH in concentrated guanidinium the protein is almost completely unfolded. Second, dilution of small aliquots from these DAGK/denaturant solutions into neutral pH detergent/lipid mixed micelles or into solutions containing synthetic liposomes results in the spontaneous insertion and folding of DAGK to its functional state (Figure 11). However, this coupled insertion and folding reaction is typically inefficient. Interestingly, nonproductive folding does not typically result in classical aggregation.192 Careful kinetic studies by Lorch and Booth revealed considerable complexity in the kinetics of these folding transitions.282
Figure 11.
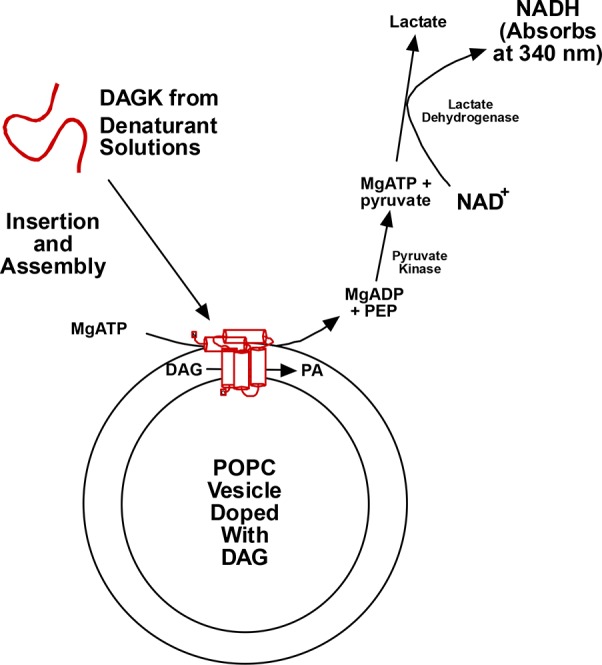
Assay to measure the efficiency of spontaneous insertion, folding, and trimerization of DAGK into preformed lipid vesicles following a many-fold dilution of a small aliquot of the pure enzyme in lipid and detergent-free urea or guanidinium solutions. Successful folding of the protein is accompanied by the appearance of enzyme activity that is monitored through a spectrophotometrically detected coupled assay system. The degree of misfolding is assessed based on comparing the final observed enzyme activity with the activity expected for 100% folding efficiency. Abbreviations: DAG, diacylglycerol; PEP, phosphenolpyruvate; NAD+ and NADH, oxidized and reduced forms of nicotinamidedinucleotide. Figure used with permission from ref (192). Copyright 2015 ACS.
The rates and efficiency of DAGK folding were typically greater when folding was initiated from detergent solutions (rather than denaturants) into vesicles, an observation that likely reflects both the preservation of structural elements in micelles and also the potential impact of submicellar detergent concentrations on the properties of the bilayer.192 Moreover, the enzyme retained an ability to assemble into its functional state when urea solutions were diluted into buffer prior to the addition of mixed micelles.283
Initial WT studies were followed by studies of the folding and insertion of the single-Cys and other mutant forms of DAGK. For a panel of ∼30 mutants, it was observed that the rate and efficiency of folding into vesicles is strongly correlated with protein stability as determined by resistance both to SDS-induced unfolding and irreversible heat inactivation.283,284 There were, however, interesting outliers. For example, the Y16C mutation does not destabilize DAGK and yet this mutant was severely folding-deficient.285 Y16C likely affects the kinetics of a key step in the DAGK insertion and folding pathway without affecting protein stability.
Altogether, the results of the folding and misfolding of DAGK led to two potentially important observations that may broadly extend to MP folding in physiological and possibly even disease conditions. First, the strong correlation between folding efficiency and protein stability led to the hypothesis that, when considering a panel of mutant forms of the same MP, the key determinant of the relative folding efficiencies for these mutants in cells is the relative thermodynamic stability of each mutant.284,286 As will be described later in this review, this hypothesis has now been tested for two disease-linked mutant forms of human proteins in cells and has, so far, held up well.183,287 Second, while the “misfolding is linked to instability” correlation is strong, it is not absolute, as revealed by the Y16C DAGK mutant, which appears to adversely alter the folding transition state but not the stability. Though they are likely to be rare, mutations that destabilize the folding transition state are likely to be represented among the large number of human mutations that promote human disease (see section 5). Successful therapeutic approaches to stabilize the transition state may be very different from those required to address destabilized mutant forms of the very same protein.
Whether MP misfolding transitions are commonly related to formation of amyloid-like assemblies remains unclear, though there is good reason to believe these phenomena may sometimes be connected. DAGK was not found to form amyloids or even classical aggregates in vitro.192 However, Vendruscolo and colleagues recently demonstrated that lactose permease is capable of forming fibrils with many characteristics of classical amyloid fibrils under certain conditions.288 This observation provides additional support for the notion that nearly any protein can form an amyloid,289 though in this case the physiological relevance is unclear.
The conversion of a membrane protein to amyloids does appear to play a direct role in the molecular basis of at least one human disease. Mature lung surfactant protein-C (SP-C) has 35 residues and, in its healthy physiological form, has a single transmembrane α-helix.290 However, this same segment, which is rich in valine, also has a strong propensity to form beta assemblies, leading to formation of amyloid structures. For this reason, nature has endowed nascent SP-C with a BRICHOS prodomain that suppresses amyloid formation, ensuring healthy SP-C function. However, any one of roughly 50 known mutations in pro-SP-C is sufficient to disrupt the protective function of the BRICHOS domain, which results in amyloid formation that causes interstitial lung disease (ILD).290
Another likely physiological connection between membranes and amyloid formation is for the amyloid-β polypeptide, which is a proteolytic fragment of the transmembrane C99 domain of the amyloid precursor protein and represents the primary component of the amyloid plaques found in Alzheimer’s patients. While amyloid-β is somewhat soluble, under some conditions it is known to spontaneously insert into membranes in a manner that promotes its homomeric assembly into pores.291−295 Moreover, even short of insertion to adopt a transbilayer structure, amyloid-β retains considerable affinity for membranes, a fact that impacts formation of amyloid-β oligomers and amyloid fibrils.296−306 The same is true for a variety of other membrane-active peptides that form amyloid fibrils (such as the α-synuclein protein involved in Parkinson’s307,308) and the islet amyloid polypeptide that may contribute to some forms of diabetes.309−311 The interactions of amyloid-forming proteins and amyloid assemblies with membranes is highly likely to be important in the etiology and pathology of disorders such as Alzheimer’s and Parkinson’s, although definitively establishing the pathophysiological relevance of phenomena observed in studies of isolated molecules (or even in model cell lines) to neurodegenerative disease in a living human being remains a daunting task.
The human prion protein (PrP), which is the root cause of several related neurodegenerative disorders, exists in both a membrane-anchored glycosylphosphatidylinositol (GPI) modified form and a form that contains a single TM helix. Interestingly, the TM form of PrP can exist in both possible topologies (review in ref (312)). The conversion of “healthy” PrP to the toxic and infectious PrPsc form of that seeds the formation of toxic aggregates appears to occur at the membrane.313 This process may also be linked to the formation of amyloid-like PrP fibrils.314 Whether these phenomena are etiologically related to the dread prion disorders is not yet established.
3.7. Proteins that Spontaneously Insert into Membranes under Native Conditions
Many microbial toxins spontaneously insert into bilayers to form multispan membrane proteins with both helical and beta-sheet secondary structure, often forming pores.315−325 Toxins differ from most other MPs in that they are usually folded and freely soluble in aqueous solution following secretion from the pathogen through specialized membrane translocation machinery.326 Membrane insertion nearly always requires the binding of a soluble toxin to a specific lipid (often cholesterol) or protein on the surface of the target cell. Surface binding is followed by structural changes on the membrane surface (often including oligomerization) prior to or during the process of membrane insertion. Toxins can be thought of as weaponized MPs. Whether there is an ancient evolutionary relationship between toxins and more conventional modern MPs is not well established, but the ability of toxins to spontaneously insert into membranes is a testament to the fact that translocation machinery is not always required for a protein to efficiently insert into a membrane.
Numerous small natural and artificial proteins/peptides, including “cell penetrating peptides” are capable of spontaneous bilayer insertion, sometimes in a pH-triggered manner.68,317,327−334 Some of these peptides insert from solution into bilayers where they remain as stable membrane proteins. Others are able to dynamically insert into and cross lipid bilayers but only transiently adopt transmembrane configurations, with their preferred states being either surface-associated or soluble.
As summarized earlier, MPs such as DAGK and the OMPs exhibit a capacity for membrane autoinsertion when diluted from detergent-free denaturant solutions into preformed lipid vesicles (other examples given in ref (335)). It is unclear how many other nontoxin polytopic helical MPs have the ability to autoinsert from denaturant solutions, but it has been shown that DsbB and GlpG cannot.336 It has been shown that some purified membrane proteins can be “delivered” to preformed lipid vesicles followed by spontaneous insertion using amphipols or membrane-noninteractive fluorinated surfactants.167,337,338
There has been considerable interest during the past 20 years in developing in vitro translation methods for producing membrane proteins.339−341 Among the approaches developed is the PURE system, which employs purified ribosomes and translation factors rather than cell extracts. This enables, among other applications, well-controlled studies of translation and cotranslational folding.342 PURE has been successfully used to synthesize membrane proteins, with evidence being presented that some MPs produced with this system are integrated and folded into preformed lipid vesicles in the absence of a translocon or other translocase system343−345 Booth and co-workers have recently adopted PURE as a platform for quantitative studies of cotranslational folding of membrane proteins. Using this system to synthesize DsbB and GlpG in the presence of preformed lipid vesicles they have shown that insertion and folding of both proteins can occur with fairly high (>60%) efficiency, with the results depending on the protein, lipid composition, and the lateral surface pressure vesicles.336 The insertion pathways were shown to involve cotranslational engagement of the nascent DsbB and GlpG emerging from the ribosome with the bilayer.
It is interesting to speculate192 that cell membranes in some ancient life forms might not have had the functional equivalent of a translocon or other modern membrane translocase systems. The existence of spontaneously inserting membrane proteins such as DAGK, the OMPs, and bacterial toxins, combined with the observation of cotranslational membrane integration of proteins such as DsbB and GlpG in the absence of a translocon supports the notion that early forms of life might have been able to persist without membrane translocation/integration systems such as Sec61/SecYEG.
4. Folding of Membrane Proteins in Eukaryotic Cells
4.1. Membrane Integration at or near the Translocon
The vast majority of MPs are integrated into cell membranes with the assistance of dedicated cellular machinery. The most common pathway involves the action of the heterotrimeric translocon complex known as Sec61 in eukaryotes or SecYEG in bacteria and archaea, which acts in concert with the ribosome to thread the nascent chain into the membrane. However, there are subsets of MPs that rely on other assembly pathways. For example, certain inner MPs in Gram negative bacteria utilize an insertase known as YidC,346 whereas the OMPs in Gram negatives are dependent on a mechanistically distinct membrane integrase known as BamA.262 Posttranslational membrane integration of some bacterial proteins can also be achieved through the actions of the SecA ATPase.347 Eukaryotic tail anchored proteins find their way into the membrane by way of the guided entry of tail-anchored protein (GET) pathway, including the membrane-bound GET1/GET2 complex.348−350 Furthermore, the recently characterized endoplasmic reticulum membrane protein complex (EMC) appears to actively facilitate the integration of a range of MPs, possibly often in concert with the Sec61 complex.351−353 Other organelles, such as mitochondria, have their own systems. Given the central importance of MP biosynthesis and assembly in life processes, it is unsurprising that nature has developed multiple mechanisms to both facilitate membrane integration and to suppress and manage misfolding during and after MP translation. For the purposes of this review, we will focus on the structure, mechanism, and activity of the Sec61 translocon complex and its associated chaperones and other folding accessory proteins
4.1.1. Structure and Function of the Translocon
MPs across all kingdoms of life are produced and integrated into the membrane through the concerted actions of the ribosome and the Sec61/YEG complex. Structural models derived from crystallographic and cryo-EM data have provided a wealth of insight into the structure and function of this complex (Figure 12). The core functional unit of the translocon is a heterotrimeric complex consisting of Sec α, β, and γ subunits in the ER membrane of eukaryotes or SecY, E, and G in the plasma membrane of bacterial and archaeal cells.354,355 In most cases, stalled ribosomes carrying the transcripts of secreted proteins or of integral MPs are delivered to this heterotrimeric complex by the signal recognition particle (SRP) and its receptor. Upon delivery, the ribosome associates with the translocon through its universal ribosomal adaptor site located within the cytosolic loops connecting TM segments 6/7 and TM segments 8/9 of Secα/Y.354,356−359 Once associated with the translocon, the ribosome resumes translation of the client protein and the nascent polypeptide chain is guided into the protein-conducting channel (PCC) by the C-terminal helix of Sec α/Y.360 The PCC is contained within the Sec α/Y subunit, which contains ten TM helices that form two pseudosymmetric lobes composed of TM segments 1–5 and 6–10.360 The interface between these lobes creates a polar, hourglass-shaped cavity that is filled with water.361,362 The cytosolic and luminal chambers are separated by a 5–6 Å pore created by a ring of hydrophobic residues, which forms a seal around the nascent chain and minimizes the exchange of ions and small molecules between the cytosol and ER lumen during translation.354,361,362 In the inactive state, ion leakage through the PCC is also prevented by the association of a plug domain (TM2a) with the luminal face of Sec α/Y.358,361,363,364 Association of the translating ribosome with the translocon causes a subtle conformational change that guides the nascent chain through the gasket into the ER lumen and displaces the plug.359 During translation, the separation of TM segments 2b/3 and 7/8b creates a lateral gate that allows the nascent chain to transiently sample the membrane environment.42,356−360,362,365 Upon entry of a hydrophobic segment of the nascent chain into the PCC, a rigid body rotation of several TM helices within the N-terminal lobe of Sec α/Y opens the lateral gate to facilitate entry of the nascent chain into the bilayer.359,366 This movement of hydrophobic segments through the lateral gate and into the membrane (topogenesis) establishes the orientation of TM helices with respect to the membrane (topology). It is widely assumed that nascent TM helices typically move from the PCC to the membrane. However, given the energetic and geometric constraints involved in topogenesis, it has also been argued that many nascent TM domains may initially partition into the membrane interface prior to topological isomerization.367 Considering the relatively weak energetic drivers involved in these reactions and the passive nature of these molecular machines, it seems likely that nascent membrane proteins may find more than one way into the membrane.
Figure 12.

(A) Structure of the ribosome-translocon complex. A 3.4 Å resolution model of a mammalian ribosome (blue and brown) bound to the Sec61 complex (red) determined by single particle Cryo-EM is shown. The absence of tRNA in the peptidyl transfer center (PTC) indicates this complex represents the inactive conformation of the complex. Subtle structural rearrangements have been observed within the active complex (not shown). Panel adapted with permission from (359). Copyright 2014, Elsevier under CC BY 3.0 https://creativecommons.org/licenses/by/3.0/. (B) Sec61 translocon and complex with the ribosome, TRAP, and the oligosaccharyl transferase. Segmented densities for the 40S (yellow) and 60S (light blue) ribosomal subunits, translation elongation factors (magenta), Sec61 (blue), TRAP (green), and OST (red) from a subtomogram average of the ER membrane associated translocon complex filtered to 9.0 Å resolution. Figure adapted with permission from ref (365). Copyright 2015 Nature under CC BY 4.0 http://creativecommons.org/licenses/by/4.0/.
Though a single monomeric Sec61/YEG complex is sufficient to mediate topogenesis, it should be noted that several ribosome-translocon complexes may occupy a single mRNA transcript at the ER membrane.355,368 Formation of these polysomes presumably increases the local concentration of nascent MPs in a manner that may help nascent MPs to form native oligomeric assemblies. Indeed, this aspect of topogenesis appears to bias the oligomerization state of AcrB in the inner membrane of E. coli.369 A minimal complex composed of Sec α/Y and Sec γ/E is sufficient to mediate basal translocation.370 Nevertheless, native translocons are associated with a wide variety of accessory subunits that serve to tune the activity of the translocon and to carry out processing of the nascent chain in order to better suit the needs of divergent client proteins.
Figure 12B depicts how some of these chaperones associate with the ribosome-translocon complex. The ∼200 kDa hetero-octameric complex known as the oligosaccharyltransferase complex (OST)371 is found in nearly half of all native translocons372,373 and catalyzes the glycosylation of asparagine side chains of N-X-S/T motifs (where X is any amino acid except Pro), which is one of the most common protein modifications in eukaryotes. Indeed, N-linked glycosylation is known to play a key role in the folding and trafficking of a wide array of MP substrates.374 N-Glycosylation can take place either cotranslationally or post-translationally.375 In many cases, maturation of the nascent chain also requires proteolytic removal of signal peptides, which are often found in MPs containing luminal/extracellular domains upstream of their TM domains. Removal of signal peptides is mediated by signal peptidases, which are a family of intramembrane proteases.376
Proper folding of translocon substrates may also hinge on interactions of the nascent chain with certain intramembrane chaperones within the translocon complex. For instance, the translocating chain-associated membrane protein (TRAM) is an abundant integral membrane glycoprotein that associates with the translocon and improves the translocation of certain substrates.377 Though its precise mechanism of action is unclear, TRAM appears to mediate the handoff of polar TM domains from the translocon into the lipid bilayer in a way that depends on the sequence context of the client protein.42,378 Similarly, the heterotetrameric translocon-associated protein (TRAP) complex379 appears to enhance the translocation of certain substrates bearing ambiguous topogenic signals.380,381 The EMC also associates with some emerging MPs cotranslationally, especially those enriched with charged residues, to facilitate proper membrane integration, to protect clients from premature degradation, and to enable interactions with chaperones.352 Correct orientation of proteins with semipolar signal peptides appears to depend on the highly abundant Sec62 subunit,382 which also mediates the posttranslational translocation of certain client proteins.383 Sec62 is associated with another Hsp40 homologue at the translocon known as Sec63, the J-domain of which facilitates BiP-mediated ratcheting of nascent polypeptides into the ER lumen.384 Thus, some of these subunits serve to connect the translocon to other components of the cellular proteostasis network.385
It should be recognized that many of the accessory subunits alluded to above are present at substoichiometric concentrations within the ER membrane, which renders native translocon complexes functionally heterogeneous.372,386 This polydispersity is clearly physiologically relevant, as mutations that alter the relative abundance and association of OST are linked to congenital glycosylation diseases.387 Furthermore, this observation implies that the cotranslational folding of integral MPs cannot be mediated by a single set of core chaperones. Rather, the chaperones available to the nascent chain are likely to be determined by the organization of its ribosome–translocon complex. Ongoing investigation into the structure and function of these subunits and other relevant chaperones will undoubtedly provide key insights into how these dynamic assemblies facilitate the production and processing of the membrane proteome.
4.1.2. Energetics of Translocon-Mediated Membrane Integration
Though the structure of the translocon complex is intricate, topogenesis itself is thought to be driven by the minimalist principles associated with the partitioning of the nascent polypeptide chain between the PCC and the membrane.42,388 Because the PCC is hydrated361 and the membrane core is hydrophobic, the energetics associated with lateral partitioning of the nascent chain between the translocon and the membrane mirrors the transfer free energies of polypeptides between oil and water,388,389 with subtle deviations potentially arising from certain kinetic constraints.390 An analysis of the contribution of non-native amino acids to the energetics of translocon-mediated membrane integration revealed that transfer free energy scales with the hydrophobic surface area of the nascent chain.391 However, biological membranes are not a uniform solvent, and the relative abundance of bulk water and other polar chemical groups varies as a function of membrane depth.392,393 Due to this transverse heterogeneity, the transfer free energy of amino acid side chains from the translocon into the bilayer (or from the aqueous phase to the bilayer)189,207 also exhibits an appreciable dependence upon depth within the membrane.394,395 Native bilayers are also asymmetric with respect to the electrostatic properties of the inner and outer leaflets, as lipids with anionic head groups are enriched on the cytosolic leaflet. The net-negative charge of the cytosolic membrane interface facilitates the formation of electrostatic interactions between lipid head groups and cationic amino acid side chains in a manner that biases the orientation of TM domains with respect to the membrane.396 This energetic bias apparently plays a key role in the stabilization of the native topology, as cationic side chains are highly enriched near the cytosolic edge of TM domains.38,397 The generality of this “positive-inside rule” in combination with current estimates for the energetics of translocon-mediated membrane integration can be used to predict MP topology from sequence with considerable accuracy.395,398
The energetics of topogenesis are sufficient to guide the cotranslational membrane integration of thousands of chemically diverse substrate proteins in the absence of their native structures. This is remarkable considering that the magnitude of the forces driving membrane integration of the nascent chain are relatively modest. Furthermore, many client proteins bear TM domains that are enriched with polar residues, which are critical for function.15,399 An analysis of the sequences of known MPs using an experimentally trained energetic algorithm revealed that ∼25% of TM domains within polytopic MPs are likely too polar to spontaneously partition into the membrane in the absence of additional stabilizing interactions.400 Polar TM segments like these introduce frustration into the nascent chain, which can promote the formation of aberrant topomers.401,402 The formation of these non-native topologies during biosynthesis is perhaps inevitable for certain client proteins considering the tertiary contacts that stabilize the native topology may be inaccessible during translation. Proper membrane integration of some topologically frustrated segments may hinge on the formation of interhelical contacts with neighboring TM domains.397,403 Strong topological preferences in neighboring TM domains can also drive polar segments into the membrane.391 For instance, the topology of the nascent form of aquaporin 1 features only four of six TM domains within the membrane, and the membrane integration of the remaining two TM domains is accomplished through a posttranslational inversion of its third TM domain.243,404 Protein–lipid interactions also appear capable of inverting the entire N-terminal domain of E. coli lactose permease.405 However, there are few examples of proteins that undergo such an extreme topological rearrangement, and it seems that many MPs are likely to remain trapped within the global topology established by the translocon.41 Nevertheless, a comparison of crystal structures and energetic predictions suggests that, in many cases, the segments selected by the translocon may only partially overlap with native TM helices.406 Thus, posttranslational folding reactions may often involve mild to moderate adjustments to the nascent topology. These sorts of topological isomerizations are likely constrained by the polarity and size of soluble loops, which influence the magnitude of the kinetic barriers involved in their movement across the bilayer.407,408
The persistence of nascent MPs within non-native topologies may potentially contribute to cellular misfolding, as aberrant topomers are rapidly degraded in the cell.409 Indeed, it has been estimated that 10–15% of mutations associated with diseases of MP misfolding enhance formation of aberrant topomers,287,410 suggesting that the fidelity of topogenesis is tied to the efficiency of MP folding. Proteins exhibiting gross topological defects are likely to remain kinetically trapped in non-native topologies. Additional investigations into the connection between cotranslational folding and membrane proteostasis are needed.
4.2. Formation of Tertiary and Quaternary Structure in the Endoplasmic Reticulum
MP folding in the cell is assisted not only by the translocon and related MP complexes but also by a series of other “quality control” proteins that assess the conformational state of the client MP. These processes ultimately facilitate correct folding in a manner that is coupled to the subsequent trafficking of the protein through the secretory pathway. These proteins also target misfolded MPs for terminal degradation. Quality control systems vary considerably between organisms, tissues, cells, and organelles. For the purposes of this review, we focus here on the quality control system of the mammalian endoplasmic reticulum. However, it must be noted that a good deal of what we know about ER quality control stems from studies of quality control in yeast, as is represented by a vast literature (cf. ref (411)). A diagram summarizing the central ER-based pathways for MP folding, misfolding, and degradation is given in Figure 13.
Figure 13.

Overview of MP folding in the early secretory pathway of mammalian cells. This figure encompasses ERAD, ERAF, and ERES, plus some components of the broader proteostatic network.
4.2.1. Endoplasmic Reticulum Quality Control
MPs begin to fold into their proper tertiary structure concurrent with their cotranslational membrane integration. While MP folding QC begins even at the ribosome,412 this process is mainly surveilled and managed by the ER quality control (ERQC) network.413−422,6,416 Beyond the translocon, ERQC can be further broken down into three overlapping subsystems: the coterie of proteins involved in facilitating ER-associated folding (ERAF), those involved in recognizing properly folded proteins and targeting them for export to the Golgi or other destinations, and those involved in recognizing and targeting misfolded proteins for degradation in a pathway known as “ER associated degradation” (ERAD). In the following sections, we will discuss these systems. We note that the ERQC network also works in collaboration with the ER-based unfolded protein response (UPR), a system that produces a transcriptional response to stress associated with the burden of protein production and folding in the lumen and membrane of the ER.415,417,423 This linkage between ERQC and the UPR is critical to the function of the overall cellular proteostasis network and for the biogenesis of the ∼35% of all cellular proteins that reside in or pass through the secretory pathway.
The extent to which MP folding occurs at or near the translocon versus after clearage from the translocon likely varies among client proteins. Nevertheless, there are two observations that suggest many MPs may complete folding only well after dissociation from the translocon complex. First, while it has long been recognized that there are spatially distinct domains of the ER—the translocon-rich “rough ER” and the translocon-depleted “smooth ER”—studies over the past decade have revealed that the architecture of the ER is actually much more complex. The ER now appears to extend throughout the cell, making direct contacts with other organelles thorough “membrane contact sites”.424 Different domains of the ER feature biased protein compositions, which are likely associated with distinct ER region-specific functions.223 This observation implies that certain aspects of MP folding may be facilitated and managed in spatially distal domains of the ER. Consistent with this notion is the fact that MPs can spend considerable time in the ER prior to export to the Golgi, much longer than the time required for completion of translation and membrane integration. For instance, mature cystic fibrosis transmembrane regulator (CFTR) begins to appear at the plasma membrane on the order of 1–2 h after the initiation of translation, while degradation of misfolded CFTR occurs with a half-life of 45 min.425−427 Similarly, the half-life of biosynthesis and trafficking of the epithelial ENaC channel is roughly 1 h, with half-lives for the degradation of misfolded protein on the order of 15–20 min.428,429 Nascent PMP22 associates with the ER-resident lectin chaperone calnexin with a half-life of 11 min, indicating nascent PMP22 molecules that engage within this chaperone spend at least minutes in the ER before trafficking on to the Golgi. By comparison, the folding-defective L16P disease mutant of PMP22 forms a complex with calnexin that exhibits a half-life of over 1 h.430 These rough kinetic estimates highlight the fact that most MPs require considerable time to clear ERQC. Further studies and advances in technology are needed to better understand how kinetic control of MP trafficking influences MP biogenesis.
4.2.2. The Calnexin Cycle as a Central Component of Endoplasmic Reticulum Quality Control for Membrane Protein Folding
An estimated 70% of proteins inserted into the ER are cotranslationally N-glycosylated on their luminal domain by the addition of one or more units of a 14-sugar complex oligosaccharide (“core N-glycan”).431,432 In mammals, this glycoform is composed of three glucoses, nine mannoses, and three N-acetylglucosamines arranged in a tree-like structure (shown in Figure 14A). Core N-glycan biosynthesis is initiated on the cytosolic face of the ER membrane and completed on the luminal side via sequential additions of uridine diphosphate-linked monosaccharides to a membrane-anchored dolichol phosphate scaffold.433,434 OST catalyzes the luminal addition of this oligosaccharide en bloc to asparagine residues of N-X-S/T sequence motifs of nascent proteins.416,433,434 In order to catalyze N-glycosylation, OST requires local structural flexibility at the glycosylation site.433 N-Glycosylation can be disrupted by tunicamycin, a compound often used to induce ER stress and that inhibits GlcNAc-1-phosphotransferase responsible for catalyzing the initial addition of N-acetylglucosamine to dolichol phosphate.435N-Glycans increase the hydrophilicity of the polypeptide chain and may potentially facilitate arrangement of the ER-luminal segments/domains in a way that could stabilize and/or alter the tertiary structure of the membrane domain. The steric properties of these glycans can also help prevent proteolytic cleavage and decrease the propensity for protein aggregation. Beyond the physical effects of these modifications on the folding process, N-glycans also serve as “folding barcodes” for components of the ERQC to track the trajectory of the nascent protein through the lectin chaperone pathway (Figure 14B).433,434,436 Modification of the core oligosaccharide through the addition or removal of sugars is coupled to the folding of the nascent protein and its binding to ERQC proteins.
Figure 14.

N-Linked glycan (A) and the calnexin cycle (B). As proteins are translocated into the ER membrane they are tagged with a 14-sugar moiety at N-X-S/T (where X is any amino acid but P) sequence motifs as shown in panel A. This post-translational modification serves a multitude of functions for the protein including increasing hydrophilicity, preventing aggregation, influencing tertiary contacts, and serving as a folding “barcode”. Through sequential cleavage of monosaccharides (gray dotted lines), the folding polypeptide can be engaged with different lectin chaperones involved in either ERAF or ERAD. The enzymes responsible for cleaving various glyosidic linkages are shown. The predominant folding pathway for glycosylated proteins is shown in panel B. Monoglucosylated proteins (1) are engaged by the membrane-anchored lectin chaperone calnexin (2, gray). Calnexin provides the protein with an isolated environment to fold, recruits essential cochaperones such as the disulfide isomerase ERp57 (cyan), and may also facilitate the proper tertiary packing of a MP by associating with exposed hydrophilic residues in the plane of the membrane. Association is transient; glucosidase II cleaves the terminal glucose residue on the glycan chain freeing the client polypeptide from engagement with calnexin. Proteins that have yet to attain their proper tertiary structure (3), are sensed by UGGT1 (orange) and reglucosylated, allowing reentry into the calnexin cycle (1). Proteins that have obtained their proper structure and post-translational modifications (particularly disulfide bonding) are sensed by UGGT1 and funneled toward the ER export machinery for ER exit (4). Polypeptides that fail to complete folding after consecutive cycles are eventually funneled out of the cycle and targeted for ERAD by the action of mannosidases.
Once transferred onto a nascent protein, the terminal glucose residue on the core N-glycan is cleaved by the membrane-anchored glucosidase I. This hydrolytic event enables the nascent glycoprotein protein to engage with the lectin chaperone malectin.416,437 Malectin displays an affinity for misfolded conformations of proteins, and its overexpression reduces the total protein trafficking out of the ER.438 Malectin may serve as the initial sensor of the conformational state of nascent chains early in their lifetime, possibly targeting certain misfolded proteins for degradation. This function would reduce the burden on downstream chaperones by reducing the flux through later branches of ERAD. Malectin may also protect against aggregation and accumulation of misfolded proteins in the ER.439 However, it is not yet clear whether malectin is involved in the surveillance of all nascent N-glycoproteins or if it exhibits selectivity.
ER luminal glycosidase II, the next component of the lectin pathway, cleaves the outermost remaining glucose to yield a monoglucosylated N-linked glycan. Cleavage of this glucose residue promotes interactions with either the membrane-anchored lectin chaperone calnexin or its soluble paralog calreticulin (Figure 14B step 2).414−417,434 Because of its colocalization in the ER membrane, most nascent integral MPs preferentially interact with calnexin.437,440 Calnexin is a Ca2+ binding protein composed of a luminal N-terminal lectin domain (275 residues) followed by an extended proline-rich “P-domain” (ca. 135 residues), a single TM segment, and a highly acidic cytosolic domain (ca. 90 residues).437,441 Calnexin and its water-soluble homologue, calreticulin, appear to be critical folding sensors of ERQC. Elegant biochemical studies have shown that calnexin interacts with misfolded, monoglycosylated glycoproteins with submicromolar affinities in vitro.434,442 Transient interactions of calnexin with partially folded proteins occur with a half-life on the order of minutes to hours depending on the substrate, with sequestration by calnexin, affording clients a protective environment during folding.434,443 Calnexin also recruits the accessory chaperones ER protein 57 (ERp57) and cyclophilin P (CycP) via interactions with its P domain, which catalyze the oxidation of free cysteines and cis/trans isomerization of proline residues, respectively.414,416,434,437 Interestingly, it has been shown that calnexin can selectively bind misfolded conformations of integral MPs, even in the absence of glycosylation.443−446 For glycoproteins, it is possible that the glycosylation status of a protein may trigger initial binding or increase the affinity of a client for calnexin but that its actual chaperone activity involves a different set of client-calnexin interactions involving their TM domains. For example, the lone TM segment of calnexin has been shown to be both necessary and sufficient to retain trafficking-defective mutants of the γ-aminobutyric acid (GABA) transporter in the ER, localizing it to concentric assemblies that can be visualized by EM (discussed further below).447 Likewise, the recognition of the misfolding-prone L16P disease mutant form of PMP22 by calnexin appears to involve direct recognition by calnexin of folding defects in the PMP22 TM domain.430,443,448 It has also been hypothesized that the TM domain of calnexin can recognize mis- or partially assembled helices of polytopic MPs or unassembled oligomers by serving as a temporary stand-in for unpaired TM domains.447,449 Thus, the chaperone activity of calnexin is multifaceted and involves both glycosylation-dependent and independent modes.
Once released from calnexin/calreticulin, the terminal glucose residue on the N-glycan of a substrate protein is cleaved by glucosidase II, which lowers the affinity of the substrate for calnexin/calreticulin. At this point, the client protein is engaged by another folding sensor, the UDP-glucose:glycoprotein glucosyltransferase-1 (UGGT1).415−417,437,450 Proteins that have failed to mature fully at this point are recognized by UGGT1, which catalyzes the readdition of a glucose residue from UDP-glucose to the N-glycan of the client protein (Figure 14B, step 3). This modification reactivates interactions of the client protein with calnexin and/or calreticulin and continued ER retention.415−417,437 Only some proteins must be cycled back through the lectin chaperone pathway in this manner. Biochemical and cellular biological assays have shown UGGT1 exhibits a preference for incompletely folded substrates over either fully folded or irreversibly misfolded proteins.451−453 Thus, folded proteins appear to escape reglucosylation of UGGT1, which allows them to proceed to engage the ER export machinery and escape the ER (see Figure 14B step 4). Knocking out UGGT1 in mice results in embryonic lethality, suggesting that this pathway is essential in higher organisms. However, studies in mouse embryonic fibroblasts show that the maturation of most proteins is unaffected by the loss of UGGT1,454 suggesting that only a subset of essential proteins need to associate with calnexin more than once to complete maturation.
How UGGT1 monitors the conformational state of integral MP substrates is unclear. UGGT1 is a soluble 170 kDa protein whose N-terminal region contains a hydrophobic pocket that could potentially detect misfolded polypeptides, with its C-terminal domain potentially catalyzing glucosylation.455 Interestingly, UGGT1 accommodates a wide variety of substrates that vary considerably in terms of both size and shape. It should also be noted that this protein catalyzes glucosylation of N-glycans as far as 40 Å from misfolded domains.456,457 Recent structural studies of UGGT1 showed that there is a high degree of flexibility between the folding sensor region of the protein and the glucose transfer region.456,457 Introduction of interdomain disulfide bridges reduced this intrinsic flexibility and decreased enzymatic activity.458 The interdomain flexibility of UGGT1 may facilitate protein substrate promiscuity and promote the ability of the enzyme to reglucosylate N-glycans located at variable distances from the misfolding-recognition site. Based on our current knowledge of UGGT1, it seems likely to interact with misfolded substrates through improperly exposed hydrophobic residues within the ER lumen. It has also been suggested that that UGGT1 is able to recognize aberrant introduction of a polar residue into a TM site.459
Terminally misfolded or slowly folding proteins are eventually funneled out of the calnexin cycle through the action of mannosidases. Cleavage of the terminal mannose residue on the A chain of the N-glycan (Figure 14A) inhibits glucose readdition and targets the protein for degradation (see below). Multiple proteins with mannosidase activity are present in the ER, including ER α-1,2 mannosidase I (ER Man I) and ER degradation enhancing α-mannosidase protein (EDEM) isoforms 1, 2, and 3.415−417,434,437 ER Man I inhibitors have been shown to selectively slow the degradation of misfolded proteins in the ER.416 Thus, these enzymes appear to convert glycoproteins into targets for the ERAD pathway (discussed below). These observations have collectively led to the “mannose timer” hypothesis for ERQC,415,434,460 which is that nascent glycoproteins only have a certain amount of time to fold and exit the ER before they are eventually cleaved and targeted to ERAD by a catalytically sluggish mannosidase. This logic may explain why degradation of nascent proteins is typically slow, even under conditions of proteotoxic stress.461 Slow folding kinetics may well constitute a key determinant of the fraction of nascent proteins that face cleavage following mannosidase action. Various aspects of the activity of these mannosidases have yet to be characterized in detail, though there is some evidence that they have the capacity to directly recognize conformational defects (see section 4.3.1).462
4.2.3. Other Mechanisms of Endoplasmic Reticulum Quality Control
Not all ERQC is dependent on protein glycosylation, and there is mounting evidence to suggest nonglycosylated proteins can also interact with ERQC proteins that were previously believed to serve in a glycosylation-dependent fashion.445,446,463 Additionally, some components of the ERQC pathway may play a role in both glycan-dependent and glycan-independent pathways. For instance, the ER-localized heat shock protein 70 (Hsp70) family member “binding immunoglobin protein” (BiP) is involved in ERAF and ERAD of both glycosylated and nonglycosylated MPs.417,464 This chaperone is found in the ER lumen where it plays a role in the recognition of misfolded MPs. BiP contains both an ATPase domain and a substrate-binding domain.465 The substrate binding domain contains a hydrophobic binding pocket that recognizes exposed hydrophobic residues. When bound to ATP, BiP exists in an open state. Upon substrate binding, BiP hydrolyzes ATP and “clamps” down on its substrate. Nucleotide exchange factors (NEFs) Sil1 and Drp170 catalyze the exchange of ADP for ATP and facilitate the release of the client protein.466 BiP also associates with a variety cochaperones including DNAJ/Hsp40 family (ERdJ1–7) proteins that facilitate oxidative folding, rearrangement of disulfide bonds, and disulfide bond reduction.467 Since BiP is soluble, it presumably interacts with misfolded MPs through recognition of exposed hydrophobic patches in luminal domains or through interactions with mis-incorporated membrane domains that become exposed to the aqueous lumen.
BiP clients and those for the calnexin cycle are not mutually exclusive; proteins may “ping-pong” between both pathways during maturation.434,464 The location of the N-glycan within the primary structure of the nascent chain may also influence its trajectory through ERQC. N-Glycans within the first ∼50 residues of a protein usually target it to the calnexin cycle, bypassing BiP.468 Indeed, BiP binding to misfolded substrates is increased when calnexin is knocked down or out.469,470 Thus, it appears that nascent MP substrates may kinetically partition between the BiP and calnexin pathways in a way that is dictated by their intrinsic conformational properties.
Another important chaperone involved in ERQC is the Hsp90 family member, glucose-regulated protein 94 (GRP94). GRP94 is involved in both ERAF and in targeting proteins to ERAD and is the only known Hsp90 member within the ER lumen.471 The ATPase activity of GRP94 is essential for its chaperoning activity in vivo. However, the exact mechanism of action of GRP94 has yet to be elucidated.472 Unlike BiP, GRP94 appears to interact with a more limited number of substrates, including the Toll-like receptors, integrins, and members of the low density lipoprotein receptor (LDLR) family.473−475 Cytosolic Hsp90s typically bind substrates after Hsp70, and it appears likely that GRP94 engages substrates released by BiP.473 GRP94 also interacts with OS-9, which is a component of the ERAD pathway. Nevertheless, how GRP94 facilitates the degradation of misfolded proteins is unclear.476 Much work is still needed to elucidate the role(s) of GRP94 in both the ERAF and ERAD pathways.
Protein disulfide isomerases (PDIs) also plays important roles in ERQC.416,464,477 PDIs are the primary oxidants of cysteine thiols in the ER, and can break (reduce), form (oxidize), or rearrange (isomerize) disulfide bonds depending on the oxidation state of its Cys-X-X-Cys active site.478 PDIs sometimes serve as chaperones that recognize exposed hydrophobic patches in misfolded proteins.477 Of particular importance is the ERdj5 protein, which has the dual function of being both a PDI and a J-domain cochaperone.479 Recent solution state NMR studies have mapped the PDI substrate binding site using unfolded ribonuclease A as substrate, and also using substrate peptides mastoparan and somatostatin.480 These studies showed that PDIs specifically recognize misfolded proteins with exposed hydrophobic patches in a manner that circumvents the kinetic barriers associated with the isomerization of disulfide bonds. This activity helps overcome kinetic entrapment of intermediate folding states.
The compartmentalization afforded by the ER is also essential for these quality control reactions. The segregation of proteins into ER subcompartments appears to help guide immature, mature, and misfolded proteins through the cell.223 Certain proteins appear to cycle between the bulk ER and so-called quality control vesicles (QCVs) that are enriched for ER Man I.481 This spatial segregation presumably protects nascent glycoproteins against premature mannose trimming by ER Man I. Misfolded proteins also appear to localize within subcompartments known as ER quality control compartments. ERAD machinery that recognize misfolded proteins, such as OS-9 and HMG-CoA reductase degradation protein 1 (Hrd1, discussed below), are enriched within this compartment. Additional quality control proteins and ERAD machinery including calnexin, calreticulin, EDEM1, and Derlin-1, are also found within this compartment in the presence of proteotoxic stress.481,482 Finally, mature proteins appear to accumulate near ER exit sites, which stem from smooth ER membranes.483 These exit sites are enriched with ER to Golgi transport machinery, as further discussed below.
Calnexin appears to play a critical role in the sorting of substrate proteins between different ER subcompartments. As noted above, the TM region of calnexin sequesters misfolded GABA receptors in distinct regions of the ER.447 In yeast, it has been shown that a model misfolded integral MP Ste6p localizes to distinct vesicular ER quality control compartments adjacent to the bulk ER.484,485 These compartments are composed of proliferated, tubular, ER membranes that are absent in cells not expressing the misfolded variant of Ste6p, and their presence was shown not to affect the trafficking of other proteins. Compartmentalization of misfolded proteins is likely to be advantageous because it restricts their diffusion and concentrates them to potentially increase the efficiency of degradation.485 Additionally, the sequestration of misfolded proteins likely minimizes their aberrant interactions with healthy proteins.
Finally, many MPs, such as the CFTR, have large soluble domains facing the cytosol. The folding of these domains is mediated by some of the same chaperones (Hsp70s and Hsp90s) and accessory proteins that interact with water-soluble cytosolic proteins (cf. ref (486)). The folding of proteins such as CFTR involves the coordinated action of both ER-resident and cytosolic chaperones and folding sensors.
4.2.4. Recognition of Misfolded Membrane Proteins and the Logic of Endoplasmic Reticulum Quality Control
Figure 15 diagrams some of the tertiary and quaternary defects in MPs that could potentially occur within the ER membrane. MP folding quality control systems must have mechanisms for the specific recognition of these defects.487 This is a tall order; native protein structures vary considerably with respect to their shapes, sizes, and conformational dynamics. What are the structural features that the folding sensors of ERQC proteins recognize and how specific are these interactions? For soluble proteins or aqueous domains of MPs within the ER lumen, ERQC seems most often to recognize exposed hydrophobic segments.414−417,477 UGGT1 and BiP utilize this mechanism, for example.465,488
Figure 15.

Some of the possible folding defects in MPs that must be recognized and managed by the folding quality control systems of all cells.
How misassembly of TM domains is recognized is poorly understood but likely involves their intrinsic structural properties.489,490 The aberrant partial or total exposure of a TM segment within the ER lumen or cytosol may represent a structural cue indicating misfolding or instability of these proteins (Figure 16A).429,491 As noted earlier, many polytopic integral MPs (and single pass integral MPs that function as dimers or higher order oligomers) have TM segments that are only marginally hydrophobic.16,492 Solvation of these helices within the membrane may be contingent on the formation of proper tertiary or quaternary contacts. Single amino acid mutations within these domains could potentially disrupt native helical packing interactions. These helices or helical hairpins could then “slip” into the ER lumen (or cytosol), thereby exposing hydrophobic patches for recognition by BiP or other folding sensors/chaperones, as has been demonstrated for the Na+/K+ ATPase.243,493,494 This pump is comprised of a 10 TM α subunit and a single TM β subunit, which heterooligomerize to form the functional transporter. In the absence of the β subunit, the C terminal TM helices fail to properly integrate into the membrane, and the TM7/8 hairpin becomes exposed to the cytosol (Figure 16A).493 Exposure of hydrophobic residues apparently results in the recognition of this domain by cytosolic sensors, leading to increased degradation of the protein within the cell. The single pass TM α subunit of the αβ T cell receptor (αβTCR) provides a second example of this phenomenon.491 In the absence of the β subunit, the α subunit of TCR slips completely out of the membrane and into the ER lumen where it is recognized by BiP and targeted for degradation. If the β subunit is present, or if the α subunit is rendered more hydrophobic by addition of Leu residues, the α subunit instead remains embedded within the ER membrane and eventually traffics to the plasma membrane.491 Thus, the marginal hydrophobicity of some TM helices seems actually to be exploited by ERQC as a means for monitoring the folding and assembly of certain MP complexes.
Figure 16.
Examples of mechanisms for ERQC detection of a misfolded integral MP. In panel A, the 10 TM α subunit (red) of the Na+, K+ ATPase is able to properly integrate into the ER membrane in the presence of the single-pass TM β subunit (blue). In the absence of the β subunit, TM helices 7 and 8 fail to properly integrate into the ER membrane and sidle into the cytoplasm where they can be recognized by the Hsp70 chaperone (black), leading to targeting for degradation. In panel B, a hypothetical dual pass TM protein (cyan) requires a salt-bridge (acidic and basic amino acids shown with stars) in the plane of the membrane in order to maintain its proper fold. If the protein is misfolded or unstable, it can expose these hydrophilic amino acids in the plane of the membrane. Hrd1 (green) contains a number of hydrophilic amino acids in the plane of the membrane that may aid in recognizing aberrantly exposed polar residues in MPs, resulting in ERAD targeting.
A second mechanism by which ERQC may monitor the folding and oligomerization status of TM domains is through the exposure of hydrophilic residues in the hydrophobic membrane core (Figure 16B). Many polytopic integral MPs contain hydrophilic amino acid residues in the membrane that are important for formation of tertiary or quaternary structure and/or functional dynamics.16,492 Misassembled proteins may expose these residues within the hydrophobic membrane phase, where they may readily form lateral interactions with MPs of ERQC. For instance, the TM segments of the E3 ubiquitin ligase Hrd1, a key player in ERAD (discussed below), appear to specifically recognize some misfolded polytopic MPs with exposed polar residues in the hydrophobic interior of the membrane.495,496 The presence of a variety of hydrophilic residues within the TM domain of Hrd1 may endow this protein with a high degree of substrate promiscuity. Calnexin may also recognize some misfolded MPs through this mechanism. The hydroxyl groups of Tyr487 and Thr490 within its lone TM domain of calnexin may mediate recognition of exposed hydrophilic sites; they appear to be involved in the QC of GABA receptors and other misfolded, nonglycosylated MPs.447,470
A third mechanism by which components of ERQC can recognize unstable or misfolded MPs may involve the recognition of “dangling strands” within polytopic MPs, or TM domains that fail to associate with their neighboring TM domains. The recognition of unstable PMP22 variants by calnexin may be based on this principle. Cell biology results have suggested that calnexin recognizes certain unstable mutants of PMP22 via a mechanism that involves recognition of the first of four TM helices present in PMP22.443,497 Structural studies later showed that this helix transiently dissociates from TM helices 2–4 (see Figure 6).182 Together, these observations raise the intriguing possibility that calnexin and other ERQC proteins may recognize dangling TM helices in polytopic proteins. Yet, it remains unclear how the single TM helix of calnexin accomplishes this feat of molecular recognition.
ERQC also appears to have the capacity to recognize aggregated membrane proteins in the ER and to mediate their disposal via ERAD.498,499 The protein(s) responsible for this recognition event are not yet known.
Finally, some MPs have short amino acid motifs referred to as degrons that are structurally buried and thereby masked from recognition by components of ERQC in folded proteins but are exposed for recognition by folding sensors upon unfolding or misfolding. Exposure of these motifs lead to their ER retention and/or degradation.500−503
As indicated above and recently reviewed elsewhere,489 the structural “symptoms” of MP misfolding as well as the manner in which they may be recognized are becoming clearer. ERQC appears to include a number of mechanisms for detecting thermodynamically unstable and/or slow folding MPs as well as misfolded conformations and aggregates. There is still much to learn about this fascinating topic; we know just enough to whet our appetite for discovery of additional classes of defects, folding sensor, and recognition mechanisms.
4.2.5. The Endoplasmic Reticulum-to-Golgi Export System
Transport of mature proteins from the ER to the Golgi complex is the productive outcome of ERQC. Transport is a highly selective process mediated by coat protein complex II (COPII) transport vesicles.504 Selectivity is maintained by two distinct, yet complementary mechanisms: selective loading of mature cargo into transport vesicles and sequestration of incompletely folded proteins away from transport vesicles. Export occurs at specific exit sites in the smooth ER, which are enriched for proteins involved in COPII mediated trafficking.505 The selective loading of cargo into transport vesicles is mediated by Sec24 in conjunction with specific cargo receptors in COPII vesicles.504,506 Sec24 binds to export signals exposed on the cytosolic side of the ER membrane present on cargo proteins or on cargo receptors. Along with its heterodimeric partner Sec23, Sec24 recruits the remaining proteins necessary for COPII vesicular transport from the ER to the Golgi.506 Eukaryotic organisms express four separate isoforms of Sec24 (A-D) and each isoform contains up to four nonoverlapping cargo recognition sites that accommodate the diverse cargo proteins that transit through this pathway.507 Properly folded and/or oligomerized MPs sometimes present export signals that promote direct interactions with Sec24. However, not all proteins bound for ER exit contain these signals; cargo can also be selected for COPII trafficking by specific TM cargo receptors.504,506 These receptors specifically bind mature proteins either within the membrane core or on the luminal face of the ER membrane, whereas Sec24 proteins tend to bind their recognition sequences on the cytosolic face of the membrane. Some of these receptors, such as ERGIC53, VIP36, and VIPL, are lectins that recognize the glycosylation state of the cargo protein in order to differentiate between mature and immature states.508 Other receptors such as Erv14, Erv26, and iRhoms interact with TM regions of cargo proteins and direct them into transport vesicles.509−511 Our understanding of how these receptors specifically recognize integral MP cargo is in its infancy.
In order to prevent the inappropriate forward trafficking of immature proteins, immature or misfolded proteins are selectively excluded from transport vesicles. The resident ER chaperones BiP, calnexin, and PDI, which interact with immature or misfolded proteins, are depleted from ER exit sites.415,504 By sequestering these chaperones away from exit sites, the probability of exporting immature proteins to the Golgi is reduced. Upon inhibition of certain chaperone interactions involved in this concentrative export pathway, misfolded MP variants that are normally retained within the ER sometimes manage to leak through the export system.512 This suggests that compartmentalization of misfolded proteins away from ER exit sites by ER chaperones helps retain these proteins in the ER.
It is worth considering whether the sequestration of MPs from the ER export machinery may be promoted by the biophysical properties of the membrane itself. The hydrophobicity of TM domains appears to influence their access to export sites.513−515 Although not definitively proven, it has been suggested that ER exit sites have a different lipid composition than the bulk ER.513 This may reflect an enrichment of these sites with lipids that are synthesized within the ER (such as sterols and ceramide) primarily for export to downstream organelle membranes (Golgi and plasma membranes).91,516 This idea is supported by the observation that the depletion of cholesterol from the ER inhibits COPII transport.517,518 Mature (well-folded) polytopic MPs may preferentially partition into these cholesterol-rich ER exit site membrane domains.513
4.2.6. Quality Control beyond the Endoplasmic Reticulum
Cellular sorting mechanisms are not 100% efficient. Furthermore, MPs presumably continue to sample non-native conformations well after they are exported from the ER provided their unfolding rates are not glacial. To prevent issues arising from misfolded molecules in the late secretory pathway and beyond, the cell has developed a number of QC mechanisms for service beyond the ER. For instance, the Rer1 and ERp44 proteins specifically recognize immature proteins in the Golgi and facilitate their retrograde trafficking to the ER.448,519,520 Rer1 is a TM protein that has been shown to specifically recognize misfolded integral MPs, most likely through contacts with hydrophilic residues in its transmembrane domain.448,519 Rer1 contains an ER retention KDEL motif, which allows it to be recognized by the KDEL (ERD2) receptor and to associate with COPI vesicles as cargo for return to the ER. ERp44 is a soluble member of the PDI family that contains only a single cysteine residue within its active site, rendering it nonfunctional as an isomerase.520,521 In the neutral environment of the ER, the substrate binding site of ERp44 is occupied by its own C-terminal tail. However, within the more acidic environment of the Golgi lumen, this C-terminus is displaced and its single reduced cysteine residue is exposed in order to facilitate mixed disulfide bond formation with immature proteins. This conformational change also exposes a KDEL motif, which enables ERp44 and its substrate to traffic back to the ER as cargo in COPI vesicles. Once in the ER, conventional PDIs can remove the substrate from ERp44, which allows it to return to the Golgi.520,521 Some immature proteins have been shown to transport out of the ER in complex with BiP.522,523 These Golgi retrieval mechanisms appear to function as an additional layer of quality control beyond the ER. There is now much evidence that even the plasma membrane has its own quality control system to monitor the structural integrity of MPs, triggering degradation of those deemed defective.524,525
4.3. The Degradative ERAD Branch of Endoplasmic Reticulum Quality Control
Proteins that are unable to pass QC in a timely manner are removed from the ER and degraded via the ERAD pathway (Figure 13). ERAD involves four coupled steps: substrate selection, substrate retrotranslocation from the ER to the cytosol, substrate ubiquitination, and substrate degradation via the 26S proteasome (Figure 17).223,490,526−528 Most of these steps are accomplished by a multiprotein complex centered around RING finger-containing membrane-embedded E3 ubiquitin ligases (E3s).529,530 Failure to degrade misfolded MPs may induce ER stress, which typically leads to activation of an additional wing of the proteostasis network, the UPR.531 Among many other effects, the activation of the UPR leads to the upregulation of a series of chaperones that help to buffer protein misfolding within the ER lumen.532 In extreme cases the failure of ERAD and consequent accumulation of misfolded protein can lead to a UPR-triggered apoptotic responses, which in some cases can contribute to human disease. This trans-acting effect of misfolded proteins may constitute a form of “toxic gain of function”. While ERAD is also responsible for degradation of water-soluble proteins that misfold in the ER lumen, we focus on MPs. Examples of MPs that appear to sometimes elude ERAD upon misfolding, and for which a loss of function is compounded by toxic gain of function include the proteolipid protein (Pelizaeus-Merzbacher disease), PMP22 (Charcot-Marie-Tooth disease, CMTD), and rhodopsin (retinitis pigmentosa).418,533,534 Here, we will briefly explain some of the known pathways and proteins involved in ERAD, as well as how the cell overcomes the energy barriers involved in the removal of integral MPs from the ER membrane.
Figure 17.

ERAD of a representative integral MP (red). In step 1, a misfolded protein (red) is recognized either through its N-glycan by the ER lectin chaperone OS9 (orange) or in the plane of the membrane by proteins such as the derlins (gray) or Hrd1 (green). Sel1L (purple) nucleates an ERAD complex in the ER membrane and also recruits ER luminal factors such as BiP and its PDI cochaperone ERdJ5 (teal). Herp (cyan) localizes the E3 ubiquitin ligase Hrd1 to ERAD sites. In step 2, the derlins may function to lower the energetic barrier for substrate retrotranslocation by partially unwinding helices in the plane of the membrane. The cytoplasmic region of Hrd1 catalyzes the addition of ubiquitin (brown) to lysine residues on the ERAD substrate. Ufd1 and NPL4 (dark orange) associate with the AAA ATPase p97 (yellow) and recognize the ubiquitinated ERAD substrate. In step 3, the ERAD substrate is retrotranslocated into the cytoplasm potentially through a pore formed by Hrd1. PNGase (light green) then removes the N-linked glycan from the ERAD substrate. The substrate is pulled out of the membrane via the energy provided by p97 ATPase activity which also functions as a retrochaperone to maintain the solubility of the ERAD substrate in the cytoplasm. The ERAD substrate is eventually degraded via the 26S proteasome (gray and navy).
4.3.1. Pathways and Proteins of ERAD
Many of the pioneering studies on the biochemical mechanisms of ERAD utilized yeast as a model organism. Although the main principles are conserved from yeast to mammals, the mammalian system is far more complicated. For example, the degradation of all ERAD substrates is mediated by only two E3s in yeast, Hrd1p and Doa10, in combination with a number of shared ERAD factors.419,526,535 Hrd1p is utilized to degrade proteins with conformational defects within the membrane or luminal domains, while Doa10 seems to mainly degrade proteins with defects within cytosolic domains. By comparison, greater than 30 E3s localize to the ER membrane in mammals, where they have been hypothesized and sometimes confirmed to play a role in ERAD.536,537 These E3s exhibit a broad range of substrate specificities: some are nonspecific while others appear to specialize in the degradation of a single substrate. Additionally, distinct combinations of ERAD factors are required for the degradation of certain client proteins.538 Beautiful work integrating proteomics, functional genomics, and gene expression data has elucidated the organization of many of these ERAD complexes in mammals.529,530 For the sake of brevity, we here focus on the most studied ERAD complex found in mammals, centered around Hrd1 and some of its more prominent auxiliary factors. Hrd1 has been implicated in the degradation of numerous integral MPs.233,448,529,535
As described in section 4.2.2, glycosylated ERAD substrates are removed from the calnexin cycle and targeted for ERAD through the trimming of mannose residues by ERManI and the EDEMs.433,434,463,477 Moreover, substrates containing anywhere from five to seven mannose residues interact with the OS9 or XTP3B lectins through a mannose-6 phosphate receptor homology (MRH) domain, which uses a double Trp motif to recognize the sugar on the ERAD substrate (Figure 17 step 1).535,539,540 Surprisingly, this pathway does not appear to exclusively handle glycosylated proteins. Overexpression of EDEM1 (with or without its carbohydrate recognition domain) increases the degradation of both glycosylated and nonglycosylated MPs.463 OS9 and XTP3B may also interact with substrates through exposed hydrophobic residues, either directly or indirectly, through interactions with BiP or GRP94.476,535,539,540 OS9 and XTP3B then link substrates to the membrane-anchored scaffolding protein Sel1L, most likely through interactions with ER lumen-exposed N-linked glycans on Sel1L.476 Sel1L also scaffolds essential reductases such as ERFAD and BiP-associated ERdj5 on the luminal side of the ER membrane in order to reduce disulfide bonds prior to removal of the substrate from the ER membrane.529 Additionally, Sel1L nucleates a complex with integral membrane ERAD components including but not limited to Herp, VIMP, Derlin1, Derlin2, Derlin3, and Hrd1 (Figure 17 step 1).526,529,530 This complex in turn recruits the cytosolic VCP/p97/Cdc48 AAA+ ATPase (hereafter referred to as p97) as well as necessary cofactors required to drive substrate retrotranslocation.526,529,530
ERAD factors located in the ER membrane and in the cytosol serve distinct functions. Herp, which is upregulated during ER stress, has been shown to localize Hrd1 to sites at the ER membrane where ERAD occurs.541 The highly tunable expression of Herp affords the cell granular control over the ERAD process. VIMP has been implicated in both substrate and p97 recruitment to the ERAD complex, although its interaction with p97 may be functionally redundant considering that Hrd1 and the Derlin proteins also contain cytosolic p97-binding motifs.299,529,542 The function of the Derlin family of proteins remains somewhat enigmatic. The Derlins are a family of inactive rhomboid pseudoproteases proposed to carry out a variety of functions including ERAD substrate recognition, retrotranslocation of misfolded proteins (passage across the ER membrane into the cytosol), or destabilization of TM helices (Figure 17 step 2).526,529,543,544 This latter function will be expanded on in the next section. Hrd1 is a polytopic protein with a cytosolic ubiquitin E3 RING finger ubiquitin ligase that ubiquitinates ERAD substrates on their cytosolic face.527,530,535
Hrd1 has also been hypothesized to serve as the major retrotranslocation channel responsible for the extrusion of misfolded ubiquitinated MPs from the ER membrane (Figure 17, step 3). Reconstitution of Hrd1 in liposomes containing a membrane-anchored ERAD substrate is sufficient to catalyze retrotranslocation of the substrate in the presence of cytosolic ATP and requisite components for ubiquitination.362 However, it is unclear from these experiments if the substrate was fully extracted from the membrane in the absence of p97. In any case, Hrd1 autoubiquitination was sufficient to initiate substrate retrotranslocation. In vitro analysis of the retrotranslocation of HMG-CoA reductase showed this process to be dependent on Hrd1 but not on other proposed retrotranslocation channels (Sec61 and Derlins).495 Interestingly, a recent cryo-EM structure of the dimeric form of yeast Hrd1 revealed an aqueous cavity bridging the ER lumen to the cytosol.545 This structure also features a “lateral seal” that could potentially accommodate entry of an integral MP substrate into the channel. This feature is reminiscent of the lateral gate of the Sec61 translocon (see section 4.1.1), which facilitates cotranslational membrane integration of nascent proteins. Once a client protein is exposed to the cytosol, Hrd1 ubiquitinates lysine residues using its RING finger domain and expands these ubiquitin chains to enhance the affinity of the substrate protein for the proteasome.233,419,490,526,527,536
The cytosolic components of the ERAD complex, which include the p97 AAA+ ATPase and the glycolytic enzyme PNGase, function to prepare the ERAD substrates for proteasomal degradation and to deliver the substrate to the 26S proteasome (Figure 17, steps 2 and 3). p97 binds to ubiquitinated substrates on the cytosolic side of the ER membrane via its cofactors Ufd1 and Npl4.546 It then removes the substrate from the membrane by using ATP hydrolysis to generate force (discussed further below).547 Once substrates are removed from the ER membrane, PNGase removes all N-linked glycans from the substrate to prepare it for proteolytic digestion.490 It was recently demonstrated that the yeast homologue of p97, Cdc48, also helps to maintain the solubility of dislocated integral MPs prior to proteasomal degradation.544 Additionally, Hsp104 has been shown to associate with ubiquitinated, and retrotranslocated substrates in complex with the p97 homologue in yeast.498 Thus, p97 and Hsp104 appear to function together as “retrochaperones” in order to prevent the cytosolic aggregation of hydrophobic substrates before delivering them to the 26S proteasome for degradation. Once an ERAD substrate is delivered to the proteasome, its ubiquitin linkages are cleaved prior to protein degradation. The proteasome appears to be tightly coupled to p97.548Figure 18 provides a space filling representations of key components of the later ERAD pathway, offering a perspective of scale.
Figure 18.
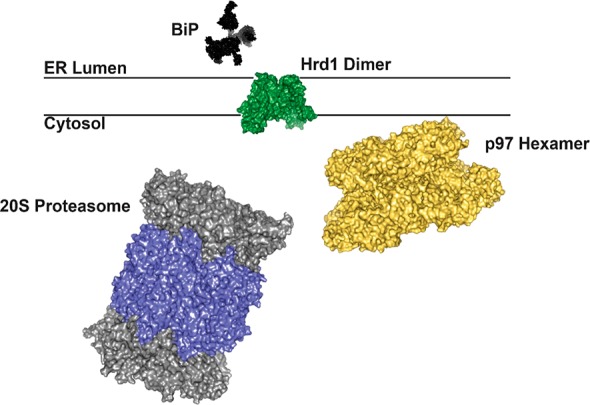
To-scale surface representation the structures of some of the key players involved in ERAD. Human BiP (black; PDB: 5E84), Hrd1 from S. cerevisiae (green; PDB: 5 V6P), human p97 (yellow; PDB: 5FTJ), and the human 20S proteasome (α subunits in gray, β subunits in navy; PDB: 5LE5).
4.3.2. Energetics of Membrane Protein Retrotranslocation
The removal of misfolded integral MPs from the membrane comes at a steep energetic cost. The free energy difference between native, membrane-integrated bacteriorhodopsin and a hydrated, unfolded state of the protein was estimated at 230 ± 40 kcal mol–1 by atomic force microscopy; an average free energy change of about 1.3 kcal mol–1 per residue!200 Although this may be an extreme case given the high stability of bacteriorhodopsin, it nevertheless serves as a useful benchmark for the magnitude of the energetic barriers the cell must overcome in order to dislocate hydrophobic proteins from the membrane. One of the machines that accomplishes this task is the E. coli protease FtsH, which forms a TM hexamer with the proteolytic/ATPase sites located in the cytosol. Elegant studies of the degradation of the helical multispan rhomboid GlpG by purified FtsH in the lab of Heedeok Hong revealed that FtsH acts by accelerating the unfolding rate of GlpG by a factor of at least 800, a process that is coupled to complete degradation and solubilization of the rhomboid fragments.210 The half-life for FtsH-mediated degradation of a single rhomboid molecule is on the order of 25 min, with the combined unfolding-degradation-translocation process being driven by consumption of 380–550 ATP molecules.210 In eukaryotes, p97 is also able to couple ATP hydrolysis to mechanical force by way of a series of conformational changes. Each p97 forms a water-soluble homohexameric complex, with each subunit containing an N-terminal domain and two conserved AAA domains that, together, form stacked rings.549,550 The p97 oligomer contains six ATP binding sites and physiological ATPase activity.550,551 The specific mechanisms by which p97 bridges its ubiquitinated substrates with the other components of the retrotranslocation complex in order to pull it through Hrd1 is not certain, but has been hypothesized to involve ATP-mediated molecular “ratcheting”.546,550
Certain aspects of the structure and function of the Derlin proteins may also shed light on how the energetic barriers to retrotranslocation can be overcome. The Derlins are a family of inactive pseudorhomboid proteases that lack the catalytic Ser-His dyad.529,544 Rhomboid proteases are believed to bind their substrates in the plane of the membrane to partially (and passively) unfold the TM helices of substrate proteins in order to expose the scissile bond for proteolysis. Since the noncatalytic pseudorhomboids retain the essential active site architecture, they may also be able to bind proteins in the plane of the membrane and partially unwind the TM helix (Figure 17, step 2). Such unwinding would lower the energy required for retrotranslocation since the per-residue free energy cost of disrupting H-bonds within the membrane is on the order of ∼4 kcal mol–1.16,552 The unique structure of the rhomboid proteins has also been proposed to reduce the thickness of the membrane bilayer, reducing the permeability barrier and potentially altering hydration within the membrane to facilitate the breaking of native hydrogen bonds.433 It was recently shown that the pseudorhomboid domain of a yeast homologue of the Derlin proteins, Dfm1, was required for retrotranslocation of multiple integral MP substrates.544 The association of ERAD substrates with Derlin proteins may therefore constitute a required precursor for retrotranslocation of integral MPs that helps to lower the energetic cost of removing them from the membrane.
Another mechanism by which cells lower the energetic cost of retrotranslocation is through intramembrane proteolysis. Cleavage of MP substrates by presenilin, the catalytic subunit of the γ-secretase complex, or by rhomboid proteases constitute two classic examples. γ-Secretase cleaves a wide variety of single-span MPs within the TM domain, which results in the formation of new polar termini and reduced summed hydrophobicity that promotes release of the remnants of the TM domain from the membrane along with any associated soluble domains. Prominent γ-secretase substrates include the 99 residue amyloid precursor protein C-terminal domain (C99, the immediate precursor of the amyloid-beta polypeptides), the Notch receptor, and receptor tyrosine kinases such as the ErbB epidermal growth factor receptors. The ER-resident rhomboid-like protein 4 protease (RHBDL4) also mediates the turnover of certain MP substrates.553 However, RHBDL4, which is upregulated in response to ER stress, cleaves both single pass and polytopic MPs.553,554 Although RHBDL4 has yet to be linked to the turnover of any endogenous ERAD substrates, proteolytic processing by this protein could very well be involved in the dissociation of misfolded MPs from the membrane.
4.3.3. Aggresomes and Autophagy
The balance between chaperone-mediated folding and degradation are typically kept in check through various lines of cellular regulation. However, the accumulation of misfolded MPs under stress conditions sometimes exceeds the capacity of the proteasomal degradation pathway. Under these conditions, the accumulation of protein aggregates can trigger disposal through orthogonal degradation pathways. As the proteasome becomes saturated, cytosolic aggregates of ERAD substrates are actively side-tracked to perinuclear foci known as aggresomes (Figure 13).555,556 This process is believed to protect cells by sequestering cytotoxic aggregates and promoting their clearance through macroautophagy. A number of disease-linked MPs are known to form aggresomes, including CFTR,211 PMP22,557−560 rhodopsin,561,562 caveolin-1,563 SIMPLE,564 ABCG2,565 and presenilin.566 However, how the cell senses proteotoxic stress, adapts, and rerouts the flux of misfolded proteins remains unclear. The nature of the intermediate states that bridge retrotanslocation from the ER membrane to aggresome deposition is also not known.
Water-soluble proteins can also be transported into aggresomes via pathways that likely overlap with those of MPs.567 Indeed, a wide variety of proteostatic stressors can promote the accumulation of soluble protein aggregates that are also picked up by common components of the proteostasis network.568,569 In some cases, aggresome formation seems to arise from specific unstable domains or signal sequences. For example, an ankyrin repeat domain within synphilin-1 is sufficient to direct it to aggresomes, though replacement of this domain with an aggregation-prone segment of the huntingtin protein is also capable targeting the protein to the aggresome network.570 Alternatively, ERAD substrates can be differentially directed to these pathways by specific ubiquitin linkages. For instance, K63-linked polyubiquitination (attachment of ubiquitin to substrate and subsequent polyubiquitination via ubiquitin’s K63 residue, K63U) appears to target certain clients for degradation through the aggresomal/lysosomal pathway, whereas K48-linked polyubiquitination targets misfolded proteins for proteasomal degradation.571 Furthermore, the specificity of these pathways appears to be linked to certain pathologies. For example, the E3 ligases parkin and TRAF6 are involved in catalyzing K63U modifications, with mutations in parkin constituting a common cause of familial Parkinson disease.572,573 Knockdown of the deubiquitinating enzyme ataxin reduces the extent to which destabilized CFTR and superoxide dismutase variants are packaged into aggresomes; the consequences of which include programmed cell death.574,575 Similarly, knockdown of PLIC1, a ubiquitin-like protein that binds to the ubiquitin-interacting motif of ataxin 3, also inhibits aggresome formation.576
The downstream recruitment of client proteins to perinuclear aggresomes requires a network of adaptor and motor proteins that come in diverse shapes and sizes. The cargo receptors p62 and NBR1 bind to ubiquitinated proteins to promote the stabilization of these aggregates.567 Other adapters, such as histone deacetylase (HDAC6) recognizes K63U-modified proteins in a way that connects them to dynein motors. These motor proteins then engage in retrograde transport along the microtubule network in order to deliver protein aggregates to the aggresome, which are then bundled into vimentin cages at the microtubule organizing center.577 It should be noted that the activity of HDAC6 is also modulated both by phosphorylation and through protein–protein interactions with various cargo receptors, chaperones, and E3 ligases.578 Finally, aggresome formation can be mediated by the Bcl-2-associated athanogene 3 (BAG3) cochaperone in response to the upregulation of Hsp70. This is mediated through formation of a ternary complex containing the molecular adaptor 14-3-3 and dynein.579,580 Taken together, the complexity of these aggresomal pathways provides an example of the multifaceted regulation of the proteostasis network. Aggresome formation is rendered tunable through gene expression, protein–protein interactions, and post-translational modifications.
Though protein aggregates are typically associated with toxicity, we emphasize that aggresomes are generally considered to be protective storage compartments for sequestering misfolded proteins until they can be safely disposed of via autophagy. Indeed, certain adaptors and cargo receptors found near aggresomes appear to seed the formation of autophagosomes that eventually fuse with lysosome to promote their degradation.581,582 Aging and/or mutations that compromise autophagy result in a failure to clear aggresomes, which potentially promotes disease states.567,583
In a related vein, it should be appreciated that misfolded proteins sometimes aggregate within the ER. Such aggregates and the associated local membrane can be targeted for autophagy and lysosomal degradation via a pathway sometimes referred to as ER-phagy or reticulophagy (see Figure 13).584−587
5. Membrane Protein Misfolding in Human Disease
5.1. The Sometimes Delicate Balance between Folding and Misfolding
While some proteins appear remarkably tolerant to single amino acid mutations,588,589 there is much evidence that the folding of nascent proteins in the cell, including some MPs, is often strikingly inefficient.418,590−597 For example, the in vivo efficiency for the folding and maturation of human wild type PMP22, as inferred from its steady-state glycosylation and cellular trafficking, has been reported by multiple groups to be only ca. 20%, which apparently is sufficient to generate the population of functional PMP22 required to maintain healthy peripheral nervous system (PNS) myelin in humans.287,598,599 Inefficient folding, which typically leads to significant levels of misfolding and/or degradation, implies that the energetic barriers involved in folding and misfolding pathways are often similar in magnitude. As we have previously treated in more detail,286 this implies that mutations that disrupt only a single hydrogen bond, ion pair, or hydrophobic interaction may significantly reduce the yield of folded protein. In these cases, pathology can arise when the level of the functional protein drops below the threshold required for normal health and/or when the accumulation of misfolded proteins becomes toxic. This often-delicate balance between folding and misfolding of wild type MPs possibly helps to explain why a diverse spectrum of mutations distributed throughout the three-dimensional structure of a misfolding-prone protein (see below) are all capable of causing the same human disease phenotype.286,591
5.2. Contributions of Pathogenic Mutations in Integral Membrane Proteins to Disease Etiology
Mutations that promote MP misfolding are known to cause or contribute to a wide variety of human diseases. While our focus will be destabilized MP variants, it should be noted that the propensity of wild-type proteins to misfold is also relevant to certain pathologies. For instance, the proteotoxic stress arising from the overexpression of WT PMP22 upon gene duplication is responsible for the most common (type 1A) form of CMTD. The misfolding of WT proteins may also exacerbate proteotoxicity arising from post-translational modifications600 or environmental stressors that include fever, oxidative stress, or defects in QC stemming from gene variations affecting components of the proteostasis network.601−603 In this regard, it is important to recognize the implications of the extensive connectivity of the proteostasis network. Sometimes the cumulative load of misfolded proteins may represent the root source of pathophysiology rather than the defects in a single protein.604 For hereditary diseases or those caused by a germline mutation, the deleterious effects of the mutation may have consequences in whichever tissues the affected protein is expressed. Alternatively, diseases can also arise from somatic mutations that occur spontaneously within a single cell.605 Somatic mutations within oncogenes can cause a single cell to proliferate into a tumor.606 It has long been hypothesized that sporadic mutations may also trigger the sporadic forms of prion disorders in which mutant PrP from a single cell adopts the toxic scrapie conformation that can seed the toxic conversions of WT protein from surrounding cells into its infectious conformation.607,608 It has also been suggested that the progression of the sporadic form of Alzheimer’s disease may also involve the propagation of toxic oligomer folds in the brain.609 Small amounts of misfolded proteins resulting from sporadic mutations could also conceivably trigger toxic autoimmune responses.
Diseases that arise from mutations in a single gene (monogenic) and that follow a Mendelian pattern of inheritance represent the best-characterized examples of diseases of MP misfolding. For many disorders, it only takes one mutation in one protein to cause disease. However, for a given inherited disorder, there may be an entire panel of proteins in which a single mutation is sufficient for causation. For example, mutations impacting the expression level or amino acid sequence of PMP22 are by far the most common cause of CMTD. Nevertheless, the same clinical pathophysiology can also arise from mutations in any one of more than 40 other proteins, many of which are likely to carry out functions on pathways linked to PMP22 function.610 However, mechanistic insights garnered from investigations of inherited disorders are likely to be relevant both to sporadic disorders and to complex disorders that arise from a combination of genetic and nongenetic risk factors. In the case of complex disorders involving the interplay and additivity of multiple disease-predisposing risk factors, MP misfolding may represent one piece of a larger puzzle.
How common are diseases arising from the pathogenic consequences of MP misfolding? A search of the UNIPROT database611 for all disease-linked human proteins returned ca. 4000 hits (not counting splice-variants), which accounts for ∼20% of the proteome. This number is perhaps unsurprising given that ca. 20% of yeast proteins are essential for viability.612 Of the 4000 disease-linked human proteins in UNIPROT, about 1100 have at least one TM segment. Given that MPs constitute ∼25% of the proteome, this seems reasonable. How many of these 1100 disease-linked MPs undergo misfolding as the primary disease-promoting defect? We suggest that there are four lines of evidence to suggest that misfolding is the most common disease mechanism. (1) Many disease-linked MPs are mistrafficked within the cell.418,613,614 While misfolding is not the only phenomenon that can cause mistrafficking, it seem likely to be the most common cause in light of what we know about the intimate linkage between folding and trafficking along the ER-to-plasma membrane pathway. (2) The pathogenic defects in most disease-linked MPs can typically be promoted by a wide variety of substitutions that typically do not cluster within a functional site or domain.590 For example, Figure 19 shows both the sites of known diabetes insipidus mutations in the human vasopressin V2 receptor, as well as a list of the specific disease-causing mutations.615 This scatter of mutation sites throughout the sequence suggests the pathogenic defect often does not directly involve perturbation of an active site or of a protein–protein binding interface, as was also found to be the case for retinitis pigmentosa mutations in rhodopsin, a related class A GPCR of known 3D structure (see Section 5.4.3).286 Instead, this distribution indicates that most mutations are likely to either disrupt the cooperative interactions between TM helices that stabilize the native fold or destabilize interaction of the protein with the membrane phase. Indeed, it is known that the vast majority of disease mutations in the V2R cause mistrafficking of the receptor, consistent with these classes of defects.616−621 (3) Pathogenic mutations in MPs are biased toward nonconservative mutations that are likely to perturb tightly packed native conformations or TM domain-membrane interactions. Of 96 sites in V2R for which there are known disease mutations, 80 of them are located in TM helices.615 Moreover, mutations that introduce charged residues, proline, or glycine for native aliphatic residues within TM domains are common among pathogenic mutations within MPs.622 (4) Rigorous experimental investigations of the effects of pathogenic mutations in disease-linked MPs have revealed that a majority of the tested disease mutations reduce the conformational stability of the protein in a way that appears to be directly linked to their cellular mistrafficking (cf. refs (183 and 287)). Similar observations have previously been made for water-soluble proteins that are linked to inherited disorders.623−627 Taken together, the available data suggest the pathogenic misfolding of MPs is of central importance to a wide variety of diseases.628 Thus, investigations into the nature of these conformational defects are needed to provide basic insight into the many ways that mutations disrupt the folding of disease-linked MPs. In the following, we outline how the tools and perspectives arising from such investigations can be applied to address emerging challenges in pharmacology and precision medicine.
Figure 19.

Documented nephrogenic diabetes insipidus mutations in the vasopressin V2 receptor. List of mutation is from.615
5.3. Membrane Proteins and Precision Medicine
Precision (or “personalized”) medicine refers to the practice of medicine in a way that is informed by knowledge of the patient’s genotype in order to deliver optimal care.629 Most genetic variants within the human genome are harmless, even when they result in changes to a consensus “wild-type” amino acid sequence. Indeed, most human proteins bear multiple common variants (frequency >1%).630 As of mid-2018, roughly 225 000 variants that cause or enhance the risk of known heritable human diseases have been identified (Figure 20A).615 However, the causal variants for approximately half of all known genetic diseases remain unidentified.631 Among known disease variants, by far the most common (∼45%) are missense variants that encode a single amino acid change in the affected protein. Nonsense mutations, which encode for premature stop codons that improperly terminate protein translation, account for 11% of pathogenic mutations. 22% of pathogenic mutations are small insertions or deletions (indels) that result either in insertion or deletion of one or more amino acid residues or cause a frameshift that scrambles the downstream C-terminal sequence of the protein.615 Considering that TM domains are less tolerant to sequence variations than their soluble counterparts,632 it is perhaps unsurprising that mutations within a wide array of functionally distinct MPs have been linked to the onset of numerous diseases. An understanding of the mechanistic effects of these mutations at the level of MP structure and function is therefore essential for efforts to elucidate and treat the molecular defects underlying disease.
Figure 20.

(A) Growth with time in the total number of identified human mutations that result in inherited (Mendelian) monogenic disorders Data from the Human Gene Mutation Database.615 (B) Growth with time in the total number of validated human genome variations (SNVs and other small scale variations) as logged in the online dbSNP Database. The small decreases in the number of variations seen in this plot for some time points reflect the consequences of changes in the reference genome and its annotation with time. Figure adapted with permission from ref (629). Copyright 2015 ACS.
Pathogenic mutations that encode for changes in amino acid sequence can promote disease by inducing either a loss of protein function (LOF) or a toxic “gain of protein function” (GOF), a term that includes the toxicity associated with amyloids and other protein aggregates. Pathogenic variants that act by inducing protein LOF appear to be much more common than GOF variants. In some cases, LOF may arise from the mutagenic perturbation of functional sites. However, the most common mechanism underlying LOF is the mutation-induced enhancement of protein misfolding.624,626,633,634 As noted earlier, “misfolding” is used broadly in this review to indicate the formation of alternative non-native/nonfunctional conformational states. Mutations that induce misfolding effectively reduce the yield of properly folded, functional protein within the cell. While there has not been a systematic examination of the many tens of thousands of known pathogenic protein variants, studies of water-soluble proteins have consistently pointed to protein misfolding as the single most common mechanism underlying pathogenic protein variants.623−626 In contrast to LOF variants, many gain of function mutations act by causing dysregulation of normal protein function. For example, certain mutations within ligand- or voltage-gated ion channels induce constitutive channel activation, which can cause atrial fibrillation and other disorders. In other cases, GOF mutations lead to the formation of cytotoxic preamyloid oligomers and/or amyloid aggregates. Overall, the mechanistic diversity associated with known disease variants, often even for a different mutations in the same protein (cf. ref (183)), significantly complicates personalized medicine diagnostics and therapeutic decision-making.
For the ca. 6000 so-called “simple” (monogenic) diseases that follow Mendelian inheritance patterns, mutations that induce a LOF or GOF of the corresponding protein are sufficient to independently manifest a disease phenotype. For dominant and fully penetrant Mendelian disorders, the disease can be caused by a single variant allele. Alternatively, both alleles must contain pathogenic variants in order to manifest a recessive disorder. Nevertheless, the most common human diseases are “complex” and arise from the interplay of multiple contributing genetic risk factors. These factors typically include a series of genes and genetic variants that individually increase disease risk by only a modest factor.635,636 A variety of nongenetic factors also influence risk for most complex diseases, such as diet, exercise, exposure to environmental risk factors, and so forth. Many diseases arise from the interplay between genetic risk factors and these stressors. For example, carriers of low density lipoprotein receptor (LDLR) variants that enhance the risk of hypercholesterolemia sometimes can avoid this disease by exercise and a healthy diet. It should also be recognized that some risk factor mutations are germline (noninherited sperm or egg mutations passed on to the zygote) or somatic (any mutation that occurs following conception in the zygote or in differentiated cells).
Our understanding of human genetic variation has been transformed since the determination of the first human genome sequence thanks in part to the subsequent acceleration of DNA sequencing technology.637 These advances enabled widespread whole genome sequencing (WGS), and its more cost-effective cousin, whole exome sequencing (WES), in which approximately 2% of the human genome that codes for proteins is specifically captured and sequenced. It is estimated that across the academic, clinical, and private sectors, more than one million human genomes have been sequenced.637 Publicly available genomic data include whole genomes from more than 2500 diverse humans from around the world (collected by the 10 000 Genomes Project) as well as more than 120 000 whole exome sequences and 15 000 whole genome sequences collected by the Genome Aggregation Database.638,639 These data provide a preview into the vast diversity of human genetic variation. A typical haploid human genome containing 3.2 × 109 total base pairs differs with respect to the reference genome at between 4 and 5 million positions. Analysis and comparison of these sequences has led to validation of more than 110 000 000 human genetic variants (Figure 20B), which now include >99% of all of the common (>1% frequency) variants in the human population. Among the insights arising from analyses of the current databases of genetic variants is an appreciation for the fact that one out of every eight coding bases has been observed to vary. It should also be noted that a typical human genome has between 10 000 to 12 000 coding variants.640 This means that each person has non-“wild type” variant alleles encoding about half of their proteins! In light of the awe-inspiring magnitude of this genetic diversity, it is perhaps unsurprising that the pharmacological profiles of individuals exhibit considerable variability. Rationalizing the functional and medical implications of the many millions of observed sequence variants recorded to date and how they interact is a formidable task.
As the clinical utility of genetic information increases, genome and exome sequencing will become even more widespread. The patterns of variation observed among sequenced genomes indicate that we currently have a near complete catalog of common variants. Nevertheless, we are likely to continue to discover novel rare variants.639 These are often referred to as “variants of unknown significance” (VUS), for which there is not yet sufficient data to decrypt whether they are benign or pathogenic. VUS that encode for amino acid substitutions in disease-linked genes are perhaps most likely to manifest adverse effects. In such cases, successful decryption of VUS in known disease-linked proteins can be “medically actionable”, meaning that treatment or prophylactic medical action is available that can then be pursued to prevent the disease or slow its progression.629 For example, patients who are known to be at risk for LQTS cardiac arrhythmias based on the fact that they carry known LQTS-predisposing mutations in certain ion channels can often avoid sudden death by pre-emptive treatment such as surgical implantation of a cardioverter defibrillator in the chest.641,642 Genotypic information may therefore inform best medical practice if the effects of relevant VUS can be inferred from experiment or predictions. The American College of Medical Genetics and Genomics has established standards for the use of genomic data in medical decision-making, in which experimental observation that a given protein variant is functionally defective under laboratory conditions is classified as “strong evidence” in support of a “call” that the variant is indeed pathogenic.643 Such information may eventually also be of utility for optimal matching of specific therapeutic compounds to certain patient populations, as the FDA has recently ruled that experimental data from in vitro assays may be employed for these purposes in certain cases.
Given that numerous disease-promoting mutations adversely impact MP folding, there is an imperative to continue basic research into how and why MPs fold and misfold. There is also a strong impetus to study the folding of actual disease-linked mutant forms of human MPs. If civilization persists, it seem likely that the cumulative observations from these studies will ultimately translate into improved disease prevention and therapy. Ongoing efforts to illuminate the molecular mechanisms underlying folding-related diseases may also eventually provide critical insight for the development of algorithms to decrypt the impacts of VUS and to determine whether they are benign or pathogenic, a central challenge in the development of precision medicine. In this regard, the recently documented failure of various existing algorithms to successfully predict the stabilities of mutant forms of membrane proteins highlights opportunities for future innovation.644 Continued study is also needed as the basis for developing methods to distinguish pathogenic mutations that induce misfolding from those that perturb the activity of the folded protein, as the efficacy of certain drugs is tied to the mechanistic effects of the mutations. In the following section, we highlight examples of MPs for which knowledge of the effects of specific pathogenic mutations may offer utility for drug discovery and implementation of therapeutic regimens in the clinic.
5.4. Examples of Membrane Proteins that Misfold and Contribute to Human Diseases
5.4.1. Voltage-Gated KCNQ1 Potassium Channel and Long QT Syndrome Cardiac Arrhythmia
KCNQ1 (Kv7.1) is a homotetrameric voltage-gated potassium channel in which each subunit consists of six TM segments, the first four of which (S1–S4) comprise a voltage sensor domain and the final two of which (S5 and S6) contribute to the central pore domain of the fully assembled channel. KCNQ1 forms heteromeric complexes with different regulatory β-subunits (KCNE family members), which tune the kinetics, voltage dependence of channel activation, and conductance as needed to mediate diverse tissue-specific physiological functions.645−651 In the heart, KCNQ1 coassembles with KCNE1 (single TM span) to form a channel complex that serves as the slow component of the delayed rectifier current (IKs) that is critical for the cardiac action potential.652,653 Genetically dominant mutations in KCNQ1 are responsible for about 50% of all cases of congenital long-QT syndrome (LQTS), an arrhythmia that is characterized by a prolonged QT interval on electrocardiograms.654,655 LQTS also predisposes children and young adults to cardiac arrest and sudden death, as is often triggered by swimming or other stressful physical activity. Recently, the structure of Xenopus KCNQ1 has been determined by cryo-EM (Figure 21).656
Figure 21.

Structure of KCNQ1 and location of mutations examined in Huang et al. (2018).183 (Left) Cryo-EM structure of Xenopus KCNQ1, highlighting one of its four voltage sensor domains. Note that some of the connecting loops between TM helices were not resolved in the EM structure and are therefore not depicted. (Center and Right) Orthogonal views of an isolated voltage sensor domain with side chains shown for sites that correspond to those experimentally characterized by Huang et al. in their study of the human KCNQ1 channel. Blue sites are those where mutations did not dramatically alter the stability or trafficking of KCNQ1. These sites are seen to be enriched on the surface of the domain. Red sites correspond to mutants that were seen to cause both mistrafficking in cells and (usually) instability of the voltage sensor domain under NMR conditions. These sites tend to cluster in the interior of the VSD.
More than 350 dominant LQTS mutations in KCNQ1 have been identified in humans.615 Such mutations may cause LOF or dysfunction by promoting misfolding, mistrafficking, assembly, and/or improper gating of the channel protein. Experimental studies have revealed that KCNQ1 mutations often impair trafficking and that the resulting reduction in the concentration of the channel at the plasma membrane occurs independently of attenuated protein synthesis rates.657−660 We and our collaborators recently completed a comprehensive characterization of a set of 51 KCNQ1 variants involving single mutation sites located in the voltage sensor domain.183 The selected variants included 20 known LQTS mutants, 18 variants of unknown significance (VUS), and 13 predicated benign variants (locations illustrated in Figure 21, center and right panels). For each variant, the channel electrophysiological function, total expression level, cell surface trafficking efficiency, protein stability, and impact of proteasome inhibition on channel levels were quantitatively compared (Figure 22A). For the 51 mutants studied, 31 exhibited loss of channel function (potassium conductance), with an additional 5 exhibiting various other forms of dysfunction.183 For the 31 LOF variants, 22 exhibited very low expression levels, which accounts for the LOF of those variants (Figure 22A). Low expression levels arise due to proteasomal degradation of the nascent channel rather than through inefficient biosynthesis. This observation, in conjunction with the fact that poorly expressed mutants also traffic inefficiently, suggests a majority of LOF mutations result in targeting of the channel for ERAD degradation. Taken together, these observations indicate that 22 LOF variants cause misfolding of the channel within the cell. Examination of the NMR spectra from 47 of these mutant voltage sensor domains revealed that the NMR spectra for 17 out of 22 low-expressing mutants clearly exhibited clear conformational defects (see example in Figure 22B).183 In contrast no conformational defects were detected by NMR among the more than 20 variants that exhibited WT-like expression and trafficking. We also observed that some mistrafficked KCNQ1 mutants, especially those completely fail to traffic to the plasma membrane, exert a dominant negative effect when coexpressed with the WT channel.183 This suggests that these mutants are still able to form heterooligomers with the WT protein, but remain sufficiently misfolded to be recognized as folding-defective by ERQC, resulting in degradation of the entire heterooligomer. While the trends summarized above from studying 51 mutant remain to be confirmed for the other >300 currently known LQTS mutant forms of KCNQ1, these results suggest that mutation-induced destabilization, mistrafficking, and degradation of the channel is the single most common mechanism underlying LQTS.
Figure 22.

Results from characterizing the channel function, trafficking, and stability of 51 mutant forms of the human KCNQ1 potassium channel. (A) KCNQ1 potassium channel cell surface expression levels versus measured K+ channel peak current density as determined in HEK293 cells. Data are color-coded: known LQTS disease mutants (red), variants of unknown significance (VUS) observed in humans but not previously classified (cyan), or predicted neutral polymorphism (black). The vertical red lines indicate values that are 65% of WT, corresponding to the approximate cutoff between “healthy” and LQTS-predisposing. These data illustrate that for a majority of disease mutants, loss of channel function is the consequence of failure of the channel to traffic to the cell surface. It is also seen that a number of the VUS mutants exhibit loss of channel function, again usually as a result of mistrafficking. These VUS are likely LQTS-predisposing. (B) 1H,15N-TROSY NMR spectrum of the WT KCNQ1 voltage sensor domain (residues 100 to 249) and (C) spectrum of a mistrafficking-prone disease mutant form of KCNQ1, E115G (red) superimposed on the WT spectrum (back). These spectra were acquired for the two forms of the VSD solubilized in lyso-myristoylphosphatidylglycerol (LMPG) micelles. The spectrum of the E115G disease mutant exhibits extensive broadening, disappearance, and shifts of peaks, indicating that its structure is destabilized relative to that of the WT protein. NMR revealed that the vast majority of the mistrafficking-prone mutants were folding-destabilized. Adapted with permission from (183). Copyright 2018 American Association for the Advancement of Science.
5.4.2. Voltage-Gated hERG (Kv11.1) Potassium Channel
The human ether-à-go-go-related gene, KCNH2, encodes the voltage-gated potassium channel hERG (also known as Kv11.1) that conducts the rapid-delayed rectifier IKr current of the cardiac action potential in cardiomyocytes. Full-length hERG1 is composed the cytosolic N-terminal Per-Arnt-Sim (PAS) domain, six TM segments that form the channel domain, followed by the cytosolic C-linker domain and C-terminal cyclic nucleotide-binding domain (cNBD). Similar to other Kv channels, the hERG channel functions as homotetramer. Mutations in hERG cause LQTS type 2 (LQT2), an arrhythmia that is commonly triggered by emotional stress.661 LQT2 may also be acquired as a side effect of certain drug treatments. Indeed, approximately 8% of all marketed drugs exert an off-target effect on hERG,662 leading to blocked hERG channel conductance, impaired protein trafficking, and/or accelerated protein degradation.663
More than 700 disease-linked mutations in hERG have been identified to date. Deficient trafficking is the most common defect of pathogenic hERG missense mutations, as confirmed by a large-scale mutational analysis.664 This study concluded that 88% (169 out of 193) of known mutations in the PAS, C-linker/CNBD, and pore domains cause mistrafficking of hERG, with 76% of pore mutations exhibit dominant negative effects on the WT channel. On the other hand, mutations in the distal C-terminus do not appear to impede cellular trafficking. Mutations that promote mistrafficking located in the PAS domain also tend to exhibit moderate to significant reductions in both thermal and thermodynamic stability.665,666 The spectrum of mutational effects on hERG parallel those in KCNQ1: while there is more than one way to break this channel, cellular mistrafficking seems to be the most common pathogenic mechanism.
The balance between ER export and retention of hERG appears to be reciprocally controlled by the cytosolic molecular chaperones Hsp70 and Hsc70, which promote its folding and degradation, respectively.667,668 It is unsurprising that much of its folding quality control is based on cytosolic chaperones since the majority of its residues are located in cytosolic domains.663 Coexpression of Hsp70 with hERG increases the levels of both immature and mature forms of hERG and decreases its ubiquitination, opposite to the case of Hsc70. Hsp70-mediated folding is assisted by Hsp90 and Hsp40 DNAJA1.669,670 Both DNAJA1 and DNAJA2 promote hERG degradation through Hsc70, which interacts with the E3 ubiquitin ligase C-terminal Hsp70-interacting protein (CHIP).670 The polyubiquitinated protein is then targeted for proteasomal degradation. How the channel domain of hERG is extruded from the ER membrane as part of this pathway does not yet seem to be known.
hERG is also subjected to post-ER folding quality control.663 Cardiac glycosides destabilize hERG at the plasma membrane in a manner that promotes chaperone and CHIP-dependent polyubiquitination. Ubiquitinated channels are then endocytosed and degraded by the lysosome. CHIP ablation partially inhibits this glycoside-induced hERG removal from the plasma membrane.671
5.4.3. Rhodopsin and Retinitis Pigmentosa
A variety of G protein-coupled receptors (GPCRs) are subject to mutations that cause or promote various human disorders. In most cases, a majority of the disease mutations result in trafficking defects that reflect misfolding. These include the V2R (NDI, see Figure 19),616−621 the melanocortin 2 receptor (MC2R, glucocorticoid deficiency),672 the melanocortin-4 receptor (MC4R, severe obesity and flaming red hair),673 and certain developmental factor receptors (linked to achondroplasia and disorders of sexual development).674 However, the most prominent of the GPCRs for which misfolding appears to be a common disease mechanism is the visual photoreceptor rhodopsin, for which there over 140 known mutations that cause retinitis pigmentosa (RP). The sites bearing pathogenic missense mutations are rather evenly distributed throughout both the sequence and the 3D structure of the receptor (Figure 23). We focus here on rhodoposin as a representative GPCR.
Figure 23.
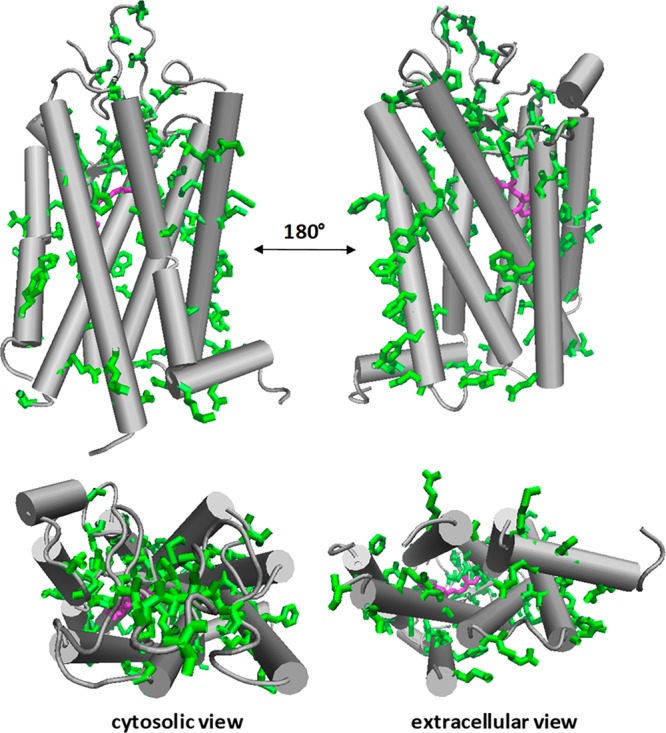
Four views of the structure of rhodopsin, illustrating the sites of known retinitis pigmentosa point mutations (green). The retinal cofactor is violet. PDB code: 1L9H.
RP involves serious visual impairment that begins in adolescence and progresses throughout adulthood.675,676 Most patients are considered legally blind by the age of 40, though the disease itself exhibits heterogeneity with respect to age of onset, symptoms, and severity.675 Like many other genetic disorders, RP is less a single disease than a panel of genetic disorders that result in the dysfunction and/or death of rod cells in the retina. Though mutations in the gene encoding rhodopsin are the most common cause of autosomal dominant RP (adRP), mutations in any one of ∼70 other genes involved in the phototransduction cascade, the retinoid cycle, or the visual sensory system have also been linked to the onset of RP and/or congenital night blindness.677 The exact pathophysiological mechanism of RP remains elusive. Nevertheless, the onset of retinal degeneration is typically marked by the accumulation of pigmented deposits known as bone spicules along the periphery of the retina.678,679 At the cellular level, retinal degeneration often coincides with the onset of chronic ER stress, which is triggered by the accumulation of misfolded rhodopsin and/or other proteins within rod cells.680,681 For this reason, many efforts to develop therapeutics for the treatment of adRP and related conditions have focused on correcting the underlying molecular defects responsible for the lapse of protein homeostasis within rod cells.682
The discovery of three seemingly unrelated missense mutations in the rhodopsin gene in adRP patients provided the first clinical evidence linking defects in the rhodopsin protein to the molecular basis of the disease.683 Subsequent investigations of RP mutations revealed that they induce a spectrum of defects in the cellular processing and/or function of the rhodopsin protein.684 Some RP variants disrupt binding of the protein’s 9-cis-retinal cofactor or decouple the photoisomerization of retinal from the activation of transducin (Class I).684,685 However, the lion’s share of RP mutations appear to compromise the stability of the protein in a manner that attenuates its export from the endoplasmic reticulum (Class II) and/or reduce its accumulation within the cell (Class III).684,685 Many class II mutants disrupt native tertiary contacts that impart stability to the native rhodopsin fold.686 Others are likely to perturb the early steps of translocon-mediated cotranslational folding.687 Folding-defective forms of rhodopsin are targeted by ERQC for degradation, with both the proteasomal and aggresome/autophagy pathways being active.562,688 The extent to which individual RP mutations destabilize the folded form of rhodopsin appears to be correlated with the age of onset as well as the severity of other pathogenic phenotypes,689 though parsing mutations according to the severity of their corresponding conformational defects remains a pressing challenge.629,682 Nevertheless, the effects of certain RP mutations in model cellular systems can be partially offset by the stabilization afforded by excess 9-cis-retinal cofactor or other retinal analogs.690−693 Gaging the true therapeutic utility of these compounds remains a challenge given both the native cellular physiology of rod cells and because trafficking of rhodopsin proteins within these cells is distinct from that in more easily studied transfected model cell lines that are amenable to screening.682 Despite this caveat, there remains considerable optimism regarding the possibility of developing adRP therapeutics that directly target the folding defects in rhodopsin.
When referring to the stability or instability of rhodopsin in the above section, the “folding stability/instability” that is being referred to has been either computationally estimated or reflects experimental measurement of thermal stability, rather than experimentally measured thermodynamic stability. A particular challenge when considering how mutations in rhodopsin and other GPCRs impact protein folding is that it has thus far not been possible to experimentally measure the true thermodynamic folding stability for any GPCR, WT or otherwise. This is not from want of trying. Rather, reversible refolding from the unfolded state has proven elusive for reasons that are not well understood. If induced unfolding is not fully reversible, thermodynamic stability cannot be measured. GPCRs represent a prominent family of proteins for which it is hoped that the emerging “next generation” methods for assessing MP stability (i.e., steric trap, optical tweezers, etc.) may lead to a welcome breakthrough.
5.4.4. Peripheral Myelin Protein 22 and Charcot-Marie-Tooth Disease
Mutations affecting the PMP22 gene are the leading cause of the debilitating peripheral neuropathy CMTD as well as the related dysmyelinating disorders Djerine-Sottas syndrome (DSS, severe) and hereditary neuropathy with liability to pressure palsies (HNPP, mild).433,694,695 Patients with CMTD suffer from clinical symptoms ranging in severity (depending on the causative mutation), including impaired tendon reflexes, progressive weakness and atrophy of the distal musculature, abnormalities of peripheral nerves and its adjacent myelin sheath, and—in the most severe cases—blindness, auditory loss, and confinement to a wheelchair.433,696,697 Disease symptoms are thought to be the consequence of abnormal myelin production and assembly by the myelin-producing Schwann cells of the PNS. PMP22 is one of the most abundant proteins in compact myelin, where it is believed to play a structural role.166 However, PMP22 is likely also involved a number of other processes within Schwann cells including cellular proliferation, differentiation, and cell death.166,433,695,698
PMP22 is a tetraspan integral MP, and was the first multispan eukaryotic MP for which thermodynamic stability of folding was measured.699 Spectroscopic studies revealed that the conformational stability of WT PMP22 is strikingly modest in detergent micelles: the native conformation is favored over the denatured ensemble by only 1.5 ± 0.1 kcal mol–1 in the presences of stabilizing osmolytes. Such marginal stability perhaps accounts for why most of the nascent protein is rapidly degraded within the ER following biosynthesis; only ∼20% of the nascent protein manages to fully mature and traffic to the plasma membrane.598,599 The rest is either degraded by the proteasome or deposited into aggresomes.433,448
The most common (type 1A) form of CMTD (CMT1A) arises from a heterozygous duplication of the chromosome 17p.11–2.12, which results in trisomy (three copies) of WT PMP22. CMT1A is a common inherited disordered (1:5,000 people).433,694,695 The exact mechanism underlying the pathogenicity associated with expression of third copy of PMP22 is not yet clearly established. However, it has been hypothesized that the elevated expression in conjunction with the instability of the PMP22 protein imposes a heavy burden on ERQC, causing proteotoxic stress and the formation of aggresomes.558,699 While aggresomes are not toxic if they are properly engaged by the autophagy pathway, the activity of the autophagy pathway is believe to decline with age,700 which could lead to chronic accumulation of PMP22 aggresomes.558,701 This may account for the fact that CMT1A patients only exhibit disease symptoms later in life even though they are born with the causitive mutations. This model for the etiology of CMT1A, if correct, provides an example of a disease related to MP misfolding that is caused by a combination of both toxicity of the misfolded protein and loss of native function.
More rare forms of Charcot-Marie-Tooth disease (recently dubbed CMT1E694) as well as the related DSS and HNPP are caused by heterozygous expression of WT PMP22 in combination with missense variants of PMP22, over 35 of which have been identified to date. Experimental correlations between conformational stability, trafficking efficiency, and disease severity have been demonstrated for a cross-section of these variants.287 The thermodynamic stability of these variants appears to be directly correlated with the efficiency of cellular trafficking: the efficiency of protein folding scales with trafficking to the plasma membrane (Figure 24). The degree of PMP22 destabilization also correlates linearly with patient nerve conduction velocities, which serve as quantitative clinical readouts of disease severity (Figure 24).287 Mutations causing the mild HNPP phenotype were the least destabilizing, while mutations causing the severe DSS were the most destabilizing. These results point to the thermodynamic stability of PMP22 as being the prime determinant of the maturation and trafficking of PMP22 in the cell. ERQC evidently recognizes some conformational trait of misfolded PMP22 in the ER that scales with the stability of the native tertiary structure. Figure 24 shows the structural locations of the PMP22 mutants whose stabilities were probed, revealing that most of the severe mutations are for sites located in the interior of the TM domain, where side chains interact mostly with other TM sites rather than with the lipid phase.702
Figure 24.

Thermodynamic destabilization of human PMP22 results in mistrafficking of the protein and Charcot-Marie Tooth disease (peripheral neuropathy). Panels A and B show the locations of the disease mutants in the sequence and modeled 3-D structure of the protein, respectively. Panel C shows a strong correlation between surface trafficking efficiency and stability across the panel of tested PMP22 mutants. Panel D shows that the extent of surface trafficking correlates well with nerve conduction velocity in humans carrying each mutant form. Healthy patients present with high conduction velocities, with reductions in conduction velocity correlating with disease severity. Panel E shows that there is also a strong correlation between the stability of PMP22 and nerve conduction velocity. Figures adapted with permission from ref (287). Copyright 2015 ACS.
Interestingly, a heterozygous deletion in the chromosome bearing the PMP22 gene also causes a mild disease phenotype (HNPP).433 Disease in these patients arises from the lack of a second allele and the resulting deficiency of WT PMP22 expression. This mild disease phenotype is actually less severe that those arising from the heterozygous expression of missense variants (CMT1A and DSS patients).695 There are two possible explanations that may contribute to this difference. First, a toxic gain-of-function due to formation of mutant PMP22 aggregates may exacerbate the partial loss of PMP22 expression in heterozygotes expressing a single mutant variant in combination with WT. Second, certain mutant forms of PMP22 expressed in heterozygotes are capable of forming nonproductive oligomers with the WT protein.703 Recognition of these non-native oligomers by ERQC may cause a dominant negative effect leading to the degradation of the WT protein, which may further reduce the abundance of the WT protein.
Exactly how the misfolding of PMP22 is managed by ERQC is not yet well understood, but there are some clues. BiP, calreticulin, and ERp57 do not appear to be important for the maturation of PMP22.430,497 However, there is evidence that calnexin may serve as both a PMP22 chaperone and folding sensor.430,443,448,497 As noted earlier, calnexin binds WT PMP22 under cellular conditions with a half-life of about 11 min, an interaction that depends on the presence of PMP22’s single N-linked glycan. However, calnexin sequesters the severely misfolded L16P PMP22 mutant (“Trembler-J”) with a half-life of more than 1 h. This interaction appears to specifically involve TM1, which includes the mutated residue. Interestingly, binding of calnexin to L16P PMP22 also appears to occur in a manner that is independent of the glycosylation state of the mutant protein.443 NMR structural studies have shown that this mutant samples a conformational state in which TM1 is dissociated from the other three TM helices, a conformational state that may be detected by calnexin (Figure 6).182 However, it cannot be ruled out that calnexin may instead recognize the swiveling kink introduced into TM1 helix by the L16P mutation. It is interesting to note that cnx – /– calnexin knockout mice are viable but display abnormalities in their peripheral nerves, potentially highlighting the role that calnexin plays in managing folding and misfolding of PMP22 and perhaps other myelin MPs.704 It is also noteworthy that misfolded PMP22 variants that escape the ER are retrieved from the Golgi complex and returned to the ER through the action of the Rer1 protein,448 while inducible cytosolic Hsp70 may be involved in shepherding misfolded PMP22 molecules through the lysosomal degradation pathway.705
5.4.5. Myelin Protein Zero and Charcot-Marie-Tooth Disease
The single-pass MP known as myelin protein zero (MPZ) is the most abundant protein found within the compact myelin of the PNS, and makes up 20–50% of the total protein content in these tissues by mass.296,706 MPZ forms a homotetramer within the myelin membrane and functions as “molecular glue” between spiraled myelin membranes.296 Adhesion is accomplished through “trans” homophilic interactions that are mediated by its extracellular domain. MPZ also interacts with juxtaposed PMP22 molecules in a similar fashion, although the role of these interactions in myelin compaction are currently unclear.497 Over 200 different mutations in MPZ that cause Charcot-Marie-Tooth Disease type B (CMT1B) have been identified to date. The severity of the neuropathy depends on the specific mutation, though CMT1B phenotypes are generally similar to those of CMT1A and range from dys- or demyelination and attenuated nerve conduction velocities to severe axonal defects.707
Most of the pathogenic mutations in MPZ reside within its extracellular domain, although a few TM domain variants have also been identified, including G163R and G167R. It has been shown that a peptide corresponding to the TMD of WT MPZ homodimerizes as driven by its GxxxG163xxxG167 glycine zipper motif. The G163R disease mutation was seen to disrupt homodimerization, which likely is the root cause of how it disrupts MPZ assembly and function in myelin.708 It is also known that many disease mutations in MPZ cause the protein to be accumulate in the ER, triggering stress and activating the UPR.709,710 Indeed, the ablation of pro-apoptotic factors that result from ER stress ameliorates the neuropathic phenotype in mouse models.711,712 Efforts to increase MPZ trafficking out of the ER or to suppress its aggregation may be useful in treating many patients with CMT1B.
5.4.6. Myelin Proteolipid Protein and Pelizaeus-Merzbacher Disease
Myelin proteolipid protein (PLP) and its shorter splice variant lacking 35 residues in its intracellular loop (DM20) make up about 50% of the total protein content in compact myelin of the central nervous system (CNS).713 PLP is expressed in oligodendrocytes, and is composed of four TM domains and two sizable extracellular loops. Its intracellular loop is post-translationally lipidated, which further increases its hydrophobicity.296 The specific function of PLP has not been fully elucidated, but seems likely that it plays a central role in both the assembly and stability of CNS myelin.714,715 Gene duplication and a variety of missense mutations in the gene encoding PLP (PLP1) lead to demyelination in the CNS, a pathology associated with Pelizaeus-Merzbacher disease (PMD).716
Much like PMP22 and MPZ, different missense mutations in PLP cause a broad range of disease phenotypes. Most disease mutations in PLP disrupt its cellular trafficking and cause accumulation of the protein in the ER.709 During myelination, oligodendrocytes produce a high quantity of both lipids and myelin proteins, including PLP, which requires the secretory pathway to operate at maximum capacity. Mutations that induce PLP misfolding or increased expression levels (e.g., due to gene duplication) can overload the secretory pathway and saturate the ERAD pathway, inducing ER stress and activation of the UPR.717,718 Perpetual UPR activation can lead to the induction of CHOP-mediated pro-apoptotic pathways that ultimately promote demyelination. PLP variants that are efficiently degraded by ERAD cause a much milder clinical phenotype than those that accumulate and aggregate within the ER.719 Collectively, the evidence suggests that efforts to improve the degradation of PLP in patients suffering from PMD may alleviate disease severity.
5.4.7. Cystic Fibrosis Transmembrane Conductance Regulator
Cystic fibrosis (CF) is a lethal monogenic LOF disease that impacts more than 85 000 individuals worldwide.720 CF is caused by an array of mutations in the CFTR, an ATP-binding cassette (ABC) transporter that functions as a Cl–/HCO3– channel.721−723 Most CF mutations disrupt the folding, trafficking, and/or activity of the CFTR channel protein.724,725 The resulting loss of CFTR function causes irregularities in the pancreas, intestines, sweat glands, and lung epithelia.726 Clinical manifestations include malnourishment, increased sweat chloride concentration, and chronic pulmonary infection, the latter of which is often fatal. An imbalance in the salinity and a corresponding thickening of the airway surface liquid within the lungs of CF patients disrupts the mucociliary escalator, which normally clears debris from the lungs.363,727 Accumulation of this debris over time eventually leads to infection, hyperinflammation, scarring, and a loss of pulmonary fitness.728 Given the underlying molecular mechanisms of CF, drug discovery efforts have mostly focused on the identification of compounds that restore function to mutant CFTR channels.
CFTR is a complex, multidomain protein comprised of a pair of membrane spanning domains (MSD1/2, 6 TM helices each), two cytosolic nucleotide binding domains (NBD1/2), and an unstructured regulatory region (R domain) that is unique among ABC transporters.729 A series of cryo-EM structures of CFTR in different functional states has recently been published (see Figure 25).729−732 The ca. 2000 known pathogenic mutations within the CFTR gene are distributed across all five domains, which is a classical predictor that most induce misfolding.733,734 Nevertheless, an examination of CF mutations in conjunction with the emerging structural insight has revealed that several of the most clinically abundant mutations cluster at the interface between NBD1 and MSD1, which highlights the importance of this interface to the conformational stability of the channel.733 CFTR mutations have been classified according to whether they compromise the biosynthesis (type I), folding (type II), function (types III and IV), expression level (type V), or turnover (type VI) of the protein.735,736 However, as the effects of these variants have been studied in greater detail, it has become clear that many CF variants induce compound defects.736 For instance, the ΔF508 mutation present in ∼90% of CF patients primarily compromises CFTR folding, yet also exhibits secondary functional deficits when exported to the plasma membrane.737
Figure 25.
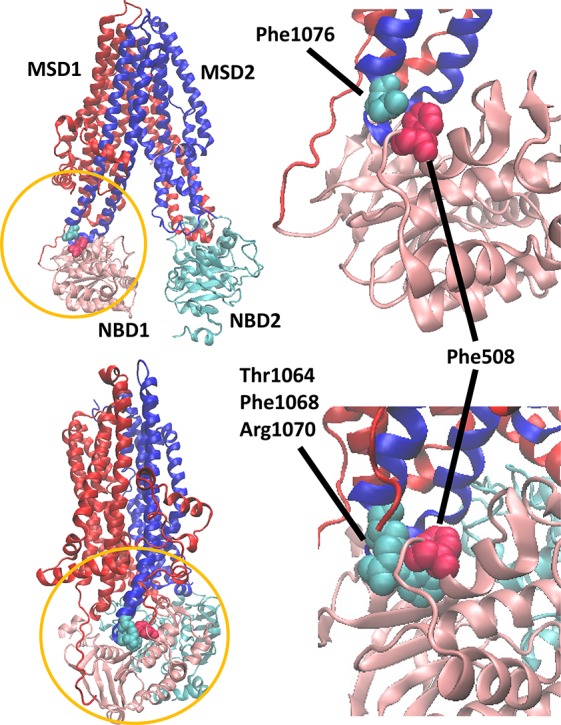
Structures of CFTR in its resting conformation (top) and in its phosphorylated+ATP-bound state (bottom). The side chain of Phe508 shown in van der Waals form (red) along with the interacting residues (cyan) from the hairpin connecting the 10th and 11th TM helices (blue). PDB: 5TSI (top) and 6MSM (bottom).
Although the exact nature of the conformational defects in CFTR have yet to be unambiguously mapped, it is clear that the fidelity of both cotranslational and posttranslational folding processes are central for the assembly and accumulation of mature CFTR at the plasma membrane.254,738−740 The formation of interdomain contacts appears to be a key mediator of CFTR folding and stability.254,741 Notably, the primary structural defect associated with the ΔF508 mutation is the decoupling of the interactions between NBD1, MSD1, and NBD2.742−744 The location of ΔF508 in two different functional states of CFTR is highlighted in Figure 25. Though the dynamics of interdomain coupling may be rapid in the context of full-length proteins, it should be noted that initial interdomain interactions must form slowly during biosynthesis and cotranslational folding of the CFTR molecule given its size (1480 residues) and the rate of eukaryotic translation (∼6 AA/s). Indeed, maturation of CFTR in the cell requires up to 2 h and hinges on the concerted activities of a wide array of molecular chaperones.425−427 Furthermore, the introduction of the ΔF508 mutation has been reported to lead to the formation of over 200 new protein–protein interactions.427,745 In addition to the formation of new QC interactions, the decoupling of interdomain interaction by the ΔF508 mutation also appears to change the stoichiometry of the interactions between CFTR and various core cytosolic chaperones including Hsp40, Hsp70, and Hsp90.746 These perturbations can have dramatic proteostatic consequences, as changes in the interaction of CFTR and Hsp70s appear to coincide with the recruitment of E3 ubiquitin ligases that tag the protein for retrotranslocation and proteasomal degradation.747 Under conditions of proteotoxic stress, misfolded CFTR molecules form large aggresomes, which are eventually cleared through autophagy.555 Though the chain of molecular events involved in the misfolding, mistrafficking, and premature degradation of pathogenic CFTR mutants is obviously complex and multifaceted, correctors and potentiators partially restore some native protein–protein interactions to certain variants (see section 6.3.6).745 Thus, approaches to correcting the underlying conformational defects may be sufficient to restore order to the complex network of interactions involved in CFTR biosynthesis. Together, these insights into the pathophysiology and pharmacology of CF provide a benchmark for ongoing efforts to identify and target other disease-linked integral MPs.
5.5. The Most Common Defect of Disease-Linked MPs Appears to Be Destabilization of Native Structure
From studies of both model and disease-linked MPs (examples above), it seems to be the case that the most common defect leading to misfolding is rooted in thermodynamics: destabilization of the native state.283,284,418,592,593 It should also, of course, be emphasized that some disease mutations operate via mechanisms that are unrelated to misfolding. For example, while studies of LQTS mutations in KCNQ1 revealed that destabilizing mutations that lead to misfolding are by far the most common single class of disease mutations, there are less common disease mutants that cause loss of channel function without altering folding and/or trafficking.183
The notion that many mutations promote disease through a reduction in protein stability may be good news from a therapeutic standpoint for two reasons. First, it implies that a single drug that acts by stabilizing protein structure could potentially be used to treat most patients carrying any one of a series of destabilizing mutations in the target protein. Second, the fact that energetic perturbations associated with the disease mutant forms are modest suggest that drugs need not be “super-stabilizers”, a modest enhancement in stability conferred by drug binding may often be all that is needed to restore native-like folding efficiency. In the following section we turn our attention to therapeutics development based on targeting membrane protein instability and misfolding.
6. Small Molecule Manipulation of Membrane Protein Folding
Roughly 30 years ago, the discovery that many diseases are mechanistically linked to defects in the MP folding and trafficking led to the first efforts to “rescue” misfolded MPs. Early on, it came to be appreciated that protein folding efficiency could be altered under cellular conditions by varying the temperature or using chemical chaperones (usually at high concentrations), approaches that are reviewed in section 6.1 below. This early work eventually led to the discovery of compounds that correct the folding defects of specific proteins through direct interactions: pharmacological chaperones (PCs, see sections 6.2 to 6.5). The concept of cellular “proteostasis” networks emerged in subsequent years, followed soon after by the development of small molecule proteostasis regulators that indirectly promote protein folding by tuning the activity of the proteostasis network (i.e., upregulation of the UPR). Proteostatic optimization can perhaps be regarded as “a high tide that lifts all boats” (see section 6.6). For the sake of completeness, we note that the destabilizing effects of certain mutations can be compensated for by suppressor mutants, though this observation offers no clinical utility.748−750 Diseases of MP misfolding could also be hypothetically repaired using CRISPR/Cas9 technology, although the timetable associated with the application of this technology remains unclear for technical and ethical reasons.
6.1. Use of Chemical Chaperones to Rescue Misfolded Membrane Proteins
Misfolded proteins that are engaged by ERQC and other cellular quality control systems are typically aggregated and/or degraded before reaching the cellular compartments in which they function. However, some misfolded MP variants, including ΔF508 CFTR, retain residual function if they manage to escape QC. Accordingly, there can be functional benefit to “rescuing” the cellular trafficking of misfolded MPs. A variety of approaches to restore trafficking of misfolding-prone variants have been evaluated in cultured cells. Temperature often has a profound impact on folding efficiency in the cell. A reduction in growth temperature from 37 °C to room temperature markedly improves the trafficking of pathogenic variants for a variety of proteins including CFTR,751 luteinizing hormone receptor (LHR),752 the hERG channel,753 and the V2R.617 Given that protein stability is typically maximal at 25 °C, at least for soluble proteins, this observation again suggests a decrease in conformational stability as a key driver of cellular mistrafficking. Furthermore, stabilizing osmolytes including dimethyl sulfoxide (DMSO), trehalose, trimethylamine-N-oxide (TMAO), and glycerol also restore cellular trafficking to certain variants. These “chemical chaperones” are cosolvents that stabilize compact protein conformations to minimize their repulsive interactions with the peptide backbone.754 It has also been suggested that osmolytes may also enhance the activity of endogenous molecular chaperones to suppress aggregation.755−757 Despite the revealing nature of these observations, they offer little to no direct clinical utility. The high concentrations of osmolytes that are needed to stabilize proteins also render them clinically bereft. It should be added that some compounds originally identified as “chemical chaperones” impact protein folding by indirect means and are better classified as proteostasis modulators (see section 6.6). These include 4-phenylbutryate and docosahexaenoic acid.
6.2. Pharmacological Chaperones and Their Mechanisms of Action
Because native binding pockets are typically disrupted in unfolded or misfolded states, the binding of small molecules usually stabilizes the native state. This intrinsic linkage between binding and folding can be exploited to restore folding and trafficking to destabilized variants. A wide variety of small molecule pharmacological chaperones (PCs), which are also sometimes referred to as “correctors” or “pharmacoperones,” have been developed as potential therapeutics.227,758−761 These molecules come in many shapes and sizes, but share the properties of being able to permeate cell membranes and specifically bind a target protein with high affinity. Indeed, even low concentrations of a potent inhibitor of native function can result in a partial recovery of activity by binding and stabilizing the folded protein to increase the yield of mature protein.762 For PCs that inhibit MP function, rescue of folding and trafficking does, of course, also require that the PC then dissociate from the correctly folded and trafficked target. This likely requires both that the dissociation rate of the PC-MP complex not be too slow and also that excess free compound be cleared from the physiological milieu following “rescue” of the target (so that saturation of binding is not chronically maintained).
To date, small molecules that behave as PCs have been identified for a variety of GPCRs, channels, enzymes, and other MPs (below). The therapeutic potential of PCs has also been evaluated in cellular assays, animal models, and, in a few cases, successful clinical trials. The development of stabilizing molecules offers an attractive and generalized approach to treat diseases of MP misfolding, some of which were previously regarded as undruggable.763 Small molecule PCs may also provide a safer alternative to riskier therapeutic approaches like gene therapy.764
PCs bind, stabilize, and promote the functional maturation of nascent MPs in the ER membrane. Depending on the location of the relevant binding pocket, some compounds may bind to the cytosolic face of the target protein, while others likely permeate the ER to reach and bind to the luminal/extracellular domain. Given that most binding pockets are formed through the association of a complete set of TM segments, it seems likely that most of these compounds act on the nascent protein after it clears the translocon. Eukaryotic proteins are thought to be synthesized on the minute time scale given the average translation rate of 5.6 amino acids per second in higher organisms.765 This is a long time relative to the kinetics of molecular association to form a complex. Nevertheless, the time required for translation typically pales in comparison to the half-lives of nascent MPs in the ER, which range from roughly 15 min to hours.620,766−768 Thus, cotranslational folding intermediates are likely short-lived in relation to later folding intermediates for the fully translated protein. In support of this notion, it has been demonstrated that certain PCs still promote the maturation of nascent MPs after inhibition of protein synthesis, suggesting that these compounds primarily act post-translationally on full length proteins.769−771
There are at least 4 mechanisms by which PCs could influence MP folding in the cell. (1) In the simplest, their efficacy effects stems from their selective binding and thermodynamic stabilization of the native state. (2) PCs could also potentially bind and stabilize on-pathway folding intermediates to accelerate the folding reaction. Indeed, some metabolites may also accelerate folding this way.772 There may be some overlap between mechanisms 1 and 2, as certain folding intermediates may contain a partially or fully formed binding pocket that is also present in the native conformation.773 (3) The PC may act to prevent conversion of the target MP into toxic oligomers or aggregates, a mechanism that could overlap with mechanisms 1 and/or 2 above. (4) The stabilization of monomeric forms by PCs could facilitate the disaggregation of toxic oligomers. The distinction between mechanisms 3 and 4 could potentially arise from the extreme kinetic barriers involved in certain aggregation processes.
Of these four mechanisms, thermodynamic stabilization of the native state is probably the most common (Figure 26). This is supported by the fact that many agonists and antagonists known to bind the native form also act as PCs. The efficacy of PCs is also typically proportional to binding affinity,774−776 which is thermodynamically linked to the folding equilibrium (Figures 8 and 26).777,778 Indeed, for at least one PC it was recently demonstrated that there is no relationship between its PC activity and its agonistic or antagonistic effects on function,779 which is consistent with the interpretation that PC activity is directly linked to binding energy. One aspect of this mechanism that is less clear is how PC-mediated stabilization plays out in the context of cellular compartmentalization. Is stabilization only relevant at the stage of maturation of nascent proteins in the ER? The conformational stability of MPs in the ER membrane could potentially be lower as a consequence of the low cholesterol content of that membrane (Figure 3). Moreover, nascent MPs are also more likely to be poked and prodded by chaperones in the ER, which may effectively destabilize these proteins by virtue of their selective interaction with misfolded conformations.780 Next-generation experiments are needed to tease out the effects of these compounds on MPs within specific cellular compartments.
Figure 26.

Hypothetical scenario of action for a PC as a drug. In this case the PC selectively binds to and stabilizes the folded form of the with target MP, tipping the balance between correct folding/trafficking and ERAD degradation in favor of folding and trafficking. The initial binding/rescue event occurs shortly after administration of the PC as a drug, at which point the PC concentration is fairly high. Once the MP reaches the plasma membrane and the total PC concentration is cleared (due to cytochrome P450 action, for example) the PC will dissociate and not be replenished, at which point the protein remains mostly folded because it is thermodynamically stable in the plasma membrane.
Compounds that reduce the kinetic barriers to folding are likely to be relatively rare given that the transition states for membrane protein folding are potentially disordered (see section 3.4). Nevertheless, this mechanism has for some cases been suggested. For instance, an allosteric agonist (NPS R-568) of the calcium-sensing receptor is believed to act on nascent proteins during or shortly after protein synthesis based on its effects on biosynthesis and stability. By comparison, an allosteric antagonist of this protein (NPS-2143) promotes protein degradation.781,782 A similar observation has been made for the nicotinic acetylcholine receptor, for which nicotine promotes a critical subunit–subunit interaction that enhances protein maturation.783 Nevertheless, the unambiguous differentiation of the first two mechanisms will likely require kinetic and thermodynamic measurements on purified proteins in the presence and absence of these compounds.
Compounds that principally serve to suppress aggregation (mechanisms 3 and 4) have long been sought for the treatment of various amyloidogenic disorders, such as Alzheimer’s disease.784−789 Interestingly, the PC known as Fe-TMPyP appears to reshape the energy landscape of the protease-resistant fragment of the prion protein.790 Force spectroscopy measurements revealed that this compound binds to non-native states to suppress aggregation, specifically, by reducing levels of a specific dimeric intermediate leading to aggregation of the prion protein. This effectively increases the stability of the native state by increasing the kinetic barriers to unfolding (Figure 8). It is unclear how common this mechanism of action may be. Nevertheless, these studies make it clear that there are multiple ways by which small molecules might correct conformational defects.
6.3. Protein-Specific Small Molecule Pharmacological Chaperones
6.3.1. P-Glycoprotein
Some of the very first PCs to be identified target the P-glycoprotein (PGP, MDR1).791 PGP, an ABC transporter, is an ATP-dependent efflux pump that exhibits little substrate specificity. PGP is notorious for its ability to promote multidrug resistance in cancer cells. Both substrates and modulators of PGP act as PCs in a manner that enhances the maturation and trafficking of destabilized PGP variants. Administration of these compounds alters the processing of the protein within a few hours. Given that the activity of PGP promotes cancer drug resistance, there is no obvious therapeutic utility for PGP PCs. Nevertheless, these early studies helped to establish the generality of the linkage between binding, folding, and maturation within the cell.
6.3.2. Vasopressin V2 Receptor and Diabetes Insipidus
As noted earlier V2R is a GPCR expressed in the kidney that, when stimulated by the vasopressin hormone, triggers water reabsorption into the bloodstream and concentration of urine to optimize blood osmolarity. A variety of mutations in V2R cause X-linked nephrogenic diabetes insipidus (see Figure 19) in which patients fail to properly reabsorb water, resulting in salty blood and watery urine. Over 90% of these disease mutations induce destabilization/misfolding of V2R.618
In cellular studies, a number of small molecule antagonists (VPA985, YM087, SR121463A, SR121463B, SR49059, OPC31260, and OPC41061) and biased agonists (MCF14, MCF18, and MCF57) have been found to enhance the folding and trafficking of pathogenic V2R mutants (Figure 27).616,619,775,792−796 Small molecule screening has identified additional molecules that act as PCs, though it is unclear how these compounds influence the activation state of V2R.799 Only one such PC has been evaluated in short-term NDI clinical trials to date.616 A 24 h administration of SR49059 in NDI patients bearing V2R mutations significantly decreased patient urine volume, increased urine osmolarity, and increased water intake into the bloodstream, all suggesting a significant clinical benefit in these patients. However, efforts to develop this particular compound were discontinued due to its unfortunate hepatic toxicity. Despite this setback, the encouraging results for SR49058 bode well for the potential utility of PC therapy for the treatment of NDI.
Figure 27.

Structures of PCs for the V2 vasopressin receptor. The apparent affinities of each ligand for the WT V2R are indicated.793−796
6.3.3. Gonadoptropin-Releasing Hormone Receptor and Hypogonadotropic Hypogonadism
The gonadotropin-releasing hormone receptor (GnRHR) is a GPCR expressed in the pituitary gland that has proven to be an excellent model for the study of PCs, largely through work of the lab of the late P. Michael Conn. Mutations in GnRHR cause hypogonadotropic hypogonadism, a condition in which patients fail to enter puberty. GnRH peptidomimetic antagonists from four different chemical classes including indoles, quinolones, thienopyrimidinediones, and erythromycin-derived macrolides, have been comprehensively studied and documented to rescue misrouted GnRHR mutants (Figure 28).774,800−804 All peptidomimetics studied that exhibited an IC50 value of <2.4 nM had measurable effects on the maturation of several different pathogenic GnRHR variants.774 Investigations in cultured cells were extended to a knock-in mouse model expressing the pathogenic E90K variant.805 Pulsed administration of the experimental PC IN3 using a catheter to directly inject the compound into the pituitary generated significant rescue of misfolded GnRHR. Furthermore, the rescued receptor exhibited a functional response to GnRH stimulation. This treatment also corrects steroidogenesis and spermatogenesis in mice, demonstrating considerable efficacy in the context of this mouse model.
Figure 28.

Structures of PCs for the gonadotropin-releasing hormone receptor. The apparent affinities of each ligand for the WT GnRHR are indicated.774,804
6.3.4. Melanocortin-4 Receptor and Morbid Obesity
The MC4R, another GPCR target, is critical for the regulation of energy homeostasis. Mutations in MC4R cause a severe form of early onset obesity.806,807 Of the many MC4R variants identified in humans to date, 45% impair the cellular maturation and trafficking of the receptor.776,808,809 An agonist and several antagonists have been found to enhance the trafficking and function of misfolded MC4R variants (Figure 29).776,810−812 The potency of these compounds appears to be related to their binding affinities. For example, ligands with high affinity such as Ipsen 5i (Ki, 2.0 nM) and Ipsen 17 (Ki, 0.96 nM) exhibit rescue EC50 values in the nanomolar range, whereas ligands with lower affinity such as ML00253764 (Ki, 0.17 μM) and DCPMP (Ki, 0.02 μM) exhibit rescue EC50 in the micromolar range.776,813−815 Of special note, Ipsen 5i exhibits broad efficacy; 21 of 23 mutants studied could be partially rescued by this compound.776,815 Only two nonsense mutants that truncate TM7 or the intracellular C-terminal domain were unresponsive. The recovered function exhibited by 14 of these mutant receptors generally scales with the increased abundance of the receptor at the plasma membrane, indicating that the rescued receptors retain their ability to signal. The efficacies exhibited by most of these PCs were independent of the cell line employed. For example, the antagonists ML00253764 and Ipsen 5i have similar effects in different cell lines, although the agonist THIQ was more effective in neuronal cell lines (rescuing 7 of 10 mutants studied) than in HEK293 cells (rescuing 3 of 10 mutants studied).776,815,816 The EC50 values associated with THIQ (IC50, 1.2 nM) also vary from 10 nM to 1 μM among MC4R mutants.816 Interestingly, THIQ also improves the function of two MC4R mutants without improving their trafficking. This peculiar observation could reflect an increased signaling efficiency for those receptors upon reaching the plasma membrane. Although these compounds show very good PC activity in model cell lines, their physiological utility has yet to be tested.
Figure 29.

Structures of PCs for the melanocortin-4 receptor. The apparent ligand affinities for the WT MC4R are indicated.810−812
6.3.5. hERG, SCN5A, and Cardiac Arrhythmias
The pore-forming subunit of the hERG voltage-gated potassium ion channel mediates the cardiac repolarizing IKr current, and a variety of LOF mutations in hERG give rise to LQTS type 2 cardiac arrhythmia (see section 5.4.2). A series of hERG channel blockers that include E-4031, astemizole, cisapride, quinidine, and ranolazine also appear to exhibit PC activity toward some hERG variants.753,770,817 However, trafficking defects exhibited by variants bearing mutations in the C-terminal domain typically fail to respond to these compounds, probably because this domain folds autonomously relative to the rest of this multidomain channel.817
Surprisingly, VX-809 (lumacaftor), a small molecule approved for the treatment of CF patients (see below), also appears to enhance the maturation of misfolded hERG mutants in induced pluripotent stem cell-derived cardiomyocytes.818 Similar observations for certain other PCs have also been reported for other pairs of proteins. For instance, 1-deoxygalactonojirimycin was developed as a PC for alpha-galactosidase but also acts as an PC for alpha-N-acetylgalactosaminidase.819 The sodium channel blocker carbamazepine also exhibits cross-reactivity toward ATP-sensitive potassium (KATP) channel mutants.691 Some degree of cross-reactivity is not surprising considering these compounds likely target native binding pockets for metabolites or signaling molecules. Depending on the nature of the screening employed, it may also be difficult to differentiate compounds that exhibit PC activity from those that actually act as proteostasis modulators.
Another arrhythmia-linked ion channel is the cardiac sodium channel Nav1.5, which is encoded by the SCN5A gene that mediates the fast depolarization of the cardiac action potential. Pathogenic LOF variants of the Nav1.5 channel are associated with Brugada syndrome and cardiac conduction disease.820 The antiarrhythmia drugs mexiletine, quinidine, and flecainide improve the functional expression and trafficking of some misfolded Nav1.5 variants.821,822 However, mexiletine and the IKr blocker cisapride also exacerbate the pathogenic phenotype associated with the torsades de pointes form of LQTS, which is associated with an augmented late inward sodium current.823,824 Such SCN5A mutants implicated in type 3 LQTS appear to induce toxic GOF. These variants cause complex variations in electrophysiology such as a coincident delay in INa and a reduction in peak INa currents.823,824 These observations highlight the fact that even the most successful PCs will not necessarily address the defects associated with all disease mutant forms they target because not all disease mutations operate by promoting misfolding. Moreover, some channelopathies feature both pathogenic LOF and aberrant GOF mutations associated with a spectrum of diseases. Optimal clinical use of PCs will therefore require data on the disease-promoting mechanism of each mutation in the target protein so that PCs are targeted to those patients carrying mutant forms likely to be responsive to PC treatment. The spectrum of disease mutations of SNC5A provides a clear example of this emerging challenge in precision medicine.
6.3.6. Cystic Fibrosis Transmembrane Conductance Regulator
The most successful PCs developed to date are those that correct the misfolding and mistrafficking of CFTR variants associated with the recessive genetic disorder CF. Though over 2000 CFTR mutations have been identified in CF patients to date, ∼90% of CF patients carry ΔF508 CFTR.725 The enrichment of the ΔF508 allele in humans of European ancestry may be the consequence of natural selection, as heterozygous expression of this mutant (WT/ΔF508 conditions) has been suggested to confer resistance to certain infectious diseases.825,826 Historically, CF treatments have been symptomatic; with saline breathing treatments, supplemental digestive enzymes, and antibiotics typically being employed to address respiratory and gastrointestinal dysfunction.726,827 However, building on pioneering early work with chemical chaperones and prototype PCs, the past decade has seen remarkable progress in the development of CFTR-targeting small molecules aimed either at repairing channel gating defects (“potentiators:, Figure 30A) or conformational/folding defects (PCs, also referred to as “correctors”). Figure 30B).737,828−834 RDR1 rescues ΔF508 CFTR at micromolar concentrations.835 Chief among these compounds are three medications developed by Vertex that have been approved for clinical use by the FDA since 2012: the potentiator Ivacaftor (VX-770, kalydeco) and the correctors Lumacaftor (VX-809) and Tezacaftor (VX-661). The administration of these and other emerging compounds is transforming treatment and management of CF.
Figure 30.

Structures of functional potentiators (A) and PCs (B) for the CFTR channel. The apparent ligand affinities or rescue potencies of each compound for the ΔF508 or G551D mutant forms of CFTR are indicated.737,829−834 RDR1 rescues ΔF508 CFTR at micromolar concentrations.835
The potentiator Ivacaftor was the first clinically approved CF therapeutic designed to treat the molecular basis of the disease. This compound has proven transformative for a subgroup of CF patients that express CFTR variants that traffic to the plasma membrane but exhibit functional defects. For example, Ivacaftor generates a lasting decrease in sweat chloride concentration, weight gain, and an improved pulmonary function as indicated by an increased forced expiratory volume in patients expressing G551D CFTR, which causes a channel gating defect.653,829,836−838 However, Ivacaftor is ineffective toward misfolded variants like ΔF508 CFTR, which fail to traffic to the plasma membrane. Furthermore, chronic administration of Ivacaftor increases the turnover of both ΔF508 and WT CFTR at the plasma membrane by stimulating interactions between CFTR and the RFFL E3-Ubiquitin ligase.525,839,840 Rescue of complex variants like ΔF508, which exhibit deficient trafficking and function, instead requires the combination of a potentiator and one or more PCs. Lumacaftor, the first clinically approved PC, corrects the misfolding and mistrafficking of ΔF508 and various other misfolded CFTR variants through stabilization of the interfaces between subdomains.841,842 Administration of the correctors Lumacaftor and/or Tezacaftor in combination with Ivacaftor serves to rescue the folding and trafficking of these variants and promotes their activity at the plasma membrane.843,844 The initially improved combination therapy of this nature (Lumacaftor + Ivacaftor: Orkambi) offered modest clinical benefits for most patients. However, a more recently approved combination therapy (Tezacaftor + Ivacaftor: Symdeko) offers a measurable improvement for a wide array of patient genotypes.737,845−848 More promising therapies also appear to be on the horizon, however, as combinations of Symdeko with the novel correctors VX-445 or VX-659 have shown remarkably positive results in recently completed phase II clinical trials.849,850 While both VX-659 and VX-445 exhibit unique mechanisms of action, each triple combination greatly increases surface expression of mature CFTR as well as chloride flux in cultured patient-derived ΔF508 homozygous and heterozygous cells. Clinically, these combinations appear to provide patients with significant and durable improvements in FEV1, sweat chloride concentration, and quality of life.199
Ongoing investigations have also evaluated the potential for synergistic effects of proteostasis regulators that alter the activity of the cellular proteostasis network.828,851 A promising example of this is the autophagy activator cysteamine in combination with the polyphenol epigallocatechin gallate, which together improve the maturation and function of ΔF508 CFTR in cell culture models.852 A proof of concept clinical trial also demonstrated this combination resulted in decreased sweat chloride concentration and an increased forced expiratory volume; promising indications of clinical efficacy.852,853
Efforts to discover, develop, and evaluate small molecule therapies for CF have highlighted several critical considerations for ongoing drug discovery efforts for other diseases of MP misfolding. Clinical trials have revealed that the mechanistic diversity associated with rare mutations in conjunction with variations in genetic background can undermine the utility of PCs such as Lumacaftor. These complex variables highlight the potential utility of emerging precision medicine platforms in the administration of clinical trials and in the interpretation of clinical data. Furthermore, of the ∼2000 known pathogenic CFTR mutations, nearly 1200 of them have been observed in 5 or fewer patients, which precludes the design of traditional clinical trials for specific patient genotypes.854 In a historic decision aimed at addressing this shortcoming, the FDA recently approved the use of in vitro cellular assays to demonstrate efficacy of a compound with respect to individual variants, a ruling that could have wide ranging implications for the pharmacology of genetic diseases.854,855 Since this ruling, a number of more pointed in vitro assays as well as genotype-oriented clinical trials have served to expand the list of druggable CFTR variants.855 These efforts to pair specific CF compounds to specific patient genotypes may serve as a template to guide the development of next-generation targeted therapeutics for other diseases of MP misfolding. CF and other such diseases are also likely to constitute a prominent target for the development of CRISPR/Cas9-based therapies to directly correct the genetic lesions within lung tissues and the gastrointestinal tract.856−859 Together, these developments highlight the role of CFTR therapies as a harbinger for both the clinical use of PCs and the future of precision medicine.
6.4. Influence of Small Molecule Pharmacological Chaperones on the Folding and Trafficking of Wild Type Membrane Proteins
As reviewed in section 5.1 the folding and trafficking of many WT MPs is often inefficient. The evolutionary mechanisms that give rise to this biochemical inefficiency remain unclear. However, it has been suggested that the inefficient folding of cellular MPs may confer a regulatory benefit.860 Indeed, the marginal free energy differences associated with many of the co- and post-translational folding processes discussed herein are likely to render ER export exquisitely tunable. Furthermore, the accumulation of a pool of mislocalized protein in the ER provides an available source of proteins that can potentially be quickly rerouted to the plasma membrane through modifications to the composition and/or activity of the proteostasis network. Changes in the concentrations of natural ligands and/or substrates that bind and stabilize immature proteins might also adjust the flux of nascent proteins through the ER exit pathway.227−229 Indeed, glutamate binding is required for glutamate receptor biogenesis, and choline enhances the maturation of nicotinic acetylcholine receptor.783,861 Conversely, certain intermediates on the mevalonate pathway involved in the biosynthesis of steroids and related compounds are known bind to the yeast Hmg2p reductase (the rate-limiting enzyme of the pathway), resulting in its destabilization.228,235 This results in ERQC targeting of Hmg2p to the ERAD pathway, downregulating biosynthetic flux through the mevalonate pathway. A somewhat analogous system is used to reduce levels of HMG-CoA reductase in higher organisms, although the details are more complex.235,862
The energetic tunability of partitioning between degradation, ER retention, and forward trafficking also has important pharmacological implications in the context of heterozygous patients carrying a single WT allele, given that PCs designed to stabilize misfolded variants typically also influence the physiological folding efficiency of WT proteins. Indeed, the biogenesis of the WT forms of the dopamine transporter,863 delta-opioid receptor,864 calcium-sensing receptor (CaSR),782,865 GnRHR,800,866 MC4R,776 and the frizzled 4 receptor763 are known to be sensitive to the effects of PCs. Thus, the administration of these compounds to heterozygous individuals must consider the effects of these compounds on both the WT and mutant forms of the protein. Of course, in many cases this may be beneficial: an increase in the folding efficiency of the WT protein may help to compensate for the loss of irreversibly misfolded variants. The ultimate relevance of these effects is likely to vary from case to case depending on the specific threshold of protein functional activity required to support normal health.
Table 1 presents a representative sampling of wild type and mutant proteins that can be pharmacologically rescued from mistrafficking and/or degradation by PCs.
Table 1. Representative Pharmacological Chaperones and Their Membrane Protein Targets.
| membrane protein | function | disease | PC | refs |
|---|---|---|---|---|
| P-glycoprotein | drug efflux | multidrug resistance and inflammatory bowel disease | capsaicin, cyclosporine, vinblastine, verapamil, rhodamine, propafenone analogs, and VRT-325 | (867−870) |
| CFTR | salt and water homeostasis | CF | corr-2b, corr-4a, CFpot-532, KM11060, RDR1, glafenine, VRT-325, VX-809, VX-661, ABBV/GLPG-2222 | (737, 830, 831, 835, 847, 870−875) |
| ABCB4 | biliary phosphatidylcholine excretion | familial intrahepatic cholestasis | cyclosporine A | (876) |
| sulfonylurea receptor-1 | insulin secretion | hyperinsulinemia hypoglycemia | sulfonylureas and carbamazepine | (874, 875, 877, 878) |
| solute carrier 6(s) | neurotransmission | epilepsy, depression, and neuropathic pain | noribogaine, bupropion, ibogaine, and PAL-1045 | (659, 863, 879, 880) |
| nicotinic acetylcholine receptor | post- and presynaptic excitation | epilepsy, Alzheimer disease, and myasthenia | nicotine | (881, 882) |
| hERG potassium channel | repolarization of cardiac action potential | LQTS | E-4031, astemizole, cisapride, quinidine, and VX-809 | (753, 770, 817, 818, 882, 883) |
| SCN1A (Nav1.1) | signal transmission in the brain | epilepsy and migraine | phenytoin and lamotrigine | (884−886) |
| SCN5A (Nav1.5) | depolarization of cardiac action potential | LQTS and Brugada syndrome | mexiletine, quinidine, flecainide, and cisapride | (770, 821−824, 882, 883) |
| rhodopsin | visual phototransduction | retinitis pigmentosa | 11-cis-7-ring retinal, β-ionone, and NSC45012 | (887−889) |
| V2R | water reabsorption | NDI | VPA985, YM087, SR121463A, SR121463B, SR49059, OPC31260, OPC41061, MCF14, MCF18, and MCF57 | (616, 775, 793) |
| GnRHR | gonadotropin release | hypogonadotropic hypogonadism | IN3, Q89, TAK-013, and A177775 | (774, 803, 890) |
| MC4R | food intake and energy expenditure | obesity | ML00253764, DCPMP, Ipsen 5i, Ipsen 17, and THIQ | (776, 813, 814, 816, 891, 892) |
| luteinizing hormone receptor | steroidogenesis and spermatogenesis | hypergonadotropic hypogonadism | Org42599 | (893) |
| follicle-stimulating hormone receptor | follicular development and spermatogenesis | hypergonadotropic hypogonadism | Org41841 | (802) |
| glucagon receptor | glucose homeostasis | type 2 diabetes | L-168,049 | (894) |
| calcium-sensing receptor | calcium homeostasis | hypocalciuric hypercalcemia and hyperparathyroidism | NPS R-568 | (782, 865, 895) |
| opioid receptors | analgesia, emotion and motor control | drug addiction, alcohol dependence, and anorexia nervosa | naltrexone, naltrindole, naloxone, norbinaltorphimine, buprenorphine, and etorphine | (864, 896, 897) |
| adenosine A1 receptor | cardio- and neuroprotection | cognitive dysfunction, sleep deprivation, and glioma | DFCPX, IBMX, CPA, and adenosine | (898, 899) |
| D4 dopamine receptor | cognition, memory, learning, and motor control | Parkinson’s disease and psychiatric disorders | quinpirole, haloperidol, butaclamol(+), clozapine, domperidone, and dopamine | (771) |
| frizzled 4 receptor | retinal vascularization | familial exudative vitreoretinopathy | FzM1, 2, and 3 | (763) |
6.5. High-Throughput Screening to Identify Novel Pharmacological Chaperones
Natural substrates, inhibitors, or ligands are often employed as the initial scaffolds for the development of PCs. For GPCRs, many of the starting PCs are orthosteric antagonists that compete with endogenous agonists, though some agonists and allosteric modulators have also been utilized as lead compounds.229,865,893 However, the classic function-altering activities of these compounds may result in detrimental side effects. For instance, the PC activity of certain agonists may be offset by their effects on activation-induced internalization, or through inhibition of receptor recycling.816,839,840,864 Thus, in some cases allosteric PCs may offer the most promising therapeutic potential. It should also be added that the cognate ligands of many orphan receptors have yet to be discovered, which significantly limits currently available avenues for medicinal chemistry. Given these caveats, unbiased high-throughput screening (HTS) may often represent an attractive approach to identify novel PCs.
HTS typically leverages cellular assays to identify small molecules that enhance the accumulation of misfolded variants at the plasma membrane (Figure 31). Carlile et al. developed a corrector-screening assay of this nature to identify PCs that influence the trafficking of ΔF508 CFTR.871 A screen of 2000 compounds identified 224 that enhanced the trafficking of ΔF508 CFTR, which were further characterized according to their ability to thermally stabilize the first nucleotide binding domain of ΔF508 CFTR (NBD1) as determined by differential scanning fluorimetry.835,900,901 Of the 224 hits, only one compound (RDR1) appreciably increased the thermal stability of NBD1 in a dose-dependent manner. Follow-up efforts revealed that a related compound (RDR3) also thermally stabilized the ΔF508 NBD1, while RDR2, which lacks the terminal phenyl ring, did not. Interestingly, RDR1 does not compete with ATP binding, which indicates this compound associates with a distinct binding pocket. Follow-up investigations also demonstrated that RDR1 partially rescues the damage of ΔF508 CFTR, which is partially dysfunctional even when folded. Thus, despite the fact that this screen was carried out to identify PCs, this molecule might also act as a weak potentiator of ΔF508 CFTR. Similar HTS studies involving measurement of the cell surface expression of tagged pathogenic variants have also been successfully performed for other MPs such as clarin-1902 and frizzled4.763
Figure 31.
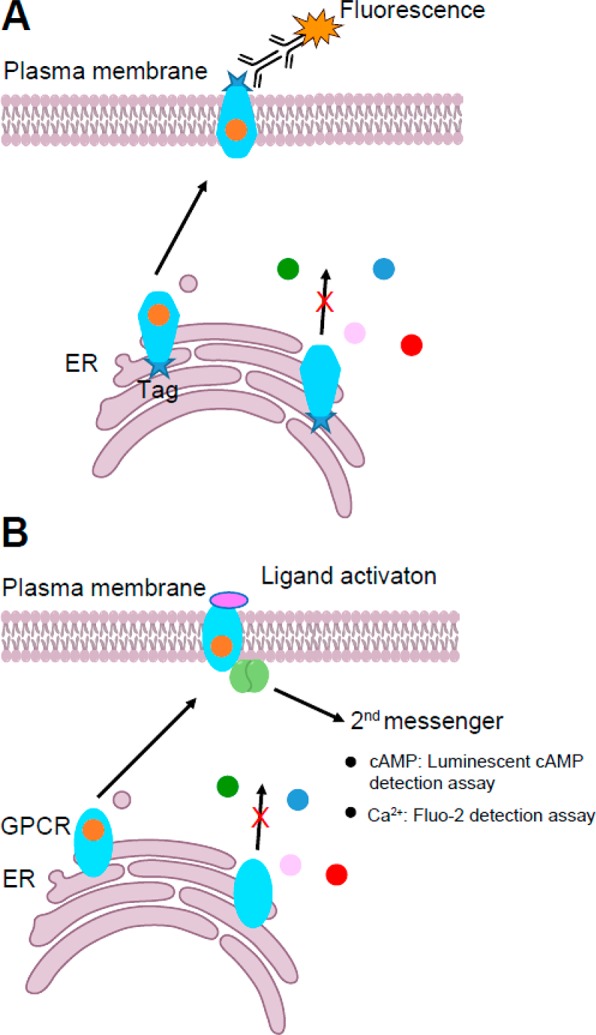
Examples of cell-based HTS for pharmacological chaperones that restore plasma membrane trafficking (A) or function (B) of a MP, in this case a GPCR. Cells stably expressing a misfolded mutant of the target MP are treated with different compounds (represented as small colored balls). Compounds that act as PCs stabilize the misfolded protein and facilitate its trafficking to the plasma membrane where its expression and/or function can be detected or reported.
An ultrahigh-throughput cellular assay that leverages changes in cellular cAMP, a downstream reporter of Gαs signaling, has been developed to identify PCs for destabilized V2R variants.797−799 This assay was employed using HeLa cells stably expressing L83Q V2R under the control of a tetracycline-controlled transactivator. The use of an inducible expression system provides a convenient means to detect false positives. A screen of 645 000 compounds identified 83 compounds that significantly increased the activity of L83Q V2R. These hits clustered into three predominant structural groups by affinity.798 A similar cell-based HTS assay to identify PCs for GnRHR has been completed using HeLa cells stably expressing E90K GnRHR, though the screen was carried out using a cytoplasmic calcium release assay to detect Gαq activation.760,903 Though effective, it should be noted that these blinded screens are likely to return numerous compounds in addition to PCs that induce a positive assay result.
When structural information for the protein of interest is available, in silico computational methods are sometimes used to identify potential interacting compounds. In silico screening can (ideally) narrow down the number of possible compounds from a large virtual library of small molecules, providing an inexpensive and fast way to identify candidate PCs.888,904−906
6.6. Modulation of Proteostasis Networks
As summarized in section 4, MPs often rely on a specific set of molecular chaperones to facilitate their folding and assembly.907 Because these chaperones are shared between a wide variety of client proteins, the folding efficiency of one protein may depend on the aggregation state of seemingly unrelated members of the proteome.604 Whether through the actions of proteostasis regulators or through direct manipulation of expression profiles, the tuning of cellular chaperone levels can have dramatic effects on the maturation and aggregation levels of misfolding-prone variants.908−911 Moreover, the accumulation of misfolded or oxidatively damaged proteins over time also enhances the collective proteomic burden on the proteostasis network, which contributes to the pathology of many diseases of aging. Thus, pathogenic defects in MP folding and assembly may sometimes arise, in part, from a wider failure of the proteostasis network. Fortunately, the interconnectedness of the proteostasis network affords a variety of approaches to indirectly tune the efficiency of MP biosynthesis. An emerging class of therapeutic compounds known as proteostasis regulators provide an alternative to small molecule pharmacological chaperones that selectively stabilize a single target protein.912 Targeting the activity of the wider proteostasis network provides considerable flexibility, as lead compounds can be discovered from general phenotypic screens.913−915 In addition to the relative ease associated with screening, compounds that target the proteostasis network may offer therapeutic potential for the treatment of multiple diseases.916 Furthermore, proteostasis regulators can also be used to synergistically enhance the effects of pharmacological chaperones on the folding and trafficking of integral MPs.916,917 Thus, this class of compounds is likely to play a key role in the pharmacology of folding disorders going forward.
A variety of proteostasis regulators have been developed for the rescue of misfolded soluble proteins. However, it is unclear whether these compounds will be equally efficacious toward misfolded MPs. Nevertheless, early indications are encouraging. Inhibition of the proteasome by MG-132 not only reduces the degradation of misfolded soluble proteins, but also seems to increase the steady-state concentrations of stable and unstable variants of CFTR and KCNQ1.183,425,916,918 Compounds that reduce the degradation of CFTR through inhibition of ubiquitination also appear to enhance the surface expression of misfolding-prone CFTR variants.919 Inhibition of E3 ubiquitin ligases using an N-aryl benzimidazole derivative also seems to reduce the toxicity associated with α-synuclein.920 Even in cases where the response of soluble proteins and MPs may differ, these compounds may provide valuable tools to identify the portions of the proteostasis network that are most relevant to pathogenic defects. For instance, beyond the direct effects of MG-132 on the activity of the proteasome, it has been shown that this compound also triggers the UPR in certain cells.916 Thus, it is likely that some of the beneficial effects of MG-132 on CFTR expression may arise from the UPR-mediated upregulation of chaperones. Compounds that modulate the UPR and associated ER stress pathways may therefore provide additional routes for the correction of MP misfolding.239,921 The heat shock responses and its associated chaperones has also proven to be a fruitful target.922−924 The HSP coinducer Arimoclomal is currently being evaluated as a possible treatment for Niemann-Pick Disease Type C (NPC), a fatal neurodegenerative disease associated with mutations in the cholesterol transporter Niemann-Pick C1 Protein (NPC1).362,925,926
The HDAC inhibitor suberoylanilide hydroxamic acid (SAHA), an FDA-approved drug also known as Vorinostat, increases the expression and partially corrects the function of epilepsy-associated GABA type A (GABAA) receptors by both enhancing receptor gene transcription and promoting the interaction of GABAA with BiP and calnexin.927 A combination of SAHA treatment and ERAD pathway inhibition produces an additive effect on rescuing the GABAA α1(A322D).928 HDAC inhibition has also been shown to enhance the proteostasis of other proteins including CFTR.929 The discovery of new proteostasis regulators that target different quality control proteins will ultimately expand our ability to elucidate the nature of protein misfolding pathways and expand potential avenues for therapeutic interventions.
7. Future Directions
Progress in our understanding of the kinetics and thermodynamics of MP folding in vitro has progressed immensely during the past three decades. These advances are underscored by impressive de novo design efforts that have yielded membrane peptides and MPs with fundamentally new structures and/or functions.930−936 Methods are now in place for enhancing the stability of MPs937,938 and totally new ways of studying MP folding continue to be devised. Moreover, the interactions of client MPs with chaperone networks are beginning to be explored with experimentally restrained systems modeling.220 However, many gaps remain in our understanding of how the conformational properties of MPs relate to their behavior within the cell. For example, in the past decade, spectacular advances in crystallography and cryo-EM have given rise to a structural revolution in GPCR biochemistry and pharmacology. Nevertheless, quantitative investigations of the folding/unfolding kinetics and thermodynamic stability of GPCRs have, to date, proven elusive. Moreover, current computational platforms are still incapable of predicting the effects of mutations on the conformational equilibria of MPs.644 Based on our collective observations, we posit that the next such revolution is unlikely to arise solely from an understanding MPs in isolation but rather from insights into how their conformational equilibria are navigated in the context of the cellular milieu.
It is imperative that emerging insight into the conformational stability of MPs is connected to a broader understanding of cellular processes. Recent progress provides considerable room for optimism. For example, the physical mechanisms associated with the Sec61-mediated cotranslational folding of nascent MPs have been outlined in considerable detail. However, the recent discovery that the EMC complex appears to work closely with Sec61 to initiate integration of MPs into the ER membrane353 suggests that there are stunning discoveries still to be made, even for systems that appeared to be reasonably well understood. Along the same lines the energetics of cotranslational folding has yet to be connected with the structural properties of the nascent ensemble or the outcomes of cellular QC. The superstructural organization of the endoplasmic reticulum throughout the cell is now appreciated to be much more complex and sophisticated than long realized, with different domains of this organelle serving as focal points for different subsystems of ERQC. Our current understanding of this superstructure and the spatial distributions of the components of ERQC is fuzzy, at best. While the pertinent biochemical activities of many central components of ERAD, such as the Hrd1 retrotranslocon, have been biochemically characterized, additional work is needed to rationalize how these activities interface with specific conformational states of client proteins. There are still numerous proteins that are believed to be ERQC factors but otherwise have unknown functions. The anticipated wave of experiments in these areas will likely yield major discoveries that merge structural biochemistry and biophysical chemistry with a broader understanding of cellular systems.
It is also essential to consolidate and establish new linkages between MP folding and misfolding in the cell and the molecular basis of disease. In many cases, recent observations have revealed the sources of the smoke—clear disease linkages for numerous membrane proteins, lists of mutations, and so forth—but have yet to elucidate the mechanistic basis of the fire. Beyond their emerging impact in the clinic, small molecule pharmacological chaperones represent a tremendous tool for biochemical and biophysical investigations of MP misfolding in the cell. Ongoing HTS efforts to identify new stabilizing molecules in conjunction with mechanistic studies of their effects are likely to provide new insights into the many ways in which MP misfolding can be curtailed in the cell. Such advances are also likely to help streamline next-generation drug discovery platforms.
Future investigations of these topics must find new ways to reckon with and utilize the emerging wave of genome sequencing data. Indeed, the opportunities for molecular scientists to contribute to personalized (or “precision”) medical diagnostics and decision-making are numerous.629 The tools and perspectives of biochemistry and biophysics are needed to interpret the effects of rare variants in disease-linked MPs. Such information may prove critical for the use of genomic information in the clinic, especially for cases in which different pathogenic mechanisms can arise from a spectrum of mutations within a single protein.183 Such advances may provide novel ways to optimally match certain medications to specific patient genotypes. Deciphering the complexity within individual genomes will require next-generation tools to enable rapid, low cost, and reliable experimental or predictive methods for these purposes. It is increasingly clear that misfolding is the most common consequence of pathogenic mutations in MPs. However, novel methods to parse the spectrum of molecular defects associated with these mutations are sorely needed.
Acknowledgments
This work was supported by U.S. NIH Grants RF1 AG056147, RO1 HL122010, and RO1 NS095989 (to C.R.S.); T32 GM008320 (J.T.M.); R01 GM129261 (J.P.S.); and R35 GM127087 (J.A.C.). This work was also supported by the Cystic Fibrosis Foundation (SCHLEB18I0 to J.P.S.). We thank the reviewers of the initial version of this paper for making many helpful comments and also Sarah Veatch of the University of Michigan and Sarah Keller of the University of Washington for patiently answering questions about critical behavior in membranes. We also thank Jean-Luc Popot of CNRS-Paris for his comments, especially with respect to the early history of the field of MP folding.
Biographies
Justin T. Marinko is a Ph.D. candidate in the laboratory of Charles Sanders at Vanderbilt University. He received his B.A. with honors in Chemistry with a concentration in Biochemistry from Boston University. At B.U., Justin worked under the guidance of Dr. Karen Allen doing X-ray crystallography. Prior to graduate school he worked at Celgene Pharmaceuticals. Justin’s research interest focuses on MP folding and trafficking in mammalian cells, and he currently works on peripheral myelin protein 22 (PMP22).
Hui Huang is a postdoctoral fellow in the Sanders lab. She obtained a Ph.D. in Biomedical Sciences from Auburn University in 2014. Her research interests are in elucidating how defects in protein structure, folding, and function are linked to human diseases, with recent work focusing on the human KCNQ1 potassium channel and long QT syndrome.
Wesley Penn received his M.S. in Molecular/Systems Biology from Purdue University in West Lafayette, Indiana. He is currently a Lab Manager and Research Associate with the Schlebach Lab at Indiana University in Bloomington, Indiana. His work at IU focuses on the application of deep mutational scanning to questions of defective MP folding in disease. His primary research interests include infectious disease, molecular diagnostics, and the application of molecular biology to the mechanisms of disease.
John A. Capra is an Assistant Professor in the Department of Biological Sciences at Vanderbilt University. He is also a member of the Vanderbilt Genetics Institute and Center for Structural Biology and has secondary appointments in Biomedical Informatics and Computer Science. He received his Ph.D. in computer science from Princeton University under the supervision of Mona Singh. He was a postdoc in the group of Katherine Pollard at the Gladstone Institutes and the University of California, San Francisco. His group uses the tools of computer science and statistics to address problems in genetics, evolution, and biomedicine.
Jonathan P. Schlebach received his Ph.D. in Medicinal Chemistry and Molecular Pharmacology from Purdue University, where he studied the kinetics and thermodynamics of MP folding in the laboratory of Chiwook Park. In 2012, he began postdoctoral studies in the laboratory of Charles R. Sanders at Vanderbilt University, where he studied the biophysical properties of disease-linked integral MPs. He joined the Department of Chemistry at Indiana University, Bloomington as an Assistant Professor in 2016. His laboratory focuses on the biochemistry and biophysics of MP folding and misfolding in the cell.
Charles R. Sanders is Associate Dean for Research, Professor of Biochemistry, and holder of the Aileen M. Lange and Annie Mary Lyle Chair in Cardiovascular Research in the Vanderbilt University School of Medicine Basic Sciences. He completed his Ph.D. in chemistry in 1988 at The Ohio State University, where he worked under the direction of Ming-Daw Tsai. Following studies as an NIH Postdoctoral Fellow in the Department of Chemistry at Yale University with James H. Prestegard, he joined the faculty of the Department of Physiology and Biophysics at Case Western Reserve University in 1991, moving to Vanderbilt in 2002. He served as an Associate Editor of Biochemistry from 2004–2016. The Sanders lab studies human MPs using biochemical, chemical biological, and biophysical approaches, with the goal of elucidating the molecular mechanisms by which these proteins contribute to various diseases.
Author Contributions
⊥ J.T.M. and H.H. contributed equally.
The authors declare no competing financial interest.
References
- Deisenhofer J.; Epp O.; Miki K.; Huber R.; Michel H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3A resolution. Nature 1985, 318, 618–624. 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- Kendrew J. C.; Bodo G.; Dintzis H. M.; Parrish R. G.; Wyckoff H.; Phillips D. C. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 1958, 181, 662–666. 10.1038/181662a0. [DOI] [PubMed] [Google Scholar]
- Huang K. S.; Bayley H.; Liao M. J.; London E.; Khorana H. G. Refolding of an integral membrane protein. Denaturation, renaturation, and reconstitution of intact bacteriorhodopsin and two proteolytic fragments. J. Biol. Chem. 1981, 256, 3802–3809. [PubMed] [Google Scholar]
- London E.; Khorana H. G. Denaturation and renaturation of bacteriorhodopsin in detergents and lipid-detergent mixtures. J. Biol. Chem. 1982, 257, 7003–7011. [PubMed] [Google Scholar]
- Popot J. L.; Gerchman S. E.; Engelman D. M. Refolding of bacteriorhodopsin in lipid bilayers. A thermodynamically controlled two-stage process. J. Mol. Biol. 1987, 198, 655–676. 10.1016/0022-2836(87)90208-7. [DOI] [PubMed] [Google Scholar]
- Popot J. L.; Trewhella J.; Engelman D. M. Reformation of crystalline purple membrane from purified bacteriorhodopsin fragments. EMBO J. 1986, 5, 3039–3044. 10.1002/j.1460-2075.1986.tb04603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth P. J.; Flitsch S. L.; Stern L. J.; Greenhalgh D. A.; Kim P. S.; Khorana H. G. Intermediates in the folding of the membrane protein bacteriorhodopsin. Nat. Struct. Mol. Biol. 1995, 2, 139–143. 10.1038/nsb0295-139. [DOI] [PubMed] [Google Scholar]
- Lau F.; Bowie J. A method for assessing the stability of a membrane protein. Biochemistry 1997, 36, 5884–5892. 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- Haber E.; Anfinsen C. B. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J. Biol. Chem. 1962, 237, 1839–1844. [PubMed] [Google Scholar]
- Anfinsen C. B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Flanagan J. M.; Hunt J. F.; Adair B. D.; Bormann B. J.; Dempsey C. E.; Engelman D. M. Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J. Biol. Chem. 1992, 267, 7683–7689. [PubMed] [Google Scholar]
- Lemmon M. A.; Flanagan J. M.; Treutlein H. R.; Zhang J.; Engelman D. M. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 1992, 31, 12719–12725. 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- Treutlein H. R.; Lemmon M. A.; Engelman D. M.; Brunger A. T. The glycophorin A transmembrane domain dimer: sequence-specific propensity for a right-handed supercoil of helices. Biochemistry 1992, 31, 12726–12732. 10.1021/bi00166a003. [DOI] [PubMed] [Google Scholar]
- Popot J. L.; Engelman D. M. Membrane protein folding and oligomerization: the two-stage model. Biochemistry 1990, 29, 4031–4037. 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- Popot J. L.; Engelman D. M. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 2000, 69, 881–922. 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- Cymer F.; von Heijne G.; White S. H. Mechanisms of integral membrane protein insertion and folding. J. Mol. Biol. 2015, 427, 999–1022. 10.1016/j.jmb.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. H.; Wimley W. C. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- Chamberlain A. K.; Faham S.; Yohannan S.; Bowie J. U. Construction of helix-bundle membrane proteins. Adv. Protein Chem. 2003, 63, 19–46. 10.1016/S0065-3233(03)63002-0. [DOI] [PubMed] [Google Scholar]
- Neumann J.; Klein N.; Otzen D. E.; Schneider D. Folding energetics and oligomerization of polytopic alpha-helical transmembrane proteins. Arch. Biochem. Biophys. 2014, 564, 281–296. 10.1016/j.abb.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Otzen D. E.; Andersen K. K. Folding of outer membrane proteins. Arch. Biochem. Biophys. 2013, 531, 34–43. 10.1016/j.abb.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Hong H. Toward understanding driving forces in membrane protein folding. Arch. Biochem. Biophys. 2014, 564, 297–313. 10.1016/j.abb.2014.07.031. [DOI] [PubMed] [Google Scholar]
- London E.; Shahidullah K. Transmembrane vs. non-transmembrane hydrophobic helix topography in model and natural membranes. Curr. Opin. Struct. Biol. 2009, 19, 464–472. 10.1016/j.sbi.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Fleming K. G. Energetics of membrane protein folding. Annu. Rev. Biophys. 2014, 43, 233–255. 10.1146/annurev-biophys-051013-022926. [DOI] [PubMed] [Google Scholar]
- Chaturvedi D.; Mahalakshmi R. Transmembrane beta-barrels: Evolution, folding and energetics. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 2467–2482. 10.1016/j.bbamem.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm L. K.; Hong H.; Liang B. Folding and assembly of beta-barrel membrane proteins. Biochim. Biophys. Acta, Biomembr. 2004, 1666, 250–263. 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Booth P. J. The trials and tribulations of membrane protein folding in vitro. Biochim. Biophys. Acta, Biomembr. 2003, 1610, 51–56. 10.1016/S0005-2736(02)00714-9. [DOI] [PubMed] [Google Scholar]
- Booth P. J.; Templer R. H.; Meijberg W.; Allen S. J.; Curran A. R.; Lorch M. In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 501–603. 10.1080/20014091074246. [DOI] [PubMed] [Google Scholar]
- Schiffrin B.; Brockwell D. J.; Radford S. E. Outer membrane protein folding from an energy landscape perspective. BMC Biol. 2017, 15, 123. 10.1186/s12915-017-0464-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A.; Engel D. E.; DeGrado W. F. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 2004, 14, 465–479. 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Mackenzie K. R. Folding and stability of alpha-helical integral membrane proteins. Chem. Rev. 2006, 106, 1931–1977. 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- Popot J. L.Membrane Proteins in Aqueous Solutions: From Detergents to Amphipols; Springer: New York, 2018. [Google Scholar]
- Bracey M. H.; Cravatt B. F.; Stevens R. C. Structural commonalities among integral membrane enzymes. FEBS Lett. 2004, 567, 159–165. 10.1016/j.febslet.2004.04.084. [DOI] [PubMed] [Google Scholar]
- Garavito R. M.; Mulichak A. M. The structure of mammalian cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 183–206. 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- Root K. T.; Plucinsky S. M.; Glover K. J. Recent progress in the topology, structure, and oligomerization of caveolin: a building block of caveolae. Curr. Top. Membr. 2015, 75, 305–336. 10.1016/bs.ctm.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Engelman D. M.; Steitz T. A.; Goldman A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 321–353. 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Sharpe H. J.; Stevens T. J.; Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169. 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 1992, 225, 487–494. 10.1016/0022-2836(92)90934-C. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7, 909–918. 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Engelman D. M.; Steitz T. A. The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell 1981, 23, 411–422. 10.1016/0092-8674(81)90136-7. [DOI] [PubMed] [Google Scholar]
- Fluman N.; Tobiasson V.; von Heijne G. Stable membrane orientations of small dual-topology membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7987–7992. 10.1073/pnas.1706905114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich S. U.; Mothes W.; Brunner J.; Rapoport T. A. The Sec61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell 2000, 102, 233–244. 10.1016/S0092-8674(00)00028-3. [DOI] [PubMed] [Google Scholar]
- Ulmschneider M. B.; Sansom M. S. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta, Biomembr. 2001, 1512, 1–14. 10.1016/S0005-2736(01)00299-1. [DOI] [PubMed] [Google Scholar]
- Ulmschneider M. B.; Sansom M. S.; Di Nola A. Properties of integral membrane protein structures: derivation of an implicit membrane potential. Proteins: Struct., Funct., Genet. 2005, 59, 252–265. 10.1002/prot.20334. [DOI] [PubMed] [Google Scholar]
- Bowie J. U. Membrane protein folding: how important are hydrogen bonds?. Curr. Opin. Struct. Biol. 2011, 21, 42–49. 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Bowie J. U. Shifting hydrogen bonds may produce flexible transmembrane helices. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8121–8126. 10.1073/pnas.1201298109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; White S. H. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 1992, 61, 434–447. 10.1016/S0006-3495(92)81849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer D. W.; Bramhall J. Permeability of lipid bilayers to water and ionic solutes. Chem. Phys. Lipids 1986, 40, 167–188. 10.1016/0009-3084(86)90069-1. [DOI] [PubMed] [Google Scholar]
- Miyano M.; Ago H.; Saino H.; Hori T.; Ida K. Internally bridging water molecule in transmembrane alpha-helical kink. Curr. Opin. Struct. Biol. 2010, 20, 456–463. 10.1016/j.sbi.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Orban T.; Gupta S.; Palczewski K.; Chance M. R. Visualizing water molecules in transmembrane proteins using radiolytic labeling methods. Biochemistry 2010, 49, 827–834. 10.1021/bi901889t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein N.; Neumann J.; O’Neil J. D.; Schneider D. Folding and stability of the aquaglyceroporin GlpF: Implications for human aqua(glycero)porin diseases. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 622–633. 10.1016/j.bbamem.2014.11.015. [DOI] [PubMed] [Google Scholar]
- Kauko A.; Illergard K.; Elofsson A. Coils in the membrane core are conserved and functionally important. J. Mol. Biol. 2008, 380, 170–180. 10.1016/j.jmb.2008.04.052. [DOI] [PubMed] [Google Scholar]
- Viklund H.; Granseth E.; Elofsson A. Structural classification and prediction of reentrant regions in alpha-helical transmembrane proteins: application to complete genomes. J. Mol. Biol. 2006, 361, 591–603. 10.1016/j.jmb.2006.06.037. [DOI] [PubMed] [Google Scholar]
- De Marothy M. T.; Elofsson A. Marginally hydrophobic transmembrane alpha-helices shaping membrane protein folding. Protein Sci. 2015, 24, 1057–1074. 10.1002/pro.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt J. F.; Rath P.; Rothschild K. J.; Engelman D. M. Spontaneous, pH-dependent membrane insertion of a transbilayer alpha-helix. Biochemistry 1997, 36, 15177–15192. 10.1021/bi970147b. [DOI] [PubMed] [Google Scholar]
- Meruelo A. D.; Samish I.; Bowie J. U. TMKink: a method to predict transmembrane helix kinks. Protein Sci. 2011, 20, 1256–1264. 10.1002/pro.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohannan S.; Faham S.; Yang D.; Whitelegge J. P.; Bowie J. U. The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 959–963. 10.1073/pnas.0306077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Richter H. T.; Cartailler J. P.; Lanyi J. K. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 1999, 291, 899–911. 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- Jorgensen C.; Darre L.; Oakes V.; Torella R.; Pryde D.; Domene C. Lateral Fenestrations in K(+)-channels explored using molecular dynamics simulations. Mol. Pharmaceutics 2016, 13, 2263–2273. 10.1021/acs.molpharmaceut.5b00942. [DOI] [PubMed] [Google Scholar]
- Lichtenegger M.; Tiapko O.; Svobodova B.; Stockner T.; Glasnov T. N.; Schreibmayer W.; Platzer D.; de la Cruz G. G.; Krenn S.; Schober R.; Shrestha N.; Schindl R.; Romanin C.; Groschner K. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018, 14, 396–404. 10.1038/s41589-018-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Zhou H.; Chi S.; Wang Y.; Wang J.; Geng J.; Wu K.; Liu W.; Zhang T.; Dong M. Q.; Wang J.; Li X.; Xiao B. Structure and mechanogating mechanism of the Piezo1 channel. Nature 2018, 554, 487–492. 10.1038/nature25743. [DOI] [PubMed] [Google Scholar]
- De Loof H.; Harvey S. C.; Segrest J. P.; Pastor R. W. Mean field stochastic boundary molecular dynamics simulation of a phospholipid in a membrane. Biochemistry 1991, 30, 2099–2113. 10.1021/bi00222a015. [DOI] [PubMed] [Google Scholar]
- Harris N. J.; Charalambous K.; Findlay H. E.; Booth P. J. Lipids modulate the insertion and folding of the nascent chains of alpha helical membrane proteins. Biochem. Soc. Trans. 2018, 46, 1355–1366. 10.1042/BST20170424. [DOI] [PubMed] [Google Scholar]
- Curnow P.; Lorch M.; Charalambous K.; Booth P. J. The reconstitution and activity of the small multidrug transporter EmrE is modulated by non-bilayer lipid composition. J. Mol. Biol. 2004, 343, 213–222. 10.1016/j.jmb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- van den Brink-van der Laan E.; Chupin V.; Killian J. A.; de Kruijff B. Stability of KcsA tetramer depends on membrane lateral pressure. Biochemistry 2004, 43, 4240–4250. 10.1021/bi036129d. [DOI] [PubMed] [Google Scholar]
- Booth P. J.; Curnow P. Folding scene investigation: membrane proteins. Curr. Opin. Struct. Biol. 2009, 19, 8–13. 10.1016/j.sbi.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey P. A.; Kinnunen P. K. Biophysical properties of lipids and dynamic membranes. Trends Cell Biol. 2006, 16, 538–546. 10.1016/j.tcb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Karabadzhak A. G.; Weerakkody D.; Deacon J.; Andreev O. A.; Reshetnyak Y. K.; Engelman D. M. Bilayer thickness and curvature influence binding and insertion of a pHLIP peptide. Biophys. J. 2018, 114, 2107–2115. 10.1016/j.bpj.2018.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrey T.; Jahnig F. Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 7457–7461. 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsch I. K.; Daste F.; Gallop J. L. Membrane curvature in cell biology: An integration of molecular mechanisms. J. Cell Biol. 2016, 214, 375–387. 10.1083/jcb.201604003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H. T.; Gallop J. L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596. 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- Itoh T.; De Camilli P. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2006, 1761, 897–912. 10.1016/j.bbalip.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Mim C.; Unger V. M. Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 2012, 37, 526–533. 10.1016/j.tibs.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Voth G. A.; Callan-Jones A.; Bassereau P. When physics takes over: BAR proteins and membrane curvature. Trends Cell Biol. 2015, 25, 780–792. 10.1016/j.tcb.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt A.; Killian J. A. Orientation and dynamics of transmembrane peptides: the power of simple models. Eur. Biophys. J. 2010, 39, 609–621. 10.1007/s00249-009-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm T. K.; Ozdirekcan S.; Killian J. A. How protein transmembrane segments sense the lipid environment. Biochemistry 2007, 46, 1457–1465. 10.1021/bi061941c. [DOI] [PubMed] [Google Scholar]
- Song Y.; Mittendorf K. F.; Lu Z.; Sanders C. R. Impact of bilayer lipid composition on the structure and topology of the transmembrane amyloid precursor C99 protein. J. Am. Chem. Soc. 2014, 136, 4093–4096. 10.1021/ja4114374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennis R. B.Biomembranes: Molecular Structure and Function; Springer-Verlag: New York, 1989. [Google Scholar]
- Dupuy A. D.; Engelman D. M. Protein area occupancy at the center of the red blood cell membrane. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2848–2852. 10.1073/pnas.0712379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucerka N.; Nieh M. P.; Pencer J.; Sachs J. N.; Katsaras J. What determines the thickness of a biological membrane. Gen. Physiol. Biophys. 2009, 28, 117–125. 10.4149/gpb_2009_02_117. [DOI] [PubMed] [Google Scholar]
- Mitra K.; Ubarretxena-Belandia I.; Taguchi T.; Warren G.; Engelman D. M. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4083–4088. 10.1073/pnas.0307332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski L. E.; de Mendoza D. Bilayer hydrophobic thickness and integral membrane protein function. Curr. Protein Pept. Sci. 2011, 12, 760–766. 10.2174/138920311798841681. [DOI] [PubMed] [Google Scholar]
- Soubias O.; Teague W. E. Jr.; Hines K. G.; Gawrisch K. Rhodopsin/lipid hydrophobic matching-rhodopsin oligomerization and function. Biophys. J. 2015, 108, 1125–1132. 10.1016/j.bpj.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho A. V.; Huber T.; Sakmar T. P.; Brown M. F. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 2006, 91, 4464–4477. 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B. W.; Kulp D. W.; Span L. M.; DeGrado J. L.; Billings P. C.; Senes A.; Bennett J. S.; DeGrado W. F. Consensus motif for integrin transmembrane helix association. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 703–708. 10.1073/pnas.0910873107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S. M.; Mueller B. K.; Lange E. J.; Senes A. Combination of Calpha-H hydrogen bonds and van der Waals packing modulates the stability of GxxxG-mediated dimers in membranes. J. Am. Chem. Soc. 2017, 139, 15774–15783. 10.1021/jacs.7b07505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg S. D.; Clinthorne G. D.; Goulian M.; DeGrado W. F. Transmembrane polar interactions are required for signaling in the Escherichia coli sensor kinase PhoQ. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8141–8146. 10.1073/pnas.1003166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogozheva I. D.; Lomize A. L. Evolution and adaptation of single-pass transmembrane proteins. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 364–377. 10.1016/j.bbamem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Oberai A.; Ihm Y.; Kim S.; Bowie J. U. A limited universe of membrane protein families and folds. Protein Sci. 2006, 15, 1723–1734. 10.1110/ps.062109706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev A. Y.; Fahy E.; Guan Z.; Kelly S.; Li X.; McDonald J. G.; Milne S.; Myers D.; Park H.; Ryan A.; Thompson B. M.; Wang E.; Zhao Y.; Brown H. A.; Merrill A. H.; Raetz C. R.; Russell D. W.; Subramaniam S.; Dennis E. A. Subcellular organelle lipidomics in TLR-4-activated macrophages. J. Lipid Res. 2010, 51, 2785–2797. 10.1194/jlr.M008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J. E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. 10.1111/tra.12230. [DOI] [PubMed] [Google Scholar]
- Bloom M.; Evans E.; Mouritsen O. G. Physical properties of the fluid lipid-bilayer component of cell membranes: a perspective. Q. Rev. Biophys. 1991, 24, 293–397. 10.1017/S0033583500003735. [DOI] [PubMed] [Google Scholar]
- Rog T.; Pasenkiewicz-Gierula M.; Vattulainen I.; Karttunen M. Ordering effects of cholesterol and its analogues. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 97–121. 10.1016/j.bbamem.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Boggs J. M. Lipid intermolecular hydrogen bonding: influence on structural organization and membrane function. Biochim. Biophys. Acta, Rev. Biomembr. 1987, 906, 353–404. 10.1016/0304-4157(87)90017-7. [DOI] [PubMed] [Google Scholar]
- Slotte J. P. The importance of hydrogen bonding in sphingomyelin’s membrane interactions with co-lipids. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 304–310. 10.1016/j.bbamem.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Kaiser H. J.; Orlowski A.; Rog T.; Nyholm T. K.; Chai W.; Feizi T.; Lingwood D.; Vattulainen I.; Simons K. Lateral sorting in model membranes by cholesterol-mediated hydrophobic matching. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16628–16633. 10.1073/pnas.1103742108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S. J.; Nicolson G. L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Goni F. M. The basic structure and dynamics of cell membranes: an update of the Singer-Nicolson model. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1467–1476. 10.1016/j.bbamem.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Kusumi A.; Fujiwara T. K.; Chadda R.; Xie M.; Tsunoyama T. A.; Kalay Z.; Kasai R. S.; Suzuki K. G. Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson’s fluid-mosaic model. Annu. Rev. Cell Dev. Biol. 2012, 28, 215–250. 10.1146/annurev-cellbio-100809-151736. [DOI] [PubMed] [Google Scholar]
- Nicolson G. L. The Fluid-Mosaic Model of Membrane Structure: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1451–1466. 10.1016/j.bbamem.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; London E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Rietveld A.; Simons K. The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim. Biophys. Acta, Rev. Biomembr. 1998, 1376, 467–479. 10.1016/S0304-4157(98)00019-7. [DOI] [PubMed] [Google Scholar]
- Quinn P. J.; Wolf C. The liquid-ordered phase in membranes. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 33–46. 10.1016/j.bbamem.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Debruin L. S.; Harauz G. White matter rafting--membrane microdomains in myelin. Neurochem. Res. 2007, 32, 213–228. 10.1007/s11064-006-9137-4. [DOI] [PubMed] [Google Scholar]
- Saher G.; Quintes S.; Nave K. A. Cholesterol: a novel regulatory role in myelin formation. Neuroscientist 2011, 17, 79–93. 10.1177/1073858410373835. [DOI] [PubMed] [Google Scholar]
- Cao X.; Surma M. A.; Simons K. Polarized sorting and trafficking in epithelial cells. Cell Res. 2012, 22, 793–805. 10.1038/cr.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton R. G. Caveolae: structure, runction, and relationship to disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 111–136. 10.1146/annurev-cellbio-100617-062737. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; London E. Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes?. Biochem. Biophys. Res. Commun. 1997, 240, 1–7. 10.1006/bbrc.1997.7575. [DOI] [PubMed] [Google Scholar]
- Delacour D.; Jacob R. Apical protein transport. Cell. Mol. Life Sci. 2006, 63, 2491–2505. 10.1007/s00018-006-6210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. J.; Jen A.; Warley A. Isolation of nano-meso scale detergent resistant membrane that has properties expected of lipid ’rafts. J. Neurochem. 2011, 116, 671–677. 10.1111/j.1471-4159.2010.07076.x. [DOI] [PubMed] [Google Scholar]
- Simons K.; Ikonen E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Levental I.; Veatch S. The continuing mystery of lipid rafts. J. Mol. Biol. 2016, 428, 4749–4764. 10.1016/j.jmb.2016.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Sezgin E.; Levental I.; Mayor S.; Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. 10.1038/nrm.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental K. R.; Levental I. Giant plasma membrane vesicles: models for understanding membrane organization. Curr. Top. Membr. 2015, 75, 25–57. 10.1016/bs.ctm.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Morigaki K.; Tanimoto Y. Evolution and development of model membranes for physicochemical and functional studies of the membrane lateral heterogeneity. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 2012–2017. 10.1016/j.bbamem.2018.03.010. [DOI] [PubMed] [Google Scholar]
- Rayermann S. P.; Rayermann G. E.; Cornell C. E.; Merz A. J.; Keller S. L. Hallmarks of Reversible Separation of living, unperturbed cell Membranes into two liquid phases. Biophys. J. 2017, 113, 2425–2432. 10.1016/j.bpj.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft M. L. Plasma membrane organization and function: moving past lipid rafts. Mol. Biol. Cell 2013, 24, 2765–2768. 10.1091/mbc.e13-03-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida P. F.; Pokorny A.; Hinderliter A. Thermodynamics of membrane domains. Biochim. Biophys. Acta, Biomembr. 2005, 1720, 1–13. 10.1016/j.bbamem.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Lorent J. H.; Levental I. Structural determinants of protein partitioning into ordered membrane domains and lipid rafts. Chem. Phys. Lipids 2015, 192, 23–32. 10.1016/j.chemphyslip.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Shah A.; Chen D.; Boda A. R.; Foster L. J.; Davis M. J.; Hill M. M. RaftProt: mammalian lipid raft proteome database. Nucleic Acids Res. 2015, 43, D335–338. 10.1093/nar/gku1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I.; Lingwood D.; Grzybek M.; Coskun U.; Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22050–22054. 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorent J. H.; Diaz-Rohrer B.; Lin X.; Spring K.; Gorfe A. A.; Levental K. R.; Levental I. Structural determinants and functional consequences of protein affinity for membrane rafts. Nat. Commun. 2017, 8, 1219. 10.1038/s41467-017-01328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson L. D.; Chiantia S.; London E. Perfringolysin O association with ordered lipid domains: implications for transmembrane protein raft affinity. Biophys. J. 2010, 99, 3255–3263. 10.1016/j.bpj.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. T.; Kiessling V.; Simmons J. A.; White J. M.; Tamm L. K. HIV gp41-mediated membrane fusion occurs at edges of cholesterol-rich lipid domains. Nat. Chem. Biol. 2015, 11, 424–431. 10.1038/nchembio.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolini C.; Baranski J.; Schlummer S.; Palomo J.; Lumbierres-Burgues M.; Kahms M.; Kuhlmann J.; Sanchez S.; Gratton E.; Waldmann H.; Winter R. Visualizing association of N-ras in lipid microdomains: influence of domain structure and interfacial adsorption. J. Am. Chem. Soc. 2006, 128, 192–201. 10.1021/ja055779x. [DOI] [PubMed] [Google Scholar]
- Weise K.; Triola G.; Brunsveld L.; Waldmann H.; Winter R. Influence of the lipidation motif on the partitioning and association of N-Ras in model membrane subdomains. J. Am. Chem. Soc. 2009, 131, 1557–1564. 10.1021/ja808691r. [DOI] [PubMed] [Google Scholar]
- Burns M.; Wisser K.; Wu J.; Levental I.; Veatch S. L. Miscibility transition temperature scales with growth temperature in a zebrafish cell line. Biophys. J. 2017, 113, 1212–1222. 10.1016/j.bpj.2017.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honerkamp-Smith A. R.; Cicuta P.; Collins M. D.; Veatch S. L.; den Nijs M.; Schick M.; Keller S. L. Line tensions, correlation lengths, and critical exponents in lipid membranes near critical points. Biophys. J. 2008, 95, 236–246. 10.1529/biophysj.107.128421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honerkamp-Smith A. R.; Veatch S. L.; Keller S. L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 53–63. 10.1016/j.bbamem.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Cicuta P.; Sengupta P.; Honerkamp-Smith A.; Holowka D.; Baird B. Critical fluctuations in plasma membrane vesicles. ACS Chem. Biol. 2008, 3, 287–293. 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- Marsh D. Liquid-ordered phases induced by cholesterol: a compendium of binary phase diagrams. Biochim. Biophys. Acta, Biomembr. 2010, 1798, 688–699. 10.1016/j.bbamem.2009.12.027. [DOI] [PubMed] [Google Scholar]
- Stone M. B.; Shelby S. A.; Nunez M. F.; Wisser K.; Veatch S. L.. Protein sorting by lipid phase-like domains supports emergent signaling function in B lymphocyte plasma membranes. eLife 2017, 6. 10.7554/eLife.19891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Seeing spots: complex phase behavior in simple membranes. Biochim. Biophys. Acta, Mol. Cell Res. 2005, 1746, 172–185. 10.1016/j.bbamcr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Frisz J. F.; Klitzing H. A.; Lou K.; Hutcheon I. D.; Weber P. K.; Zimmerberg J.; Kraft M. L. Sphingolipid domains in the plasma membranes of fibroblasts are not enriched with cholesterol. J. Biol. Chem. 2013, 288, 16855–16861. 10.1074/jbc.M113.473207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wustner D.; Modzel M.; Lund F. W.; Lomholt M. A. Imaging approaches for analysis of cholesterol distribution and dynamics in the plasma membrane. Chem. Phys. Lipids 2016, 199, 106–135. 10.1016/j.chemphyslip.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Frisz J. F.; Lou K.; Klitzing H. A.; Hanafin W. P.; Lizunov V.; Wilson R. L.; Carpenter K. J.; Kim R.; Hutcheon I. D.; Zimmerberg J.; Weber P. K.; Kraft M. L. Direct chemical evidence for sphingolipid domains in the plasma membranes of fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E613–622. 10.1073/pnas.1216585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft M. L. Sphingolipid organization in the plasma membrane and the mechanisms that influence it. Front. Cell Dev. Biol. 2017, 4, 154. 10.3389/fcell.2016.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X.; Liu Y.; Liu W.; Liang X.; Laganowsky A. Allosteric modulation of protein-protein interactions by individual lipid binding events. Nat. Commun. 2017, 8, 2203. 10.1038/s41467-017-02397-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Li J.; Liko I.; Gault J.; Bechara C.; Wu D.; Hopper J. T. S.; Giles K.; Benesch J. L. P.; Robinson C. V. Identifying key membrane protein lipid interactions using mass spectrometry. Nat. Protoc. 2018, 13, 1106–1120. 10.1038/nprot.2018.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta, Biomembr. 2004, 1666, 62–87. 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Jaipuria G.; Leonov A.; Giller K.; Vasa S. K.; Jaremko L.; Jaremko M.; Linser R.; Becker S.; Zweckstetter M. Cholesterol-mediated allosteric regulation of the mitochondrial translocator protein structure. Nat. Commun. 2017, 8, 14893. 10.1038/ncomms14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna M.; Niemela M.; Tynkkynen J.; Javanainen M.; Kulig W.; Muller D. J.; Rog T.; Vattulainen I.. Mechanism of allosteric regulation of beta2-adrenergic receptor by cholesterol. eLife 2016, 5. 10.7554/eLife.18432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liko I.; Degiacomi M. T.; Lee S.; Newport T. D.; Gault J.; Reading E.; Hopper J. T. S.; Housden N. G.; White P.; Colledge M.; Sula A.; Wallace B. A.; Kleanthous C.; Stansfeld P. J.; Bayley H.; Benesch J. L. P.; Allison T. M.; Robinson C. V. Lipid binding attenuates channel closure of the outer membrane protein OmpF. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 6691–6696. 10.1073/pnas.1721152115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner F. J.; Fernandez-Ballester G.; Fernandez-Carvajal A.; Ferrer-Montiel A. TRP channels interaction with lipids and its implications in disease. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1818–1827. 10.1016/j.bbamem.2015.03.022. [DOI] [PubMed] [Google Scholar]
- Hansen S. B. Lipid agonism: The PIP2 paradigm of ligand-gated ion channels. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2015, 1851, 620–628. 10.1016/j.bbalip.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. C.; Sanders C. R. Regulation of KCNQ/Kv7 family voltage-gated K(+) channels by lipids. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 586–597. 10.1016/j.bbamem.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. G. Lipid-protein interactions. Biochem. Soc. Trans. 2011, 39, 761–766. 10.1042/BST0390761. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Hutchison J. M. Membrane properties that shape the evolution of membrane enzymes. Curr. Opin. Struct. Biol. 2018, 51, 80–91. 10.1016/j.sbi.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Donlan J. A. C.; Hopper J. T. S.; Uzdavinys P.; Landreh M.; Struwe W. B.; Drew D.; Baldwin A. J.; Stansfeld P. J.; Robinson C. V. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 2017, 541, 421–424. 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C. Specific protein-lipid interactions in membrane proteins. Biochem. Soc. Trans. 2005, 33, 938–942. 10.1042/BST0330938. [DOI] [PubMed] [Google Scholar]
- Landreh M.; Marty M. T.; Gault J.; Robinson C. V. A sliding selectivity scale for lipid binding to membrane proteins. Curr. Opin. Struct. Biol. 2016, 39, 54–60. 10.1016/j.sbi.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L.; Sharpe M. A.; Garavito R. M.; Ferguson-Miller S. Conserved lipid-binding sites in membrane proteins: a focus on cytochrome c oxidase. Curr. Opin. Struct. Biol. 2007, 17, 444–450. 10.1016/j.sbi.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeagle P. L. Non-covalent binding of membrane lipids to membrane proteins. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1548–1559. 10.1016/j.bbamem.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Bogdanov M.; Dowhan W. Lipid-assisted protein folding. J. Biol. Chem. 1999, 274, 36827–36830. 10.1074/jbc.274.52.36827. [DOI] [PubMed] [Google Scholar]
- Bogdanov M.; Dowhan W.; Vitrac H. Lipids and topological rules governing membrane protein assembly. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843, 1475–1488. 10.1016/j.bbamcr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon A. M.; Lorch M.; Ces O.; Templer R. H.; Macrae F.; Booth P. J. Phosphatidylglycerol lipids enhance folding of an alpha helical membrane protein. J. Mol. Biol. 2008, 380, 548–556. 10.1016/j.jmb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Skorko-Glonek J.; Lipinska B.; Krzewski K.; Zolese G.; Bertoli E.; Tanfani F. HtrA heat shock protease interacts with phospholipid membranes and undergoes conformational changes. J. Biol. Chem. 1997, 272, 8974–8982. 10.1074/jbc.272.14.8974. [DOI] [PubMed] [Google Scholar]
- Debnath D. K.; Basaiawmoit R. V.; Nielsen K. L.; Otzen D. E. The role of membrane properties in mistic folding and dimerisation. Protein Eng., Des. Sel. 2011, 24, 89–97. 10.1093/protein/gzq095. [DOI] [PubMed] [Google Scholar]
- Dewald A. H.; Hodges J. C.; Columbus L. Physical determinants of beta-barrel membrane protein folding in lipid vesicles. Biophys. J. 2011, 100, 2131–2140. 10.1016/j.bpj.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymer F.; Veerappan A.; Schneider D. Transmembrane helix-helix interactions are modulated by the sequence context and by lipid bilayer properties. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 963–973. 10.1016/j.bbamem.2011.07.035. [DOI] [PubMed] [Google Scholar]
- Allen S. J.; Curran A. R.; Templer R. H.; Meijberg W.; Booth P. J. Controlling the folding efficiency of an integral membrane protein. J. Mol. Biol. 2004, 342, 1293–1304. 10.1016/j.jmb.2004.07.041. [DOI] [PubMed] [Google Scholar]
- Sanders M. R.; Findlay H. E.; Booth P. J. Lipid bilayer composition modulates the unfolding free energy of a knotted alpha-helical membrane protein. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E1799–E1808. 10.1073/pnas.1714668115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M.; Mileykovskaya E.; Dowhan W. Lipids in the assembly of membrane proteins and organization of protein supercomplexes: implications for lipid-linked disorders. Subcell. Biochem. 2008, 49, 197–239. 10.1007/978-1-4020-8831-5_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittendorf K. F.; Marinko J. T.; Hampton C. M.; Ke Z. L.; Hadziselimovic A.; Schlebach J. P.; Law C. L.; Li J.; Wright E. R.; Sanders C. R.; Ohi M. D.. Peripheral myelin protein 22 alters membrane architecture. Sci. Adv. 2017, 3, e1700220. 10.1126/sciadv.1700220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot J. L.; Engelman D. M. Membranes do not tell proteins how to fold. Biochemistry 2016, 55, 5–18. 10.1021/acs.biochem.5b01134. [DOI] [PubMed] [Google Scholar]
- Ulrih N. P.; Gmajner D.; Raspor P. Structural and physicochemical properties of polar lipids from thermophilic archaea. Appl. Microbiol. Biotechnol. 2009, 84, 249–260. 10.1007/s00253-009-2102-9. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Jefferys E. E.; Sansom M. S. Multiscale simulations reveal conserved patterns of lipid interactions with aquaporins. Structure 2013, 21, 810–819. 10.1016/j.str.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K. Dispensable nature of phosphatidylglycerol in Escherichia coli: dual roles of anionic phospholipids. Mol. Microbiol. 2001, 39, 1427–1433. 10.1046/j.1365-2958.2001.02320.x. [DOI] [PubMed] [Google Scholar]
- Shibuya I. Metabolic regulations and biological functions of phospholipids in Escherichia coli. Prog. Lipid Res. 1992, 31, 245–299. 10.1016/0163-7827(92)90010-G. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Mittendorf K. F. Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry 2011, 50, 7858–7867. 10.1021/bi2011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane T.; Rappaport F.; Popot J. L. Amphipol-assisted folding of bacteriorhodopsin in the presence or absence of lipids: functional consequences. Eur. Biophys. J. 2013, 42, 85–101. 10.1007/s00249-012-0839-z. [DOI] [PubMed] [Google Scholar]
- Gorzelle B. M.; Hoffman A. K.; Keyes M. H.; Gray D. N.; Ray D. G.; Sanders C. R. Amphipols can support the activity of a membrane enzyme. J. Am. Chem. Soc. 2002, 124, 11594–11595. 10.1021/ja027051b. [DOI] [PubMed] [Google Scholar]
- Dill K. A.; Chan H. S. From Levinthal to pathways to funnels. Nat. Struct. Mol. Biol. 1997, 4, 10–19. 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Mayne L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 15873–15880. 10.1073/pnas.1411798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.; Marqusee S. Probing the High Energy States in Protein by Proteolysis. J. Mol. Biol. 2004, 343, 1467–1476. 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- Bibow S.; Hiller S. A guide to quantifying membrane protein dynamics in lipids and other native-like environments by solution-state NMR spectroscopy. FEBS J. 2018, 10.1111/febs.14639. [DOI] [PubMed] [Google Scholar]
- Dutta A.; Altenbach C.; Mangahas S.; Yanamala N.; Gardner E.; Hubbell W. L.; Klein-Seetharaman J. Differential dynamics of extracellular and cytoplasmic domains in denatured States of rhodopsin. Biochemistry 2014, 53, 7160–7169. 10.1021/bi401557e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A.; Tirupula K. C.; Alexiev U.; Klein-Seetharaman J. Characterization of membrane protein non-native states. 1. Extent of unfolding and aggregation of rhodopsin in the presence of chemical denaturants. Biochemistry 2010, 49, 6317–6328. 10.1021/bi100338e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafer H.; Hiller S.; Hilty C.; Fernandez C.; Wuthrich K. Nonrandom structure in the urea-unfolded Escherichia coli outer membrane protein X (OmpX). Biochemistry 2004, 43, 860–869. 10.1021/bi0356606. [DOI] [PubMed] [Google Scholar]
- Sakakura M.; Hadziselimovic A.; Wang Z.; Schey K. L.; Sanders C. R. Structural basis for the trembler-J phenotype of Charcot-Marie-Tooth disease. Structure 2011, 19, 1160–1169. 10.1016/j.str.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Kuenze G.; Smith J. A.; Taylor K. C.; Duran A. M.; Hadziselimovic A.; Meiler J.; Vanoye C. G.; George A. L. Jr.; Sanders C. R. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv. 2018, 4, eaar2631. 10.1126/sciadv.aar2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfield C. NMR studies of partially folded molten-globule states. Methods Mol. Biol. 2004, 278, 233–254. 10.1385/1-59259-809-9:233. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt J. H.; Popot J. L. Folding and stability of integral membrane proteins in amphipols. Arch. Biochem. Biophys. 2014, 564, 327–343. 10.1016/j.abb.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt J. H. Folding of beta-barrel membrane proteins in lipid bilayers - Unassisted and assisted folding and insertion. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1927–1943. 10.1016/j.bbamem.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Barrera F.; Renart M.; Molina M.; Poveda J.; Encinar J.; Fernández A.; Neira J.; González-Ros J. Unfolding and refolding in vitro of a tetrameric, alpha-helical membrane protein: the prokaryotic potassium channel KcsA. Biochemistry 2005, 44, 14344–14352. 10.1021/bi050845t. [DOI] [PubMed] [Google Scholar]
- Hong H.; Tamm L. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4065–4070. 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C. P.; Fleming K. G. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10174–10177. 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay H. E.; Rutherford N. G.; Henderson P. J.; Booth P. J. Unfolding free energy of a two-domain transmembrane sugar transport protein. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18451–18456. 10.1073/pnas.1005729107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris N. J.; Findlay H. E.; Simms J.; Liu X.; Booth P. J. Relative domain folding and stability of a membrane transport protein. J. Mol. Biol. 2014, 426, 1812–1825. 10.1016/j.jmb.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Nagy J. K.; Lonzer W. L.; Sanders C. R. Kinetic study of folding and misfolding of diacylglycerol kinase in model membranes. Biochemistry 2001, 40, 8971–8980. 10.1021/bi010202n. [DOI] [PubMed] [Google Scholar]
- Otzen D. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. J. Mol. Biol. 2003, 330, 641–649. 10.1016/S0022-2836(03)00624-7. [DOI] [PubMed] [Google Scholar]
- Paslawski W.; Lillelund O. K.; Kristensen J. V.; Schafer N. P.; Baker R. P.; Urban S.; Otzen D. E. Cooperative folding of a polytopic alpha-helical membrane protein involves a compact N-terminal nucleus and nonnative loops. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 7978–7983. 10.1073/pnas.1424751112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach J. P.; Peng D.; Kroncke B. M.; Mittendorf K. F.; Narayan M.; Carter B. D.; Sanders C. R. Reversible folding of human peripheral myelin protein 22, a tetraspan membrane protein. Biochemistry 2013, 52, 3229–3241. 10.1021/bi301635f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. C.; Bowie J. U. Measuring membrane protein stability under native conditions. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 219–224. 10.1073/pnas.1318576111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R.; Gaffney K.; Yang Z.; Kim M.; Sungsuwan S.; Huang X.; Hubbell W. L.; Hong H. Steric trapping reveals a cooperativity network in the intramembrane protease GlpG. Nat. Chem. Biol. 2016, 12, 353–360. 10.1038/nchembio.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson R. E.; Min D.; Corin K.; Wang J. Y.; Bowie J. U. Applications of single-molecule methods to membrane protein folding studies. J. Mol. Biol. 2018, 430, 424–437. 10.1016/j.jmb.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D.; Jefferson R. E.; Qi Y.; Wang J. Y.; Arbing M. A.; Im W.; Bowie J. U. Unfolding of a ClC chloride transporter retains memory of its evolutionary history. Nat. Chem. Biol. 2018, 14, 489–496. 10.1038/s41589-018-0025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D. J.; Kessler M.; Oesterhelt F.; Moller C.; Oesterhelt D.; Gaub H. Stability of bacteriorhodopsin alpha-helices and loops analyzed by single-molecule force spectroscopy. Biophys. J. 2002, 83, 3578–3588. 10.1016/S0006-3495(02)75358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortle D. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 1996, 10, 27–34. 10.1096/fasebj.10.1.8566543. [DOI] [PubMed] [Google Scholar]
- Chen G.; Gouaux E. Probing the folding and unfolding of wild-type and mutant forms of bacteriorhodopsin in micellar solutions: evaluation of reversible unfolding conditions. Biochemistry 1999, 38, 15380–15387. 10.1021/bi9909039. [DOI] [PubMed] [Google Scholar]
- Burgess N.; Dao T.; Stanley A.; Fleming K. Beta-barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. J. Biol. Chem. 2008, 283, 26748–26758. 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C. P.; Zaccai N. R.; Fleming P. J.; Gessmann D.; Fleming K. G. Membrane protein thermodynamic stability may serve as the energy sink for sorting in the periplasm. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4285–4290. 10.1073/pnas.1212527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakelar J.; Buchanan S. K.; Noinaj N. The structure of the beta-barrel assembly machinery complex. Science 2016, 351, 180–186. 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdbrook D. A.; Burmann B. M.; Huber R. G.; Petoukhov M. V.; Svergun D. I.; Hiller S.; Bond P. J. A spring-loaded mechanism governs the clamp-like dynamics of the Skp chaperone. Structure 2017, 25, 1079–1088e3. 10.1016/j.str.2017.05.018. [DOI] [PubMed] [Google Scholar]
- McDonald S. K.; Fleming K. G. Aromatic side chain water-to-lipid transfer free energies Show a depth dependence across the membrane normal. J. Am. Chem. Soc. 2016, 138, 7946–7950. 10.1021/jacs.6b03460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx D. C.; Fleming K. G. Influence of protein scaffold on side-chain transfer free energies. Biophys. J. 2017, 113, 597–604. 10.1016/j.bpj.2017.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Rinehart D.; Tamm L. K. Membrane depth-dependent energetic contribution of the tryptophan side chain to the stability of integral membrane proteins. Biochemistry 2013, 52, 4413–4421. 10.1021/bi400344b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Guo R.; Gaffney K.; Kim M.; Muhammednazaar S.; Tian W.; Wang B.; Liang J.; Hong H. Folding-degradation relationship of a membrane protein mediated by the universally conserved ATP-dependent protease FtsH. J. Am. Chem. Soc. 2018, 140, 4656–4665. 10.1021/jacs.8b00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito R. R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. 10.1016/S0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Krishnamani V.; Hegde B. G.; Langen R.; Lanyi J. K. Secondary and tertiary Structure of bacteriorhodopsin in the SDS denatured state. Biochemistry 2012, 51, 1051–1060. 10.1021/bi201769z. [DOI] [PubMed] [Google Scholar]
- Curnow P.; Booth P. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18970–18975. 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faham S.; Yang D.; Bare E.; Yohannan S.; Whitelegge J.; Bowie J. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 2004, 335, 297–305. 10.1016/j.jmb.2003.10.041. [DOI] [PubMed] [Google Scholar]
- Cao Z.; Schlebach J.; Park C.; Bowie J. U. Thermodynamic stability of bacteriorhodopsin mutants measured relative to the bacterioopsin unfolded state. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 1049. 10.1016/j.bbamem.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker R. P.; Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nat. Chem. Biol. 2012, 8, 759–768. 10.1038/nchembio.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faerch M.; Christensen J. H.; Rittig S.; Johansson J. O.; Gregersen N.; de Zegher F.; Corydon T. J. Diverse vasopressin V2 receptor functionality underlying partial congenital nephrogenic diabetes insipidus. Am. J. Physiol. Renal Physiol. 2009, 297, F1518–1525. 10.1152/ajprenal.00331.2009. [DOI] [PubMed] [Google Scholar]
- Wiseman R. L.; Powers E. T.; Buxbaum J. N.; Kelly J. W.; Balch W. E. An adaptable standard for protein export from the endoplasmic reticulum. Cell 2007, 131, 809–821. 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- Powers E. T.; Powers D. L.; Gierasch L. M. FoldEco: a model for proteostasis in E. coli. Cell Rep. 2012, 1, 265–276. 10.1016/j.celrep.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello S. M.; Plummer A. M.; Fleming P. J.; Fleming K. G. Dynamic periplasmic chaperone reservoir facilitates biogenesis of outer membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4794–4800. 10.1073/pnas.1601002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra M.; Farrell D. W.; Dill K. A. Bacterial proteostasis balances energy and chaperone utilization efficiently. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E2654–E2661. 10.1073/pnas.1620646114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson R. E.; Blois T. M.; Bowie J. U. Membrane proteins can have high kinetic stability. J. Am. Chem. Soc. 2013, 135, 15183–15190. 10.1021/ja407232b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyair R.; Ogen-Shtern N.; Lederkremer G. Z. Glycan regulation of ER-associated degradation through compartmentalization. Semin. Cell Dev. Biol. 2015, 41, 99–109. 10.1016/j.semcdb.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Otzen D. E. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. J. Mol. Biol. 2003, 330, 641–649. 10.1016/S0022-2836(03)00624-7. [DOI] [PubMed] [Google Scholar]
- Sehgal P.; Otzen D. E. Thermodynamics of unfolding of an integral membrane protein in mixed micelles. Protein Sci. 2006, 15, 890–899. 10.1110/ps.052031306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curnow P.; Booth P. J. The contribution of a covalently bound cofactor to the folding and thermodynamic stability of an integral membrane protein. J. Mol. Biol. 2010, 403, 630–642. 10.1016/j.jmb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Leidenheimer N. J. Pharmacological chaperones: beyond conformational disorders. Handb. Exp. Pharmacol. 2017, 245, 135–153. 10.1007/164_2017_68. [DOI] [PubMed] [Google Scholar]
- Shearer A. G.; Hampton R. Y. Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. EMBO J. 2005, 24, 149–159. 10.1038/sj.emboj.7600498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y. X.; Conn P. M. Chaperoning G protein-coupled receptors: from cell biology to therapeutics. Endocr. Rev. 2014, 35, 602–647. 10.1210/er.2013-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. B.; Vivithanaporn P.; Swanson G. T. Glutamate binding and conformational flexibility of ligand-binding domains are critical early determinants of efficient kainate receptor biogenesis. J. Biol. Chem. 2009, 284, 14503–14512. 10.1074/jbc.M900510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodohl A.; Volkmer T.; Finger C.; Schneider D. Defining the structural basis for assembly of a transmembrane cytochrome. J. Mol. Biol. 2005, 350, 744–756. 10.1016/j.jmb.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Engelman D. M.; Chen Y.; Chin C. N.; Curran A. R.; Dixon A. M.; Dupuy A. D.; Lee A. S.; Lehnert U.; Matthews E. E.; Reshetnyak Y. K.; Senes A.; Popot J. L. Membrane protein folding: beyond the two stage model. FEBS Lett. 2003, 555, 122–125. 10.1016/S0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Xu Y.; Liu Y.; Ye Y. gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell 2015, 26, 4438–4450. 10.1091/mbc.E15-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H.; Sakmar T. P.; Huber T. The energetics of chromophore binding in the visual photoreceptor rhodopsin. Biophys. J. 2017, 113, 60–72. 10.1016/j.bpj.2017.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangeline M. A.; Vashistha N.; Hampton R. Y. Proteostatic tactics in the strategy of sterol regulation. Annu. Rev. Cell Dev. Biol. 2017, 33, 467–489. 10.1146/annurev-cellbio-111315-125036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R. G.; Hampton R. Y. A ’distributed degron’ allows regulated entry into the ER degradation pathway. EMBO J. 1999, 18, 5994–6004. 10.1093/emboj/18.21.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinthal C. Are there pathways for protein folding?. J. Chim. Phys. Phys.-Chim. Biol. 1968, 65, 44–45. 10.1051/jcp/1968650044. [DOI] [Google Scholar]
- Bai Y.; Sosnick T. R.; Mayne L.; Englander S. W. Protein folding intermediates: native-state hydrogen exchange. Science 1995, 269, 192–197. 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R.; Parra V.; Gatica D.; Rodriguez A. E.; Torrealba N.; Paredes F.; Wang Z. V.; Zorzano A.; Hill J. A.; Jaimovich E.; Quest A. F.; Lavandero S. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Hummer G. Microscopic interpretation of folding varphi-values using the transition path ensemble. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3263–3268. 10.1073/pnas.1520864113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J. Solving the membrane protein folding problem. Nature 2005, 438, 581–589. 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- Riley M.; Wallace B.; Flitsch S.; Booth P. Slow alpha helix formation during folding of a membrane protein. Biochemistry 1997, 36, 192–196. 10.1021/bi962199r. [DOI] [PubMed] [Google Scholar]
- Beguin P.; Hasler U.; Staub O.; Geering K. Endoplasmic reticulum quality control of oligomeric membrane proteins: Topogenic determinants involved in the degradation of the unassembled Na,K-ATPase alpha subunit and in its stabilization by beta subunit assembly. Mol. Biol. Cell 2000, 11, 1657–1672. 10.1091/mbc.11.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curnow P.; Di Bartolo N. D.; Moreton K. M.; Ajoje O. O.; Saggese N. P.; Booth P. J. Stable folding core in the folding transition state of an alpha-helical integral membrane protein. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14133–14138. 10.1073/pnas.1012594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. R.; Matouschek A.; Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 1992, 224, 771–782. 10.1016/0022-2836(92)90561-W. [DOI] [PubMed] [Google Scholar]
- Schlebach J. P.; Cao Z.; Bowie J. U.; Park C. Revisiting the folding kinetics of bacteriorhodopsin. Protein Sci. 2012, 21, 97–106. 10.1002/pro.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach J. P.; Woodall N. B.; Bowie J. U.; Park C. Bacteriorhodopsin folds through a poorly organized transition state. J. Am. Chem. Soc. 2014, 136, 16574–16581. 10.1021/ja508359n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxco K. W.; Simons K. T.; Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998, 277, 985–994. 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- Gruenhagen T. C.; Ziarek J. J.; Schlebach J. P. Bicelle size modulates the rate of bacteriorhodopsin folding. Protein Sci. 2018, 27, 1109–1112. 10.1002/pro.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otzen D. E. Mapping the folding pathway of the transmembrane protein DsbB by protein engineering. Protein Eng., Des. Sel. 2011, 24, 139–149. 10.1093/protein/gzq079. [DOI] [PubMed] [Google Scholar]
- Nam H. J.; Han S. K.; Bowie J. U.; Kim S. Rampant exchange of the structure and function of extramembrane domains between membrane and water soluble proteins. PLoS Comput. Biol. 2013, 9, e1002997. 10.1371/journal.pcbi.1002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrey T.; Jahnig F. Kinetics of folding and membrane insertion of a beta-barrel membrane protein. J. Biol. Chem. 1995, 270, 28199–28203. 10.1074/jbc.270.47.28199. [DOI] [PubMed] [Google Scholar]
- Surrey T.; Schmid A.; Jahnig F. Folding and membrane insertion of the trimeric beta-barrel protein OmpF. Biochemistry 1996, 35, 2283–2288. 10.1021/bi951216u. [DOI] [PubMed] [Google Scholar]
- Du K.; Lukacs G. L. Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 2009, 20, 1903–1915. 10.1091/mbc.e08-09-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt J. H.; den Blaauwen T.; Driessen A. J.; Tamm L. K. Outer membrane protein A of Escherichia coli inserts and folds into lipid bilayers by a concerted mechanism. Biochemistry 1999, 38, 5006–5016. 10.1021/bi982465w. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt J. H.; Tamm L. K. Secondary and tertiary structure formation of the beta-barrel membrane protein OmpA is synchronized and depends on membrane thickness. J. Mol. Biol. 2002, 324, 319–330. 10.1016/S0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; Arora A.; Kleinschmidt J. H. Structure and assembly of beta-barrel membrane proteins. J. Biol. Chem. 2001, 276, 32399–32402. 10.1074/jbc.R100021200. [DOI] [PubMed] [Google Scholar]
- Huysmans G. H.; Radford S. E.; Baldwin S. A.; Brockwell D. J. Malleability of the folding mechanism of the outer membrane protein PagP: parallel pathways and the effect of membrane elasticity. J. Mol. Biol. 2012, 416, 453–464. 10.1016/j.jmb.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huysmans G. H.; Baldwin S. A.; Brockwell D. J.; Radford S. E. The transition state for folding of an outer membrane protein. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4099–4104. 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danoff E. J.; Fleming K. G. Membrane defects accelerate outer membrane beta-barrel protein folding. Biochemistry 2015, 54, 97–99. 10.1021/bi501443p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danoff E. J.; Fleming K. G. Novel Kinetic Intermediates Populated along the Folding Pathway of the Transmembrane beta-Barrel OmpA. Biochemistry 2017, 56, 47–60. 10.1021/acs.biochem.6b00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voulhoux R.; Bos M. P.; Geurtsen J.; Mols M.; Tommassen J. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 2003, 299, 262–265. 10.1126/science.1078973. [DOI] [PubMed] [Google Scholar]
- Plummer A. M.; Fleming K. G. BamA alone accelerates outer membrane protein folding in vitro through a catalytic mechanism. Biochemistry 2015, 54, 6009–6011. 10.1021/acs.biochem.5b00950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton T. A.; Sandoval C. M.; Fowler C. A.; Pardi A.; Sousa M. C. The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1772–1777. 10.1073/pnas.0809275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blois T. M.; Hong H.; Kim T. H.; Bowie J. U. Protein unfolding with a steric trap. J. Am. Chem. Soc. 2009, 131, 13914–13915. 10.1021/ja905725n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach J. P.; Sanders C. R. The safety dance: biophysics of membrane protein folding and misfolding in a cellular context. Q. Rev. Biophys. 2015, 48, 1–34. 10.1017/S0033583514000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Blois T. M.; Cao Z.; Bowie J. U. Method to measure strong protein-protein interactions in lipid bilayers using a steric trap. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19802–19807. 10.1073/pnas.1010348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Bowie J. U. Dramatic destabilization of transmembrane helix interactions by features of natural membrane environments. J. Am. Chem. Soc. 2011, 133, 11389–11398. 10.1021/ja204524c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D.; Jefferson R. E.; Bowie J. U.; Yoon T. Y. Mapping the energy landscape for second-stage folding of a single membrane protein. Nat. Chem. Biol. 2015, 11, 981–987. 10.1038/nchembio.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot J. L. Integral membrane-protein structure - transmembrane alpha-helices as autonomous folding domains. Curr. Opin. Struct. Biol. 1993, 3, 532–540. 10.1016/0959-440X(93)90079-Z. [DOI] [Google Scholar]
- King C.; Raicu V.; Hristova K. Understanding the FRET signatures of interacting membrane proteins. J. Biol. Chem. 2017, 292, 5291–5310. 10.1074/jbc.M116.764282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C.; Stoneman M.; Raicu V.; Hristova K. Fully quantified spectral imaging reveals in vivo membrane protein interactions. Integr. Biol. (Camb.) 2016, 8, 216–229. 10.1039/C5IB00202H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarabipour S.; Ballmer-Hofer K.; Hristova K. VEGFR-2 conformational switch in response to ligand binding. eLife 2016, 5, e13876. 10.7554/eLife.13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comar W. D.; Schubert S. M.; Jastrzebska B.; Palczewski K.; Smith A. W. Time-resolved fluorescence spectroscopy measures clustering and mobility of a G protein-coupled receptor opsin in live cell membranes. J. Am. Chem. Soc. 2014, 136, 8342–8349. 10.1021/ja501948w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Bharill S.; Karandur D.; Peterson S. M.; Marita M.; Shi X.; Kaliszewski M. J.; Smith A. W.; Isacoff E. Y.; Kuriyan J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. eLife 2016, 5, e14107. 10.7554/eLife.14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Horn W. D.; Sanders C. R. Prokaryotic diacylglycerol kinase and undecaprenol kinase. Annu. Rev. Biophys. 2012, 41, 81–101. 10.1146/annurev-biophys-050511-102330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Lyons J. A.; Pye V. E.; Vogeley L.; Aragao D.; Kenyon C. P.; Shah S. T.; Doherty C.; Aherne M.; Caffrey M. Crystal structure of the integral membrane diacylglycerol kinase. Nature 2013, 497, 521–524. 10.1038/nature12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Stansfeld P. J.; Sansom M. S.; Keogh A.; Vogeley L.; Howe N.; Lyons J. A.; Aragao D.; Fromme P.; Fromme R.; Basu S.; Grotjohann I.; Kupitz C.; Rendek K.; Weierstall U.; Zatsepin N. A.; Cherezov V.; Liu W.; Bandaru S.; English N. J.; Gati C.; Barty A.; Yefanov O.; Chapman H. N.; Diederichs K.; Messerschmidt M.; Boutet S.; Williams G. J.; Marvin Seibert M.; Caffrey M. Ternary structure reveals mechanism of a membrane diacylglycerol kinase. Nat. Commun. 2015, 6, 10140. 10.1038/ncomms10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J.; Chen X.; Bowie J. U. Exploring the allowed sequence space of a membrane protein. Nat. Struct. Biol. 1996, 3, 141–148. 10.1038/nsb0296-141. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Wen J.; Bowie J. U. A passive transmembrane helix. Nat. Struct. Biol. 1997, 4, 986–990. 10.1038/nsb1297-986. [DOI] [PubMed] [Google Scholar]
- Gorzelle B. M.; Nagy J. K.; Oxenoid K.; Lonzer W. L.; Cafiso D. S.; Sanders C. R. Reconstitutive refolding of diacylglycerol kinase, an integral membrane protein. Biochemistry 1999, 38, 16373–16382. 10.1021/bi991292n. [DOI] [PubMed] [Google Scholar]
- Lorch M.; Booth P. J. Insertion kinetics of a denatured alpha helical membrane protein into phospholipid bilayer vesicles. J. Mol. Biol. 2004, 344, 1109–1121. 10.1016/j.jmb.2004.09.090. [DOI] [PubMed] [Google Scholar]
- Mi D.; Kim H. J.; Hadziselimovic A.; Sanders C. R. Irreversible misfolding of diacylglycerol kinase is independent of aggregation and occurs prior to trimerization and membrane association. Biochemistry 2006, 45, 10072–10084. 10.1021/bi060887x. [DOI] [PubMed] [Google Scholar]
- Nagy J. K.; Sanders C. R. Destabilizing mutations promote membrane protein misfolding. Biochemistry 2004, 43, 19–25. 10.1021/bi035918s. [DOI] [PubMed] [Google Scholar]
- Nagy J. K.; Sanders C. R. A critical residue in the folding pathway of an integral membrane protein. Biochemistry 2002, 41, 9021–9025. 10.1021/bi020318z. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Myers J. K. Disease-related misassembly of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 25–51. 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- Schlebach J. P.; Narayan M.; Alford C.; Mittendorf K. F.; Carter B. D.; Li J.; Sanders C. R. Conformational stability and pathogenic misfolding of the integral membrane protein PMP22. J. Am. Chem. Soc. 2015, 137, 8758–8768. 10.1021/jacs.5b03743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroobants K.; Kumita J. R.; Harris N. J.; Chirgadze D. Y.; Dobson C. M.; Booth P. J.; Vendruscolo M. Amyloid-like fibrils from an alpha-helical transmembrane protein. Biochemistry 2017, 56, 3225–3233. 10.1021/acs.biochem.7b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Webster P.; Taddei N.; Clark A.; Stefani M.; Ramponi G.; Dobson C. M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 3590–3594. 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz A.; Presto J.; Lara P.; Akinyi-Oloo L.; Garcia-Fojeda B.; Nilsson I.; Johansson J.; Casals C. Folding and intramembraneous BRICHOS binding of the prosurfactant protein C transmembrane segment. J. Biol. Chem. 2015, 290, 17628–17641. 10.1074/jbc.M114.630343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kim Y. H.; Arce F. T.; Gillman A. L.; Jang H.; Kagan B. L.; Nussinov R.; Yang J.; Lal R. Amyloid beta Ion channels in a membrane comprising brain total lipid extracts. ACS Chem. Neurosci. 2017, 8, 1348–1357. 10.1021/acschemneuro.7b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Batiste M.; Ninot-Pedrosa M.; Bayoumi M.; Gairi M.; Maglia G.; Carulla N. Abeta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 10866–10871. 10.1073/pnas.1605104113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Batiste M.; Tolchard J.; Giusti F.; Zoonens M.; Carulla N. Stabilization of a membrane-associated amyloid-beta oligomer for Its validation in Alzheimer’s disease. Front. Mol. Biosci. 2018, 5, 38. 10.3389/fmolb.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan B. L. Membrane pores in the pathogenesis of neurodegenerative disease. Prog. Mol. Biol. Transl. Sci. 2012, 107, 295–325. 10.1016/B978-0-12-385883-2.00001-1. [DOI] [PubMed] [Google Scholar]
- Jang H.; Arce F. T.; Ramachandran S.; Kagan B. L.; Lal R.; Nussinov R. Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs. Chem. Soc. Rev. 2014, 43, 6750–6764. 10.1039/C3CS60459D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrom R.; Aisenbrey C.; Borowik T.; Bokvist M.; Lindstrom F.; Sani M. A.; Olofsson A.; Grobner G. Disordered proteins: biological membranes as two-dimensional aggregation matrices. Cell Biochem. Biophys. 2008, 52, 175–189. 10.1007/s12013-008-9033-4. [DOI] [PubMed] [Google Scholar]
- Dikiy I.; Eliezer D. Folding and misfolding of alpha-synuclein on membranes. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 1013–1018. 10.1016/j.bbamem.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbenko G. P.; Kinnunen P. K. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 2006, 141, 72–82. 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Habchi J.; Chia S.; Galvagnion C.; Michaels T. C. T.; Bellaiche M. M. J.; Ruggeri F. S.; Sanguanini M.; Idini I.; Kumita J. R.; Sparr E.; Linse S.; Dobson C. M.; Knowles T. P. J.; Vendruscolo M. Cholesterol catalyses Abeta42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem. 2018, 10, 673–683. 10.1038/s41557-018-0031-x. [DOI] [PubMed] [Google Scholar]
- Kazlauskaite J.; Pinheiro T. J. Aggregation and fibrillization of prions in lipid membranes. Biochem. Soc. Symp. 2005, 72, 211–222. 10.1042/bss0720211. [DOI] [PubMed] [Google Scholar]
- Stefani M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010, 277, 4602–4613. 10.1111/j.1742-4658.2010.07889.x. [DOI] [PubMed] [Google Scholar]
- Straub J. E.; Thirumalai D. Membrane-protein interactions are key to understanding amyloid formation. J. Phys. Chem. Lett. 2014, 5, 633–635. 10.1021/jz500054d. [DOI] [PubMed] [Google Scholar]
- Terakawa M. S.; Lin Y.; Kinoshita M.; Kanemura S.; Itoh D.; Sugiki T.; Okumura M.; Ramamoorthy A.; Lee Y. H. Impact of membrane curvature on amyloid aggregation. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1741. 10.1016/j.bbamem.2018.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K.; Kato K.; Yanagisawa K. Abeta polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2010, 1801, 868–877. 10.1016/j.bbalip.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Rangachari V.; Dean D. N.; Rana P.; Vaidya A.; Ghosh P. Cause and consequence of Abeta - lipid interactions in Alzheimer disease pathogenesis. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1652. 10.1016/j.bbamem.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock M.; Stanga S.; Octave J. N.; Dewachter I.; Smith S. O.; Constantinescu S. N.; Kienlen-Campard P. Glycines from the APP GXXXG/GXXXA transmembrane motifs promote formation of pathogenic Abeta oligomers in cells. Front. Aging Neurosci. 2016, 8, 107. 10.3389/fnagi.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvagnion C. The role of lipids Interacting with alpha-synuclein in the pathogenesis of Parkinson’s disease. J. Parkinson's Dis. 2017, 7, 433–450. 10.3233/JPD-171103. [DOI] [PubMed] [Google Scholar]
- West A.; Brummel B. E.; Braun A. R.; Rhoades E.; Sachs J. N. Membrane remodeling and mechanics: Experiments and simulations of alpha-Synuclein. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1594–1609. 10.1016/j.bbamem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Winter R. The Effects of lipid membranes, crowding and osmolytes on the aggregation, and fibrillation propensity of human IAPP. J. Diabetes Res. 2015, 2015, 849017. 10.1155/2015/849017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat A.; Langen R.; Varkey J. Membranes as modulators of amyloid protein misfolding and target of toxicity. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1863. 10.1016/j.bbamem.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahara K. Membrane-mediated amyloid deposition of human islet amyloid polypeptide. Biophys. Rev. 2018, 10, 453–462. 10.1007/s12551-017-0351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnataro D.; Pepe A.; Zurzolo C. Cell biology of prion protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 57–82. 10.1016/bs.pmbts.2017.06.018. [DOI] [PubMed] [Google Scholar]
- Goold R.; Rabbanian S.; Sutton L.; Andre R.; Arora P.; Moonga J.; Clarke A. R.; Schiavo G.; Jat P.; Collinge J.; Tabrizi S. J. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2011, 2, 281. 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadi Thody S.; Mathew M. K.; Udgaonkar J. B. Mechanism of aggregation and membrane interactions of mammalian prion protein. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1927. 10.1016/j.bbamem.2018.02.031. [DOI] [PubMed] [Google Scholar]
- Dal Peraro M.; van der Goot F. G. Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. 10.1038/nrmicro.2015.3. [DOI] [PubMed] [Google Scholar]
- Ladokhin A. S. Cellular entry of binary and pore-forming bacterial toxins. Toxins 2018, 10, 11. 10.3390/toxins10010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W. C. Energetics of peptide and protein binding to lipid membranes. Adv. Exp. Med. Biol. 2010, 677, 14–23. 10.1007/978-1-4419-6327-7_2. [DOI] [PubMed] [Google Scholar]
- Friebe S.; van der Goot F. G.; Burgi J. The ins and outs of anthrax toxin. Toxins 2016, 8, 69. 10.3390/toxins8030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojko N.; Anderluh G. How lipid membranes affect pore forming toxin activity. Acc. Chem. Res. 2015, 48, 3073–3079. 10.1021/acs.accounts.5b00403. [DOI] [PubMed] [Google Scholar]
- Savinov S. N.; Heuck A. P. Interaction of cholesterol with perfringolysin O: What Have We Learned from Functional Analysis?. Toxins 2017, 9, 381. 10.3390/toxins9120381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweten R. K.; Hotze E. M.; Wade K. R. The unique molecular choreography of giant pore formation by the cholesterol-dependent cytolysins of gram-positive bacteria. Annu. Rev. Microbiol. 2015, 69, 323–340. 10.1146/annurev-micro-091014-104233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. B.; Heuck A. P. Perfringolysin O structure and mechanism of pore formation as a paradigm for cholesterol-dependent cytolysins. Subcell. Biochem. 2014, 80, 63–81. 10.1007/978-94-017-8881-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyanarayana P.; Maurya S.; Behera A.; Ravichandran M.; Visweswariah S. S.; Ayappa K. G.; Roy R. Cholesterol promotes cytolysin A activity by stabilizing the intermediates during pore formation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7323–E7330. 10.1073/pnas.1721228115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert E.; Vetter I. R.; Prumbaum D.; Penczek P. A.; Raunser S. Membrane insertion of alpha-xenorhabdolysin in near-atomic detail. eLife 2018, 7, e38017. 10.7554/eLife.38017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pee K.; Neuhaus A.; D’Imprima E.; Mills D. J.; Kuhlbrandt W.; Yildiz O.. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin. eLife 2017, 6. 10.7554/eLife.23644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland I. B. The extraordinary diversity of bacterial protein secretion mechanisms. Methods Mol. Biol. 2010, 619, 1–20. 10.1007/978-1-60327-412-8_1. [DOI] [PubMed] [Google Scholar]
- Deacon J. C.; Engelman D. M.; Barrera F. N. Targeting acidity in diseased tissues: mechanism and applications of the membrane-inserting peptide, pHLIP. Arch. Biochem. Biophys. 2015, 565, 40–48. 10.1016/j.abb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbiers M. C.; Spruijt R. B.; Demel R. A.; Hemminga M. A.; Wolfs C. J. Spontaneous insertion of gene 9 minor coat protein of bacteriophage M13 in model membranes. Biochim. Biophys. Acta, Biomembr. 2001, 1511, 309–316. 10.1016/S0005-2736(01)00288-7. [DOI] [PubMed] [Google Scholar]
- Meijberg W.; Booth P. J. The activation energy for insertion of transmembrane alpha-helices is dependent on membrane composition. J. Mol. Biol. 2002, 319, 839–853. 10.1016/S0022-2836(02)00342-X. [DOI] [PubMed] [Google Scholar]
- Nguyen V. P.; Alves D. S.; Scott H. L.; Davis F. L.; Barrera F. N. A novel soluble peptide with pH-responsive membrane insertion. Biochemistry 2015, 54, 6567–6575. 10.1021/acs.biochem.5b00856. [DOI] [PubMed] [Google Scholar]
- Ridder A. N.; van de Hoef W.; Stam J.; Kuhn A.; de Kruijff B.; Killian J. A. Importance of hydrophobic matching for spontaneous insertion of a single-spanning membrane protein. Biochemistry 2002, 41, 4946–4952. 10.1021/bi0158674. [DOI] [PubMed] [Google Scholar]
- Kauffman W. B.; Fuselier T.; He J.; Wimley W. C. Mechanism Matters: A taxonomy of cell penetrating peptides. Trends Biochem. Sci. 2015, 40, 749–764. 10.1016/j.tibs.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert R. J.; Dalla Serra M.; Froelich C. J.; Wallace M. I.; Anderluh G. Membrane pore formation at protein-lipid interfaces. Trends Biochem. Sci. 2014, 39, 510–516. 10.1016/j.tibs.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Sani M. A.; Separovic F. How membrane-active peptides get into lipid membranes. Acc. Chem. Res. 2016, 49, 1130–1138. 10.1021/acs.accounts.6b00074. [DOI] [PubMed] [Google Scholar]
- Popot J. L. Folding membrane proteins in vitro: a table and some comments. Arch. Biochem. Biophys. 2014, 564, 314–326. 10.1016/j.abb.2014.06.029. [DOI] [PubMed] [Google Scholar]
- Harris N. J.; Reading E.; Ataka K.; Grzegorzewski L.; Charalambous K.; Liu X.; Schlesinger R.; Heberle J.; Booth P. J. Structure formation during translocon-unassisted co-translational membrane protein folding. Sci. Rep. 2017, 7, 8021. 10.1038/s41598-017-08522-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrychenko A.; Rodnin M. V.; Posokhov Y. O.; Holt A.; Pucci B.; Killian J. A.; Ladokhin A. S. Thermodynamic measurements of bilayer insertion of a single transmembrane helix chaperoned by fluorinated surfactants. J. Mol. Biol. 2012, 416, 328–334. 10.1016/j.jmb.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy J. K.; Kuhn Hoffmann A.; Keyes M. H.; Gray D. N.; Oxenoid K.; Sanders C. R. Use of amphipathic polymers to deliver a membrane protein to lipid bilayers. FEBS Lett. 2001, 501, 115–120. 10.1016/S0014-5793(01)02627-8. [DOI] [PubMed] [Google Scholar]
- Focke P. J.; Hein C.; Hoffmann B.; Matulef K.; Bernhard F.; Dotsch V.; Valiyaveetil F. I. Combining in vitro folding with cell free protein synthesis form membrane protein expression. Biochemistry 2016, 55, 4212–4219. 10.1021/acs.biochem.6b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A.; Shin K.; Patterson R. E.; Liu X. Q.; Rainey J. K. Current strategies for protein production and purification enabling membrane protein structural biology. Biochem. Cell Biol. 2016, 94, 507–527. 10.1139/bcb-2015-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos C.; Kai L.; Proverbio D.; Ghoshdastider U.; Filipek S.; Dotsch V.; Bernhard F. Co-translational association of cell-free expressed membrane proteins with supplied lipid bilayers. Mol. Membr. Biol. 2013, 30, 75–89. 10.3109/09687688.2012.693212. [DOI] [PubMed] [Google Scholar]
- Matsubayashi H.; Ueda T. Purified cell-free systems as standard parts for synthetic biology. Curr. Opin. Chem. Biol. 2014, 22, 158–162. 10.1016/j.cbpa.2014.09.031. [DOI] [PubMed] [Google Scholar]
- Nishiyama K.; Maeda M.; Abe M.; Kanamori T.; Shimamoto K.; Kusumoto S.; Ueda T.; Tokuda H. A novel complete reconstitution system for membrane integration of the simplest membrane protein. Biochem. Biophys. Res. Commun. 2010, 394, 733–736. 10.1016/j.bbrc.2010.03.061. [DOI] [PubMed] [Google Scholar]
- Kuruma Y.; Ueda T. The PURE system for the cell-free synthesis of membrane proteins. Nat. Protoc. 2015, 10, 1328–1344. 10.1038/nprot.2015.082. [DOI] [PubMed] [Google Scholar]
- Gessesse B.; Nagaike T.; Nagata K.; Shimizu Y.; Ueda T. G-protein coupled receptor protein synthesis on a lipid bilayer using a reconstituted cell-free protein synthesis system. Life (Basel, Switz.) 2018, 8, 54. 10.3390/life8040054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson J. C.; Chen M.; Jiang F.; Moller I.; Wiedmann M.; Kuhn A.; Phillips G. J.; Dalbey R. E. YidC mediates membrane protein insertion in bacteria. Nature 2000, 406, 637–641. 10.1038/35020586. [DOI] [PubMed] [Google Scholar]
- Cabelli R. J.; Chen L.; Tai P. C.; Oliver D. B. SecA protein is required for secretory protein translocation into E. coli membrane vesicles. Cell 1988, 55, 683–692. 10.1016/0092-8674(88)90227-9. [DOI] [PubMed] [Google Scholar]
- Chio U. S.; Cho H.; Shan S. O. Mechanisms of tail-anchored membrane protein targeting and Insertion. Annu. Rev. Cell Dev. Biol. 2017, 33, 417–438. 10.1146/annurev-cellbio-100616-060839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde R. S.; Keenan R. J. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2011, 12, 787–798. 10.1038/nrm3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateja A.; Keenan R. J. A structural perspective on tail-anchored protein biogenesis by the GET pathway. Curr. Opin. Struct. Biol. 2018, 51, 195–202. 10.1016/j.sbi.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guna A.; Volkmar N.; Christianson J. C.; Hegde R. S. The ER membrane protein complex is a transmembrane domain insertase. Science 2018, 359, 470–473. 10.1126/science.aao3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff M. J.; Itzhak D. N.; Hussmann J. A.; Schirle Oakdale N. T.; Costa E. A.; Jonikas M.; Weibezahn J.; Popova K. D.; Jan C. H.; Sinitcyn P.; Vembar S. S.; Hernandez H.; Cox J.; Burlingame A. L.; Brodsky J. L.; Frost A.; Borner G. H.; Weissman J. S. The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. eLife 2018, 7, e37018. 10.7554/eLife.37018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood P. J.; Juszkiewicz S.; Guna A.; Shao S.; Hegde R. S. EMC Is required to initiate accurate membrane protein topogenesis. Cell 2018, 175, 1507–1519. 10.1016/j.cell.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T.; Bhushan S.; Jarasch A.; Armache J. P.; Funes S.; Jossinet F.; Gumbart J.; Mielke T.; Berninghausen O.; Schulten K.; Westhof E.; Gilmore R.; Mandon E. C.; Beckmann R. Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 2009, 326, 1369–1373. 10.1126/science.1178535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S.; Brandt F.; Hrabe T.; Lang S.; Eibauer M.; Zimmermann R.; Forster F. Structure and 3D arrangement of endoplasmic reticulum membrane-associated ribosomes. Structure 2012, 20, 1508–1518. 10.1016/j.str.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Frauenfeld J.; Gumbart J.; Sluis E. O.; Funes S.; Gartmann M.; Beatrix B.; Mielke T.; Berninghausen O.; Becker T.; Schulten K.; Beckmann R. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat. Struct. Mol. Biol. 2011, 18, 614–621. 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg-Hua Y.; Freudenberg J.; Vacic V.; Abhyankar A.; Emde A. K.; Ben-Avraham D.; Barzilai N.; Oschwald D.; Christen E.; Koppel J.; Greenwald B.; Darnell R. B.; Germer S.; Atzmon G.; Davies P. Disease variants in genomes of 44 centenarians. Mol. Genet. Genomic Med. 2014, 2, 438–450. 10.1002/mgg3.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogala M.; Becker T.; Beatrix B.; Armache J. P.; Barrio-Garcia C.; Berninghausen O.; Beckmann R. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 2014, 506, 107–110. 10.1038/nature12950. [DOI] [PubMed] [Google Scholar]
- Voorhees R. M.; Fernandez I. S.; Scheres S. H.; Hegde R. S. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 2014, 157, 1632–1643. 10.1016/j.cell.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea P. F.; Stroud R. M. Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17182–17187. 10.1073/pnas.1012556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg B.; Clemons W. M.; Collinson I.; Modis Y.; Hartmann E.; Harrison S. C.; Rapoport T. A. X-ray structure of a protein-conducting channel. Nature 2004, 427, 36–44. 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- Baldridge R. D.; Rapoport T. A. Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell 2016, 166, 394–407. 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher R. C. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 2007, 58, 157–170. 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- Saparov S. M.; Erlandson K.; Cannon K.; Schaletzky J.; Schulman S.; Rapoport T. A.; Pohl P. Determining the conductance of the SecY protein translocation channel for small molecules. Mol. Cell 2007, 26, 501–509. 10.1016/j.molcel.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.; Burbaum L.; Unverdorben P.; Pech M.; Chen Y.; Zimmermann R.; Beckmann R.; Forster F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403. 10.1038/ncomms9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees R. M.; Hegde R. S. Structure of the Sec61 channel opened by a signal sequence. Science 2016, 351, 88–91. 10.1126/science.aad4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymer F.; von Heijne G.; White S. H. Mechanisms of integral membrane protein insertion and folding. J. Mol. Biol. 2015, 427, 999–1022. 10.1016/j.jmb.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron J. W.; Hunt K. C.; Frydman J. Cotranslational signal-independent SRP preloading during membrane targeting. Nature 2016, 536, 224–228. 10.1038/nature19309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W.; Chai Q.; Zhong M.; Yu L.; Fang J.; Wang T.; Li H.; Zhu H.; Wei Y. Assembling of AcrB trimer in cell membrane. J. Mol. Biol. 2012, 423, 123–134. 10.1016/j.jmb.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage L.; Hendrick J. P.; Schiebel E.; Driessen A. J.; Wickner W. The purified E. coli integral membrane protein SecY/E is sufficient for reconstitution of SecA-dependent precursor protein translocation. Cell 1990, 62, 649–657. 10.1016/0092-8674(90)90111-Q. [DOI] [PubMed] [Google Scholar]
- Braunger K.; Pfeffer S.; Shrimal S.; Gilmore R.; Berninghausen O.; Mandon E. C.; Becker T.; Forster F.; Beckmann R. Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 2018, 360, 215–219. 10.1126/science.aar7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S.; Dudek J.; Gogala M.; Schorr S.; Linxweiler J.; Lang S.; Becker T.; Beckmann R.; Zimmermann R.; Forster F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. 10.1038/ncomms4072. [DOI] [PubMed] [Google Scholar]
- Harada Y.; Li H.; Li H.; Lennarz W. J. Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6945–6949. 10.1073/pnas.0812489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozman R.; Okiyoneda T.; Mulvihill C. M.; Rini J. M.; Barriere H.; Lukacs G. L. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 2009, 184, 847–862. 10.1083/jcb.200808124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimal S.; Cherepanova N. A.; Gilmore R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol. 2015, 41, 71–78. 10.1016/j.semcdb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 1986, 14, 4683–4690. 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D.; Hartmann E.; Prehn S.; Rapoport T. A. A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature 1992, 357, 47–52. 10.1038/357047a0. [DOI] [PubMed] [Google Scholar]
- Sauri A.; McCormick P. J.; Johnson A. E.; Mingarro I. Sec61alpha and TRAM are sequentially adjacent to a nascent viral membrane protein during its ER integration. J. Mol. Biol. 2007, 366, 366–374. 10.1016/j.jmb.2006.11.052. [DOI] [PubMed] [Google Scholar]
- Hartmann E.; Gorlich D.; Kostka S.; Otto A.; Kraft R.; Knespel S.; Burger E.; Rapoport T. A.; Prehn S. A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur. J. Biochem. 1993, 214, 375–381. 10.1111/j.1432-1033.1993.tb17933.x. [DOI] [PubMed] [Google Scholar]
- Fons R. D.; Bogert B. A.; Hegde R. S. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 2003, 160, 529–539. 10.1083/jcb.200210095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer N.; Junne T.; Kalies K. U.; Spiess M.; Hartmann E. TRAP assists membrane protein topogenesis at the mammalian ER membrane. Biochim. Biophys. Acta, Mol. Cell Res. 2013, 1833, 3104–3111. 10.1016/j.bbamcr.2013.08.018. [DOI] [PubMed] [Google Scholar]
- Tyedmers J.; Lerner M.; Bies C.; Dudek J.; Skowronek M. H.; Haas I. G.; Heim N.; Nastainczyk W.; Volkmer J.; Zimmermann R. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7214–7219. 10.1073/pnas.97.13.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reithinger J. H.; Kim J. E.; Kim H. Sec62 protein mediates membrane insertion and orientation of moderately hydrophobic signal anchor proteins in the endoplasmic reticulum (ER). J. Biol. Chem. 2013, 288, 18058–18067. 10.1074/jbc.M113.473009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlack K. E.; Misselwitz B.; Plath K.; Rapoport T. A. BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell 1999, 97, 553–564. 10.1016/S0092-8674(00)80767-9. [DOI] [PubMed] [Google Scholar]
- Zahedi R. P.; Volzing C.; Schmitt A.; Frien M.; Jung M.; Dudek J.; Wortelkamp S.; Sickmann A.; Zimmermann R. Analysis of the membrane proteome of canine pancreatic rough microsomes identifies a novel Hsp40, termed ERj7. Proteomics 2009, 9, 3463–3473. 10.1002/pmic.200800722. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.; Dudek J.; Zimmermann R.; Forster F. Organization of the native ribosome-translocon complex at the mammalian endoplasmic reticulum membrane. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860, 2122–2129. 10.1016/j.bbagen.2016.06.024. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.; Dudek J.; Schaffer M.; Ng B. G.; Albert S.; Plitzko J. M.; Baumeister W.; Zimmermann R.; Freeze H. H.; Engel B. D.; Forster F. Dissecting the molecular organization of the translocon-associated protein complex. Nat. Commun. 2017, 8, 14516. 10.1038/ncomms14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T.; Kim H.; Bihlmaier K.; Lundin C.; Boekel J.; Andersson H.; Nilsson I.; White S. H.; von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 2005, 433, 377–381. 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; Creamer T. P.; White S. H. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry 1996, 35, 5109–5124. 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- Gumbart J. C.; Teo I.; Roux B.; Schulten K. Reconciling the roles of kinetic and thermodynamic factors in membrane-protein insertion. J. Am. Chem. Soc. 2013, 135, 2291–2297. 10.1021/ja310777k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojemalm K.; Halling K. K.; Nilsson I.; von Heijne G. Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol. Cell 2012, 45, 529–540. 10.1016/j.molcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. H.; Wimley W. C. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta, Rev. Biomembr. 1998, 1376, 339–352. 10.1016/S0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- Joh N.; Min A.; Faham S.; Whitelegge J.; Yang D.; Woods V.; Bowie J. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature 2008, 453, 1266–1270. 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Sato Y.; Hessa T.; von Heijne G.; Lee J. K.; Kodama I.; Sakaguchi M.; Uozumi N. Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 8263–8268. 10.1073/pnas.0611007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar A.; Weinstein J.; Biran I.; Fridman Y.; Bibi E.; Fleishman S. J.. Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. eLife 2016, 5. 10.7554/eLife.12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojemalm K.; Higuchi T.; Lara P.; Lindahl E.; Suga H.; von Heijne G. Energetics of side-chain snorkeling in transmembrane helices probed by nonproteinogenic amino acids. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 10559–10564. 10.1073/pnas.1606776113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meindl-Beinker N. M.; Lundin C.; Nilsson I.; White S. H.; von Heijne G. Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep. 2006, 7, 1111–1116. 10.1038/sj.embor.7400818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernsel A.; Viklund H.; Falk J.; Lindahl E.; von Heijne G.; Elofsson A. Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7177–7181. 10.1073/pnas.0711151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illergard K.; Kauko A.; Elofsson A. Why are polar residues within the membrane core evolutionary conserved?. Proteins: Struct., Funct., Genet. 2011, 79, 79–91. 10.1002/prot.22859. [DOI] [PubMed] [Google Scholar]
- Hessa T.; Meindl-Beinker N. M.; Bernsel A.; Kim H.; Sato Y.; Lerch-Bader M.; Nilsson I.; White S. H.; von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 2007, 450, 1026–1030. 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- Gafvelin G.; von Heijne G. Topological ″frustration″ in multispanning E. coli inner membrane proteins. Cell 1994, 77, 401–412. 10.1016/0092-8674(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Moss K.; Helm A.; Lu Y.; Bragin A.; Skach W. R. Coupled translocation events generate topological heterogeneity at the endoplasmic reticulum membrane. Mol. Biol. Cell 1998, 9, 2681–2697. 10.1091/mbc.9.9.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermansson M.; von Heijne G. Inter-helical hydrogen bond formation during membrane protein integration into the ER membrane. J. Mol. Biol. 2003, 334, 803–809. 10.1016/j.jmb.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Virkki M. T.; Agrawal N.; Edsbacker E.; Cristobal S.; Elofsson A.; Kauko A. Folding of Aquaporin 1: Multiple evidence that helix 3 can shift out of the membrane core. Protein Sci. 2014, 23, 981–992. 10.1002/pro.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M.; Heacock P. N.; Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002, 21, 2107–2116. 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauko A.; Hedin L. E.; Thebaud E.; Cristobal S.; Elofsson A.; von Heijne G. Repositioning of transmembrane alpha-helices during membrane protein folding. J. Mol. Biol. 2010, 397, 190–201. 10.1016/j.jmb.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Van Lehn R. C.; Zhang B.; Miller T. F. 3rd. Regulation of multispanning membrane protein topology via post-translational annealing. eLife 2015, 4, e08697. 10.7554/eLife.08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall N. B.; Hadley S.; Yin Y.; Bowie J. U. Complete topology inversion can be part of normal membrane protein biogenesis. Protein Sci. 2017, 26, 824–833. 10.1002/pro.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck T. M.; Skach W. R. Differential stability of biogenesis intermediates reveals a common pathway for aquaporin-1 topological maturation. J. Biol. Chem. 2005, 280, 261–269. 10.1074/jbc.M409920200. [DOI] [PubMed] [Google Scholar]
- Schlebach J. P.; Sanders C. R. Influence of Pathogenic Mutations on the energetics of translocon-mediated bilayer integration of transmembrane helices. J. Membr. Biol. 2015, 248, 371–381. 10.1007/s00232-014-9726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham P. G.; Brodsky J. L. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim. Biophys. Acta, Mol. Cell Res. 2013, 1833, 2447–2457. 10.1016/j.bbamcr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O.; Hegde R. S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtley S.; Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 1989, 5, 277–307. 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- Ellgaard L.; Helenius A. ER quality control: towards an understranding at the molecular level. Curr. Opin. Cell Biol. 2001, 13, 431–437. 10.1016/S0955-0674(00)00233-7. [DOI] [PubMed] [Google Scholar]
- Anelli T.; Sitia R. Protein quality control in the early secratory pathway. EMBO J. 2008, 27, 315–327. 10.1038/sj.emboj.7601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K.; Nagata K. Protein folding and quality control in the ER. Cold Spring Harbor Perspect. Biol. 2011, 3, 1–25. 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey K.; Braakman I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235. 10.1042/EBC20160003. [DOI] [PubMed] [Google Scholar]
- Guerriero C. J.; Brodsky J. L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar S. S.; Brodsky J. L. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky J. L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A.; Bukau B.; Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol. Cell 2010, 40, 238–252. 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Vincenz-Donnelly L.; Hipp M. S. The endoplasmic reticulum: A hub of protein quality control in health and disease. Free Radical Biol. Med. 2017, 108, 383–393. 10.1016/j.freeradbiomed.2017.03.031. [DOI] [PubMed] [Google Scholar]
- Hetz C.; Chevet E.; Oakes S. A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Carvalho P.; Voeltz G. K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. 10.1126/science.aan5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha C. M.; Amaral M. D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J.; Skach W. R. Mechanisms of CFTR folding at the endoplasmic reticulum. Front. Pharmacol. 2012, 3, 201. 10.3389/fphar.2012.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S.; Bamberger C.; Calzolari D.; Martinez-Bartolome S.; Lavallee-Adam M.; Balch W. E.; Yates J. R. 3rd. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. 10.1038/nature15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O.; Gautschi I.; Ishikawa T.; Breitschopf K.; Ciechanover A.; Schild L.; Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck T. M.; Jordahl A. S.; Yates M. E.; Preston G. M.; Cook E.; Kleyman T. R.; Brodsky J. L. Interactions between intersubunit transmembrane domains regulate the chaperone-dependent degradation of an oligomeric membrane protein. Biochem. J. 2017, 474, 357–376. 10.1042/BCJ20160760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson K. M.; Bergeron J. J.; Shames I.; Colby J.; Nguyen D. T.; Chevet E.; Thomas D. Y.; Snipes G. J. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for ″gain-of-function″ ER diseases. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9852–9857. 10.1073/pnas.152621799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R.; Hermjakob H.; Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta, Gen. Subj. 1999, 1473, 4–8. 10.1016/S0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- Zielinska D. F.; Gnad F.; Wisniewski J. R.; Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequnce constraints. Cell 2010, 141, 897–907. 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Breitling J.; Aebi M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harbor Perspect. Biol. 2013, 5, 1–14. 10.1101/cshperspect.a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo J. J.; Parodi A. J. A sweet code for glycoprotein folding. FEBS Lett. 2015, 589, 3379–3387. 10.1016/j.febslet.2015.07.021. [DOI] [PubMed] [Google Scholar]
- Lehle L.; Tanner W. The specific site of tunicamycin Inhibition in the formation of dolichol-bound N-acetylglucosamine derivatives. FEBS Lett. 1976, 71, 167–170. 10.1016/0014-5793(76)80922-2. [DOI] [PubMed] [Google Scholar]
- Lamriben L.; Graham J. B.; Adams B. M.; Hebert D. N. N-Glycan-based ER molecular chaperone and protein quality control system: the calnexin binding cycle. Traffic 2016, 17, 308–326. 10.1111/tra.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous A.; Pisoni G. B.; Hebert D. N.; Molinari M. N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 2015, 41, 79–89. 10.1016/j.semcdb.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli C.; Bernasconi R.; Solda T.; Calanca V.; Molinari M. Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PLoS One 2011, 6, e16304. 10.1371/journal.pone.0016304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Hu D.; Yabe R.; Tateno H.; Qin S. Y.; Matsumoto N.; Hirabayashi J.; Yamamoto K. Role of malectin in Glc(2)Man(9)GlcNAc(2)-dependent quality control of alpha1-antitrypsin. Mol. Biol. Cell 2011, 22, 3559–3570. 10.1091/mbc.e11-03-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada I.; Imai S.-i.; Kai M.; Sakane F.; Kanoh H. chaperone function of calreticulin When expressed in the endoplasmic reticulum as the membrane-anchored and soluble forms. J. Biol. Chem. 1995, 270, 20298–20304. 10.1074/jbc.270.35.20298. [DOI] [PubMed] [Google Scholar]
- Schrag J. D.; Bergeron J. J. M.; Li Y.; Borisova S.; Hahn M.; Thomas D. Y.; Cygler M. The structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol. Cell 2001, 8, 633–644. 10.1016/S1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- Vassilakos A.; Michalak M.; Lehrman M. A.; Williams D. B. Oligosacharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry 1998, 37, 3480–3490. 10.1021/bi972465g. [DOI] [PubMed] [Google Scholar]
- Fontanini A.; Chies R.; Snapp E. L.; Ferrarini M.; Fabrizi G. M.; Brancolini C. Glycan-independent role of calnexin in the intracellular retention of Charcot-Marie Tooth 1A Gas3/PMP22 mutants. J. Biol. Chem. 2005, 280, 2378–2387. 10.1074/jbc.M405104200. [DOI] [PubMed] [Google Scholar]
- Danilczyk U. G.; Williams D. B. The lectin chaperone calnexin utilizes polypeptide-based Interactions to associate with many of Its substrates in vivo. J. Biol. Chem. 2001, 276, 25532–25540. 10.1074/jbc.M100270200. [DOI] [PubMed] [Google Scholar]
- Swanton E.; High S.; Woodman P. Role of calnexin in the glycan-independent quality control of proteolipid protein. EMBO J. 2003, 22, 2948–2958. 10.1093/emboj/cdg300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. B. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. 10.1242/jcs.02856. [DOI] [PubMed] [Google Scholar]
- Korkhov V. M.; Milan-Lobo L.; Zuber B.; Farhan H.; Schmid J. A.; Freissmuth M.; Sitte H. H. Peptide-based Interactions with calnexin target misassembled membrane proteins into endoplasmic reticulum-derived multilamellar bodies. J. Mol. Biol. 2008, 378, 337–352. 10.1016/j.jmb.2008.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T.; Hashimoto Y.; Akuzawa T.; Hirai R.; Kobayashi H.; Sato K. Rer1 and calnexin regulate endoplasmic reticulum retention of a peripheral myelin protein 22 mutant that causes type 1A Charcot-Marie-Tooth disease. Sci. Rep. 2015, 4, 6992. 10.1038/srep06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanamaker C. P.; Green W. N. N-linked Glycosylation is required for nicotinic receptor assemby but not for subunit associations with calnexin. J. Biol. Chem. 2005, 280, 33800–33810. 10.1074/jbc.M501813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio C.; Caramelo J. J.; Parodi A. J. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin. Cell Dev. Biol. 2010, 21, 491–499. 10.1016/j.semcdb.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta E. S.; Helenius A. Conformational Requirements for Glycoprotein Reglucosylation in the Endoplasmic Reticulum. J. Cell Biol. 2000, 148, 1123–1129. 10.1083/jcb.148.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo J. J.; Castro O. A.; Prat-Gay d.; Parodi A. J. The Endoplasmic Reticulum Glucosyltransferase Recognizes Nearly Native Glycoprotein Folding Intermediates. J. Biol. Chem. 2004, 279, 46280–46285. 10.1074/jbc.M408404200. [DOI] [PubMed] [Google Scholar]
- Ito Y.; Takeda Y.; Seko A.; Izumi M.; Kajihara Y. Functional analysis of endoplasmic reticulum glucosyltransferase (UGGT): Synthetic chemistry’s initiative in glycobiology. Semin. Cell Dev. Biol. 2015, 41, 90–98. 10.1016/j.semcdb.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Solda T.; Galli C.; Kaufman R. J.; Molinari M. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol. Cell 2007, 27, 238–249. 10.1016/j.molcel.2007.05.032. [DOI] [PubMed] [Google Scholar]
- Takeda Y.; Seko A.; Fujikawa K.; Izumi M.; Kajihara Y.; Ito Y. Effects of domain composition of catalytic activity of human glucose:glycoprotein glucosyltransferases. Glycobiology 2016, 26, 999–1006. 10.1093/glycob/cww069. [DOI] [PubMed] [Google Scholar]
- Calles-Garcia D.; Yang M.; Soya N.; Melero R.; Menade M.; Ito Y.; Vargas J.; Lukacs G. L.; Kollman J. M.; Kozlov G.; Gehring K. Single-particle electron microscopy structure of UDP-glucose:glycoprotein glucosyltransferase suggests a selectivity mechanism for misfolded proteins. J. Biol. Chem. 2017, 292, 11499–11507. 10.1074/jbc.M117.789495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T.; Song C.; Zhu T.; Toshimori T.; Murata K.; Hayashi Y.; Kamikubo H.; Uchihashi T.; Kato K. Visualisation of a flexible modular structure of the ER folding-sensor enzyme UGGT. Sci. Rep. 2017, 7, 12142. 10.1038/s41598-017-12283-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roversi P.; Marti L.; Caputo A. T.; Alonzi D. S.; Hill J. C.; Dent K. C.; Kumar A.; Levasseur M. D.; Lia A.; Waksman T.; Basu S.; Albrecht Y. S.; Qian K.; McIvor J. P.; Lipp C. B.; Siliqi D.; Vasiljevic S.; Mohammed S.; Lukacik P.; Walsh M. A.; Santino a.; Zitzmann N. Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8544–8549. 10.1073/pnas.1703682114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merulla J.; Solda T.; Molinari M. A novel UGGT1 and p97-dependent checkpoint for native ectodomains with ionizable intramembrane residue. Mol. Biol. Cell 2015, 26, 1532–1542. 10.1091/mbc.E14-12-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagioli C.; Sitia R. Mannose trimming by endoplasmic reticulum mannosidase I times the proteasomal degradation of unassembled immunoglobulin subunits. J. Biol. Chem. 2001, 276, 12885–12892. 10.1074/jbc.M009603200. [DOI] [PubMed] [Google Scholar]
- Vabulas R. M.; Hartl F. U. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 2005, 310, 1960–1963. 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- Roth J.; Zuber C. Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochem. Cell Biol. 2017, 147, 269–284. 10.1007/s00418-016-1513-9. [DOI] [PubMed] [Google Scholar]
- Shenkman M.; Groisman B.; Ron E.; Avezov E.; Hendershot L. M.; Lederkremer G. Z. A shared Endoplasmic Reticulum-associated Degradation Pathway Involving the EDEM Protein for Glycosylated and Nonglycosylated Proteins. J. Biol. Chem. 2013, 288, 2167–2178. 10.1074/jbc.M112.438275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L.; McCaul N.; Chatsisvili A.; Braakman I. Co- and post-translational protein folding in the ER. Traffic 2016, 17, 615–638. 10.1111/tra.12392. [DOI] [PubMed] [Google Scholar]
- Hendershot L. M. The ER function BiP is a master regulator of ER runction. Mount Sinai J. Med. 2004, 71, 289–297. [PubMed] [Google Scholar]
- Behnke J.; Feige M. J.; Hendershot L. M. BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. 10.1016/j.jmb.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk A.; Rieger H.; Zimmermann R.. Co-chaperones of the mammalian endoplasmic reticulum. In The Networking of Chaperones by Co-Chaperones; Blatch G., Edkins A. L., Eds.; Springer; New York, 2014; pp 179–200. [Google Scholar]
- Molinari M.; Helenius A. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 2000, 288, 331–333. 10.1126/science.288.5464.331. [DOI] [PubMed] [Google Scholar]
- Vanoni O.; Paganetti P.; Molinari M. Consequences of individual N-glycan deletions and of proteasomal inhibition on secretion of active BACE. Mol. Biol. Cell 2008, 19, 4086–4098. 10.1091/mbc.e08-05-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokhtaeva E.; Sachs G.; Vagin O. Diverse pathways for maturation of the Na,K-ATPase beta1 and beta2 subunits in the endoplasmic reticulum of Madin-Darby canine kidney cells. J. Biol. Chem. 2010, 285, 39289–39302. 10.1074/jbc.M110.172858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletto D.; Dersh D.; Argon Y. GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 2010, 21, 479–485. 10.1016/j.semcdb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrovsky O.; Makarewich C. A.; Snapp E. L.; Argon Y. An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 11600–11605. 10.1073/pnas.0902626106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick J.; Dul J. L.; Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 1994, 370, 373–375. 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- Weekes M. P.; Antrobus R.; Talbot S.; Hor S.; Simecek N.; Smith D. L.; Bloor S.; Randow F.; Lehner P. J. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J. Proteome Res. 2012, 11, 1475–1484. 10.1021/pr201135e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Hong F.; Gewirth D.; Guo B.; Liu B.; Li Z. The molecular chaperone gp96/GRP94 interacts with Toll-like receptors and integrins via its C-terminal hydrophobic domain. J. Biol. Chem. 2012, 287, 6735–6742. 10.1074/jbc.M111.309526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson J. C.; Shaler T. A.; Tyler R. E.; Kopito R. R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maattanen P.; Gehring K.; Bergeron J. J.; Thomas D. Y. Protein quality control in the ER: the recognition of misfolded proteins. Semin. Cell Dev. Biol. 2010, 21, 500–511. 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Okumura M.; Kadokura H.; Inaba K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radical Biol. Med. 2015, 83, 314–322. 10.1016/j.freeradbiomed.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Hagiwara M.; Nagata K. Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation. Antioxid. Redox Signaling 2012, 16, 1119–1128. 10.1089/ars.2011.4495. [DOI] [PubMed] [Google Scholar]
- Denisov A. Y.; Maattanen P.; Dabrowski C.; Kozlov G.; Thomas D. Y.; Gehring K. Solution structure of the bb’ domains of human protein disulfide isomerase. FEBS J. 2009, 276, 1440–1449. 10.1111/j.1742-4658.2009.06884.x. [DOI] [PubMed] [Google Scholar]
- Benyair R.; Ogen-Shtern N.; Mazkereth N.; Shai B.; Ehrlich M.; Lederkremer G. Z. Mammalian ER mannosidase I resides in quality control vesicles, where it encounters its glycoprotein substrates. Mol. Biol. Cell 2015, 26, 172–184. 10.1091/mbc.E14-06-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman J.; Shenkman M.; Gofman Y.; Shtern N. O.; Ben-Tal N.; Hendershot L. M.; Lederkremer G. Z. Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 2014, 25, 1050–1060. 10.1091/mbc.e13-06-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A.; Letourneur F.; Garcia-Estefania D.; Carpentier J. L.; Orci L.; Paccaud J. P. Sec24 proteins and sorting at the endoplasmic reticulum. J. Biol. Chem. 1999, 274, 7833–7840. 10.1074/jbc.274.12.7833. [DOI] [PubMed] [Google Scholar]
- Huyer G.; Longsworth G. L.; Mason D. L.; Mallampalli M. P.; McCaffery J. M.; Wright R. L.; Michaelis S. A striking quality control subcompartment in Saccharomyces cerevisiae: The endoplasmic reticulum-associated compartment. Mol. Biol. Cell 2004, 15, 908–921. 10.1091/mbc.e03-07-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E. M.; Vonk W. I. M.; Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr. Opin. Cell Biol. 2014, 26, 139–146. 10.1016/j.ceb.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J. C. The role of the cytosolic HSP70 chaperone system in diseases caused by misfolding and aberrant trafficking of ion channels. Dis. Models & Mech. 2014, 7, 319–329. 10.1242/dmm.014001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S.; Hegde R. S. Target Selection during protein quality control. Trends Biochem. Sci. 2016, 41, 124–137. 10.1016/j.tibs.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Caramelo J. J.; Castro O. A.; Alonso L. G.; De Prat-Gay G.; Parodi A. J. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 86–91. 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guna A.; Hegde R. S. Transmembrane domain recognition during membrane protein biogenesis and Quality Control. Curr. Biol. 2018, 28, R498–R511. 10.1016/j.cub.2018.02.004. [DOI] [PubMed] [Google Scholar]
- Smith M. H.; Ploegh H. L.; Weissman J. S. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige M. J.; Hendershot L. M. Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol. Cell 2013, 51, 297–309. 10.1016/j.molcel.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyden M.; Freites J. A.; Ulmschneider M. B.; White S. H.; Tobias D. J. Assembly and stability of alpha-helical membrane proteins. Soft Matter 2012, 8, 7742–7752. 10.1039/c2sm25402f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P.; Hasler U.; Beggah A.; Horisberger J. D.; Geering K. Membrane integration of Na,K-ATPase alpha-subunits and beta-subunit assembly. J. Biol. Chem. 1998, 273, 24921–24931. 10.1074/jbc.273.38.24921. [DOI] [PubMed] [Google Scholar]
- Beggah A. T.; Beguin P.; Bamberg K.; Sachs G.; Geering K. beta-subunit assembly is essential for the correct packing and the stable membrane insertion of the H,K-ATPase alpha-subunit. J. Biol. Chem. 1999, 274, 8217–8223. 10.1074/jbc.274.12.8217. [DOI] [PubMed] [Google Scholar]
- Garza R. M.; Sato B. K.; Hampton R. Y. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J. Biol. Chem. 2009, 284, 14710–14722. 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato B. K.; Schulz D.; Do P. H.; Hampton R. Y. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol. Cell 2009, 34, 212–222. 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.; Coe H.; Michalak M. Specialization of endoplasmic reticulum chaperones for the folding and function of myelin glycoproteins P0 and PMP22. FASEB J. 2011, 25, 3929–3937. 10.1096/fj.11-184911. [DOI] [PubMed] [Google Scholar]
- Preston G. M.; Guerriero C. J.; Metzger M. B.; Michaelis S.; Brodsky J. L. Substrate insolubility dictates Hsp104-dependent endoplasmic-reticulum-associated degradation. Mol. Cell 2018, 70, 242–253e6. 10.1016/j.molcel.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Brodsky J. L. The degradation pathway of a model misfolded protein is determined by aggregation propensity. Mol. Biol. Cell 2018, 29, 1422–1434. 10.1091/mbc.E18-02-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T.; Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690. 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Sinha A.; Cross B. C.; Mironov A.; Wiertz E.; High S. Endoplasmic reticulum-associated degradation of a degron-containing polytopic membrane protein. Mol. Membr. Biol. 2009, 26, 448–464. 10.3109/09687680903333839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck G.; Ebner F. A.; Shimada-Kreft H.; Kreft S. G. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J. Cell Biol. 2015, 209, 621. 10.1083/jcb.20140808804292015c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira I.; Charuvi D.; Elkabetz Y.; Hirschberg K.; Bar-Nun S. Distinguishing between retention signals and degrons acting in ERAD. J. Cell Sci. 2007, 120, 4377–4387. 10.1242/jcs.011247. [DOI] [PubMed] [Google Scholar]
- Barlowe C.; Helenius A. Cargo capture and bulk flow in the early secretory pathway. Annu. Rev. Cell Dev. Biol. 2016, 32, 197–222. 10.1146/annurev-cellbio-111315-125016. [DOI] [PubMed] [Google Scholar]
- Budnik A.; Stephens D. J. ER exit sites--localization and control of COPII vesicle formation. FEBS Lett. 2009, 583, 3796–3803. 10.1016/j.febslet.2009.10.038. [DOI] [PubMed] [Google Scholar]
- Lord C.; Ferro-Novick S.; Miller E. A. The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the golgi. Cold Spring Harbor Perspect. Biol. 2013, 5, a013367. 10.1101/cshperspect.a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeler M. W.; Paccaud J. P.; Hauri H. P. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 2007, 8, 258–264. 10.1038/sj.embor.7400893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya Y.; Kamiya D.; Yamamoto K.; Nyfeler B.; Hauri H. P.; Kato K. Molecular basis of sugar recognition by the human L-type lectins ERGIC-53, VIPL, and VIP36. J. Biol. Chem. 2008, 283, 1857–1861. 10.1074/jbc.M709384200. [DOI] [PubMed] [Google Scholar]
- Pagant S.; Wu A.; Edwards S.; Diehl F.; Miller E. A. Sec24 is a coincidence detector that simultaneously binds two signals to drive ER export. Curr. Biol. 2015, 25, 403–412. 10.1016/j.cub.2014.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bue C. A.; Barlowe C. Molecular dissection of Erv26p identifies separable cargo binding and coat protein sorting activities. J. Biol. Chem. 2009, 284, 24049–24060. 10.1074/jbc.M109.022590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrain C.; Zettl M.; Christova Y.; Taylor N.; Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. 10.1126/science.1214400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus N. Y.; Perlmutter D. H. Glucosidase and mannosidase inhibitors mediate increased secretion of mutant alpha1 antitrypsin Z. J. Biol. Chem. 2000, 275, 1987–1992. 10.1074/jbc.275.3.1987. [DOI] [PubMed] [Google Scholar]
- Borgese N. Getting membrane proteins on and off the shuttle bus between the endoplasmic reticulum and the Golgi complex. J. Cell Sci. 2016, 129, 1537–1545. 10.1242/jcs.183335. [DOI] [PubMed] [Google Scholar]
- Dukhovny A.; Yaffe Y.; Shepshelovitch J.; Hirschberg K. The length of cargo-protein transmembrane segments drives secretory transport by facilitating cargo concentration in export domains. J. Cell Sci. 2009, 122, 1759–1767. 10.1242/jcs.039339. [DOI] [PubMed] [Google Scholar]
- Ronchi P.; Colombo S.; Francolini M.; Borgese N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 2008, 181, 105–118. 10.1083/jcb.200710093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H.; Riezman H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005, 15, 312–318. 10.1016/j.tcb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Ridsdale A.; Denis M.; Gougeon P. Y.; Ngsee J. K.; Presley J. F.; Zha X. Cholesterol is required for efficient endoplasmic reticulum-to-Golgi transport of secretory membrane proteins. Mol. Biol. Cell 2006, 17, 1593–1605. 10.1091/mbc.e05-02-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H.; Miura K.; Weiss M.; Pepperkok R. Sterols regulate ER-export dynamics of secretory cargo protein ts-O45-G. EMBO J. 2006, 25, 2953–2965. 10.1038/sj.emboj.7601205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant K.; Johnson N.; Swanton E. Transmembrane domain quality control systems operate at the endoplasmic reticulum and Golgi apparatus. PLoS One 2017, 12, e0173924. 10.1371/journal.pone.0173924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavassori S.; Cortini M.; Masui S.; Sannino S.; Anelli T.; Caserta I. R.; Fagioli C.; Mossuto M. F.; Fornili A.; van Anken E.; Degano M.; Inaba K.; Sitia R. A pH-regulated quality control cycle for surveillance of secretory protein assembly. Mol. Cell 2013, 50, 783–792. 10.1016/j.molcel.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannino S.; Anelli T.; Cortini M.; Masui S.; Degano M.; Fagioli C.; Inaba K.; Sitia R. Progressive quality control of secretory proteins in the early secretory compartment by ERp44. J. Cell Sci. 2014, 127, 4260–4269. 10.1242/jcs.153239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C.; Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 1994, 126, 41–52. 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.; Fujii R.; Toyofuku Y.; Saito T.; Koseki H.; Hsu V. W.; Aoe T. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001, 20, 3082–3091. 10.1093/emboj/20.12.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M. Quality control: quality control at the plasma membrane: one mechanism does not fit all. J. Cell Biol. 2014, 205, 11–20. 10.1083/jcb.201310113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T.; Veit G.; Sakai R.; Aki M.; Fujihara T.; Higashi M.; Susuki-Miyata S.; Miyata M.; Fukuda N.; Yoshida A.; Xu H.; Apaja P. M.; Lukacs G. L. Chaperone-independent peripheral quality control of CFTR by RFFL E3 ligase. Dev. Cell 2018, 44, 694–708e7. 10.1016/j.devcel.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J. A.; Kopito R. R.; Christianson J. C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harbor Perspect. Biol. 2013, 5, a013185. 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.; Wei H.; Rajagopalan C.; Jiang H.; Wu Q.; Zaman K.; Xie Y.; Sun F. Dissection of the role of VIMP in endoplasmic reticulum-associated eegradation of CFTRDeltaF508. Sci. Rep. 2018, 8, 4764. 10.1038/s41598-018-23284-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printsev I.; Curiel D.; Carraway K. L. 3rd. Membrane protein quantity control at the endoplasmic reticulum. J. Membr. Biol. 2017, 250, 379–392. 10.1007/s00232-016-9931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson J. C.; Olzmann J. A.; Shaler T. A.; Sowa M. E.; Bennett E. J.; Richter C. M.; Tyler R. E.; Greenblatt E. J.; Harper J. W.; Kopito R. R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2012, 14, 93–105. 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J.; Walczak C. P.; Shaler T. A.; Olzmann J. A.; Zhang L.; Elias J. E.; Kopito R. R. Characterization of protein complexes of the endoplasmic reticulum-associated degradation E3 ubiquitin ligase Hrd1. J. Biol. Chem. 2017, 292, 9104–9116. 10.1074/jbc.M117.785055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J.; Qi L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. 10.1016/j.tibs.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders M. D.; Ryno L. M.; Genereux J. C.; Moresco J. J.; Tu P. G.; Wu C.; Yates J. R. 3rd; Su A. I.; Kelly J. W.; Wiseman R. L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H.; Weihl C. C. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. 10.1242/jcs.093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M. Visiting the ER: the endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv. Drug Delivery Rev. 2007, 59, 759–781. 10.1016/j.addr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Kanehara K.; Xie W.; Ng D. T. Modularity of the Hrd1 ERAD complex underlies its diverse client range. J. Cell Biol. 2010, 188, 707–716. 10.1083/jcb.200907055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M.; Sommer T.; Jarosch E. ERAD ubiquitin ligases: multifunctional tools for protein quality control and waste disposal in the endoplasmic reticulum. BioEssays 2010, 32, 905–913. 10.1002/bies.201000046. [DOI] [PubMed] [Google Scholar]
- Neutzner A.; Neutzner M.; Benischke A. S.; Ryu S. W.; Frank S.; Youle R. J.; Karbowski M. A systematic search for endoplasmic reticulum (ER) membrane-associated RING finger proteins identifies Nixin/ZNRF4 as a regulator of calnexin stability and ER homeostasis. J. Biol. Chem. 2011, 286, 8633–8643. 10.1074/jbc.M110.197459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky J. L.; Wojcikiewicz R. J. Substrate-specific mediators of ER associated degradation (ERAD). Curr. Opin. Cell Biol. 2009, 21, 516–521. 10.1016/j.ceb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R.; Pertel T.; Luban J.; Molinari M. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J. Biol. Chem. 2008, 283, 16446–16454. 10.1074/jbc.M802272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N.; Wada I.; Nagasawa K.; Moriyama T.; Okawa K.; Nagata K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008, 283, 20914–20924. 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman J.; Shenkman M.; Gofman Y.; Shtern N. O.; Ben-Tal N.; Hendershot L. M.; Lederkremer G. Z. Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 2014, 25, 1050–1060. 10.1091/mbc.e13-06-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morreale G.; Conforti L.; Coadwell J.; Wilbrey A. L.; Coleman M. P. Evolutionary divergence of valosin-containing protein/cell division cycle protein 48 binding interactions among endoplasmic reticulum-associated degradation proteins. FEBS J. 2009, 276, 1208–1220. 10.1111/j.1742-4658.2008.06858.x. [DOI] [PubMed] [Google Scholar]
- Stolz A.; Schweizer R. S.; Schafer A.; Wolf D. H. Dfm1 forms distinct complexes with Cdc48 and the ER ubiquitin ligases and is required for ERAD. Traffic 2010, 11, 1363–1369. 10.1111/j.1600-0854.2010.01093.x. [DOI] [PubMed] [Google Scholar]
- Neal S.; Jaeger P. A.; Duttke S. H.; Benner C.; Glass C. K.; Ideker T.; Hampton R. Y. The Dfm1 Derlin Is Required for ERAD Retrotranslocation of Integral Membrane Proteins. Mol. Cell 2018, 69, 306–320e4. 10.1016/j.molcel.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoebel S.; Mi W.; Stein A.; Ovchinnikov S.; Pavlovicz R.; DiMaio F.; Baker D.; Chambers M. G.; Su H.; Li D.; Rapoport T. A.; Liao M. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 2017, 548, 352–355. 10.1038/nature23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A.; Hilt W.; Buchberger A.; Wolf D. H. Cdc48: a power machine in protein degradation. Trends Biochem. Sci. 2011, 36, 515–523. 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Rouiller I.; DeLaBarre B.; May A. P.; Weis W. I.; Brunger A. T.; Milligan R. A.; Wilson-Kubalek E. M. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat. Struct. Biol. 2002, 9, 950–957. 10.1038/nsb872. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K.; Brodsky J. L.; Kamura T. A stalled retrotranslocation complex reveals physical linkage between substrate recognition and proteasomal degradation during ER-associated degradation. Mol. Biol. Cell 2013, 24, 1765–1775. 10.1091/mbc.e12-12-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye V. E.; Dreveny I.; Briggs L. C.; Sands C.; Beuron F.; Zhang X.; Freemont P. S. Going through the motions: the ATPase cycle of p97. J. Struct. Biol. 2006, 156, 12–28. 10.1016/j.jsb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Xia D.; Tang W. K.; Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583, 64–77. 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. M.; Brunger A. T.; Weis W. I. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure 2008, 16, 715–726. 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Cao Z.; Hutchison J. M.; Sanders C. R.; Bowie J. U. Backbone hydrogen bond strengths can vary widely in transmembrane Helices. J. Am. Chem. Soc. 2017, 139, 10742–10749. 10.1021/jacs.7b04819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig L.; Bergbold N.; Sahasrabudhe P.; Geiger B.; Kaltak L.; Lemberg M. K. Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol. Cell 2012, 47, 558–569. 10.1016/j.molcel.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Avci D.; Lemberg M. K. Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol. 2015, 25, 611–622. 10.1016/j.tcb.2015.07.003. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R.; Gao Y. S.; Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic 2002, 3, 388–396. 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- Johnston J. A.; Ward C. L.; Kopito R. R. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J.; Dunn W. A. Jr.; Joy S.; Li J.; Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J.; Verrier J. D.; Go J. C.; Madorsky I.; Dunn W. A.; Notterpek L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol. Dis. 2007, 25, 252–265. 10.1016/j.nbd.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L.; Ryan M. C.; Tobler A. R.; Shooter E. M. PMP22 accumulation in aggresomes: implications for CMT1A pathology. Neurobiol. Dis. 1999, 6, 450–460. 10.1006/nbdi.1999.0274. [DOI] [PubMed] [Google Scholar]
- Ryan M. C.; Shooter E. M.; Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol. Dis. 2002, 10, 109–118. 10.1006/nbdi.2002.0500. [DOI] [PubMed] [Google Scholar]
- Chen Y. F.; Wang I. J.; Lin L. L.; Chen M. S. Examining rhodopsin retention in endoplasmic reticulum and intracellular localization in vitro and in vivo by using truncated rhodopsin fragments. J. Cell. Biochem. 2011, 112, 520–530. 10.1002/jcb.22942. [DOI] [PubMed] [Google Scholar]
- Saliba R. S.; Munro P. M.; Luthert P. J.; Cheetham M. E. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 2002, 115, 2907–2918. [DOI] [PubMed] [Google Scholar]
- Tiwari A.; Copeland C. A.; Han B.; Hanson C. A.; Raghunathan K.; Kenworthy A. K. Caveolin-1 is an aggresome-inducing protein. Sci. Rep. 2016, 6, 38681. 10.1038/srep38681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. M.; Olzmann J. A.; Chin L. S.; Li L. Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J. Cell Sci. 2011, 124, 3319–3331. 10.1242/jcs.087114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleophas M. C.; Joosten L. A.; Stamp L. K.; Dalbeth N.; Woodward O. M.; Merriman T. R. ABCG2 polymorphisms in gout: insights into disease susceptibility and treatment approaches. Pharmacogenomics Pers. Med. 2017, 10, 129–142. 10.2147/PGPM.S105854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs I.; Lentini K. M.; Ingano L. M.; Kovacs D. M. Presenilin 1 forms aggresomal deposits in response to heat shock. J. Mol. Neurosci. 2006, 29, 9–19. 10.1385/JMN:29:1:9. [DOI] [PubMed] [Google Scholar]
- Hyttinen J. M.; Amadio M.; Viiri J.; Pascale A.; Salminen A.; Kaarniranta K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. 10.1016/j.arr.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Driscoll J. J.; Chowdhury R. D. Molecular crosstalk between the proteasome, aggresomes and autophagy: translational potential and clinical implications. Cancer Lett. 2012, 325, 147–154. 10.1016/j.canlet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Wojcik S. Crosstalk between autophagy and proteasome protein degradation systems: possible implications for cancer therapy. Folia Histochem. Cytobiol. 2014, 51, 249–264. 10.5603/FHC.2013.0036. [DOI] [PubMed] [Google Scholar]
- Zaarur N.; Meriin A. B.; Gabai V. L.; Sherman M. Y. Triggering aggresome formation. Dissecting aggresome-targeting and aggregation signals in synphilin 1. J. Biol. Chem. 2008, 283, 27575–27584. 10.1074/jbc.M802216200. [DOI] [PubMed] [Google Scholar]
- Pickart C. M.; Fushman D. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Lim K. L.; Dawson V. L.; Dawson T. M. Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Neurobiol. Aging 2006, 27, 524–529. 10.1016/j.neurobiolaging.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Moscat J.; Diaz-Meco M. T.; Wooten M. W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 2007, 32, 95–100. 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Burnett B. G.; Pittman R. N. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4330–4335. 10.1073/pnas.0407252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Ying Z.; Wang G. Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J. Biol. Chem. 2012, 287, 28576–28585. 10.1074/jbc.M111.299990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heir R.; Ablasou C.; Dumontier E.; Elliott M.; Fagotto-Kaufmann C.; Bedford F. K. The UBL domain of PLIC-1 regulates aggresome formation. EMBO Rep. 2006, 7, 1252–1258. 10.1038/sj.embor.7400823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y.; Kovacs J. J.; McLaurin A.; Vance J. M.; Ito A.; Yao T. P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. 10.1016/S0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Tan S.; Wong E. Kinetics of protein aggregates disposal by aggrephagy. Methods Enzymol. 2017, 588, 245–281. 10.1016/bs.mie.2016.09.084. [DOI] [PubMed] [Google Scholar]
- Gamerdinger M.; Kaya A. M.; Wolfrum U.; Clement A. M.; Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Graham K.; Foote M.; Liang F.; Rizkallah R.; Hurt M.; Wang Y.; Wu Y.; Zhou Y. 14–3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 2013, 126, 4173–4186. 10.1242/jcs.126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J.; Emr S. D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan D. G.; Dikic I. The three musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011, 21, 195–201. 10.1016/j.tcb.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan L.; Zhang X.; Li R. Recent insights into the cellular and molecular determinants of aging. J. Cell Sci. 2018, 131, jcs210831. 10.1242/jcs.210831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi M.; Fregno I.; Guerra C.; Molinari M. Eat it right: ER-phagy and recovER-phagy. Biochem. Soc. Trans. 2018, 46, 699–706. 10.1042/BST20170354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H.; Mochida K. Reticulophagy and nucleophagy: New findings and unsolved issues. Autophagy 2015, 11, 2377–2378. 10.1080/15548627.2015.1106665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.; Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. 10.1042/EBC20170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S.; Tan J.; Miao Y.; Zhang Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. 10.1002/jcp.26137. [DOI] [PubMed] [Google Scholar]
- Bershtein S.; Segal M.; Bekerman R.; Tokuriki N.; Tawfik D. S. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 2006, 444, 929–932. 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- Taverna D. M.; Goldstein R. A. Why are proteins so robust to site mutations?. J. Mol. Biol. 2002, 315, 479–484. 10.1006/jmbi.2001.5226. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Myers J. K. Disease-Related Misassembly of Membrane Proteins. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 25–51. 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Nagy J. K. Misfolding of membrane proteins in health and disease: the lady or the tiger?. Curr. Opin. Struct. Biol. 2000, 10, 438–442. 10.1016/S0959-440X(00)00112-3. [DOI] [PubMed] [Google Scholar]
- Qian S. B.; Princiotta M. F.; Bennink J. R.; Yewdell J. W. Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J. Biol. Chem. 2006, 281, 392–400. 10.1074/jbc.M509126200. [DOI] [PubMed] [Google Scholar]
- Van Craenenbroeck K.; Clark S. D.; Cox M. J.; Oak J. N.; Liu F.; Van Tol H. H. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J. Biol. Chem. 2005, 280, 19350–19357. 10.1074/jbc.M414043200. [DOI] [PubMed] [Google Scholar]
- Medicherla B.; Goldberg A. L. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell Biol. 2008, 182, 663–673. 10.1083/jcb.200803022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U.; Anton L. C.; Gibbs J.; Norbury C. C.; Yewdell J. W.; Bennink J. R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Yen H. C.; Xu Q.; Chou D. M.; Zhao Z.; Elledge S. J. Global protein stability profiling in mammalian cells. Science 2008, 322, 918–923. 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- Yewdell J. W.; Lacsina J. R.; Rechsteiner M. C.; Nicchitta C. V. Out with the old, in with the new? Comparing methods for measuring protein degradation. Cell Biol. Int. 2011, 35, 457–462. 10.1042/CBI20110055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek S.; Notterpek L.; Snipes G. J.; Naef R.; Sossin W.; Laliberte J.; Iacampo S.; Suter U.; Shooter E. M.; Murphy R. A. Neurone promote the translocation of peripheral myelin protein 22 into myelin. J. Neurosci. 1997, 17, 7754–7762. 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek S.; Suter U.; Snipes G. J.; Welcher A. A.; Shooter E. M.; Murphy R. A. Detection and processing of peripheral myelin protein Pmp22 in cultured Schwann-cells. J. Biol. Chem. 1993, 268, 10372–10379. [PubMed] [Google Scholar]
- Terragni B.; Scalmani P.; Franceschetti S.; Cestele S.; Mantegazza M. Post-translational dysfunctions in channelopathies of the nervous system. Neuropharmacology 2018, 132, 31–42. 10.1016/j.neuropharm.2017.05.028. [DOI] [PubMed] [Google Scholar]
- Doyle K. M.; Kennedy D.; Gorman A. M.; Gupta S.; Healy S. J.; Samali A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 2011, 15, 2025–2039. 10.1111/j.1582-4934.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niforou K.; Cheimonidou C.; Trougakos I. P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014, 2, 323–332. 10.1016/j.redox.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Calderwood S. K. Autophagy, protein aggregation and hyperthermia: a mini-review. Int. J. Hyperthermia 2011, 27, 409–414. 10.3109/02656736.2011.552087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T.; Ben-Zvi A.; Ho K. H.; Brignull H. R.; Morimoto R. I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006, 311, 1471–1474. 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Vijg J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. 10.1016/j.gde.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I.; Campbell P. J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. Molecular biology and transgenetics of prion diseases. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 397–438. 10.3109/10409239109086789. [DOI] [PubMed] [Google Scholar]
- Verheijen B. M.; Vermulst M.; van Leeuwen F. W. Somatic mutations in neurons during aging and neurodegeneration. Acta Neuropathol. 2018, 135, 811–826. 10.1007/s00401-018-1850-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R.; Callegari K.; Soto C. Prion-like features of misfolded Abeta and tau aggregates. Virus Res. 2015, 207, 106–112. 10.1016/j.virusres.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Ramchandren S. Charcot-Marie-Tooth disease and other genetic polyneuropathies. Continuum (Minneap Minn.) 2017, 23, 1360–1377. 10.1212/CON.0000000000000529. [DOI] [PubMed] [Google Scholar]
- The UniProt C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Ren Q. Why are essential genes essential? - The essentiality of Saccharomyces genes. Microb. Cell 2015, 2, 280–287. 10.15698/mic2015.08.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M.; Hannan L. A. Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic 2000, 1, 836–851. 10.1034/j.1600-0854.2000.011104.x. [DOI] [PubMed] [Google Scholar]
- Aridor M.; Hannan L. A. Traffic jams II: an update of diseases of intracellular transport. Traffic 2002, 3, 781–790. 10.1034/j.1600-0854.2002.31103.x. [DOI] [PubMed] [Google Scholar]
- Stenson P. D.; Mort M.; Ball E. V.; Evans K.; Hayden M.; Heywood S.; Hussain M.; Phillips A. D.; Cooper D. N. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier V.; Morello J. P.; Zarruk A.; Debrand N.; Salahpour A.; Lonergan M.; Arthus M. F.; Laperriere A.; Brouard R.; Bouvier M.; Bichet D. G. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2006, 17, 232–243. [DOI] [PubMed] [Google Scholar]
- Cheong H. I.; Cho H. Y.; PARK H. W.; Ha I. S.; Choi Y. Molecular genetic study of congenital nephrogenic diabetes insipidus and rescue of mutant vasopressin V2 receptor by chemical chaperones. Nephrology 2007, 12, 113–117. 10.1111/j.1440-1797.2006.00759.x. [DOI] [PubMed] [Google Scholar]
- Moeller H. B.; Rittig S.; Fenton R. A. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr. Rev. 2013, 34, 278–301. 10.1210/er.2012-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillac B.; Mendre C.: Pharmacological chaperones as potential therapeutic strategies for misfolded mutant vasopressin receptors. In Targeting Trafficking in Drug Development; Ulloa-Aguirre A., Tao Y.-X., Eds.; Springer International Publishing: New York, 2018; pp 63–83. [DOI] [PubMed] [Google Scholar]
- Robben J. H.; Knoers N. V. A. M.; Deen P. M. T. Characterization of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus in a polarized cell model. Am. J. Physiol.-Renal 2005, 289, F265–F272. 10.1152/ajprenal.00404.2004. [DOI] [PubMed] [Google Scholar]
- Bichet D. G. V2R mutations and nephrogenic diabetes insipidus. Prog. Mol. Biol. Transl. Sci. 2009, 89, 15–29. 10.1016/S1877-1173(09)89002-9. [DOI] [PubMed] [Google Scholar]
- Partridge A. W.; Therien A. G.; Deber C. M. Polar mutations in membrane proteins as a biophysical basis for disease. Biopolymers 2002, 66, 350–358. 10.1002/bip.10313. [DOI] [PubMed] [Google Scholar]
- Casadio R.; Vassura M.; Tiwari S.; Fariselli P.; Luigi Martelli P. Correlating disease-related mutations to their effect on protein stability: a large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. 10.1002/humu.21555. [DOI] [PubMed] [Google Scholar]
- Shi Z.; Moult J. Structural and functional impact of cancer-related missense somatic mutations. J. Mol. Biol. 2011, 413, 495–512. 10.1016/j.jmb.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl S.; Nishi H.; Petukh M.; Panchenko A. R.; Alexov E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. 10.1016/j.jmb.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P.; Li Z.; Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Sekijima Y.; Wiseman R. L.; Matteson J.; Hammarstrom P.; Miller S. R.; Sawkar A. R.; Balch W. E.; Kelly J. W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Ng D. P.; Poulsen B. E.; Deber C. M. Membrane protein misassembly in disease. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 1115–1122. 10.1016/j.bbamem.2011.07.046. [DOI] [PubMed] [Google Scholar]
- Kroncke B. M.; Vanoye C. G.; Meiler J.; George A. L. Jr.; Sanders C. R. Personalized biochemistry and biophysics. Biochemistry 2015, 54, 2551–2559. 10.1021/acs.biochem.5b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C.; Abecasis G. R.; Altshuler D.; Auton A.; Brooks L. D.; Durbin R. M.; Gibbs R. A.; Hurles M. E.; McVean G. A.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J. X.; Buckingham K. J.; Jhangiani S. N.; Boehm C.; Sobreira N.; Smith J. D.; Harrell T. M.; McMillin M. J.; Wiszniewski W.; Gambin T.; Coban Akdemir Z. H.; Doheny K.; Scott A. F.; Avramopoulos D.; Chakravarti A.; Hoover-Fong J.; Mathews D.; Witmer P. D.; Ling H.; Hetrick K.; Watkins L.; Patterson K. E.; Reinier F.; Blue E.; Muzny D.; Kircher M.; Bilguvar K.; Lopez-Giraldez F.; Sutton V. R.; Tabor H. K.; Leal S. M.; Gunel M.; Mane S.; Gibbs R. A.; Boerwinkle E.; Hamosh A.; Shendure J.; Lupski J. R.; Lifton R. P.; Valle D.; Nickerson D. A.; Bamshad M. J. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A.; Pierce L. C.; Biggs W. H.; di Iulio J.; Wong E. H.; Fabani M. M.; Kirkness E. F.; Moustafa A.; Shah N.; Xie C.; Brewerton S. C.; Bulsara N.; Garner C.; Metzker G.; Sandoval E.; Perkins B. A.; Och F. J.; Turpaz Y.; Venter J. C. Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 11901–11906. 10.1073/pnas.1613365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allali-Hassani A.; Wasney G. A.; Chau I.; Hong B. S.; Senisterra G.; Loppnau P.; Shi Z.; Moult J.; Edwards A. M.; Arrowsmith C. H.; Park H. W.; Schapira M.; Vedadi M. A survey of proteins encoded by non-synonymous single nucleotide polymorphisms reveals a significant fraction with altered stability and activity. Biochem. J. 2009, 424, 15–26. 10.1042/BJ20090723. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Moult J. SNPs, protein structure, and disease. Hum. Mutat. 2001, 17, 263–270. 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Manolio T. A.; Collins F. S.; Cox N. J.; Goldstein D. B.; Hindorff L. A.; Hunter D. J.; McCarthy M. I.; Ramos E. M.; Cardon L. R.; Chakravarti A.; Cho J. H.; Guttmacher A. E.; Kong A.; Kruglyak L.; Mardis E.; Rotimi C. N.; Slatkin M.; Valle D.; Whittemore A. S.; Boehnke M.; Clark A. G.; Eichler E. E.; Gibson G.; Haines J. L.; Mackay T. F.; McCarroll S. A.; Visscher P. M. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher P. M.; Wray N. R.; Zhang Q.; Sklar P.; McCarthy M. I.; Brown M. A.; Yang J. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J.; Balasubramanian S.; Church G. M.; Gilbert W.; Rogers J.; Schloss J. A.; Waterston R. H. DNA sequencing at 40: past, present and future. Nature 2017, 550, 345–353. 10.1038/nature24286. [DOI] [PubMed] [Google Scholar]
- Genomes Project C.; Auton A.; Brooks L. D.; Durbin R. M.; Garrison E. P.; Kang H. M.; Korbel J. O.; Marchini J. L.; McCarthy S.; McVean G. A.; Abecasis G. R.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M.; Karczewski K. J.; Minikel E. V.; Samocha K. E.; Banks E.; Fennell T.; O’Donnell-Luria A. H.; Ware J. S.; Hill A. J.; Cummings B. B.; Tukiainen T.; Birnbaum D. P.; Kosmicki J. A.; Duncan L. E.; Estrada K.; Zhao F.; Zou J.; Pierce-Hoffman E.; Berghout J.; Cooper D. N.; Deflaux N.; DePristo M.; Do R.; Flannick J.; Fromer M.; Gauthier L.; Goldstein J.; Gupta N.; Howrigan D.; Kiezun A.; Kurki M. I.; Moonshine A. L.; Natarajan P.; Orozco L.; Peloso G. M.; Poplin R.; Rivas M. A.; Ruano-Rubio V.; Rose S. A.; Ruderfer D. M.; Shakir K.; Stenson P. D.; Stevens C.; Thomas B. P.; Tiao G.; Tusie-Luna M. T.; Weisburd B.; Won H. H.; Yu D.; Altshuler D. M.; Ardissino D.; Boehnke M.; Danesh J.; Donnelly S.; Elosua R.; Florez J. C.; Gabriel S. B.; Getz G.; Glatt S. J.; Hultman C. M.; Kathiresan S.; Laakso M.; McCarroll S.; McCarthy M. I.; McGovern D.; McPherson R.; Neale B. M.; Palotie A.; Purcell S. M.; Saleheen D.; Scharf J. M.; Sklar P.; Sullivan P. F.; Tuomilehto J.; Tsuang M. T.; Watkins H. C.; Wilson J. G.; Daly M. J.; MacArthur D. G. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian A. J. Challenges in medical applications of whole exome/genome sequencing discoveries. Trends Cardiovasc. Med. 2012, 22, 219–223. 10.1016/j.tcm.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz P. J. My approach to the long QT syndrome (LQTS). Trends Cardiovasc. Med. 2015, 25, 376–377. 10.1016/j.tcm.2014.09.017. [DOI] [PubMed] [Google Scholar]
- Ackerman M. J. Genetic purgatory and the cardiac channelopathies: Exposing the variants of uncertain/unknown significance issue. Heart Rhythm 2015, 12, 2325–2331. 10.1016/j.hrthm.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Richards S.; Aziz N.; Bale S.; Bick D.; Das S.; Gastier-Foster J.; Grody W. W.; Hegde M.; Lyon E.; Spector E.; Voelkerding K.; Rehm H. L. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke B. M.; Duran A. M.; Mendenhall J. L.; Meiler J.; Blume J. D.; Sanders C. R. Documentation of an imperative to improve methods for predicting nembrane protein stability. Biochemistry 2016, 55, 5002–5009. 10.1021/acs.biochem.6b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C.; Tian C.; Sonnichsen F. D.; Smith J. A.; Meiler J.; George A. L. Jr.; Vanoye C. G.; Kim H. J.; Sanders C. R. Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry 2008, 47, 7999–8006. 10.1021/bi800875q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye C. G.; Welch R. C.; Daniels M. A.; Manderfield L. J.; Tapper A. R.; Sanders C. R.; George A. L. Jr. Distinct subdomains of the KCNQ1 S6 segment determine channel modulation by different KCNE subunits. J. Gen. Physiol. 2009, 134, 207–217. 10.1085/jgp.200910234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Horn W. D.; Vanoye C. G.; Sanders C. R. Working model for the structural basis for KCNE1 modulation of the KCNQ1 potassium channel. Curr. Opin. Struct. Biol. 2011, 21, 283–291. 10.1016/j.sbi.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott G. W. Biology of the KCNQ1 potassium channel. New J. Sci. 2014, 2014, 1–26. 10.1155/2014/237431. [DOI] [Google Scholar]
- Sahu I. D.; Kroncke B. M.; Zhang R.; Dunagan M. M.; Smith H. J.; Craig A.; McCarrick R. M.; Sanders C. R.; Lorigan G. A. Structural investigation of the transmembrane domain of KCNE1 in proteoliposomes. Biochemistry 2014, 53, 6392–6401. 10.1021/bi500943p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D.; Kim J. H.; Kroncke B. M.; Law C. L.; Xia Y.; Droege K. D.; Van Horn W. D.; Vanoye C. G.; Sanders C. R. Purification and structural study of the voltage-sensor domain of the human KCNQ1 potassium ion channel. Biochemistry 2014, 53, 2032–2042. 10.1021/bi500102w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke B. M.; Van Horn W. D.; Smith J.; Kang C.; Welch R. C.; Song Y.; Nannemann D. P.; Taylor K. C.; Sisco N. J.; George A. L. Jr.; Meiler J.; Vanoye C. G.; Sanders C. R. Structural basis for KCNE3 modulation of potassium recycling in epithelia. Sci. Adv. 2016, 2, e1501228. 10.1126/sciadv.1501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J.; Lesage F.; Guillemare E.; Fink M.; Lazdunski M.; Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996, 384, 78–80. 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- McKone E. F.; Borowitz D.; Drevinek P.; Griese M.; Konstan M. W.; Wainwright C.; Ratjen F.; Sermet-Gaudelus I.; Plant B.; Munck A.; Jiang Y.; Gilmartin G.; Davies J. C. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2014, 2, 902–910. 10.1016/S2213-2600(14)70218-8. [DOI] [PubMed] [Google Scholar]
- Splawski I.; Tristani-Firouzi M.; Lehmann M. H.; Sanguinetti M. C.; Keating M. T. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 1997, 17, 338–340. 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- Crotti L.; Celano G.; Dagradi F.; Schwartz P. J. Congenital long QT syndrome. Orphanet J. Rare Dis. 2008, 3, 18. 10.1186/1750-1172-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; MacKinnon R. Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 2017, 169, 1042–1050e9. 10.1016/j.cell.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. J.; Quinn K. V.; Graves F. M.; Bitner-Glindzicz M.; Tinker A. Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1). Cardiovasc. Res. 2005, 67, 476–486. 10.1016/j.cardiores.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Smith J. A.; Vanoye C. G.; George A. L. Jr.; Meiler J.; Sanders C. R. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 2007, 46, 14141–14152. 10.1021/bi701597s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asjad H. M. M.; Kasture A.; El-Kasaby A.; Sackel M.; Hummel T.; Freissmuth M.; Sucic S. Pharmacochaperoning in a Drosophila model system rescues human dopamine transporter variants associated with infantile/juvenile parkinsonism. J. Biol. Chem. 2017, 292, 19250–19265. 10.1074/jbc.M117.797092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroz D.; Dahimene S.; Baro I.; Loussouarn G.; Merot J. LQT1-associated mutations increase KCNQ1 proteasomal degradation independently of Derlin-1. J. Biol. Chem. 2009, 284, 5250–5256. 10.1074/jbc.M806459200. [DOI] [PubMed] [Google Scholar]
- Schwartz P. J.; Priori S. G.; Spazzolini C.; Moss A. J.; Vincent G. M.; Napolitano C.; Denjoy I.; Guicheney P.; Breithardt G.; Keating M. T.; Towbin J. A.; Beggs A. H.; Brink P.; Wilde A. A.; Toivonen L.; Zareba W.; Robinson J. L.; Timothy K. W.; Corfield V.; Wattanasirichaigoon D.; Corbett C.; Haverkamp W.; Schulze-Bahr E.; Lehmann M. H.; Schwartz K.; Coumel P.; Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. 10.1161/01.CIR.103.1.89. [DOI] [PubMed] [Google Scholar]
- Lounkine E.; Keiser M. J.; Whitebread S.; Mikhailov D.; Hamon J.; Jenkins J. L.; Lavan P.; Weber E.; Doak A. K.; Cote S.; Shoichet B. K.; Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo B.; Williamson B.; Young J. C.; Lukacs G.; Shrier A. hERG quality control and the long QT syndrome. J. Physiol. 2016, 594, 2469–2481. 10.1113/JP270531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C. L.; Kuzmicki C. E.; Childs R. R.; Hintz C. J.; Delisle B. P.; January C. T. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 2014, 5, 5535. 10.1038/ncomms6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley C. A.; Jesus C. S.; Carvalho R.; Brito R. M.; Morais-Cabral J. H. Changes in channel trafficking and protein stability caused by LQT2 mutations in the PAS domain of the HERG channel. PLoS One 2012, 7, e32654. 10.1371/journal.pone.0032654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y.; Ng C. A.; Hunter M. J.; Mann S. A.; Heide J.; Hill A. P.; Vandenberg J. I. Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem. J. 2013, 454, 69–77. 10.1042/BJ20130328. [DOI] [PubMed] [Google Scholar]
- Li P.; Ninomiya H.; Kurata Y.; Kato M.; Miake J.; Yamamoto Y.; Igawa O.; Nakai A.; Higaki K.; Toyoda F.; Wu J.; Horie M.; Matsuura H.; Yoshida A.; Shirayoshi Y.; Hiraoka M.; Hisatome I. Reciprocal control of hERG stability by Hsp70 and Hsc70 with implication for restoration of LQT2 mutant stability. Circ. Res. 2011, 108, 458–468. 10.1161/CIRCRESAHA.110.227835. [DOI] [PubMed] [Google Scholar]
- Ficker E.; Dennis A.; Kuryshev Y.; Wible B. A.; Brown A. M. HERG channel trafficking. Novartis Found. Symp. 2008, 266, 57–69. 10.1002/047002142X.ch6. [DOI] [PubMed] [Google Scholar]
- Ficker E.; Dennis A. T.; Wang L.; Brown A. M. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ. Res. 2003, 92, e87–100. 10.1161/01.RES.0000079028.31393.15. [DOI] [PubMed] [Google Scholar]
- Walker V. E.; Wong M. J.; Atanasiu R.; Hantouche C.; Young J. C.; Shrier A. Hsp40 chaperones promote degradation of the HERG potassium channel. J. Biol. Chem. 2010, 285, 3319–3329. 10.1074/jbc.M109.024000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaja P. M.; Foo B.; Okiyoneda T.; Valinsky W. C.; Barriere H.; Atanasiu R.; Ficker E.; Lukacs G. L.; Shrier A. Ubiquitination-dependent quality control of hERG K+ channel with acquired and inherited conformational defect at the plasma membrane. Mol. Biol. Cell 2013, 24, 3787–3804. 10.1091/mbc.e13-07-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung T. T.; Webb T. R.; Chan L. F.; Cooray S. N.; Metherell L. A.; King P. J.; Chapple J. P.; Clark A. J. The majority of adrenocorticotropin receptor (melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J. Clin. Endocrinol. Metab. 2008, 93, 4948–4954. 10.1210/jc.2008-1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y. X. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr. Rev. 2010, 31, 506–543. 10.1210/er.2009-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa-Aguirre A.; Zarinan T.; Dias J. A.; Conn P. M. Mutations in G protein-coupled receptors that impact receptor trafficking and reproductive function. Mol. Cell. Endocrinol. 2014, 382, 411–423. 10.1016/j.mce.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartong D. T.; Berson E. L.; Dryja T. P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Jacobson S. G.; Cideciyan A. V. Treatment possibilities for retinitis pigmentosa. N. Engl. J. Med. 2010, 363, 1669–1671. 10.1056/NEJMcibr1007685. [DOI] [PubMed] [Google Scholar]
- Dias M. F.; Joo K.; Kemp J. A.; Fialho S. L.; da Silva Cunha A. Jr.; Woo S. J.; Kwon Y. J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retinal Eye Res. 2018, 63, 107–131. 10.1016/j.preteyeres.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Jaissle G. B.; May C. A.; van de Pavert S. A.; Wenzel A.; Claes-May E.; Giessl A.; Szurman P.; Wolfrum U.; Wijnholds J.; Fisher M. D.; Humphries P.; Seeliger M. W. Bone spicule pigment formation in retinitis pigmentosa: insights from a mouse model. Graefe's Arch. Clin. Exp. Ophthalmol. 2010, 248, 1063–1070. 10.1007/s00417-009-1253-9. [DOI] [PubMed] [Google Scholar]
- Bunt-Milam A. H.; Kalina R. E.; Pagon R. A. Clinical-ultrastructural study of a retinal dystrophy. Invest. Ophthalmol. Vis. Sci. 1983, 24, 458–469. [PubMed] [Google Scholar]
- Griciuc A.; Aron L.; Ueffing M. ER stress in retinal degeneration: a target for rational therapy?. Trends Mol. Med. 2011, 17, 442–451. 10.1016/j.molmed.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Tzekov R.; Stein L.; Kaushal S. Protein misfolding and retinal degeneration. Cold Spring Harbor Perspect. Biol. 2011, 3, a007492. 10.1101/cshperspect.a007492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiou D.; Aguila M.; Bellingham J.; Li W.; McCulley C.; Reeves P. J.; Cheetham M. E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retinal Eye Res. 2018, 62, 1–23. 10.1016/j.preteyeres.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja T. P.; McGee T. L.; Hahn L. B.; Cowley G. S.; Olsson J. E.; Reichel E.; Sandberg M. A.; Berson E. L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307. 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- Sung C. H.; Schneider B. G.; Agarwal N.; Papermaster D. S.; Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 8840–8844. 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S.; Khorana H. G. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry 1994, 33, 6121–6128. 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- Rader A. J.; Anderson G.; Isin B.; Khorana H. G.; Bahar I.; Klein-Seetharaman J. Identification of core amino acids stabilizing rhodopsin. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7246–7251. 10.1073/pnas.0401429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach J. P.; Sanders C. R. Influence of pathogenic mutations on the energetics of translocon-mediated bilayer integration of transmembrane Helices. J. Membr. Biol. 2015, 248, 371–281. 10.1007/s00232-014-9726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt D. A.; Cheetham M. E. Targeting the proteostasis network in rhodopsin retinitis pigmentosa. Adv. Exp. Med. Biol. 2016, 854, 479–484. 10.1007/978-3-319-17121-0_64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeone R.; Wikstrom M.; Kiel C.; Rakoczy P. E. Assessing the correlation between mutant rhodopsin stability and the severity of retinitis pigmentosa. Mol. Vis 2014, 20, 183–199. [PMC free article] [PubMed] [Google Scholar]
- Noorwez S. M.; Malhotra R.; McDowell J. H.; Smith K. A.; Krebs M. P.; Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J. Biol. Chem. 2004, 279, 16278–16284. 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- Chen P. C.; Olson E. M.; Zhou Q.; Kryukova Y.; Sampson H. M.; Thomas D. Y.; Shyng S. L. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J. Biol. Chem. 2013, 288, 20942–20954. 10.1074/jbc.M113.470948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Chen Y.; Jastrzebska B.; Golczak M.; Gulati S.; Tang H.; Seibel W.; Li X.; Jin H.; Han Y.; Gao S.; Zhang J.; Liu X.; Heidari-Torkabadi H.; Stewart P. L.; Harte W. E.; Tochtrop G. P.; Palczewski K. A novel small molecule chaperone of rod opsin and its potential therapy for retinal degeneration. Nat. Commun. 2018, 9, 1976. 10.1038/s41467-018-04261-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgane K.; Dodo K.; Hashimoto Y. Retinobenzaldehydes as proper-trafficking inducers of folding-defective P23H rhodopsin mutant responsible for retinitis pigmentosa. Bioorg. Med. Chem. 2010, 18, 7022–7028. 10.1016/j.bmc.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Li J.; Parker B.; Martyn C.; Natarajan C.; Guo J. S. The PMP22 Gene and Its Related Diseases. Mol. Neurobiol. 2013, 47, 673–698. 10.1007/s12035-012-8370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten A. M.; Suter U. The peripheral myelin protein 22 and epithelial membrane protein family. Prog. Nucleic Acid Res. Mol. Biol. 2000, 64, 97–129. 10.1016/S0079-6603(00)64003-5. [DOI] [PubMed] [Google Scholar]
- El-Abassi R.; England J. D.; Carter G. T. Charcot-Marie-Tooth disease: an overview of genotypes, phenotypes, and clinical management Strategies. PM&R 2014, 6, 342–355. 10.1016/j.pmrj.2013.08.611. [DOI] [PubMed] [Google Scholar]
- Vallat J. M.; Mathis S.; Funalot B. The various Charcot-Marie-Tooth diseases. Curr. Opin. Neurol. 2013, 26, 473–480. 10.1097/WCO.0b013e328364c04b. [DOI] [PubMed] [Google Scholar]
- Lee S.; Bazick H.; Chittoor-Vinod V.; Al Salihi M. O.; Xia G.; Notterpek L. Elevated peripheral Myelin Protein 22, reduced mitotic potential, and proteasome impairment in dermal fibroblasts from Charcot-Marie-Tooth disease type 1A patients. Am. J. Pathol. 2018, 188, 728–738. 10.1016/j.ajpath.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach J. P.; Peng D. G.; Kroncke B. M.; Mittendorf K. F.; Narayan M.; Carter B. D.; Sanders C. R. Reversible folding of human peripheral myelin protein 22, a tetraspan membrane protein. Biochemistry 2013, 52, 3229–3241. 10.1021/bi301635f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuanalo-Contreras K.; Mukherjee A.; Soto C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 1–10. 10.1155/2013/638083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J.; Go J. C.; Li J.; Amici S. A.; Dunn W. A. Jr.; Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol. Dis. 2006, 22, 153–164. 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Mittendorf K. F.; Kroncke B. M.; Meiler J.; Sanders C. R. The homology model of PMP22 suggests mutations resulting in peripheral neuropathy disrupt transmembrane helix packing. Biochemistry 2014, 53, 6139–6141. 10.1021/bi500809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C. R.; Ismail-Beigi F.; McEnery M. W. Mutations of peripheral myelin protein 22 result in defective trafficking through mechanisms which may be common to diseases involving tetraspan membrane proteins. Biochemistry 2001, 40, 9453–9459. 10.1021/bi010894f. [DOI] [PubMed] [Google Scholar]
- Kraus A.; Groenendyk J.; Bedard K.; Baldwin T. A.; Krause K. H.; Dubois-Dauphin M.; Dyck J.; Rosenbaum E. E.; Korngut L.; Colley N. J.; Gosgnach S.; Zochodne D.; Todd K.; Agellon L. B.; Michalak M. Calnexin deficiency leads to dysmyelination. J. Biol. Chem. 2010, 285, 18928–18938. 10.1074/jbc.M110.107201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittoor-Vinod V. G.; Lee S.; Judge S. M.; Notterpek L. Inducible HSP70 is critical in preventing the aggregation and enhancing the processing of PMP22. ASN Neuro 2015, 7, 175909141556990. 10.1177/1759091415569909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichberg J.; Myelin P-0. New knowledge and new roles. Neurochem. Res. 2002, 27, 1331–1340. 10.1023/A:1021619631869. [DOI] [PubMed] [Google Scholar]
- Sanmaneechai O.; Feely S.; Scherer S. S.; Herrmann D. N.; Burns J.; Muntoni F.; Li J.; Siskind C. E.; Day J. W.; Laura M.; Sumner C. J.; Lloyd T. E.; Ramchandren S.; Shy R. R.; Grider T.; Bacon C.; Finkel R. S.; Yum S. W.; Moroni I.; Piscosquito G.; Pareyson D.; Reilly M. M.; Shy M. E. Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain 2015, 138, 3180–3192. 10.1093/brain/awv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkowski M. L.; Kim S.; Phillips M. L.; Partridge A. W.; Deber C. M.; Bowie J. U. Transmembrane domain of myelin protein zero can form dimers: possible implications for myelin construction. Biochemistry 2007, 46, 12164–12173. 10.1021/bi701066h. [DOI] [PubMed] [Google Scholar]
- Volpi V. G.; Touvier T.; D’Antonio M. Endoplasmic reticulum protein quality control failure in myelin disorders. Front. Mol. Neurosci. 2017, 9, 162. 10.3389/fnmol.2016.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Wu X.; Brennan K. M.; Wang D. S.; D’Antonio M.; Moran J.; Svaren J.; Shy M. E. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann. Clin. Transl. Neurol. 2018, 5, 445–455. 10.1002/acn3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M.; Tinelli E.; Malaguti M.; Del Carro U.; D’Antonio M.; Ron D.; Quattrini A.; Feltri M. L.; Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1 B mice. Neuron 2008, 57, 393–405. 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta M. A. C.; Shy B. R.; Patzko A.; Bai Y. H.; Pennuto M.; Ferri C.; Tinelli E.; Saveri P.; Kirschner D.; Crowther M.; Southwood C.; Wu X. Y.; Gow A.; Feltri M. L.; Wrabetz L.; Shy M. E. MpzR98C arrests Schwann cell development in a mouse model of early-onset Charcot-Marie-Tooth disease type 1B. Brain 2012, 135, 2032–2047. 10.1093/brain/aws140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern J. Y. Pelizaeus-Merzbacher disease: pathogenic mechanisms and insights into the roles of proteolipid protein 1 in the nervous system. J. Neurol. Sci. 2005, 228, 201–203. 10.1016/j.jns.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J.; Nave K. A.; Mierzwa A.; Schiff R. Subtle myelin defects in PLP-null mice. Glia 2006, 54, 172–182. 10.1002/glia.20370. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J.; Schiff R.; Lam P. Effects of osmolality on PLP-null myelin structure: Implications re axon damage. Brain Res. 2009, 1253, 191–197. 10.1016/j.brainres.2008.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward K. J. The molecular and cellular defects underlying Perlizaeus-Merzbacher disease. Expert Rev. Mol. Med. 2008, 10, e14. 10.1017/S1462399408000677. [DOI] [PubMed] [Google Scholar]
- Inoue K. Cellular pathology of pelizaeus-merzbacher cisease involving chaperones associated with endoplasmic reticulum stress. Front. Mol. Biosci. 2017, 4, 7. 10.3389/fmolb.2017.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwood C. M.; Garbern J.; Jiang W.; Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 2002, 36, 585–596. 10.1016/S0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roboti P.; Swanton E.; High S. Differences in endoplasmic-reticulum quality control determine the cellular response to disease-associated mutants of proteolipid protein. J. Cell Sci. 2009, 122, 3942–3953. 10.1242/jcs.055160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck K.; Amaral M. D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. 10.1016/S2213-2600(16)00023-0. [DOI] [PubMed] [Google Scholar]
- Jilling T.; Kirk K. L. The biogenesis, traffic, and function of the cystic fibrosis transmembrane conductance regulator. Int. Rev. Cytol. 1997, 172, 193–241. 10.1016/S0074-7696(08)62361-X. [DOI] [PubMed] [Google Scholar]
- Ishiguro H.; Steward M. C.; Naruse S.; Ko S. B.; Goto H.; Case R. M.; Kondo T.; Yamamoto A. CFTR functions as a bicarbonate channel in pancreatic duct cells. J. Gen. Physiol. 2009, 133, 315–326. 10.1085/jgp.200810122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L.; Fatehi M.; Linsdell P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J. Cystic Fibrosis 2009, 8, 115–121. 10.1016/j.jcf.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Cheung J. C.; Deber C. M. Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry 2008, 47, 1465–1473. 10.1021/bi702209s. [DOI] [PubMed] [Google Scholar]
- Farinha C. M.; Canato S. From the endoplasmic reticulum to the plasma membrane: mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. 10.1007/s00018-016-2387-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan B. P.; Freedman S. D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- Tilley A. E.; Walters M. S.; Shaykhiev R.; Crystal R. G. Cilia dysfunction in lung disease. Annu. Rev. Physiol. 2015, 77, 379–406. 10.1146/annurev-physiol-021014-071931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Criq V.; Gray M. A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. 10.1007/s00018-016-2391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Zhang Z.; Csanady L.; Gadsby D. C.; Chen J. Molecular structure of the human CFTR ion channel. Cell 2017, 169, 85. 10.1016/j.cell.2017.02.024. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Chen J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 2016, 167, 1586. 10.1016/j.cell.2016.11.014. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Liu F.; Chen J. Conformational changes of CFTR upon phosphorylation and ATP binding. Cell 2017, 170, 483. 10.1016/j.cell.2017.06.041. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Liu F.; Chen J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 12757. 10.1073/pnas.1815287115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinski S. V.; Shahani V. M.; Subramanian A. S.; MacKinnon S. S.; Woollard G.; Laforet M.; Laselva O.; Morayniss L. D.; Bear C. E.; Windemuth A. Comprehensive mapping of cystic fibrosis mutations to CFTR protein identifies mutation clusters and molecular docking predicts corrector binding site. Proteins: Struct., Funct., Genet. 2018, 86, 833–843. 10.1002/prot.25496. [DOI] [PubMed] [Google Scholar]
- Sanders C.; Myers J. Disease-related misassembly of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 25–51. 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- Welsh M. J.; Smith A. E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. 10.1016/0092-8674(93)90353-R. [DOI] [PubMed] [Google Scholar]
- Veit G.; Avramescu R. G.; Chiang A. N.; Houck S. A.; Cai Z.; Peters K. W.; Hong J. S.; Pollard H. B.; Guggino W. B.; Balch W. E.; Skach W. R.; Cutting G. R.; Frizzell R. A.; Sheppard D. N.; Cyr D. M.; Sorscher E. J.; Brodsky J. L.; Lukacs G. L. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. 10.1091/mbc.e14-04-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Stack J. H.; Straley K. S.; Decker C. J.; Miller M.; McCartney J.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. A. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 18843–18848. 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleizen B.; van Vlijmen T.; de Jonge H. R.; Braakman I. Folding of CFTR is predominantly cotranslational. Mol. Cell 2005, 20, 277–287. 10.1016/j.molcel.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Yoon J. S.; Shishido H.; Yang Z.; Rooney L. A.; Barral J. M.; Skach W. R. Protein folding. Translational tuning optimizes nascent protein folding in cells. Science 2015, 348, 444–448. 10.1126/science.aaa3974. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T.; Barriere H.; Bagdany M.; Rabeh W. M.; Du K.; Hohfeld J.; Young J. C.; Lukacs G. L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza J. L.; Schmidt A.; Li Q.; Nuvaga E.; Barrett T.; Bridges R. J.; Feranchak A. P.; Brautigam C. A.; Thomas P. J. Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 2012, 148, 164–174. 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K.; Sharma M.; Lukacs G. L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- He L.; Aleksandrov L. A.; Cui L.; Jensen T. J.; Nesbitt K. L.; Riordan J. R. Restoration of domain folding and interdomain assembly by second-site suppressors of the DeltaF508 mutation in CFTR. FASEB J. 2010, 24, 3103–3112. 10.1096/fj.09-141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serohijos A. W.; Hegedus T.; Aleksandrov A. A.; He L.; Cui L.; Dokholyan N. V.; Riordan J. R. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3256–3261. 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt D. M.; Loguercio S.; Campos A. R.; Balch W. E. A Proteomic Variant Approach (ProVarA) for Personalized Medicine of Inherited and Somatic Disease. J. Mol. Biol. 2018, 430, 2951–2973. 10.1016/j.jmb.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppinger J. A.; Hutt D. M.; Razvi A.; Koulov A. V.; Pankow S.; Yates J. R. 3rd; Balch W. E. A chaperone trap contributes to the onset of cystic fibrosis. PLoS One 2012, 7, e37682. 10.1371/journal.pone.0037682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham G. C.; Patterson C.; Zhang W.; Younger J. M.; Cyr D. M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Schulein R.; Zuhlke K.; Krause G.; Rosenthal W. Functional rescue of the nephrogenic diabetes insipidus-causing vasopressin V2 receptor mutants G185C and R202C by a second site suppressor mutation. J. Biol. Chem. 2001, 276, 8384–8392. 10.1074/jbc.M007045200. [DOI] [PubMed] [Google Scholar]
- Katada S.; Tanaka M.; Touhara K. Structural determinants for membrane trafficking and G protein selectivity of a mouse olfactory receptor. J. Neurochem. 2004, 90, 1453–1463. 10.1111/j.1471-4159.2004.02619.x. [DOI] [PubMed] [Google Scholar]
- Lin X.; Janovick J. A.; Brothers S.; Blomenrohr M.; Bogerd J.; Conn P. M. Addition of catfish gonadotropin-releasing hormone (GnRH) receptor intracellular carboxyl-terminal tail to rat GnRH receptor alters receptor expression and regulation. Mol. Endocrinol. 1998, 12, 161–171. 10.1210/mend.12.2.0056. [DOI] [PubMed] [Google Scholar]
- Denning G. M.; Anderson M. P.; Amara J. F.; Marshall J.; Smith A. E.; Welsh M. J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761. 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Jaquette J.; Segaloff D. L. Temperature sensitivity of some mutants of the lutropin/choriogonadotropin receptor1. Endocrinology 1997, 138, 85–91. 10.1210/endo.138.1.4902. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Gong Q.; January C. T. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem. 1999, 274, 31123–31126. 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]
- Street T. O.; Bolen D. W.; Rose G. D. A molecular mechanism for osmolyte-induced protein stability. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13997–14002. 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter D. H. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 2002, 52, 832–836. 10.1203/00006450-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Ejima D.; Kita Y.; Tsumoto K. Small molecule pharmacological chaperones: from thermodynamic stabilization to pharmaceutical drugs. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1677–1687. 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Babcock J. J.; Li M. Inside job: ligand-receptor pharmacology beneath the plasma membrane. Acta Pharmacol. Sin. 2013, 34, 859–869. 10.1038/aps.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidenheimer N. J.; Ryder K. G. Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol. Res. 2014, 83, 10–19. 10.1016/j.phrs.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y. X.; Conn P. M. Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol. Rev. 2018, 98, 697–725. 10.1152/physrev.00029.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. M.; Smithson D. C.; Hodder P. S.; Stewart M. D.; Behringer R. R.; Smith E.; Ulloa-Aguirre A.; Janovick J. A. Transitioning pharmacoperones to therapeutic use: in vivo proof-of-principle and design of high throughput screens. Pharmacol. Res. 2014, 83, 38–51. 10.1016/j.phrs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. M.; Spicer T. P.; Scampavia L.; Janovick J. A. Assay strategies for identification of therapeutic leads that target protein trafficking. Trends Pharmacol. Sci. 2015, 36, 498–505. 10.1016/j.tips.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J. Q.; Ishii S.; Asano N.; Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- Generoso S. F.; Giustiniano M.; La Regina G.; Bottone S.; Passacantilli S.; Di Maro S.; Cassese H.; Bruno A.; Mallardo M.; Dentice M.; Silvestri R.; Marinelli L.; Sarnataro D.; Bonatti S.; Novellino E.; Stornaiuolo M. Pharmacological folding chaperones act as allosteric ligands of Frizzled4. Nat. Chem. Biol. 2015, 11, 280–286. 10.1038/nchembio.1770. [DOI] [PubMed] [Google Scholar]
- Hunter P. J. Receptor redemption - Skirting gene therapy to correct genetic defects. Scientist 2004, 18, 30–30. [Google Scholar]
- Ingolia N. T.; Lareau L. F.; Weissman J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L.; Mohamed A.; Kartner N.; Chang X. B.; Riordan J. R.; Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroz D.; Dahimene S.; Baro I.; Loussouarn G.; Merot J. LQT1-associated mutations increase KCNQ1 proteasomal degradation independently of Derlin-1. J. Biol. Chem. 2009, 284, 5250–5256. 10.1074/jbc.M806459200. [DOI] [PubMed] [Google Scholar]
- Pietila E. M.; Tuusa J. T.; Apaja P. M.; Aatsinki J. T.; Hakalahti A. E.; Rajaniemi H. J.; Petaja-Repo U. E. Inefficient maturation of the rat luteinizing hormone receptor. A putative way to regulate receptor numbers at the cell surface. J. Biol. Chem. 2005, 280, 26622–26629. 10.1074/jbc.M413815200. [DOI] [PubMed] [Google Scholar]
- Janovick J. A.; Brothers S. P.; Cornea A.; Bush E.; Goulet M. T.; Ashton W. T.; Sauer D. R.; Haviv F.; Greer J.; Conn P. M. Refolding of misfolded mutant GPCR: post-translational pharmacoperone action in vitro. Mol. Cell. Endocrinol. 2007, 272, 77–85. 10.1016/j.mce.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. L.; Reloj A. R.; Nataraj P. S.; Bartos D. C.; Schroder E. A.; Moss A. J.; Ohno S.; Horie M.; Anderson C. L.; January C. T.; Delisle B. P. Pharmacological correction of long QT-linked mutations in KCHN2 (hERG) increases the trafficking of Kv11.1 channels stored in the transitional ER. Am. J. Physiol. Cell Physiol. 2013, 305, C919–C930. 10.1152/ajpcell.00406.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Craenenbroeck K.; Clark S. D.; Cox M. J.; Oak J. N.; Liu F.; Van Tol H. H. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J. Biol. Chem. 2005, 280, 19350–19357. 10.1074/jbc.M414043200. [DOI] [PubMed] [Google Scholar]
- Liu P. F.; Park C. Selective stabilization of a partially unfolded protein by a metabolite. J. Mol. Biol. 2012, 422, 403–413. 10.1016/j.jmb.2012.05.044. [DOI] [PubMed] [Google Scholar]
- Kasper J. R.; Park C. Ligand binding to a high-energy partially unfolded protein. Protein Sci. 2015, 24, 129–137. 10.1002/pro.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick J. A.; Goulet M.; Bush E.; Greer J.; Wettlaufer D. G.; Conn P. M. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J. Pharmacol. Exp. Ther. 2003, 305, 608–614. 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- Robben J. H.; Sze M.; Knoers N. V. A. M.; Deen P. M. T. Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus. Am. J. Physiol.-Renal 2007, 292, F253–F260. 10.1152/ajprenal.00247.2006. [DOI] [PubMed] [Google Scholar]
- Huang H.; Wang W.; Tao Y. X. Pharmacological chaperones for the misfolded melanocortin-4 receptor associated with human obesity. Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863, 2496–2507. 10.1016/j.bbadis.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; McGrath T. Substrate stabilization of lysozyme to thermal and guanidine hydrochloride denaturation. J. Biol. Chem. 1980, 255, 3862–3865. [PubMed] [Google Scholar]
- Park C.; Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2005, 2, 207–212. 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- Janovick J. A.; Spicer T. P.; Smith E.; Bannister T. D.; Kenakin T.; Scampavia L.; Conn P. M. Receptor antagonism/agonism can be uncoupled from pharmacoperone activity. Mol. Cell. Endocrinol. 2016, 434, 176–185. 10.1016/j.mce.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.; Pattamatta A.; Hildago R.; Pace M. C.; Brown H.; Borchelt D. R. Vulnerability of newly synthesized proteins to proteostasis stress. J. Cell Sci. 2016, 129, 1892–1901. 10.1242/jcs.176479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh A.; McKenna J.; Stepanchick A.; Breitwieser G. E. Calcium-sensing receptor biosynthesis includes a cotranslational conformational checkpoint and endoplasmic reticulum retention. J. Biol. Chem. 2010, 285, 19854–19864. 10.1074/jbc.M110.124792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Breitwieser G. E. Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J. Biol. Chem. 2007, 282, 9517–9525. 10.1074/jbc.M609045200. [DOI] [PubMed] [Google Scholar]
- Sallette J.; Pons S.; Devillers-Thiery A.; Soudant M.; Prado de Carvalho L.; Changeux J. P.; Corringer P. J. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron 2005, 46, 595–607. 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Cisek K.; Cooper G. L.; Huseby C. J.; Kuret J. Structure and mechanism of action of tau aggregation inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. 10.2174/1567205011666141107150331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway K. A.; Rochet J. C.; Bieganski R. M.; Lansbury P. T. Jr. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294, 1346–1349. 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Hamada Y.; Miyamoto N.; Kiso Y. Novel beta-amyloid aggregation inhibitors possessing a turn mimic. Bioorg. Med. Chem. Lett. 2015, 25, 1572–1576. 10.1016/j.bmcl.2015.02.016. [DOI] [PubMed] [Google Scholar]
- Makley L. N.; McMenimen K. A.; DeVree B. T.; Goldman J. W.; McGlasson B. N.; Rajagopal P.; Dunyak B. M.; McQuade T. J.; Thompson A. D.; Sunahara R.; Klevit R. E.; Andley U. P.; Gestwicki J. E. Pharmacological chaperone for alpha-Crystallin partially restores transparency in cataract models. Science 2015, 350, 674–677. 10.1126/science.aac9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Seyb K. I.; Huang M.; Schuman E. R.; Shi P.; Zhu H.; Glicksman M. A. A high-throughput screening method for small-molecule inhibitors of the aberrant mutant SOD1 and dynein complex interaction. J. Biomol. Screening 2012, 17, 314–326. 10.1177/1087057111429595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L. M.; Saunders J. C.; Mahood R. A.; Revill C. H.; Foster R. J.; Ashcroft A. E.; Radford S. E. ESI-IMS-MS: A method for rapid analysis of protein aggregation and its inhibition by small molecules. Methods 2016, 95, 62–69. 10.1016/j.ymeth.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A. N.; Neupane K.; Rezajooei N.; Cortez L. M.; Sim V. L.; Woodside M. T. Pharmacological chaperone reshapes the energy landscape for folding and aggregation of the prion protein. Nat. Commun. 2016, 7, 12058. 10.1038/ncomms12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem. 1997, 272, 709–712. 10.1074/jbc.272.2.709. [DOI] [PubMed] [Google Scholar]
- Morello J. P.; Salahpour A.; Laperriere A.; Bernier V.; Arthus M. F.; Lonergan M.; Petaja-Repo U.; Angers S.; Morin D.; Bichet D. G.; Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest. 2000, 105, 887–895. 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Alphonse F.; Perkovska S.; Frantz M. C.; Durroux T.; Mejean C.; Morin D.; Loison S.; Bonnet D.; Hibert M.; Mouillac B.; Mendre C. Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2009, 20, 2190–2203. 10.1681/ASN.2008121289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serradeillegal C.; Wagnon J.; Garcia C.; Lacour C.; Guiraudou P.; Christophe B.; Villanova G.; Nisato D.; Maffrand J. P.; Lefur G. Biochemical and pharmacological properties of SR-49059, a new, potent, nonpeptide antagonist of rat and human vasopressin V1R receptors. J. Clin. Invest. 1993, 92, 224–231. 10.1172/JCI116554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara A.; Saito M.; Sugimoto T.; Tomura Y.; Wada K.; Kusayama T.; Tsukada J.; Ishii N.; Yatsu T.; Uchida W.; Tanaka A. Pharmacological characterization of YM087, a potent, nonpeptide human vasopressin V1A and V2 receptor antagonist. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997, 357, 63–69. 10.1007/PL00005139. [DOI] [PubMed] [Google Scholar]
- Yamamura Y.; Nakamura S.; Itoh S.; Hirano T.; Onogawa T.; Yamashita T.; Yamada Y.; Tsujimae K.; Aoyama M.; Kotosai K.; Ogawa H.; Yamashita H.; Kondo K.; Tominaga M.; Tsujimoto G.; Mori T. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J. Pharmacol. Exp. Ther. 1998, 287, 860–867. [PubMed] [Google Scholar]
- Conn P. M.; Smith E.; Hodder P.; Janovick J. A.; Smithson D. C. High-throughput screen for pharmacoperones of the vasopressin type 2 receptor. J. Biomol. Screening 2013, 18, 930–937. 10.1177/1087057113483559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.; Janovick J. A.; Bannister T. D.; Shumate J.; Scampavia L.; Conn P. M.; Spicer T. P. Identification of Potential Pharmacoperones Capable of Rescuing the Functionality of Misfolded Vasopressin 2 Receptor Involved in Nephrogenic Diabetes Insipidus. J. Biomol. Screening 2016, 21, 824–831. 10.1177/1087057116653925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithson D. C.; Janovick J. A.; Conn P. M. Therapeutic rescue of misfolded/mistrafficked mutants: automation-friendly high-throughput assays for identification of pharmacoperone drugs of GPCRs. Methods Enzymol. 2013, 521, 3–16. 10.1016/B978-0-12-391862-8.00001-6. [DOI] [PubMed] [Google Scholar]
- Janovick J. A.; Maya-Nunez G.; Conn P. M. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J. Clin. Endocrinol. Metab. 2002, 87, 3255–3262. 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A.; Ulloa-Aguirre A.; Janovick J. A.; Conn P. M. In vitro coexpression and pharmacological rescue of mutant gonadotropin-releasing hormone receptors causing hypogonadotropic hypogonadism in humans expressing compound heterozygous alleles. J. Clin. Endocrinol. Metab. 2005, 90, 3001–3008. 10.1210/jc.2004-2071. [DOI] [PubMed] [Google Scholar]
- Janovick J. A.; Maya-Nunez G.; Ulloa-Aguirre A.; Huhtaniemi I. T.; Dias J. A.; Verbost P.; Conn P. M. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol. Cell. Endocrinol. 2009, 298, 84–88. 10.1016/j.mce.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. M.; Ulloa-Aguirre A. Pharmacological chaperones for misfolded gonadotropin-releasing hormone receptors. Adv. Pharmacol. 2011, 62, 109–141. 10.1016/B978-0-12-385952-5.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S.; Cho N.; Nara Y.; Harada M.; Endo S.; Suzuki N.; Furuya S.; Fujino M. Discovery of a thieno[2,3-d]pyrimidine-2,4-dione bearing a p-methoxyureidophenyl moiety at the 6-position: a highly potent and orally bioavailable non-peptide antagonist for the human luteinizing hormone-releasing hormone receptor. J. Med. Chem. 2003, 46, 113–124. 10.1021/jm020180i. [DOI] [PubMed] [Google Scholar]
- Janovick J. A.; Stewart M. D.; Jacob D.; Martin L. D.; Deng J. M.; Stewart C. A.; Wang Y.; Cornea A.; Chavali L.; Lopez S.; Mitalipov S.; Kang E.; Lee H. S.; Manna P. R.; Stocco D. M.; Behringer R. R.; Conn P. M. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 21030–21035. 10.1073/pnas.1315194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huszar D.; Lynch C. A.; Fairchild-Huntress V.; Dunmore J. H.; Fang Q.; Berkemeier L. R.; Gu W.; Kesterson R. A.; Boston B. A.; Cone R. D.; Smith F. J.; Campfield L. A.; Burn P.; Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Farooqi I. S.; Keogh J. M.; Yeo G. S.; Lank E. J.; Cheetham T.; O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 2003, 348, 1085–1095. 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Tao Y. X.; Huang H.; Wang Z. Q.; Yang F.; Williams J. N.; Nikiforovich G. V. Constitutive activity of neural melanocortin receptors. Methods Enzymol. 2010, 484, 267–279. 10.1016/B978-0-12-381298-8.00014-9. [DOI] [PubMed] [Google Scholar]
- Huang H.; Tao Y. X. Pleiotropic functions of the transmembrane domain 6 of human melanocortin-4 receptor. J. Mol. Endocrinol. 2012, 49, 237–248. 10.1530/JME-12-0161. [DOI] [PubMed] [Google Scholar]
- Poitout L.; Brault V.; Sackur C.; Bernetiere S.; Camara J.; Plas P.; Roubert P. Identification of a novel series of benzimidazoles as potent and selective antagonists of the human melanocortin-4 receptor. Bioorg. Med. Chem. Lett. 2007, 17, 4464–4470. 10.1016/j.bmcl.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Sebhat I. K.; Martin W. J.; Ye Z. X.; Barakat K.; Mosley R. T.; Johnston D. B. R.; Bakshi R.; Palucki B.; Weinberg D. H.; MacNeil T.; Kalyani R. N.; Tang R.; Stearns R. A.; Miller R. R.; Tamvakopoulos C.; Strack A. M.; McGowan E.; Cashen D. E.; Drisko J. E.; Hom G. J.; Howard A. D.; MacIntyre D. E.; van der Ploeg L. H. T.; Patchett A. A.; Nargund R. P. Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium-3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist. J. Med. Chem. 2002, 45, 4589–4593. 10.1021/jm025539h. [DOI] [PubMed] [Google Scholar]
- Vos T. J.; Caracoti A.; Che J. L.; Dai M.; Farrer C. A.; Forsyth N. E.; Drabic S. V.; Horlick R. A.; Lamppu D.; Yowe D. L.; Balani S.; Li P.; Zeng H.; Joseph I. B. J. K.; Rodriguez L. E.; Maguire M. P.; Patane M. A.; Claiborne C. F. Identification of 2-{2-[2-(5-bromo-2-methoxyphenyl)-ethyl]-3-fluoro-phenyl}-4,5-dihydro-1H-imidazole (ML00253764), a small molecule melanocortin 4 receptor antagonist that effectively reduces tumor-induced weight loss in a mouse model. J. Med. Chem. 2004, 47, 1602–1604. 10.1021/jm034244g. [DOI] [PubMed] [Google Scholar]
- Rene P.; Le Gouill C.; Pogozheva I. D.; Lee G.; Mosberg H. I.; Farooqi I. S.; Valenzano K. J.; Bouvier M. Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J. Pharmacol. Exp. Ther. 2010, 335, 520–532. 10.1124/jpet.110.172098. [DOI] [PubMed] [Google Scholar]
- Wang X. H.; Wang H. M.; Zhao B. L.; Yu P.; Fan Z. C. Rescue of the mutant MC4R cell surface expression and signaling by a novel pharmacoperone Ipsen 17. J. Mol. Endocrinol. 2014, 53, 17–29. 10.1530/JME-14-0005. [DOI] [PubMed] [Google Scholar]
- Tao Y. X.; Huang H. Ipsen 5i is a novel potent pharmacoperone for intracellularly retained melanocortin-4 receptor mutants. Front. Endocrinol. 2014, 5, 131. 10.3389/fendo.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Tao Y. X. A small molecule agonist THIQ as a novel pharmacoperone for intracellularly retained melanocortin-4 receptor mutants. Int. J. Biol. Sci. 2014, 10, 817–824. 10.7150/ijbs.9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E.; Obejero-Paz C. A.; Zhao S.; Brown A. M. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J. Biol. Chem. 2002, 277, 4989–4998. 10.1074/jbc.M107345200. [DOI] [PubMed] [Google Scholar]
- Mehta A.; Ramachandra C. J. A.; Singh P.; Chitre A.; Lua C. H.; Mura M.; Crotti L.; Wong P.; Schwartz P. J.; Gnecchi M.; Shim W. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Heart J. 2018, 39, 1446. 10.1093/eurheartj/ehx394. [DOI] [PubMed] [Google Scholar]
- Clark N. E.; Metcalf M. C.; Best D.; Fleet G. W.; Garman S. C. Pharmacological chaperones for human α-N-acetylgalactosaminidase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17400–17405. 10.1073/pnas.1203924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Y.; Liu N.; Priori S. G. Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 2009, 6, 337–348. 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- Valdivia C. R.; Tester D. J.; Rok B. A.; Porter C. B.; Munger T. M.; Jahangir A.; Makielski J. C.; Ackerman M. J. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 2004, 62, 53–62. 10.1016/j.cardiores.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Moreau A.; Keller D. I.; Huang H.; Fressart V.; Schmied C.; Timour Q.; Chahine M. Mexiletine differentially restores the trafficking defects caused by two brugada syndrome mutations. Front. Pharmacol. 2012, 3, 62. 10.3389/fphar.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Yang T.; Viswanathan P. C.; Roden D. M. New mechanism contributing to drug-induced arrhythmia: rescue of a misprocessed LQT3 mutant. Circulation 2005, 112, 3239–3246. 10.1161/CIRCULATIONAHA.105.564008. [DOI] [PubMed] [Google Scholar]
- Valdivia C. R.; Ackerman M. J.; Tester D. J.; Wada T.; McCormack J.; Ye B.; Makielski J. C. A novel SCN5A arrhythmia mutation, M1766L, with expression defect rescued by mexiletine. Cardiovasc. Res. 2002, 55, 279–289. 10.1016/S0008-6363(02)00445-5. [DOI] [PubMed] [Google Scholar]
- Cassano W. F. Cystic fibrosis and the plague. Med. Hypotheses 1985, 18, 51–52. 10.1016/0306-9877(85)90119-7. [DOI] [PubMed] [Google Scholar]
- Gallego Romero I.; Ober C. CFTR mutations and reproductive outcomes in a population isolate. Hum. Genet. 2008, 122, 583–588. 10.1007/s00439-007-0432-1. [DOI] [PubMed] [Google Scholar]
- Pittman J. E.; Ferkol T. W. The evolution of cystic fibrosis care. Chest 2015, 148, 533–542. 10.1378/chest.14-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltshe S. L.; Cogen J.; Ramos K. J.; Goss C. H. Cystic fibrosis: the dawn of a new therapeutic era. Am. J. Respir. Crit. Care Med. 2017, 195, 979–984. 10.1164/rccm.201606-1250PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Cao D.; Neuberger T.; Turnbull A.; Singh A.; Joubran J.; Hazlewood A.; Zhou J.; McCartney J.; Arumugam V.; Decker C.; Yang J.; Young C.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18825–18830. 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Straley K. S.; Cao D.; Gonzalez J.; Hadida S.; Hazlewood A.; Joubran J.; Knapp T.; Makings L. R.; Miller M.; Neuberger T.; Olson E.; Panchenko V.; Rader J.; Singh A.; Stack J. H.; Tung R.; Grootenhuis P. D.; Negulescu P. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1117–1130. 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Pedemonte N.; Lukacs G. L.; Du K.; Caci E.; Zegarra-Moran O.; Galietta L. J.; Verkman A. S. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Shelat A. A.; Guy R. K.; Gopinath V. S.; Ma T. H.; Du K.; Lukacs G. L.; Taddei A.; Folli C.; Pedemonte N.; Galietta L. J. V.; Verkman A. S. Nanomolar affinity small molecule correctors of defective ΔF508-CFTR chloride channel gating. J. Biol. Chem. 2003, 278, 35079–35085. 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- Wang X. Q.; Liu B.; Searle X.; Yeung C.; Bogdan A.; Greszler S.; Singh A.; Fan Y. H.; Swensen A. M.; Vortherms T.; Balut C.; Jia Y.; Desino K.; Gao W. Q.; Yong H.; Tse C.; Kym P. Discovery of 4-[(2r,4r)-4-({[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2h-chromen-2-yl]benzoic acid (abbv/glpg-2222), a potent cystic fibrosis transmembrane conductance regulator (CFTR) corrector for the treatment of cystic fibrosis. J. Med. Chem. 2018, 61, 1436–1449. 10.1021/acs.jmedchem.7b01339. [DOI] [PubMed] [Google Scholar]
- Van Goor F.; Grootenhuis P.; Hadida S.; Burton B.; Young T.; Selkirk J.; Powe A.; De La Rosa O.; Jiang L.; Zhou J.; Yu H.; Negulescu P. Nonclinical profile of the CFTR corrector VX-661. Pediatr. Pulm. 2016, 51, 274–274. [Google Scholar]
- Sampson H. M.; Robert R.; Liao J.; Matthes E.; Carlile G. W.; Hanrahan J. W.; Thomas D. Y. Identification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTR. Chem. Biol. 2011, 18, 231–242. 10.1016/j.chembiol.2010.11.016. [DOI] [PubMed] [Google Scholar]
- Accurso F. J.; Rowe S. M.; Clancy J. P.; Boyle M. P.; Dunitz J. M.; Durie P. R.; Sagel S. D.; Hornick D. B.; Konstan M. W.; Donaldson S. H.; Moss R. B.; Pilewski J. M.; Rubenstein R. C.; Uluer A. Z.; Aitken M. L.; Freedman S. D.; Rose L. M.; Mayer-Hamblett N.; Dong Q.; Zha J.; Stone A. J.; Olson E. R.; Ordonez C. L.; Campbell P. W.; Ashlock M. A.; Ramsey B. W. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey B. W.; Davies J.; McElvaney N. G.; Tullis E.; Bell S. C.; Drevinek P.; Griese M.; McKone E. F.; Wainwright C. E.; Konstan M. W.; Moss R.; Ratjen F.; Sermet-Gaudelus I.; Rowe S. M.; Dong Q.; Rodriguez S.; Yen K.; Ordonez C.; Elborn J. S. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D. G.; Balasubramanian S.; Frankish A.; Huang N.; Morris J.; Walter K.; Jostins L.; Habegger L.; Pickrell J. K.; Montgomery S. B.; Albers C. A.; Zhang Z. D.; Conrad D. F.; Lunter G.; Zheng H.; Ayub Q.; DePristo M. A.; Banks E.; Hu M.; Handsaker R. E.; Rosenfeld J. A.; Fromer M.; Jin M.; Mu X. J.; Khurana E.; Ye K.; Kay M.; Saunders G. I.; Suner M. M.; Hunt T.; Barnes I. H.; Amid C.; Carvalho-Silva D. R.; Bignell A. H.; Snow C.; Yngvadottir B.; Bumpstead S.; Cooper D. N.; Xue Y.; Romero I. G.; Genomes Project C.; Wang J.; Li Y.; Gibbs R. A.; McCarroll S. A.; Dermitzakis E. T.; Pritchard J. K.; Barrett J. C.; Harrow J.; Hurles M. E.; Gerstein M. B.; Tyler-Smith C. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828. 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholon D. M.; Quinney N. L.; Fulcher M. L.; Esther C. R. Jr.; Das J.; Dokholyan N. V.; Randell S. H.; Boucher R. C.; Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra296. 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G.; Avramescu R. G.; Perdomo D.; Phuan P. W.; Bagdany M.; Apaja P. M.; Borot F.; Szollosi D.; Wu Y. S.; Finkbeiner W. E.; Hegedus T.; Verkman A. S.; Lukacs G. L. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra297. 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laselva O.; Molinski S.; Casavola V.; Bear C. E. Correctors of the major cystic fibrosis mutant interact through membrane-spanning domains. Mol. Pharmacol. 2018, 93, 612–618. 10.1124/mol.118.111799. [DOI] [PubMed] [Google Scholar]
- Ren H. Y.; Grove D. E.; De La Rosa O.; Houck S. A.; Sopha P.; Van Goor F.; Hoffman B. J.; Cyr D. M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. 10.1091/mbc.e13-05-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis P. B. Another deginning for cystic fibrosis therapy. N. Engl. J. Med. 2015, 373, 2. 10.1056/NEJMe1504059. [DOI] [PubMed] [Google Scholar]
- Grasemann H. CFTR modulator therapy for cystic fibrosis. N. Engl. J. Med. 2017, 377, 2085–2088. 10.1056/NEJMe1712335. [DOI] [PubMed] [Google Scholar]
- Pranke I. M.; Hatton A.; Simonin J.; Jais J. P.; Le Pimpec-Barthes F.; Carsin A.; Bonnette P.; Fayon M.; Stremler-Le Bel N.; Grenet D.; Thumerel M.; Mazenq J.; Urbach V.; Mesbahi M.; Girodon-Boulandet E.; Hinzpeter A.; Edelman A.; Sermet-Gaudelus I. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. 10.1038/s41598-017-07504-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright C. E.; Elborn J. S.; Ramsey B. W. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. 10.1056/NEJMc1510466. [DOI] [PubMed] [Google Scholar]
- Taylor-Cousar J. L.; Munck A.; McKone E. F.; van der Ent C. K.; Moeller A.; Simard C.; Wang L. T.; Ingenito E. P.; McKee C.; Lu Y.; Lekstrom-Himes J.; Elborn J. S. Tezacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. 10.1056/NEJMoa1709846. [DOI] [PubMed] [Google Scholar]
- Rowe S. M.; Daines C.; Ringshausen F. C.; Kerem E.; Wilson J.; Tullis E.; Nair N.; Simard C.; Han L.; Ingenito E. P.; McKee C.; Lekstrom-Himes J.; Davies J. C. Tezacaftor-Ivacaftor in residual-function heterozygotes with cystic fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. C.; Moskowitz S. M.; Brown C.; Horsley A.; Mall M. A.; McKone E. F.; Plant B. J.; Prais D.; Ramsey B. W.; Taylor-Cousar J. L.; Tullis E.; Uluer A.; McKee C. M.; Robertson S.; Shilling R. A.; Simard C.; Van Goor F.; Waltz D.; Xuan F.; Young T.; Rowe S. M. VX-659-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1599–1611. 10.1056/NEJMoa1807119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating D.; Marigowda G.; Burr L.; Daines C.; Mall M. A.; McKone E. F.; Ramsey B. W.; Rowe S. M.; Sass L. A.; Tullis E.; McKee C. M.; Moskowitz S. M.; Robertson S.; Savage J.; Simard C.; Van Goor F.; Waltz D.; Xuan F.; Young T.; Taylor-Cousar J. L. VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1612–1620. 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijnders M.; Kleizen B.; Braakman I. Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr. Opin. Pharmacol. 2017, 34, 83–90. 10.1016/j.coph.2017.09.014. [DOI] [PubMed] [Google Scholar]
- De Stefano D.; Villella V. R.; Esposito S.; Tosco A.; Sepe A.; De Gregorio F.; Salvadori L.; Grassia R.; Leone C. A.; De Rosa G.; Maiuri M. C.; Pettoello-Mantovani M.; Guido S.; Bossi A.; Zolin A.; Venerando A.; Pinna L. A.; Mehta A.; Bona G.; Kroemer G.; Maiuri L.; Raia V. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. 10.4161/15548627.2014.973737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito S.; Tosco A.; Villella V. R.; Raia V.; Kroemer G.; Maiuri L. Manipulating proteostasis to repair the F508del-CFTR defect in cystic fibrosis. Mol. Cell. Pediatr. 2016, 3, 13. 10.1186/s40348-016-0040-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmowicz A. G.; Lim R.; Rogers H.; Rosebraugh C. J.; Chowdhury B. A. The U.S. Food and Drug Administration’s experience with Ivacaftor in cystic fibrosis. Establishing efficacy using nn vitro data in lieu of a clinical trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. 10.1513/AnnalsATS.201708-668PS. [DOI] [PubMed] [Google Scholar]
- Kingwell K. FDA OKs first in vitro route to expanded approval. Nat. Rev. Drug Discovery 2017, 16, 591–592. 10.1038/nrd.2017.140. [DOI] [PubMed] [Google Scholar]
- Colemeadow J.; Joyce H.; Turcanu V. Precise treatment of cystic fibrosis – current treatments and perspectives for using CRISPR. Exp. Rev. Precis. Med. Drug Dev. 2016, 1, 12. 10.1080/23808993.2016.1146077. [DOI] [Google Scholar]
- Schwank G.; Koo B. K.; Sasselli V.; Dekkers J. F.; Heo I.; Demircan T.; Sasaki N.; Boymans S.; Cuppen E.; van der Ent C. K.; Nieuwenhuis E. E.; Beekman J. M.; Clevers H. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Firth A. L.; Menon T.; Parker G. S.; Qualls S. J.; Lewis B. M.; Ke E.; Dargitz C. T.; Wright R.; Khanna A.; Gage F. H.; Verma I. M. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015, 12, 1385–1390. 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz D. J.; Hollywood J. A.; Scallan M. F.; Harrison P. T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS One 2017, 12, e0184009. 10.1371/journal.pone.0184009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. M.; Knollman P. E.; Brothers S. P.; Janovick J. A. Protein folding as posttranslational regulation: evolution of a mechanism for controlled plasma membrane expression of a G protein-coupled receptor. Mol. Endocrinol. 2006, 20, 3035–3041. 10.1210/me.2006-0066. [DOI] [PubMed] [Google Scholar]
- Fleck M. W. Glutamate receptors and endoplasmic reticulum quality control: looking beneath the surface. Neuroscientist 2006, 12, 232–244. 10.1177/1073858405283828. [DOI] [PubMed] [Google Scholar]
- Jo Y.; Debose-Boyd R. A. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 185–198. 10.3109/10409238.2010.485605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerepoot P.; Lam V. M.; Salahpour A. Pharmacological chaperones of the dopamine transporter rescue dopamine transporter deficiency syndrome mutations in heterologous cells. J. Biol. Chem. 2016, 291, 22053–22062. 10.1074/jbc.M116.749119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petäjä-Repo U. E.; Hogue M.; Bhalla S.; Laperrière A.; Morello J. P.; Bouvier M. Ligands act as pharmacological chaperones and increase the efficiency of δ opioid receptor maturation. EMBO J. 2002, 21, 1628–1637. 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A.; Hotsubo T.; Kobayashi K.; Mochizuki H.; Ishizu K.; Tajima T. Loss-of-function and gain-of-function mutations of calcium-sensing receptor: functional analysis and the effect of allosteric modulators NPS R-568 and NPS 2143. J. Clin. Endocrinol. Metab. 2013, 98, E1692–E1701. 10.1210/jc.2013-1974. [DOI] [PubMed] [Google Scholar]
- Conn P. M.; Ulloa-Aguirre A.; Ito J.; Janovick J. A. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol. Rev. 2007, 59, 225–250. 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002, 277, 44332–44338. 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Loo T. W.; Bartlett M. C.; Clarke D. M. Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol. Pharmacol. 2006, 71, 751–758. 10.1124/mol.106.029926. [DOI] [PubMed] [Google Scholar]
- Spork M.; Sohail M. I.; Schmid D.; Ecker G. F.; Freissmuth M.; Chiba P.; Stockner T. Folding correction of ABC-transporter ABCB1 by pharmacological chaperones: a mechanistic concept. Pharmacol. Res. Perspect. 2017, 5, e00325. 10.1002/prp2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem. 1997, 272, 709–712. 10.1074/jbc.272.2.709. [DOI] [PubMed] [Google Scholar]
- Carlile G. W.; Robert R.; Zhang D.; Teske K. A.; Luo Y.; Hanrahan J. W.; Thomas D. Y. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. ChemBioChem 2007, 8, 1012–1020. 10.1002/cbic.200700027. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu B.; Searle X.; Yeung C.; Bogdan A.; Greszler S.; Singh A.; Fan Y.; Swensen A. M.; Vortherms T.; Balut C.; Jia Y.; Desino K.; Gao W.; Yong H.; Tse C.; Kym P. Discovery of 4-[(2R,4R)-4-({[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)- 7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic Acid (ABBV/GLPG-2222), a potent cystic fibrosis transmembrane conductance regulator (CFTR) corrector for the Treatment of Cystic Fibrosis. J. Med. Chem. 2018, 61, 1436–1449. 10.1021/acs.jmedchem.7b01339. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Bartlett M. C.; Loo T. W.; Clarke D. M. Specific rescue of cystic fibrosis transmembrane conductance regulator processing mutants using pharmacological chaperones. Mol. Pharmacol. 2006, 70, 297–302. [DOI] [PubMed] [Google Scholar]
- Robert R.; Carlile G. W.; Liao J.; Balghi H.; Lesimple P.; Liu N.; Kus B.; Rotin D.; Wilke M.; de Jonge H. R.; Scholte B. J.; Thomas D. Y.; Hanrahan J. W. Correction of the Delta phe508 cystic fibrosis transmembrane conductance regulator trafficking defect by the bioavailable compound glafenine. Mol. Pharmacol. 2010, 77, 922–930. 10.1124/mol.109.062679. [DOI] [PubMed] [Google Scholar]
- Robert R.; Carlile G. W.; Pavel C.; Liu N.; Anjos S. M.; Liao J.; Luo Y.; Zhang D.; Thomas D. Y.; Hanrahan J. W. Structural analog of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Mol. Pharmacol. 2007, 73, 478–489. 10.1124/mol.107.040725. [DOI] [PubMed] [Google Scholar]
- Gautherot J.; Durand-Schneider A. M.; Delautier D.; Delaunay J. L.; Rada A.; Gabillet J.; Housset C.; Maurice M.; Ait-Slimane T. Effects of cellular, chemical, and pharmacological chaperones on the rescue of a trafficking-defective mutant of the ATP-binding cassette transporter proteins ABCB1/ABCB4. J. Biol. Chem. 2012, 287, 5070–5078. 10.1074/jbc.M111.275438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F.; Lin C. W.; Weisiger E.; Cartier E. A.; Taschenberger G.; Shyng S. L. Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J. Biol. Chem. 2004, 279, 11096–11105. 10.1074/jbc.M312810200. [DOI] [PubMed] [Google Scholar]
- Yan F. F.; Casey J.; Shyng S. L. Sulfonylureas correct trafficking defects of disease-causing ATP-sensitive potassium channels by binding to the channel complex. J. Biol. Chem. 2006, 281, 33403–33413. 10.1074/jbc.M605195200. [DOI] [PubMed] [Google Scholar]
- El-Kasaby A.; Just H.; Malle E.; Stolt-Bergner P. C.; Sitte H. H.; Freissmuth M.; Kudlacek O. Mutations in the carboxyl-terminal SEC24 binding motif of the serotonin transporter impair folding of the transporter. J. Biol. Chem. 2010, 285, 39201–39210. 10.1074/jbc.M110.118000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S.; Hasenhuetl P. S.; Kasture A.; El-Kasaby A.; Baumann M. H.; Blough B. E.; Sucic S.; Sandtner W.; Freissmuth M. Conformational state interactions provide clues to the pharmacochaperone potential of serotonin transporter partial substrates. J. Biol. Chem. 2017, 292, 16773–16786. 10.1074/jbc.M117.794081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A.; Luo J.; Cooper J.; Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human α4β2 acetylcholine receptors. Mol. Pharmacol. 2005, 68, 1839–1851. 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- Srinivasan R.; Pantoja R.; Moss F. J.; Mackey E. D.; Son C. D.; Miwa J.; Lester H. A. Nicotine up-regulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J. Gen. Physiol. 2011, 137, 59–79. 10.1085/jgp.201010532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q.; Jones M. A.; Zhou Z. Mechanisms of pharmacological rescue of trafficking-defective hERG mutant channels in human long QT syndrome. J. Biol. Chem. 2006, 281, 4069–4074. 10.1074/jbc.M511765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi R.; Scalmani P.; Cassulini R. R.; Giunti G.; Gambardella A.; Franceschetti S.; Annesi G.; Wanke E.; Mantegazza M. Modulatory proteins can rescue a trafficking defective epileptogenic Na(v)1.1 Na+ channel mutant. J. Neurosci. 2007, 27, 11037–11046. 10.1523/JNEUROSCI.3515-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. H.; Porter J. C.; Kahlig K. M.; Daniels M. A.; George A. L. Nontruncating scn1a mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J. Biol. Chem. 2012, 287, 42001–42008. 10.1074/jbc.M112.421883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechi G.; Rusconi R.; Cestele S.; Striano P.; Franceschetti S.; Mantegazza M. Rescuable folding defective Na(V)1.1 (SCN1A) mutants in epilepsy: Properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol. Dis. 2015, 75, 100–114. 10.1016/j.nbd.2014.12.028. [DOI] [PubMed] [Google Scholar]
- Noorwez S. M.; Kuksa V.; Imanishi Y.; Zhu L.; Filipek S.; Palczewski K.; Kaushal S. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J. Biol. Chem. 2003, 278, 14442–14450. 10.1074/jbc.M300087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorwez S. M.; Ostrov D. A.; McDowell J. H.; Krebs M. P.; Kaushal S. A high-throughput screening method for small-molecule pharmacologic chaperones of misfolded rhodopsin. Invest. Ophthalmol. Visual Sci. 2008, 49, 3224–3230. 10.1167/iovs.07-1539. [DOI] [PubMed] [Google Scholar]
- Krebs M. P.; Holden D. C.; Joshi P.; Clark C. L. 3rd; Lee A. H.; Kaushal S. Molecular mechanisms of rhodopsin retinitis pigmentosa and the efficacy of pharmacological rescue. J. Mol. Biol. 2010, 395, 1063–1078. 10.1016/j.jmb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Ashton W. T.; Sisco R. M.; Kieczykowski G. R.; Yang Y. T.; Yudkovitz J. B.; Cui J.; Mount G. R.; Ren R. N.; Wu T. J.; Shen X.; Lyons K. A.; Mao A. H.; Carlin J. R.; Karanam B. V.; Vincent S. H.; Cheng K.; Goulet M. T. Orally bioavailable, indole-based nonpeptide GnRH receptor antagonists with high potency and functional activity. Bioorg. Med. Chem. Lett. 2001, 11, 2597–2602. 10.1016/S0960-894X(01)00512-1. [DOI] [PubMed] [Google Scholar]
- Fan Z. C.; Tao Y. X. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J. Cell. Mol. Med. 2009, 13, 3268–3282. 10.1111/j.1582-4934.2009.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y. X.; Huang H. Ipsen 5i is a novel potent pharmacoperone for intracellularly retained melanocortin-4 receptor mutants. Front. Endocrinol. (Lausanne, Switz.) 2014, 5, 131. 10.3389/fendo.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton C. L.; Whay A. M.; McArdle C. A.; Zhang M.; van Koppen C. J.; van de Lagemaat R.; Segaloff D. L.; Millar R. P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7172–7176. 10.1073/pnas.1015723108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.; Chen C. R.; Liu X.; Kodra J. T. Rescue of a pathogenic mutant human glucagon receptor by pharmacological chaperones. J. Mol. Endocrinol. 2012, 49, 69–78. 10.1530/JME-12-0051. [DOI] [PubMed] [Google Scholar]
- White E.; McKenna J.; Cavanaugh A.; Breitwieser G. E. Pharmacochaperone-mediated rescue of calcium-sensing receptor loss-of-function mutants. Mol. Endocrinol. 2009, 23, 1115–1123. 10.1210/me.2009-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaipatikul V.; Erickson-Herbrandson L. J.; Loh H. H.; Law P. Y. Rescuing the traffic-deficient mutants of rat mu-opioid receptors with hydrophobic ligands. Mol. Pharmacol. 2003, 64, 32–41. 10.1124/mol.64.1.32. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Chen C.; Wang Y.; Liu-Chen L. Y. Ligands regulate cell surface level of the human kappa opioid receptor by activation-induced down-regulation and pharmacological chaperone-mediated enhancement: differential effects of nonpeptide and peptide agonists. J. Pharmacol. Exp. Ther. 2006, 319, 765–775. 10.1124/jpet.106.107987. [DOI] [PubMed] [Google Scholar]
- Málaga-Diéguez L.; Yang Q.; Bauer J.; Pankevych H.; Freissmuth M.; Nanoff C. Pharmacochaperoning of the A1 adenosine receptor is contingent on the endoplasmic reticulum. Mol. Pharmacol. 2010, 77, 940–952. 10.1124/mol.110.063511. [DOI] [PubMed] [Google Scholar]
- Kusek J.; Yang Q.; Witek M.; Gruber C. W.; Nanoff C.; Freissmuth M. Chaperoning of the A1-adenosine receptor by endogenous adenosine - an extension of the retaliatory metabolite concept. Mol. Pharmacol. 2015, 87, 39–51. 10.1124/mol.114.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Lavinder J. J.; Hari S. B.; Sullivan B. J.; Magliery T. J. High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. J. Am. Chem. Soc. 2009, 131, 3794–3795. 10.1021/ja8049063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagramam K. N.; Gopal S. R.; Geng R. S.; Chen D. H. C.; Nemet I.; Lee R.; Tian G. L.; Miyagi M.; Malagu K. F.; Lock C. J.; Esmieu W. R. K.; Owens A. P.; Lindsay N. A.; Ouwehand K.; Albertus F.; Fischer D. F.; Burli R. W.; MacLeod A. M.; Harte W. E.; Palczewski K.; Imanishi Y. A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat. Chem. Biol. 2016, 12, 444–U108. 10.1038/nchembio.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. M.; Smith E.; Spicer T.; Chase P.; Scampavia L.; Janovick J. A. A phenotypic high throughput screening assay for the identification of pharmacoperones for the gonadotropin releasing hormone receptor. Assay Drug Dev. Technol. 2014, 12, 238–246. 10.1089/adt.2014.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi V. J.; Berman D. E.; Simoes S.; Vetanovetz C.; Awal M. R.; Patel V. M.; Schneider R. T.; Petsko G. A.; Ringe D.; Small S. A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalid O.; Mense M.; Fischman S.; Shitrit A.; Bihler H.; Ben-Zeev E.; Schutz N.; Pedemonte N.; Thomas P. J.; Bridges R. J.; Wetmore D. R.; Marantz Y.; Senderowitz H. Small molecule correctors of F508del-CFTR discovered by structure-based virtual screening. J. Comput.-Aided Mol. Des. 2010, 24, 971–991. 10.1007/s10822-010-9390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. H.; Lim H. S. Screening methods for identifying pharmacological chaperones. Mol. BioSyst. 2017, 13, 638–647. 10.1039/C6MB00866F. [DOI] [PubMed] [Google Scholar]
- Pankow S.; Bamberger C.; Calzolari D.; Martinez-Bartolome S.; Lavallee-Adam M.; Balch W. E.; Yates J. R. III ΔF508 CFTR interactome remodelling promotes rescue of cystic ribrosis. Nature 2015, 528, 510–516. 10.1038/nature15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo-Kang L. R.; Zeitlin P. L. Induction of HSP70 promotes ΔF508 CFTR trafficking. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L58–L68. 10.1152/ajplung.2001.281.1.L58. [DOI] [PubMed] [Google Scholar]
- Norez C.; Antigny F.; Becq F.; Vandebrouck C. Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic 2006, 7, 562–573. 10.1111/j.1600-0854.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk M. S.; Knox T.; LaVail M. M.; Gorbatyuk O. S.; Noorwez S. M.; Hauswirth W. W.; Lin J. H.; Muzyczka N.; Lewin A. S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5961–5966. 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimaridou E.; Gooljar S. B.; Ramnarace N.; Anthonypillai L.; Clark A. J.; Chapple J. P. The cytosolic chaperone Hsc70 promotes traffic to the cell surface of intracellular retained melanocortin-4 receptor mutants. Mol. Endocrinol. 2011, 25, 1650–1660. 10.1210/me.2011-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W. E.; Morimoto R. I.; Dillin A.; Kelly J. W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Peterson R. T. Chemical biology and the limits of reductionism. Nat. Chem. Biol. 2008, 4, 635–638. 10.1038/nchembio1108-635. [DOI] [PubMed] [Google Scholar]
- Tardiff D. F.; Tucci M. L.; Caldwell K. A.; Caldwell G. A.; Lindquist S. Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem. 2012, 287, 4107–4120. 10.1074/jbc.M111.308668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S.; Xu Y. M.; Wijeratne E. M.; Kontnik R.; Rooney C.; Perley C. C.; Kwon H.; Clardy J.; Kesari S.; Whitesell L.; Lindquist S.; Gunatilaka A. A. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem. Biol. 2012, 7, 340–349. 10.1021/cb200353m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey S.; Nickson A. A.; Clarke J. Studying the folding of multidomain proteins. HFSP J. 2008, 2, 365–377. 10.2976/1.2991513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu C. B.; Bridges R. J.; Pena-Rasgado C.; Lacerda A. E.; Bordwell C.; Sewell A.; Nichols A. J.; Chandran S.; Lonkar P.; Picarella D.; Ting A.; Wensley A.; Yeager M.; Liu F. Fatty Acid Cysteamine Conjugates as Novel and Potent Autophagy Activators That Enhance the Correction of Misfolded F508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). J. Med. Chem. 2017, 60, 458–473. 10.1021/acs.jmedchem.6b01539. [DOI] [PubMed] [Google Scholar]
- Lee S.; Henderson M. J.; Schiffhauer E.; Despanie J.; Henry K.; Kang P. W.; Walker D.; McClure M. L.; Wilson L.; Sorscher E. J.; Zeitlin P. L. Interference with ubiquitination in CFTR modifies stability of core glycosylated and cell surface pools. Mol. Cell. Biol. 2014, 34, 2554–2565. 10.1128/MCB.01042-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure M. L.; Barnes S.; Brodsky J. L.; Sorscher E. J. Trafficking and function of the cystic fibrosis transmembrane conductance regulator: a complex network of posttranslational modifications. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L719–L733. 10.1152/ajplung.00431.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff D. F.; Jui N. T.; Khurana V.; Tambe M. A.; Thompson M. L.; Chung C. Y.; Kamadurai H. B.; Kim H. T.; Lancaster A. K.; Caldwell K. A.; Caldwell G. A.; Rochet J. C.; Buchwald S. L.; Lindquist S. Yeast reveal a ″druggable″ Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 2013, 342, 979–983. 10.1126/science.1245321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryno L. M.; Wiseman R. L.; Kelly J. W. Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr. Opin. Chem. Biol. 2013, 17, 346–352. 10.1016/j.cbpa.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calamini B.; Silva M. C.; Madoux F.; Hutt D. M.; Khanna S.; Chalfant M. A.; Saldanha S. A.; Hodder P.; Tait B. D.; Garza D.; Balch W. E.; Morimoto R. I. Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2012, 8, 185–196. 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl F. U.; Bracher A.; Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Kieran D.; Kalmar B.; Dick J. R.; Riddoch-Contreras J.; Burnstock G.; Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004, 10, 402–405. 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Carstea E. D.; Morris J. A.; Coleman K. G.; Loftus S. K.; Zhang D.; Cummings C.; Gu J.; Rosenfeld M. A.; Pavan W. J.; Krizman D. B.; Nagle J.; Polymeropoulos M. H.; Sturley S. L.; Ioannou Y. A.; Higgins M. E.; Comly M.; Cooney A.; Brown A.; Kaneski C. R.; Blanchette-Mackie E. J.; Dwyer N. K.; Neufeld E. B.; Chang T. Y.; Liscum L.; Strauss J. F. 3rd; Ohno K.; Zeigler M.; Carmi R.; Sokol J.; Markie D.; O’Neill R. R.; van Diggelen O. P.; Elleder M.; Patterson M. C.; Brady R. O.; Vanier M. T.; Pentchev P. G.; Tagle D. A. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Kwon H. J.; Abi-Mosleh L.; Wang M. L.; Deisenhofer J.; Goldstein J. L.; Brown M. S.; Infante R. E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di X. J.; Han D. Y.; Wang Y. J.; Chance M. R.; Mu T. W. SAHA enhances Proteostasis of epilepsy-associated alpha1(A322D)beta2gamma2 GABA(A) receptors. Chem. Biol. 2013, 20, 1456–1468. 10.1016/j.chembiol.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D. Y.; Di X. J.; Fu Y. L.; Mu T. W. Combining valosin-containing protein (VCP) inhibition and suberanilohydroxamic acid (SAHA) treatment additively enhances the folding, trafficking, and function of epilepsy-associated gamma-aminobutyric acid, type A (GABAA) receptors. J. Biol. Chem. 2015, 290, 325–337. 10.1074/jbc.M114.580324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt D. M.; Herman D.; Rodrigues A. P.; Noel S.; Pilewski J. M.; Matteson J.; Hoch B.; Kellner W.; Kelly J. W.; Schmidt A.; Thomas P. J.; Matsumura Y.; Skach W. R.; Gentzsch M.; Riordan J. R.; Sorscher E. J.; Okiyoneda T.; Yates J. R. 3rd; Lukacs G. L.; Frizzell R. A.; Manning G.; Gottesfeld J. M.; Balch W. E. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010, 6, 25–33. 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joh N. H.; Wang T.; Bhate M. P.; Acharya R.; Wu Y.; Grabe M.; Hong M.; Grigoryan G.; DeGrado W. F. De novo design of a transmembrane Zn(2)(+)-transporting four-helix bundle. Science 2014, 346, 1520–1524. 10.1126/science.1261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Min D.; DiMaio F.; Wei K. Y.; Vahey M. D.; Boyken S. E.; Chen Z.; Fallas J. A.; Ueda G.; Sheffler W.; Mulligan V. K.; Xu W.; Bowie J. U.; Baker D. Accurate computational design of multipass transmembrane proteins. Science 2018, 359, 1042–1046. 10.1126/science.aaq1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt L. C.; Lewis J. S.; Andreev O. A.; Reshetnyak Y. K.; Engelman D. M. Applications of pHLIP technology for cancer imaging and therapy. Trends Biotechnol. 2017, 35, 653–664. 10.1016/j.tibtech.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Slusky J. S.; Berger B. W.; Walters R. S.; Vilaire G.; Litvinov R. I.; Lear J. D.; Caputo G. A.; Bennett J. S.; DeGrado W. F. Computational design of peptides that target transmembrane helices. Science 2007, 315, 1817–1822. 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- Slusky J. S. Outer membrane protein design. Curr. Opin. Struct. Biol. 2017, 45, 45–52. 10.1016/j.sbi.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman W. B.; Guha S.; Wimley W. C. Synthetic molecular evolution of hybrid cell penetrating peptides. Nat. Commun. 2018, 9, 2568. 10.1038/s41467-018-04874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Kim S. Y.; Pittman A. E.; King G. M.; Wimley W. C.; Hristova K. Potent Macromolecule-sized poration of lipid bilayers by the macrolittins, a synthetically evolved family of pore-forming peptides. J. Am. Chem. Soc. 2018, 140, 6441–6447. 10.1021/jacs.8b03026. [DOI] [PubMed] [Google Scholar]
- Vaidehi N.; Grisshammer R.; Tate C. G. How can mutations thermostabilize G-protein-coupled receptors?. Trends Pharmacol. Sci. 2016, 37, 37–46. 10.1016/j.tips.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Bowie J. U. Building a thermostable membrane protein. J. Biol. Chem. 2000, 275, 6975–6979. 10.1074/jbc.275.10.6975. [DOI] [PubMed] [Google Scholar]