

Abstract
RAS genotyping is mandatory to predict anti-EGFR monoclonal antibodies (mAbs) therapy resistance and BRAF genotyping is a relevant prognosis marker in patients with metastatic colorectal cancer. Although the role of hotspot mutations is well defined, the impact of uncommon mutations is still unknown. In this study, we aimed to discuss the potential utility of detecting uncommon RAS and BRAF mutation profiles with next-generation sequencing. A total of 779 FFPE samples from patients with metastatic colorectal cancer with valid NGS results were screened and 22 uncommon mutational profiles of KRAS, NRAS and BRAF genes were selected. In silico prediction of mutation impact was then assessed by 2 predictive scores and a structural protein modelling. Three samples carry a single KRAS non-hotspot mutation, one a single NRAS non-hotspot mutation, four a single BRAF non-hotspot mutation and fourteen carry several mutations. This in silico study shows that some non-hotspot RAS mutations seem to behave like hotspot mutations and warrant further examination to assess whether they should confer a resistance to anti-EGFR mAbs therapy for patients bearing these non-hotspot RAS mutations. For BRAF gene, non-V600E mutations may characterise a novel subtype of mCRC with better prognosis, potentially implying a modification of therapeutic strategy.
Subject terms: Colorectal cancer, Predictive markers
Introduction
Combination of targeted therapies like anti-EGFR monoclonal antibodies (anti-EGFR mAbs) with chemotherapy regimen (FOLFOX or FOLFIRI) improves progression-free (PFS) and overall survival (OS) in patients with metastatic colorectal cancer (mCRC)1. Tumor mutation hotspots associated with clinical resistance to anti-EGFR mAbs like KRAS exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), exon 4 (codons 117 and 146) and NRAS exon 2 (codons 12 and 13), exon 3 (codons 59 and 61) and exon 4 (codons 117 and 146) are now well identified and are systematically assessed prior to anti-EGFR mAbs prescription1–5. KRAS and NRAS are both Ras serine-threonine kinases, located downstream of EGFR in the Ras/Raf/MAPK pathway. Mutations in these codons cause constitutive activation of the RAS-MAPK pathway. KRAS and NRAS mutations are reported in 40–50% and 5–8% of patients with mCRC, respectively6.
Tumor KRAS and NRAS mutational statuses are usually assessed using polymerase chain reaction (PCR)-based assays designed for detection of major hotspot mutations. Most PCR-based assays, by their design, are not able to detect non-hotspot mutations; thus, patients with a tumor bearing a non-hotspot mutation are labeled as “wild-type” even though a tumor mutation exists. Next generation sequencing (NGS) assays allow the analysis of full exons and are able to detect uncommon mutational profiles. The impact of non-hotspot mutations of KRAS, NRAS and BRAF on anti-EGFR mAbs resistance is still unclear and it may be useful to study their impact in patients with mCRC.
BRAF is a serine-threonine kinase, located downstream of EGFR in the Ras/Raf/MAPK pathway7. BRAF mutations are reported in 5–10% of patients with mCRC3,6,8. p.(Val600Glu) (V600E) is the main BRAF hotspot mutation9. This mutation, located on exon 15 of the BRAF protein kinase activation domain leads to an increase of BRAF activity, 130 to 700 times higher than in wild-type (WT) BRAF10,11. In mCRC, this hotspot mutation is recognized as a poor prognosis factor9. Non-hotspot mutations have also an impact on BRAF protein activity, leading to high, intermediate or impaired kinase activity12,13.
The aim of this study was to identify isolated or concomitant non-hotspot RAS and BRAF mutations detected during routine sequencing by NGS and discuss their potential role in treatment resistance and prognosis for patients with mCRC using in silico prediction tools.
Results
We retrospectively collected data from 857 mCRC samples including 779 samples with valid NGS results. DNA quality was suitable for 91% of the samples for NGS and uncommon mutational profiles were reported in 22 (2.7% of total) samples.
The histological subtypes were: adenocarcinoma for 11 samples, liberkunhian adenocarcinoma for 9 samples, mucinous carcinoma for 1 sample, and ductal carcinoma for 1 sample. Twenty samples were from primary tumors and 2 were from liver metastases (Table 1).
Table 1.
Uncommon mutational profiles found in our study and tumor features.
| # | Primary tumor localization | Tumor lesion analyzed | Histological type | Gene | Exon | Nucleotidic variation | Protein variatio | COSMIC ID.a | Significance | Coverage | MAF (%)d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | Left colon junction | Primary | Lieberkuhnian adenocarcinoma | KRAS | 2 | c.34G > A | p.(Gly12Ser) | COSM517 | Missense | 2623 | 32.0 |
| NRAS | 3 | c.181C > A | p.(Gln61Lys) | COSM580 | Missense | 4133 | 14.4 | ||||
| #2 | Sigmoid Colon | Primary | Mucinous carcinoma | KRAS | 2 | c.34G > A | p.(Gly12Ser) | COSM517 | Missense | 3412 | 25.4 |
| NRAS | 2 | c.38G > T | p.(Gly13Val) | COSM574 | Missense | 3009 | 12.3 | ||||
| #3 | Right colon | Primary | Ductal carcinoma | KRAS | 2 | c.37G > T | p.(Gly13Cys) | COSM527 | Missense | 2694 | 57.9 |
| #4 | Sigmoid colon | Primary | Lieberkuhnian adenocarcinoma | KRAS | 2 | c.37G > T | p.(Gly13Cys) | COSM527 | Missense | 800 | 30.6 |
| #5 | Colon | Metastasis (liver) | Lieberkuhnian adenocarcinoma | KRAS | 2 | c.24A > G | p.(Val8Val) | COSM1360891 | Silent mutation | 1117 | 70.6 |
| #6 | Sigmoid colon | Primary | Adenocarcinoma | NRAS | 4 | c.360G > A | p.(Leu120Leu) | Not described* | Silent mutation | 907 | 40.6 |
| #7 | Right colon | Primary | Lieberkuhnian adenocarcinoma | KRAS | 4 | c.360G > A | p.(Leu120Leu) | Not described* | Silent mutation | 2688 | 48.0 |
| KRAS | 4 | c.353G > A | p.(Cys118Tyr) | Not described* | Missense | 2688 | 14.4 | ||||
| KRAS | 4 | c.418C > T | p.(Pro140Ser) | COSM4169136 | Missense | 2688 | 6.0 | ||||
| #8 | Right colon | Primary | Adenocarcinoma | KRAS | 4 | c.344G > A | p.(Gly115Glu) | Not described* | Missense | 1634 | 4.3 |
| NRAS | 2 | c.69A > G | p.(Leu23Leu) | rs771113899 # | Silent mutation | 1819 | 1.8 | ||||
| #9 | Sigmoid colon | Primary | Lieberkuhnian adenocarcinoma | KRAS | 3 | c.281G > A | p.(Arg68Arg) | Not described* | Silent mutation | 1849 | 25.0 |
| KRAS | 4 | c.394G > A | p.(Asp132Asn) | Not described* | Missense | 1836 | 4.0 | ||||
| NRAS | 2 | c.64C > T | p.(Gln22)* | Not described* | Stop mutation | 2913 | 8.0 | ||||
| #10 | NA | Metastasis (liver) | Adenocarcinoma | NRAS | 2 | c.99T > G | p.(Asp33Glu) | Not describedb | Missense | 8161 | 22.5 |
| KRAS | 2 | c.35G > T | p.(Gly12Val) | COSM520c | Missense | 2397 | 17.6 | ||||
| #11 | Rectum | Primary | Adenocarcinoma | BRAF | 15 | c.1742A > G | p.(Asn581Ser) | COSM462 | Missense | 4048 | 27.6 |
| NRAS | 2 | c.34G > T | p.(Gly12Cys) | COSM562c | Missense | 2398 | 15.7 | ||||
| #12 | Right colon | Primary | Lieberkuhnian adenocarcinoma | KRAS | 2 | c.40G > A | p.(Val14Ile) | COSM12722 | Missense | 2887 | 14.5 |
| BRAF | 15 | c.1805C > T | p.(Ser602Phe) | Not describedb | Missense | 5208 | 22.6 | ||||
| HRAS | 3 | c.217C > T | p.(Arg73Cys) | Not describedb | Missense | 2825 | 19 | ||||
| MET | 14 | c.3050A > C | p.(Glu1017Ala) | Not describedb | Missense | 11344 | 14.3 | ||||
| #13 | Colon | Primary | Lieberkuhnian adenocarcinoma | BRAF | 11 | c.1396G > A | p.(Gly466Arg) | COSM253328 | Missense | 5991 | 21.2 |
| #14 | Colon | Primary | Adenocarcinoma | BRAF | 15 | c.1781A > G | p.(Asp594Gly) | COSM467 | Missense | 11967 | 31.5 |
| #15 | Rectum | Primary | Lieberkuhnian adenocarcinoma | BRAF | 11 | c.1406G > T | p.(Gly469Val) | COSM469 | Missense | 6910 | 46.2 |
| #16 | Duodenum | Primary | Adenocarcinoma | BRAF | 15 | c.1780G > A | p.(Asp594Asn) | COSM27639 | Missense | 8067 | 21 |
| KRAS | 2 | c.38G > A | p.(Gly13Asp) | COSM532 | Missense | 3651 | 19.9 | ||||
| #17 | Caecum | Primary | Lieberkuhnian adenocarcinoma | BRAF | 11 | c.1406G > C | p.(Gly469Ala) | COSM460 | Missense | 5570 | 31.4 |
| KRAS | 2 | c.35G > A | p.(Gly12Asp) | COSM521 | Missense | 736 | 50.8 | ||||
| #18 | Rectum | Primary | Adenocarcinoma | BRAF | 11 | c.1397G > A | p.(Gly466Glu) | COSM453 | Missense | 12029 | 7.9 |
| KRAS | 2 | c.57G > T | p.(Leu19Phe) | COSM20818 | Missense | 12363 | 6.5 | ||||
| #19 | Colon | Primary | Adenocarcinoma | KRAS | 2 | c.38G > A | p.(Gly13Asp) | COSM532 | Missense | 2083 | 25.6 |
| KRAS | 4 | c.436G > A | p.(Ala146Thr) | COSM19404 | Missense | 5445 | 25.6 | ||||
| KRAS | 3 | c.264A > C | p.(Lys88Asn) | Not describedb | Missense | 6549 | 28.2 | ||||
| #20 | Rectum | Primary | Adenocarcinoma | BRAF | 11 | c.1406G > C | p.(Gly469Ala) | COSM460 | Missense | 20393 | 31.3 |
| KRAS | 4 | c.351A > T | p.(Lys117Asn) | COSM28519 | Missense | 27295 | 16.8 | ||||
| #21 | Rectosigmoid | Primary | Adenocarcinoma | BRAF | 15 | c.1801A > G | p.(Lys601Glu) | COSM478 | Missense | 9254 | 23.6 |
| #22 | Caecum | Primary | Adenocarcinoma | BRAF | 15 | c.1799T > A | p.(Val600Glu) | COSM476 | Missense | 12127 | 32.5 |
| KRAS | 2 | c.35G > T | p.(Gly12Val) | COSM520 | Missense | 9389 | 49.6 |
aAs described in the Catalogue of Somatic Mutations in Cancer (COSMIC), available online at.
bNot described as somatic nor as single nucleotide polymorphism (SNP) in databases.
cKRAS and NRAS hotspot mutation.
dMutant allele fraction (MAF).
The range of coverage for rare KRAS, NRAS or BRAF mutations was 700 to 27 000x. Mutant allele fraction was between 1.8% and 40.6% for non-hotspot NRAS mutations, between 4.0% and 14.5% for non-hotspot KRAS mutations and between 7.9% and 46.2% for non-hotspot BRAF mutations. Thirty-five mutations were missense mutations, 5 were silent mutations and one was a stop mutation.
All observed mutations are described in Table 1. Among 22 samples, 3 carried an isolated non-hotspot KRAS mutation (#3, #4, #5), 1 a NRAS mutation (#6) and 4 a BRAF non-hotspot mutation (#13, #14, #15, #21). Seven concomitant RAS mutations (#1, #2, #7, #8, #9, #10, #19) and 7 concomitant RAS and BRAF mutations (#11, #12, #16, #17, #18, #20, #22) were detected. One sample (#12) bore an HRAS mutation not previously described and no sample carried MAP2K1 mutations. Twelve of these mutations are not yet described as somatic nor as single nucleotide polymorphism (SNP) in public database.
For each mutation, the PolyPhen-2 score, SIFT score, their interpretation, and protein domain impacted are described in Table 2. All KRAS and NRAS mutations observed in this study are located in the catalytic domain. The majority of these mutations are localized in the GTP binding site. For the BRAF gene, all mutations described in our study are found in the protein kinase domain. Predictions of the impact of each mutation are given in Table 2.
Table 2.
SIFT, PolyPhen-2 score and protein localization of each identified mutation.
| # | Gene | Exon | Nucleotidic variation | Protein variation | COSMIC ID.a | Significance | SIFT Score | SIFT score interpretation Predicted to be | PolyPhen-2 score | PolyPhen-2 score interpretation Predicted to be | FoldX ∆∆G (kcal/mol)e | Protein localization |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | KRAS | 2 | c.34G > A | p.(Gly12Ser) | COSM517 | Missense | 0.988 | Tolerated | 0.644 | Possibly damaging | −0.085 | GTP binding site |
| NRAS | 3 | c.181C > A | p.(Gln61Lys) | COSM580 | Missense | 0.991 | Tolerated | 0.76 | Possibly damaging | −0.159 | GTP binding site | |
| #2 | KRAS | 2 | c.34G > A | p.(Gly12Ser) | COSM517 | Missense | 0.988 | Tolerated | 0.644 | Possibly damaging | −0.085 | GTP binding site |
| NRAS | 2 | c.38G > T | p.(Gly13Val) | COSM574 | Missense | 0.975 | Tolerated | 0.975 | Damaging | 4.662 | GTP binding site | |
| #3 | KRAS | 2 | c.37G > T | p.(Gly13Cys) | COSM527 | Missense | 1 | Tolerated | 0.997 | Damaging | 2.540 | GTP binding site |
| #4 | KRAS | 2 | c.37G > T | p.(Gly13Cys) | COSM527 | Missense | 1 | Tolerated | 0.997 | Damaging | 2.540 | GTP binding site |
| #5 | KRAS | 2 | c.24A > G | p.(Val8Val) | COSM1360891 | Silent mutation | NA | NA | NA | NA | NA | Catalytic domain |
| #6 | NRAS | 4 | c.360G > A | p.(Leu120Leu) | Not described* | Silent mutation | NA | NA | NA | NA | NA | Catalytic domain |
| #7 | KRAS | 4 | c.360G > A | p.(Leu120Leu) | Not described* | Silent mutation | NA | NA | NA | NA | NA | Catalytic domain |
| KRAS | 4 | c.353G > A | p.(Cys118Tyr) | Not described* | Missense | 0.988 | Tolerated | 0.047 | Benign | 0.516 | GTP binding site | |
| KRAS | 4 | c.418C > T | p.(Pro140Ser) | COSM4169136 | Missense | NA | NA | NA | NA | 2.970 | Catalytic domain | |
| #8 | KRAS | 4 | c.344G > A | p.(Gly115Glu) | Not described* | Missense | 1 | Tolerated | 0.998 | Damaging | 6.967 | GTP binding site |
| NRAS | 2 | c.69A > G | p.(Leu23Leu) | rs771113899 # | Silent mutation | NA | NA | NA | NA | NA | ||
| #9 | KRAS | 3 | c.281G > A | p.(Arg68Arg) | Not described* | Silent mutation | NA | NA | NA | NA | NA | Catalytic domain |
| KRAS | 4 | c.394G > A | p.(Asp132Asn) | Not described* | Missense | 0.77 | Tolerated | 0.004 | Benign | 0.202 | Catalytic domain | |
| NRAS | 2 | c.64C > T | p.(Gln22)* | Not described* | Stop mutation | NA | NA | NA | NA | NA | Catalytic domain | |
| #10 | NRAS | 2 | c.99T > G | p.(Asp33Glu) | Not describedb | Missense | 0.939 | Tolerated | 0.952 | Damaging | 0.196 | Effector binding site |
| KRAS | 2 | c.35G > T | p.(Gly12Val) | COSM520c | Missense | 0.993 | Tolerated | 0.978 | Damaging | −0.451 | GTP binding site | |
| #11 | BRAF | 15 | c.1742A > G | p.(Asn581Ser) | COSM462 | Missense | 0.954 | Tolerated | 0.998 | Damaging | 0.595 | Protein kinase domain |
| NRAS | 2 | c.34G > T | p.(Gly12Cys) | COSM562c | Missense | 0.935 | Tolerated | 0.605 | Possibly damaging | −0.209 | GTP binding site | |
| #12 | KRAS | 2 | c.40G > A | p.(Val14Ile) | COSM12722 | Missense | 0.999 | Tolerated | 0.968 | Damaging | 1.971 | GTP binding site |
| BRAF | 15 | c.1805C > T | p.(Ser602Phe) | Not described | Missense | 0.999 | Tolerated | 0.916 | Damaging | −1.341 | Protein kinase domain | |
| HRAS | 3 | c.217C > T | p.(Arg73Cys) | Not described | Missense | 1 | Tolerated | 0.997 | Damaging | NAf | Catalytic domain | |
| MET | 14 | c.3050A > C | p.(Glu1017Ala) | Not described | Missense | 0.889 | Tolerated | 0.742 | Possibly damaging | NAf | ||
| #13 | BRAF | 11 | c.1396G > A | p.(Gly466Arg) | COSM253328 | Missense | 1 | Tolerated | 0.969 | Damaging | 3.722 | ATP binding site |
| #14 | BRAF | 15 | c.1781A > G | p.(Asp594Gly) | COSM467 | Missense | 0.998 | Tolerated | 0.983 | Damaging | 1.560 | Protein kinase domain |
| #15 | BRAF | 11 | c.1406G > T | p.(Gly469Val) | COSM469 | Missense | 1 | Tolerated | 0.999 | Damaging | −3.553 | ATP binding site |
| #16 | BRAF | 15 | c.1780G > A | p.(Asp594Asn) | COSM27639 | Missense | 0.999 | Tolerated | 0.998 | Damaging | −0.876 | Protein kinase domain |
| KRAS | 2 | c.38G > A | p.(Gly13Asp) | COSM532 | Missense | 0.988 | Tolerated | 0.506 | Possibly damaging | 3.455 | GTP binding site | |
| #17 | BRAF | 11 | c.1406G > C | p.(Gly469Ala) | COSM460 | Missense | 1 | Tolerated | 0.83 | Possibly damagin | −1.59 | ATP binding site |
| KRAS | 2 | c.35G > A | p.(Gly12Asp) | COSM521 | Missense | 0.99 | Tolerated | 0.361 | Possibly damaging | −0.443 | GTP binding site | |
| #18 | BRAF | 11 | c.1397G > A | p.(Gly466Glu) | COSM453 | Missense | 1 | Tolerated | 0.969 | Damaging | 3.938 | ATP binding site |
| KRAS | 2 | c.57G > T | p.(Leu19Phe) | COSM20818 | Missense | 0.999 | Tolerated | 0.999 | Damaging | 5.653 | Catalytic domain | |
| #19 | KRAS | 2 | c.38G > A | p.(Gly13Asp) | COSM532 | Missense | 0.988 | Tolerated | 0.506 | Possibly damaging | 3.455 | GTP binding site |
| KRAS | 4 | c.436G > A | p.(Ala146Thr) | COSM19404 | Missense | 0.993 | Tolerated | 0.987 | Damaging | 4.757 | GTP binding site | |
| KRAS | 3 | c.264A > C | p.(Lys88Asn) | Not describedb | Tolerated | 0.937 | Tolerated | 0.094 | Benign | 0.183 | Catalytic domain | |
| #20 | BRAF | 11 | c.1406G > C | p.(Gly469Ala) | COSM460 | Tolerated | 1.0 | Tolerated | 0.835 | Possibly damaging | −1.595 | ATP binding site |
| KRAS | 4 | c.351A > T | p.(Lys117Asn) | COSM28519 | Tolerated | 0.989 | Tolerated | 0.998 | Damaging | 0.317 | GTP binding site | |
| #21 | BRAF | 15 | c.1801A > G | p.(Lys601Glu) | COSM478 | Missense | 1.0 | Tolerated | 0.626 | Damaging | −0.276 | Protein kinase domain |
| #22 | BRAF | 15 | c.1799T > A | p.(Val600Glu) | COSM476 | Missense | 0.999 | Tolerated | 0.943 | Damaging | 0.930 | Protein kinase domain |
| KRAS | 2 | c.35G > T | p.(Gly12Val) | COSM520 | Missense | 0.993 | Tolerated | 0.978 | Damaging | −0.451 | GTP binding site |
aAs described in the Catalogue of Somatic Mutations in Cancer (COSMIC), available online at.
bNot described as somatic nor as single nucleotide polymorphism (SNP) in databases.
cKRAS and NRAS hotspot mutation.
dMutant allele fraction (MAF).
eImpacts of observed mutations on protein stability, as predicted by FoldX. Negative values of ∆∆G indicate that a mutation stabilizes the protein structure relative to the WT, and positive values indicate destabilization.
fHRAS and MET mutations were not modelled.
Discussion
In this study, we assessed the value of NGS-based testing to identify uncommon KRAS, NRAS and BRAF mutational profiles associated to in silico projection to evaluate their therapeutic and clinical implications in 779 samples of patients with mCRC. We identified 22 uncommon mutational profiles with a few missense variants that have not been previously reported in the literature, to our knowledge. Structural modelling of the observed missense variants in BRAF, KRAS and NRAS shows that most of the observed mutations can be accommodated in the protein structures without clearly adverse impacts on protein stability (Table 2). In a few cases, FoldX predicts the observed mutation to be highly destabilizing, primarily due to the introduction of interatomic clashes (e.g. KRAS Gly115Glu). While some such mutations may indeed lead to destabilization and inhibition of protein folding, it should be noted that the modelling procedure in FoldX does not consider backbone conformational changes. It is possible that some such mutations could be accommodated in the protein structure once backbone conformational changes are considered; an example is the RAS Gln61Leu mutation (not observed in this study)14. Despite this limitation, FoldX provides a quick, relatively accurate and parsimonious means to evaluate the impacts of mutations on protein stability.
We now briefly describe the results of modelling the previously unreported mutations. Modelling the Gly115Glu mutation in KRAS (Fig. 1) suggests that this mutation has an adverse impact on protein stability. The three point mutations in KRAS that have not been previously reported (Lys88Asn, Cys118Tyr and Asp132Asn) are located on the protein surface and lead to minimal impacts on stability when KRAS is considered in isolation. The same is true of the NRAS Asp33Glu mutation, although this is located in the Switch 1 loop and is not directly involved in substrate binding. Examination of structures of RAS proteins in complex with RASGAP and SOS (PDB identifiers 1WQ1 and 1XD2, respectively) showed that most of these residues in KRAS and NRAS were not directly involved in the interaction with RASGAP and SOS. The exception is NRAS Asp33Glu, which may affect the interaction with SOS (Fig. 2)15. The BRAF Ser602Phe mutation leads to moderate stabilization relative to WT BRAF. This residue is located on the surface of the protein and is not in the BRAF dimer interface.
Figure 1.
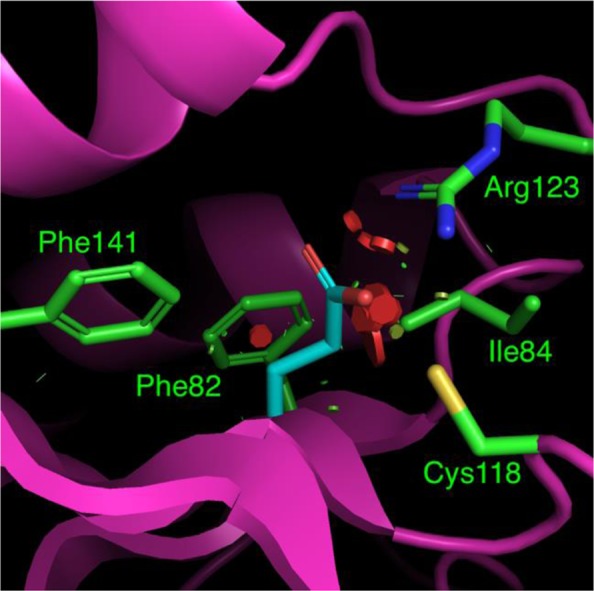
Structural model of the KRAS Gly115Glu variant, with key residue sidechains depicted as sticks. The Glu residue in position 115 is shown in cyan, along with neighboring residues. For clarity, only residues whose atoms contact Glu115 are shown. The mutation to Glu at this position causes severe atomic clashes, primarily with residues Ile84 and Arg123, although there is potential for hydrogen bond formation with Arg123. Clashes are indicated by colored discs drawn between atoms. The color and size of the disc reflects the severity of the clash, with wider, redder discs indicating the most severe clashes.
Figure 2.

Close-up view of a section of the interface between Ras (green) and SOS (magenta) at the “catalytic” Ras-binding site of SOS26, with Ras residue 33 shown in cyan. Hydrogen bonds are calculated using PyMOL and shown as dashed yellow lines. The left panel shows the wild-type Asp33 residue and the right panel shows the model of Asp33Glu. The mutation causes a change in the hydrogen bonding pattern at this site. Modelling this variant at the “distal” Ras-binding site of SOS shows a similar pattern of change in the hydrogen bonding pattern. No significant interatomic clashes are created.
BRAF non-V600E mutations were detected in 1.43% of patients tested, which is consistent with previous published studies16. The predictive value of these mutations in the context of mCRC is indeterminate. However, exon 11 codon 469 BRAF mutations are located on the gene region coding for protein kinase function11. For exon 11 codon 466 mutations, the amino acid change is located within the glycine-rich loop in the kinase domain17. For samples #14 and #16, BRAF exon 15 codon 594 mutations are located on the gene region coding for protein kinase function and lead to impaired kinase activity, potentially conferring a favorable prognosis18,19. BRAF p.(Lys601Glu) (sample #21) is described as pathogenic but without published data on response to anti-EGFR mAbs therapy.
We now turn our attention to the 14 mutational profiles in our dataset with concomitant RAS and/or BRAF mutations. KRAS and BRAF mutations have been frequently described as mutually exclusive in CRC and concomitant KRAS and BRAF mutations are rare, occurring in less than 0.001% of cases20. In our study, in 10 samples with uncommon BRAF mutations, 6 are concomitant with a RAS mutation, and 1 BRAF hotspot mutation had a concomitant RAS mutation. The percentage of concomitant KRAS and BRAF mutations in our dataset is higher than previously described21–27. Since these studies only assessed BRAF codon 600 hotspot mutations, we may infer that hotspot KRAS mutations are more frequently associated with rare BRAF mutations than with hotspot mutations. This inference agrees with a previous study which found that patients with BRAF non-V600E mutations were more likely to have concomitant RAS mutations than patients with the BRAF V600E mutation16. Due to the infrequent observation of this phenomenon, it is actually not clear whether or not these doubly mutated tumors have a different biology and natural history than KRAS or BRAF mutant tumors. We should illustrate the potential significance of this concomitant mutation on sample #12. Sample #12 bears a KRAS p.(Val14Ile) mutation described as pathogenic. Whether resistance to anti-EGFR mAbs is conferred by this mutation is unknown, however codon 14 belongs to the same domain as codons 12 and 13. As with mutations on codons 12 and 13, this mutation may be associated with a clinical resistance to anti-EGFR antibodies2. In this case, the knowledge of this rare mutation may be significant in guiding a therapeutic decision and to explain potential resistance to anti-EGFR antibodies. However, the presence of a BRAF non-V600 mutation could modify response to therapy and may even lead to potential treatment resistance. Even if first observations show that BRAF plays only a slight role in resistance to anti-EGFR mAbs, some BRAF non-hotspot mutations might contribute to reduced efficacy of anti-EGFR mAbs18,28.
In silico prediction of the functional effects of the observed mutations using both sequence- and structure-based approaches suggests possible biochemical mechanisms related to uncommon mutational profile and relevant in cancer. These in silico results warrant further in vivo examination to assess the relevance of detection of non-hotspot RAS mutations and their implication in resistance to anti-EGFR mAbs therapy. For the BRAF gene, non-V600E mutations may describe a novel subtype of mCRC with better prognosis, implying potentially different treatment management strategies.
Methods
Samples
Data from 188 formalin-fixed paraffin embedded (FFPE) samples of histologically proven colorectal cancer tumor tissue previously published were pooled with new data from 669 FFPE samples from patients with mCRC routinely analyzed for KRAS, NRAS and BRAF mutations from May 2017 to May 2018 in Institut de Cancérologie de Lorraine (France)14. All samples were FFPE tissues from mCRC primary tumor or metastases. Determination of percentage of tumor tissue content and area for macrodissection were based on examination of hematoxylin-eosin stained sections by a senior pathologist. All patients involved in this study gave their informed consent for the research of KRAS, NRAS and BRAF mutation. The experimental protocols of these study have been approved by the ethical and scientific board of Institut de Cancérologie de Lorraine. All methods were performed in accordance with the relevant guidelines and regulations. All data were anonymized prior to analysis.
DNA extraction and quality assessment
For all samples, DNA was extracted as previously described using QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany)14. After extraction of the 669 new samples, TruSeq FFPE DNA Library Prep QC kit (Illumina, San Diego, USA) and qPCR using Cobas z480 (Roche Diagnostics, Meylan, France) were used for quality DNA assessment. Cycle quantification (Cq) values were calculated using LightCycler® 480 Software W UDF 2.0.0 (Roche Diagnostics). For samples showing a ΔQC score lower than 6, DNA libraries were then prepared using TruSeq® Custom Amplicon Library Preparation Kit v1.5 (Illumina). Fifty-three samples failed to yield sufficient DNA quality and 616 samples qualified for DNA library preparation.
DNA library preparation and sequencing
For the first 188 samples, library preparation and DNA sequencing were performed using the GS Junior deep pyrosequencing system as previously described14. Library preparation was not possible for 11 samples.
For the 616 new samples, libraries were prepared using the TruSeq® Custom Amplicon Library Preparation Kit v1.5. This kit consists of two separate oligo pools (CATA and CATB) and allow the full exon analysis of 16 genes: AKT1 (exon3), ALK (exons 23 to 25), BRAF (exons 11 and 15), EGFR (exons 18 to 21), ERBB2 (exon 20), ERBB4 (exons 10 and 12), FGFR2 (exons 7, 12 and 14), FGFR3 (exons 7, 9 and 14), HRAS (exons 2, 3 and 4), KIT (exons 8, 9, 11, 13, 17 and 18), KRAS (exons 2, 3 and 4), MAP2K1 (exon2), MET (exons 2 and 14 to 20), NRAS (exons 2, 3 and 4), PDGFRA (exons 12, 14 and 18) and PIK3CA (exons 10 and 21). The two oligo pools were hybridized to DNA samples. The specific hybridized targets were ligated, extended and PCR amplified with adaptors containing index with specific barcode sequences. Two complementary libraries were generated by targeting the forward and reverse DNA strands. The PCR-amplified amplicon libraries obtained were then purified using AMPure XP beads in order to remove non-specific products and reaction components.
Library DNA concentrations were quantified using Qubit 3.0 Fluorometer (ThermoFisher Scientific Inc, Massachusetts, USA) and their quality was assessed on Fragment Analyzer (Advanced Analytical, Ankeny, USA) using the Standard Sensitivity NGS Fragment Analysis Kit (Advanced Analytical). PCR product sizes have to be around 260 base pairs in length. All 616 validated libraries were normalized to enable similar amplification and sequencing levels for each sample library in the same run. Sequencing was performed according to the manufacturer’s instructions. All libraries were pooled before sequencing on the MiSeq instrument (Illumina). Sequencing data analysis was performed on Sophia DDM® software (Sophia genetics, Saint Sulpice, Switzerland). Reference sequences NM_033360.2 for KRAS, NM_002525.4 for NRAS and NM_004333.5 for BRAF were used for alignment and variant calling. Fourteen samples had insufficient coverage to be interpretable and 602 samples had interpretable sequencing data.
Uncommon mutational profiles
Uncommon mutational profiles were defined as i) a concomitant KRAS and NRAS hotspot mutations, or ii) a KRAS, NRAF or BRAF non-hotspot mutation (associated or not associated with other mutations). A threshold of 1% allele frequency has been reported clinically relevant for KRAS mutations linked with lack of response to anti-EGFR therapy29.
For samples with an uncommon mutational profile, mutational status of MAP2K1 and HRAS genes (available in our gene panel and implicated in the MAPkinase pathway) have been also identified.
In silico prediction of mutation impact
We used PolyPhen-2 (Polymorphism Phenotyping) and SIFT (Sorting Intolerant from Tolerant) scores to predict mutation impact on the protein, as well as FoldX to model the observed variants in protein structures30–34. PolyPhen-2 score predicts the possible impact of an amino acid substitution on the structure and function of a human protein. PolyPhen combines amino acid composition analysis in multiple sequence alignments with information from solved protein structures (where available). Sequence composition is evaluated using the Position Specific Independent Counts (PSIC) tool, which calculates sequence profiles35. Differences in the profiles calculated by PSIC for the different variants at a site are indicative of damaging impact. In cases where structural data are available, the impact of a mutation is assessed by considering physicochemical properties such as residue size and hydrophobicity, and the maintenance of contacts with ligands, metals or other interacting proteins. Sequence- and structure-based features are combined to produce predictions using a naïve Bayes classifier. PolyPhen-2 produces scores between 0 and 1 along with annotations of whether the mutation is predicted to be benign or damaging.
SIFT also predicts whether an amino acid substitution affects protein function. SIFT scores are used to predict the damaging effect of nucleotide substitutions and frame shifts (insertions/deletions) on protein function based on the maintenance of amino acid composition in alignments of the target sequence with closely related sequences. The SIFT server assigns scores for each residue from 0 to 1, where mutations with a score of ≤0.05 are predicted to not be tolerated, and mutations with score >0.05 are predicted to be tolerated36.
SIFT and PolyPhen-2 scores were determined using Sophia DDM® software (version 5.0.7). Structural modelling of observed missense mutations was carried out using FoldX version 433,34. FoldX estimates the impact of point mutations on the folding energy or stability of the protein using rigid-backbone modelling and a classical forcefield whose parameters are trained to reproduce experimental observations of mutational impacts on folding energy. Starting from the wildtype structures for KRAS, BRAF and NRAS (PDB identifiers 4OBE, 5VAM and 5UHV, respectively), the missense mutations observed in these proteins were modeled using the FoldX BuildModel tool, and the structural and energetic impacts of each mutation were assessed as the predicted change in folding energy (∆∆G). Synonymous (silent) and stop mutations were not modeled. All FoldX modelling results are available at 10.5281/zenodo.1467311.
Protein domain impacted
KRAS and NRAS protein domains impacted by the different mutations described were identified using UCSF Chimera37. The COSMIC database was used to identify protein domains impacted by BRAF mutations38.
Acknowledgements
No specific funding was received for this work.
Author Contributions
C. Franczak and S.M. Kandathil collected the data and wrote the paper, S.M. Kandathil conducted structural modelling. P. Gilson, M. Husson, M. Rouyer and J. Demange analyzed the samples and data. A. Leroux qualified and validated the samples. J.L. Merlin and A. Harlé analyzed the data, corrected the paper and gave expert guidance.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article. The FoldX modelling data that support the findings of this study are available at 10.5281/zenodo.1467311.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lievre A, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 2.Van Cutsem E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 3.Douillard JY, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 4.Amado RG, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 5.Taniguchi H, et al. Japanese Society of Medical Oncology Clinical Guidelines: RAS (KRAS/NRAS) mutation testing in colorectal cancer patients. Cancer Sci. 2015;106:324–327. doi: 10.1111/cas.12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50:307–12. doi: 10.1002/gcc.20854. [DOI] [PubMed] [Google Scholar]
- 7.Roskoski R. RAF protein-serine/threonine kinases: Structure and regulation. Biochem Biophys Res Commun. 2010;399:313–317. doi: 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- 8.Kawazoe A, et al. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer. 2015;15:258. doi: 10.1186/s12885-015-1276-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 10.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 11.Andreadi C, et al. Regulation of MEK/ERK pathway output by subcellular localization of B-Raf. Biochem Soc Trans. 2012;40:67–72. doi: 10.1042/BST20110621. [DOI] [PubMed] [Google Scholar]
- 12.Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 13.Shinozaki E, et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: the Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br J Cancer. 2017;117:1450–1458. doi: 10.1038/bjc.2017.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buhrman G, Kumar VSS, Cirit M, Haugh JM, Mattos C. Allosteric Modulation of Ras-GTP Is Linked to Signal Transduction through RAF Kinase. J Biol Chem. 2011;286:3323–3331. doi: 10.1074/jbc.M110.193854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sondermann H, et al. Structural Analysis of Autoinhibition in the Ras Activator Son of Sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Jones JC, et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J Clin Oncol. 2017;35:2624–2630. doi: 10.1200/JCO.2016.71.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar R, Angelini S, Hemminki K. Activating BRAF and N-Ras mutations in sporadic primary melanomas: an inverse association with allelic loss on chromosome 9. Oncogene. 2003;22:9217–24. doi: 10.1038/sj.onc.1206909. [DOI] [PubMed] [Google Scholar]
- 18.Harlé A, et al. Rare RAS Mutations in Metastatic Colorectal Cancer Detected During Routine RAS Genotyping Using Next Generation Sequencing. Target Oncol. 2016;11:363–70. doi: 10.1007/s11523-015-0404-7. [DOI] [PubMed] [Google Scholar]
- 19.Cremolini C, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015;26:2092–7. doi: 10.1093/annonc/mdv290. [DOI] [PubMed] [Google Scholar]
- 20.Sahin IH, et al. Rare though not mutually exclusive: a report of three cases of concomitant kras and braf mutation and a review of the literature. Journal of Cancer. 2013;4:320–322. doi: 10.7150/jca.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Cutsem E, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as firstline treatment for metastaticcolorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAFmutation status. J Clin Oncol. 2011;29:2011–9. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 22.Lamy A, et al. Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls. Mod Pathol. 2011;24:1090–100. doi: 10.1038/modpathol.2011.60. [DOI] [PubMed] [Google Scholar]
- 23.Price TJ, et al. Impact of KRAS and BRAF Gene Mutation Status on Outcomes From the Phase III AGITG MAXTrial of Capecitabine Alone or in Combination With Bevacizumab and Mitomycin in AdvancedColorectal Cancer. J Clin Oncol. 2011;29:2675–82. doi: 10.1200/JCO.2010.34.5520. [DOI] [PubMed] [Google Scholar]
- 24.Maughan TS, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–14. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li HT, Lu YY, An YX, Wang X, Zhao QC. KRAS, BRAF and PIK3CA mutations in human colorectal cancer: relationship with metastatic colorectal cancer. Oncology reports. 2011;25:1691–7. doi: 10.3892/or.2011.1217. [DOI] [PubMed] [Google Scholar]
- 26.Bokemeyer C, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Annals of oncology. 2011;22:1535–46. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 27.De Roock W, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 28.Rizzo S, et al. Prognostic vs predictive molecular biomarkers in colorectal cancer: is KRAS and BRAF wild type status required for anti-EGFR therapy? Cancer Treat Rev. 2010;36:S56–61. doi: 10.1016/S0305-7372(10)70021-9. [DOI] [PubMed] [Google Scholar]
- 29.Laurent-Puig P, et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin Cancer Res. 2015;21:1087–1097. doi: 10.1158/1078-0432.CCR-14-0983. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 7, Unit 7 20 (2013). [DOI] [PMC free article] [PubMed]
- 32.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 33.Schymkowitz J, et al. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–87. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 35.Sunyaev SR, et al. PSIC: Profile extraction from sequence alignments with positionspecific counts of independent observations. Protein Eng. 1999;12:387–394. doi: 10.1093/protein/12.5.387. [DOI] [PubMed] [Google Scholar]
- 36.Hassan, M. M. et al. Bioinformatics Approach for Prediction of Functional Coding/Noncoding Simple Polymorphisms (SNPs/Indels) in Human BRAF Gene. Adv Bioinformatics. 2632917 (2016). [DOI] [PMC free article] [PubMed]
- 37.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 38.BRAF protein conformation. Available at, https://cancer.sanger.ac.uk/cosmic3d/protein/BRAF (accessed on 22/07/2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article. The FoldX modelling data that support the findings of this study are available at 10.5281/zenodo.1467311.