

Abstract
Mitochondrial complex V (CV) generates cellular energy as adenosine triphosphate (ATP). Mitochondrial disease caused by the m.8993T>G pathogenic variant in CV subunit gene, MT-ATP6, was among the first described human mitochondrial DNA (mtDNA) diseases. Due to a lack of clinically-available functional assays, validating the definitive pathogenicity of additional MT-ATP6 variants remains challenging. We reviewed all reported MT-ATP6 disease cases (n=218) to date, to assess for MT-ATP6 variants, heteroplasmy levels, and inheritance correlation with clinical presentation and biochemical findings. We further describe the clinical and biochemical features of a new cohort of 14 kindreds with MT-ATP6 variants. Despite extensive overlap in the heteroplasmy levels of MT-ATP6 variant carriers with and without a wide range of clinical symptoms, previously reported symptomatic subjects had significantly higher heteroplasmy load (p=1.6×10−39). Pathogenic MT-ATP6 variants resulted in diverse biochemical features. The most common findings were reduced ATP synthesis rate, preserved ATP hydrolysis capacity, and abnormally increased mitochondrial membrane potential. However, no single biochemical feature was universally observed. Extensive heterogeneity exists among both clinical and biochemical features of distinct MT-ATP6 variants. Improved mechanistic understanding and development of consistent biochemical diagnostic analyses are needed to permit accurate pathogenicity assessment of variants of uncertain significance in MT-ATP6.
Keywords: Mitochondria, genotype-phenotype correlation, Leigh Syndrome, NARP, neurogenic ataxia and retinitis pigmentosa, heteroplasmy
INTRODUCTION
The core function of mitochondria is to generate cellular energy in the chemical form of phosphate bonds known as adenosine triphosphate (ATP). This process is dependent on the activity of complex V (CV, ATP synthase), a bioenergetic pump located within the mitochondrial inner membrane that enzymatically converts adenosine diphosphate (ADP) and inorganic phosphate (Pi) to form ATP. This process is driven by CV releasing the mitochondrial membrane potential difference across the inner mitochondrial membrane that is created by proton pumping coupled to electron transport in complexes I through IV. CV is composed of 15 structural and 2 assembly subunits, encoded by both the mitochondrial DNA (ATP6, ATP8) and nuclear genome (15 subunits). Human mitochondrial disease has been associated to date with mutations in 6 of the 17 CV subunits. The most common gene in which mutations cause CV deficiency is MT-ATP6, which encodes the CV “a” subunit that contains the proton pore that releases the proton gradient established across the inner mitochondrial membrane. Additional pathogenic variants have further been reported in MT-ATP8 encoding subunit A6L (A I Jonckheere et al., 2007), as well as nuclear genes ATP5A1 encoding subunit alpha (An I Jonckheere et al., 2013), ATP5E encoding subunit epsilon (Mayr et al., 2010), and the CV assembly factors ATPAF2 (De Meirleir, 2004) and TMEM70 (Cízková et al., 2008).
The MT-ATP6 pathogenic variant, m.8993T>G, was one of the first discovered mtDNA diseases three decades ago (Holt, Harding, Petty, & Morgan-Hughes, 1990), and subsequently has been reported in over 100 patients. Affected patients have a variable and often severe multi-system disease that can variably manifest as Leigh syndrome, stroke, cardiomyopathy, or “NARP” (neuropathy, ataxia and retinitis pigmentosa) syndrome. Despite its frequency, there has been little systematic exploration of the clinical presentation of different MT-ATP6 variants. In addition to extensive symptom variability, CV deficiency has been reported with extensively varied biochemical findings. Biochemical understanding of different variants has been limited by the absence of a CLIA-approved functional assay. This deficiency has further contributed to the challenge of determining accurate pathogenicity assertions for the large number of variants of uncertain significance (VUS) being identified in MT-ATP6.
Here, we review all reported MT-ATP6 pathogenic variants in the literature in light of their associated clinical phenotypes and present a new clinical case series of 14 additional MT-ATP6 kindreds. Results are also reviewed of reported biochemical testing performed for each MT-ATP6 variant, including ATP level, ATP synthetic rate, ATP hydrolytic rate, mitochondrial membrane potential, and function of the other complexes of the electron transport chain (ETC).
PREVIOUSLY REPORTED GENOTYPES AND PHENOTYPES
Over 200 patients have been reported with mitochondrial disease due to pathogenic variants in MT-ATP6 (JEŠINA et al., 2004; Schon, Santra, Pallotti, & Girvin, 2001). Just 4 point mutations comprise over 82% of reported disease (Childs et al., 2007; Mäkelä-Bengs et al., 1995; Morava et al., 2006; Pfeffer et al., 2012; Pitceathly et al., 2012a; Uziel et al., 1997; Verny et al., 2011). In the remaining subset of patients, an additional 15 pathogenic variants have been reported, many of which have been described in only a single kindred (Abu-Amero & Bosley, 2005; Alila-Fersi et al., 2017; Aure et al., 2013; Duno et al., 2013; Hao, Liu, Wu, Hao, & Chen, 2015; JEŠINA et al., 2004; Lopez-Gallardo et al., 2014; López-Gallardo et al., 2009; Sikorska et al., 2009). In the absence of a large affected pedigree, proving causality of an mtDNA variant in a single pedigree can be difficult. The traditional techniques of proving pathogenicity for mtDNA variants are: (1) finding biochemical alterations that correlate with the mutation, (2) identifying the mtDNA variant to be present in symptomatic patients in a heteroplasmic state rather than homoplasmy that would be suggestive of a fixed haplogroup lineage marker, and (3) mtDNA variant heteroplasmy level in the affected patient being higher than in asymptomatic relatives. However, each of these approaches may be particularly problematic in the specific case of MT-ATP6 variants. Even definite pathogenic MT-ATP6 variants, as established by their recurrence in multiple kindreds, may have standard biochemical findings that can be subtle or inconsistent (Table 1). Further, due to rapid heteroplasmy shifts that may occur in the level of MT-ATP6 mutation load, pathogenic variants may appear to be homoplasmic. Conversely, the heteroplasmy threshold for MT-ATP6 variant -- that is the point at which heteroplasmic mutations cause clinical symptoms -- seems to be quite high (Table 1). In addition, carrier patients may express symptoms which can be rather subtle. As a result, apparently unaffected relatives may have MT-ATP6 variant heteroplasmy levels that are high as that seen in clinically affected members of the same family (Campos et al., 1997; de Vries, van Engelen, Gabreëls, Ruitenbeek, & van Oost, 1993; Lopez-Gallardo et al., 2014; Moslemi, Darin, Tulinius, Oldfors, & Holme, 2005; Pitceathly et al., 2012a). For all of these reasons, the mainstay approach to determining MT-ATP6 variant pathogenicity has been the identification of a variant in multiple unrelated affected patients, thereby providing a challenge to evaluate novel MT-ATP6 variants.
Table 1.
MT-ATP6 mutation subjects’ reported biochemical abnormalities and previously proposed mechanisms of disease.
| Biochemical anomalies | Proposed mechanism | |
|---|---|---|
| m.8528T>C | Decreased ATP synthesis in response to substrates (1/1) | |
| m.8618insT | Decreased CV holoenzyme assembly (1/1), decreased ATPase activity (1/1) | Decreased subunit a production, which is quickly degraded |
| m.8691A>G | Decreased CV enzymology (no details) (1/1) | |
| m.8839G>C | Decreased mitochondrial membrane potential (1/1), normal ATP synthesis (1/1) | Decreased coupling of proton flow with ATP production |
| m.8851T>C | Mildly decrease holoenzyme assembly (1/1) | |
| m.8950G>A | Decreased mitochondrial respiratory activity (1/1) | |
| m.8969G>A | Decreased oligomycin sensitivity (1/1), higher ROS (1/1), decreased uncoupled rate (2/2), reduced ATP synthesis (1/1), slightly reduced ATP hydrolysis (1/1), increased mitochondrial membrane potential (1/1), no proton pumping (1/1), slightly decreased holoenzyme assembly (1/1), decreased oligomycin sensitive respiration (1/1), decreased basal rate (1/1) | Abrogation of the proton channel |
| m.8989G>C | Decreased CV enzymology (no details) (1/1) | |
| m.8993T>G | Decreased ATP synthesis (36/38), increased ATP synthesis (2/34), increased ADP (2/2), normal
proton translocation (6/7) & ATP hydrolysis (19/22). Decreased holocomplex V assembly (14/18), unstable holocomplex V
(1/2) Lowered ATP/2e- ratio (1/1), decreased sensitivity to oligomycin (6/8), increased sensitivity to oligomycin (5/12), abnormal other ETC activity (7/7), increased mitochondrial membrane potential (7/8), decreased mitochondrial membrane potential (1/8), increased ROS generation (2/2), normal uncoupled rate (5/5) |
Anomalous salt bridge formation between subunits a & c result in no rotation of rotor after proton translocation, abnormal holocomplex V production OR increased ROS generation OR decreased ATP synthesis OR abnormal coupling from Fo to F1 |
| m.8993T>C | Decreased ATP synthesis (8/12) -- more mild than 8993T>G in all 8; normal ATP hydrolysis (6/6) | abnormal holocomplex V production OR increased ROS generation OR abnormal structure of proton pore causing partial reduction of ATP synthesis11 |
| m.9025G>A | Decreased baseline respiration(1/1), increased ROS(1/1) | Abnormal hydrogen bonding preventing conformational changes |
| m.9029A>G | Decreased baseline respiration(1/1), increased mitochondrial membrane potential(1/1), increased ROS (1/1) | Prevent proton translocation |
| m.9032T>C | Decreased baseline respiration and uncoupled respiration (1/1), increased mitochondrial membrane potential (1/1) | Destabilization of proton pore |
| m.9035T>C | Lower steady state ATP levels (1/1), decreased ATP hydrolysis (1/1), increased ROS (1/1), increased sensitivity to glucose deprivation (1/1) | |
| m.9101T>C | Lowered ATP/2e- ratio (1/1), normal ATP synthesis & hydrolysis (1/1) | Decreased coupling of proton flow with ATP production |
| m.9134A>G | Decreased ATP synthesis & ATP hydrolysis (1/1) | |
| m.9176T>C | Normal ATP hydrolysis rate (1/3), normal ATP production (1/7), lower ATP synthetic rate (6/7)4, reduced ATP/O efficiency (3/5), normal proton-pumping coupled to ATP hydrolysis (1/1), incomplete complex V assemblies, increased sensitivity to oligomycin, increased superoxide production (2/5), abnormal holocomplex formation (2/5) | Impaired CV stability, but controversial |
| m.9176T>G | Decreased ATP synthesis malate > succinate (1/1), normal response to oligo (1/1), increased mito membrane potential (1/1) | Impaired proton pumping efficiency with normal holocomplex |
| m.9185T>C | Decreased mitochondrial membrane potential (3/3), impaired CV holoenzyme assembly (2/4), Decreased ATP hydrolysis (3/5), normal ATP synthesis (1/1) | Impairment of proton pump |
The numerator in parentheses represents the fraction of cases manifesting the listed feature over the number of cases in which the feature was assessed. The reference sequence used was NC_012920.1
We conducted a systematic review by searching Pubmed for all papers published by July 2017 with the terms “ATP6”, “ATPase 6”, or “8993.” Papers with clinical or biochemical details of individual cases were included in the analysis, for a total of 66 publications. We reviewed the reported variants for phenotypic association, median heteroplasmy level in affected patients and their asymptomatic relatives, and their de novo or familial occurrence (Table 2). The reference sequence used for MT-ATP6 was NC_012920.1.
Table 2.
Reported pathogenic mutations in MT-ATP6, with number of reported cases, associated phenotypes and inheritance.
| Mutation | Number of reported patients (kindreds) |
Leigh Syndrome |
NARP Syndrome |
Charcot Marie Tooth |
Spinocerebellar ataxia |
Other | Symptomatic Heteroplasmy in any tissue (median, IQ1–3) |
Asymptomatic heteroplasmy in any tissue (Median, IQ1–3) |
Number de novo if maternal testing done (%) |
|---|---|---|---|---|---|---|---|---|---|
| m.8528T>C | 4* (4) | Hypertrophic cardiomyopathy | 100 (100–100) | 26 | 0/1 (0) | ||||
| m.8597T>C | 1(1) | + | 95 | ||||||
| m.8618insT | 1(1) | + | 85 | 0 | 1/1 (100) | ||||
| m.8839G>C | 1(1) | + | 88 | 31 (27–36) | 0/1 (0) | ||||
| m.8851T>C | 3 (2) | + | + | 97 (96–98) | 72 (66–79) | 2/2 (100) | |||
| m.8969G>A | 2 (2) | IgA nephropathy; Mitochondrial Lactic Acidosis and Sideroblastic Anemia (MLASA) | 93 (92–94) | 60 | 1/2 (50) | ||||
| m.8989G>C | 1 (1) | + | NARP | 94 | 0 | 1/1 (100) | |||
| m.8993T>C | 34 (23) | + | + | + | Neuropathy and ataxia; periodic paralysis; migraine and diabetes; isolated migraine muscle atrophy; | 90 (84–95) | 45 (25–59) | 0/17 (0) | |
| m.8993T>G | 72 (57) | + | + | Primary lactic acidosis, Infantile epileptic encephalopathy; Ataxia and retinitis pigmentosa; isolated weakness; retinitis pigmentosa and migraine; isolated retinitis pigmentosa; isolated migraine; isolated nyctalopia; progressive external ophthalmoplegia; | 90 (78–95) | 44 (37–63) | 5/65 (22) | ||
| m.9025G>A | 1 (1) | 3-methylglutaconic aciduria | 100 | 100 (100–100) | 0/1 (0) | ||||
| m.9029A>G | 1 (1) | Atypical LHON | 100 | 95 (90–95) | 0/1 (0) | ||||
| m.9032T>C | 1 (1) | + | 96 | 73 | 0/1 (0) | ||||
| m.9035T>C | 13 (10) | + | Short stature and speech delay | 100 (100–100) | 3/3 (100) | ||||
| m.9134A>G | 1 (1) | Primary lactic acidosis | NA | ||||||
| m.9176T>C | 28 (15) | + | + | + | Hereditary spastic paraplegia, Neuropathy and ataxia, periodic paralysis | 100 (95–100) | 54 | 0/24 (0) | |
| m.9176T>G | 3 (2) | + | 95 (82–95) | 34.5 (28–42) | 0/3 (0) | ||||
| m.9185T>C | 51 (18) | + | + | + | Upper motor neuro disease, neuropathy and ataxia, periodic paralysis and neuropathy, isolated learning differences | 100 (99–100) | 63 (42–76) | 0/44 (0) | |
| m.9191T>C | 1 (1) | + | 94 | 0/1 (0) | |||||
| m.9205delTA | 2 (2) | + | Primary lactic acidosis | 99 (98–99) | 54 (35–73) | 1/2 (50) | |||
| Total | 221 (144) | 95 (90–95) | 50 (31–73) | 14/170 |
Number of confirmed cases – additional cases by family history exist. NARP = Neuropathy, ataxia and retinitis pigmentosa; NA = not assessed. The reference sequence used was NC_012920.1
Meta-analysis of these reported data demonstrated that while overlap existed on a patient level between MT-ATP6 variant heteroplasmy levels reported in symptomatic and asymptomatic individuals, heteroplasmy load was significantly higher in affected patients than their asymptomatic relatives on a population level (p=3.2×10^−45, student’s T-test). The same result was found for recurrent pathogenic variants, where sufficient patients were identified for analysis (Fig 1A). In addition, MT-ATP6 variant heteroplasmy level correlated with phenotype, whereby an increased median heteroplasmy level was seen in earlier-onset phenotypes. For this analysis, we selected the two most common MT-ATP6 clinical phenotypes, Leigh Syndrome (a more severe, early-onset disease) and NARP Syndrome (a later-onset disease). A significantly higher heteroplasmy level was seen in MT-ATP6 patients manifesting with Leigh syndrome as compared to NARP syndrome (Fig 1B, p=0.037 by Student’s T-test). Across all MT-ATP6 disease phenotypes, a correlation was evident between a younger age of onset and a higher MT-ATP6 variant heteroplasmy level (Fig 1C, Pearson correlation coefficient=−0.37, p=1.6E-07).
Figure 1. MT-ATP6 Heteroplasmy Level Analysis.
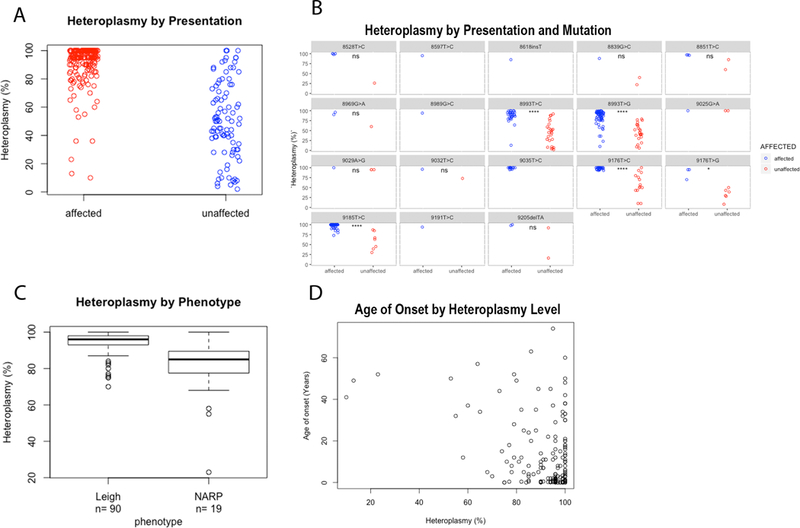
A. Average tissue heteroplasmy (averaged across all tissues analyzed) among symptomatic patients (red) and their asymptomatic relatives (blue) across all shows that there is a significantly higher heteroplasmy level in affected patients (p< 2.2e-16 by Mann-Whitney U test) B. Average tissue heteroplasmy (averaged across all tissues analyzed) among symptomatic patients (red) and their asymptomatic relatives (blue) by variant site. (*=p< 0.05; **** = p<0.00001). Heteroplasmy level is significantly higher in Leigh Syndrome compared to NARP patients across all pathogenic MT-ATP6 variants (p= 5.989e-06 by Mann-Whitney U nonparametric analysis). D. Heteroplasmy level shows a significant negative association with patient age at presentation (Pearson correlation coefficient=−0.37, p=1.6E-07).
In addition to the variability in MT-ATP6 pathogenic variant patients’ clinical phenotypes, extensive variability has been reported in the biochemical markers of patients with MT-ATP6 variants (Table 3). Distinct reasons likely underlie this observed variability on standard biochemical tests. Some findings are suggestive that differences exist in the pathophysiologic mechanisms of different MT-ATP6 variants. For instance, the m.8993T>G variant most frequently results in increased mitochondrial membrane potential, suggesting that the proton pore is not allowing a discharge of the proton gradient and leads to impaired ATP synthesis. In contrast, patients with the m.9185T>C variant have decreased mitochondrial membrane potential, suggesting there is an unregulated release of protons occurring through the proton pore. The most commonly observed biochemical abnormalities seen across all pathogenic MT-ATP6 variants include decreased CV holoenzyme assembly; abnormal mitochondrial membrane potential (increased or decreased); reduced ATP synthetic rate, ATP hydrolytic rate and/or ATP steady state levels; and abnormal sensitivity to the CV inhibitor oligomycin (increased or decreased). However, none of these individual biochemical markers was universally seen. Of note, the most commonly used biochemical indicator used to assess “CV enzymatic rate” actually measures ATP hydrolysis (i.e, the CV ‘reverse’ reaction of ATP degradation, rather than ATP synthesis), which is a function that is not dependent on the CV proton pumping function and is thus less sensitive to pathogenic MT-ATP6 variants than would be direct measures of ATP synthesis.
Table 3.
Summary of previously unreported with MT-ATP6 variants detected in patients with suspected mitochondrial disease
| # | Mutation | CNS | Neuropathy | Cardiac/ autonomic | Other | Age at evaluation (age at onset) | Heteroplasmy in proband | Familial heteroplasmy level | Mitomap allele frequency | SIFT | Conservation | Biochemical findings | Pathogenicity classification per ACMG criteria17 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | m.8573G>A p.Gly16Asp |
Autism with regression, intellectual disability, hypotonia | Heat intolerance and increased sweating | Inability to gain weight, fatigue, hypersensitivity to anesthesia | 12 (1) | 100% (blood) | 100% asymptomatic mother (blood) | 49/45,494 | 0.01 damaging |
Primates | Normal blood lactate, elevated urine lactate | Benign (BS1, BS2) |
|
| 2 | m.8608C>T p.Pro28Ser and single deletion (detected by miSeq NGS) |
CPEO, myopathy | Exercise intolerance | 56 (19) | 100% (blood) | Not done | Not present | 0.41 tolerated |
Humans | Muscle biopsy: ragged red fibers, cytochrome c oxidase deficiency, Serum: elevated lactate |
VUS (PM2, BP4, BP5) |
||
| 3 | m.8612T>C p.Leu29Pro |
Abnormal signal intensity of the putamen, hypotonia, intellectual disability | Bicuspid aortic valve | Acute decompensation with illness, hypersensitivity to anesthesia, short stature | 15 (birth) | 100% blood | Not known (adopted) | Not present | 0.03 damaging |
Mammals | Intermittent lactic acidemia, elevated alanine and proline | VUS (PM2, PP4) |
|
| 4 | m.8723G>T p.Arg66Leu |
CPEO, myopathy, mild gait ataxia | Exercise intolerance, hepatomegaly | 56 (19) | 99% (muscle) | Not done | Not present | 0.20 tolerated |
Vertebrates | Muscle biopsy: some ragged red fibers Serum: normal lactate |
VUS (PM2) |
||
| 5 | m.8843T>C p.Ile106Thr |
none | labile heart rate and blood pressure, transient ventricular dysfunction | Gastric dysmotility, joint effusions, arthritis, scotoma, mast cell activation syndrome with immune candidate gene | 23 (10) | 100% blood | Not done | 155/45,494 | 0.03 damaging |
Mammals | Normal muscle by electron microscopy, immune histochemistry, histology, CI-CIV enzymology | Likely benign (BS1, BP5) |
|
| 6 | m.8881T>C p.Ser119Pro |
Ataxia, pyramidal signs, mild intellectual disability | Sensorimotor axonal | 38 (child) | Unknown | Not done | 1/45,494 | 0.21 tolerated |
Human | Not done | VUS (BP4) |
||
| 7 | m.8921G>A p.Gly132Asp |
Agenesis of the corpus callosum, optic nerve atrophy, epilepsy | Right bundle branch block, ventricular septal defect, atrial septal defect, aortic insufficiency | Unbalanced translocation, central apnea, G-tube dependent, | 1 (birth) | 100% blood | 100% mother (blood, unclear if symptomatic) | 6/45,494 | 0.00 damaging |
Vertebrates | Normal lactate | VUS (PP3, BS1, BP5) |
|
| 8 | m.8938A>G p.Ile138Val |
none | Small fiber | Reduced sweating, abnormal autonomic reflex resting, septal hypokinesia | Exercise intolerance, gastric dysmotility, nyctalopia, ERG shows reduced central response |
67 (20) | 100% blood | Not done | 45/45,494 | 0.41 tolerated |
Vertebrates | Exertional lacticemia with normal alanine; intermittent 3-methylglutaconic aciduria.
Normal muscle by electron microscopy, immune 5histochemistry, histology, CI-CIV enzymology x 2 |
VUS (BS1) |
| 9 | m.8999T>C p.Val158Ala and m.9115A>G p.Ile197Val |
Developmental delay, left-sided weakness, Brain MRI: chronic cystic encephalomalacia in the left temporoparietal occipital lobes | tachycardia | Neurogenic colon, exercise intolerance, fatigue | 16 (7) | 100% blood | Not done | 7/45,494 | 0.02 damaging |
Animals | Normal lactate | VUS (PM1, PP3) |
|
| 10 | m.9026G>A p.Gly167Lys |
Intellectual disability | dysautonomia | Headaches, myalgias, fatigue | 11 (6) | 17.7–19.8% blood, 22.6% skin 16.4% muscle |
4–8% symptomatic mother (blood,below) Not-detected asymptomatic brother (blood) |
3/45,494 | 0.00 damaging |
Animals | Normal muscle by histology, immune histochemistry, CI-CIV enzymology x 2 Fibroblast respirometry: a defect in the phosphorylation system that limits oxidative phosphorylation with complex I, II, and III, as well as fatty acid substrates. when measured as the uncoupled or maximal rate, the combined oxidation of complex I, II, and III move into the control range. These data suggest that the phosphorylation system is limiting as the reason the ADP-stimulated rates are low (suppl fig 1) |
Likely pathogenic (PS3, PM5, PP1, PP3) | |
| (mother of above) | Small fiber | Headaches, fatigue | 46 (4) | 4.6% (blood) 8.1% (urine) |
See above | Normal lactate | |||||||
| 11 | m.9041A>G p.His172Arg |
Muscle weakness, exercise intolerance, congenital cataracts | 14 (3) | 85% (blood) 88% (saliva) |
adopted | 1/45,494 | 0.79 tolerated |
Vertebrates | Normal CI-IV Elevated lactate x 1 |
VUS | |||
| 12 | m.9058A>G p.Thr178Ala and m.9133G>A p.Glu203Lys |
Autism, intellectual disability, epilepsy and normal brain MRI | Congenital cataracts | 13 (3) | 100% (blood) | 100% asymptomatic mother (blood) | 21/45,595 | 0.41 tolerated |
Humans | Not done | Benign (BS1, BS2, BP4) |
||
| 13 | m.9088T>C p.Ser188Pro |
Unilateral facial weakness, | Postural orthostatic tachycardia | Myalgias, heat intolerance, fatigue, migraines, | 15 (15) | 66% (blood) 52% (skin) |
33% symptomatic mother (blood, below) | 19/45,494 | 0.21 tolerated |
Humans | Normal lactate Fibroblast respirometry: normal (Fig S1) |
Likely benign (BP4, BS3) |
|
| (mother of above) | Depression | Hypotension | Fatigue, myalgias, irritable bowel, frequent choking | 37 (36) | 33% (blood) | See above | 19/45,494 | Normal lactate | |||||
| 14 | m.9115A>G p.Ile197Val |
(see 8999 for patient information) | 100% (blood) | Not done | 21/45,494 | 0.19 tolerated |
Primates | Normal lactate | VUS (BP4) |
||||
| 15 | m.9133G>A p.Glu203Lys |
(see 9058 for patient information) | 100% (blood) | 100% asymptomatic mother (blood) | 4/45,494 | 0.00 damaging |
Vertebrates | Not done | VUS (BS2, PM5, PP3) |
||||
| 16 | m.9152T>C p.Ile209Thr |
Autism with cognitive development to high functioning by adulthood, fatigue | GERD, constipation | 20(3) | 23% (blood) | 10% asymptomatic mother | 14/45,494 | 0.00 damaging |
Vertebrates | Normal lactate | VUS (PP3) |
||
The reference sequence used was NC_012920.1
MT-ATP6 GENOTYPE-PHENOTYPE CORRELATION
CV subunit a, encoded by MT-ATP6, is composed of six trans-membrane domains spanning the inner mitochondrial membrane. The CV proton pore occurs between transmembrane helix 5 and subunit c (encoded by a different gene) and perhaps transmembrane helix 6. As might be expected, the majority of known pathogenic variants in MT-ATP6 occur in transmembrane helices 5 and 6 (Fig 2A). Contact between CV subunits a and c is particularly essential to couple proton translocation with rotation of the CV enzymatic subunit. Pathogenic variants at m.8993, which are involved in nearly 50% of reported MT-ATP6 disease cases, result in the formation of an abnormal salt bridge between these two subunits, which inhibits this rotation (Baracca, Barogi, Carelli, Lenaz, & Solaini, 2000). On average, MT-ATP6 variants at position m.8993 appear to result in the earliest-onset and most severe clinical disease, where m.8993T>G causes a more severe disease phenotype than does m.8993T>C (Morava et al., 2006).
Figure 2. Variant map of variants in MT-ATP6 mapped by protein domain.
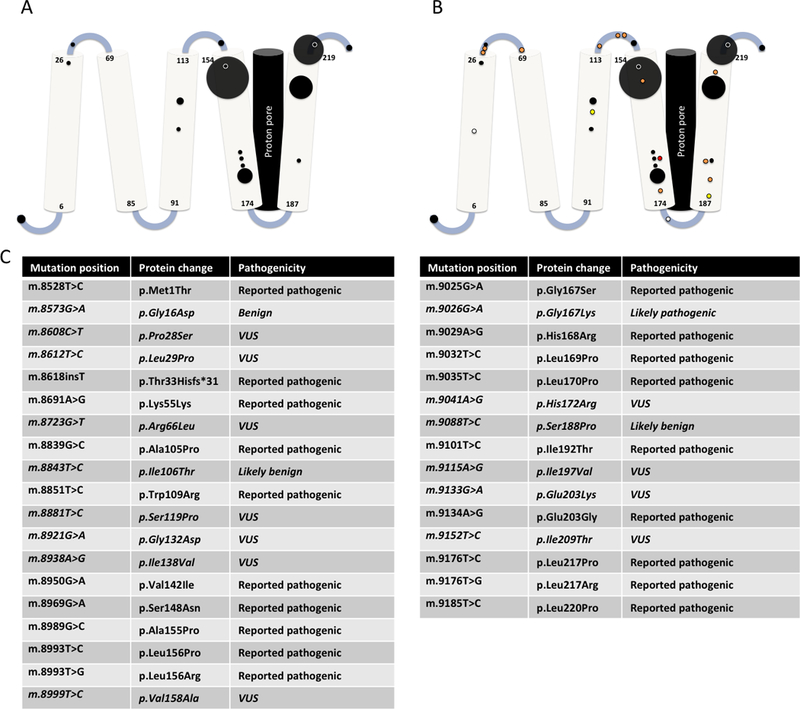
A. Reported pathogenic variants in MT-ATP6 mapped across its five transmembrane domains. The area of each circle is proportional to the number of reported patients. B. Reported MT-ATP6 variants in a new cohort reported in this publication (n=16). White circles represent variants re-classified as benign, yellow circles represent variants re-classified as likely benign, orange circles represent VUS, and red circles represent variants re-classified as likely pathogenic. C: List of all mutations including protein changes and classification.
MT-ATP6 VARIANTS OF UNCERTAIN SIGNIFICANCE IN A NEW CLINICAL COHORT OF 14 PROBANDS
Multiple variants of uncertain significance (VUS) in MT-ATP6 have been reported in patients deemed to have ‘possible’ mitochondrial disease. It is difficult to fully assess these variants’ contribution to clinical disease manifestations considering the absence of clinically-available CV activity testing, the possibility that incomplete penetrance of pathogenic MT-ATP6 variants or high tissue variability for MT-ATP6 variant heteroplasmy levels within a given individual may limit the utility of pedigree interpretation, and the occurrence of many non-specific clinical symptoms in some patients. Therefore, the only reliable way for clinicians to currently determine the likely clinical significance of a VUS identified in MT-ATP6 is to identify recurrence in similarly affected, unrelated patients.
We ascertained subjects enrolled in the IRB-approved study “Metabolic Consequences of Mitochondrial Disease” at the Children’s Hospital of Philadelphia (CHOP) or enrolled in the German mitoNET registry who had MT-ATP6 VUS identified on mtDNA genome sequence performed for suspected mitochondrial disease (Table 3, n=16 variants, 14 kindreds, 16 subjects). All variants have been submitted to the Mitochondrial Disease Sequence Data Resource Consortisum (https://mseqdr.org)(Shen et al., 2016). Clinical presentations among carriers of both pathogenic variants and VUS were highly variable, ranging from pediatric-onset Leigh syndrome, to adult-onset multisystemic disease. Among patients with VUS, the recurrent reported symptoms occurring in over 50% of subjects with variants of uncertain significance were largely non-specific, including headaches, fatigue, and exercise intolerance. Muscle weakness, peripheral neuropathy, heart rate or blood pressure variability, and gastrointestinal dysmotility were also recurrent in multiple subjects. These variants were located throughout the MT-ATP6 gene, including some variants that occur proximal to known pathogenic MT-ATP6 variants (Fig 2B). However nearly half of these variants (7/16) were located in the linker regions between the transmembrane domains of ATP6, whereas only two of the known pathologic variants are located in these regions.
Through consideration of clinical phenotype data, five of the MT-ATP6 VUS (m.8573G>A, m.8843T>C, m.9026G>A, m.9058A>G and m.9088T>C) identified in our CHOP cohort were able to be reclassified according to the ACMG classification criteria (Richards et al., 2015); a fourth VUS, (m.8612T>C), which is clinically highly suspicious, did not meet ACMG criteria to be reclassified, as discussed below.
The MT-ATP6 m.8573G>A variant was seen in apparent homoplasmy in a 12-year-old patient with regressive autism, intellectual disability and hypotonia. Her unaffected mother was also apparently homoplasmic. This variant occurs in 49 of 45,494 healthy individuals in MITOMAP (Lott et al., 2013) and is conserved only among primates. Because of the high frequency in the general population and her mother’s lack of symptoms, it was reclassified as ‘benign.’ This re-classification is not without caveats: other MT-ATP6 variants are not fully penetrant even at homoplasmy(Aure et al., 2013; Lopez-Gallardo et al., 2014; Pitceathly et al., 2012b). However, most of the known pathogenic MT-ATP6 variants that were also homoplasmic in healthy relatives cause a Charcot Marie Tooth or upper motor neuron disease-like phenotype, which was not seen in this patient.
The MT-ATP6 m.8843T>C variant was seen in apparent homoplasmy in a 22-year-old subject with dysautonomia, gastric dysmotility, and multiple inflammatory symptoms who was ultimately found to have an immunogenetic condition causing mast cell activation syndrome that was felt to fully explain her symptoms (Table 3, subject 2). Further, the m.8843T>C variant has been shown to occur in 155 of 45,494 healthy individuals in MITOMAP (Lott et al., 2013), and is conserved only among mammals. Overall, since this patient has an alternate genetic explanation, it was appropriate per ACMG criteria to reclassify it as ‘likely benign’.
The MT-ATP6 m.9026G>A variant was identified at low heteroplasmy levels in all tissues studied (blood, buccal, urine and muscle) of a 6-year-old proband with intellectual disability, dysautonomia, headaches, myalgias, and fatigue (Table 3, subject 6). His mother (Table 3, subject 7) was also found to harbor low heteroplasmy in blood and urine for the m.9026G>A variant, with subsequent clinical evaluation notable for her having lifelong complaints of fatigue, pain from small fiber neuropathy, and headaches. Fibroblasts from the proband had very low integrated oxygen consumption capacity in response to substrates for complex I, II and III that was relieved by uncoupling, consistent with a complex V deficiency. The m.9026G>A variant is highly conserved, predicted damaging by SIFT, and reported in MITOMAP to occur in only 0.006% of controls. Based on these considerations, we reclassified this variant as ‘likely pathogenic’. It is not definitely responsible for the symptoms observed; however, since the heteroplasmy levels measured in readily accessible tissues seen in both the proband and his mother were much lower than that reported with all other symptomatic variants in MT-ATP6, uncertainty of its definite role in causing symptoms remains.
The MT-ATP6 m.9058A>G variant was seen in a 13-year-old subject with autism, intellectual disability, epilepsy and a brain MRI that did not show evidence of Leigh Syndrome. The variant was apparently homoplasmic in this subject and her asymptomatic mother, and both also had an additional homoplasmic MT-ATP6 variant (m.9133G>A). The m.9058A>G variant was predicted benign by SIFT, affects a poorly conserved residue that varies from humans in gorillas, and occurs in 21/45,494 healthy individuals in MITOMAP. Therefore, it was determined to be benign, with similar caveats as with the m.8573 variant. It is of interest that congenital cataracts were seen both in this subject and in a subject with a nearby variant (m.9041A>G).
The MT-ATP6 m.9088T>C variant was identified at 52% heteroplasmy in skin and 66% heteroplasmy in blood in an 18-year-old proband with unilateral facial weakness, dysautonomia, myalgias, heat intolerance, fatigue and migraines (Table 3, subject 9). Her mother (Table 3, subject 10) also carried the variant at 33% heteroplasmy in blood and experienced depression, fatigue, myalgias, irritable bowel syndrome and occasional hypotension. This variant is predicted tolerated by SIFT and PolyPhen and is reported in MITOMAP to occur in 0.04% of controls. In addition, fibroblasts from the proband had a normal oxygen consumption capacity. Based on these considerations, we reclassified this variant as ‘likely benign’. However, one outstanding factor to consider that may change this current classification is whether fibroblast respirometry would be impaired at higher heteroplasmy levels, which may occur in some of the subject’s tissues that were not accessible for testing. Functional evaluation of transmitochondrial cybrids (Vithayathil, Ma, & Kaipparettu, 2012) carrying this variant on a research basis is underway in our group.
The MT-ATP6 m.8612T>C VUS was identified in a 15-year-old patient with suspected mitochondrial disease, brain MRI consistent with Leigh syndrome, illness-induced neurodevelopmental regression, and volatile anesthetic hypersensitivity (Table 3, subject 1). Although this variant did not meet ACMG criteria for likely pathogenic status as no familial information (e.g. maternal heteroplasmy) was available for analysis and no functional studies have yet been performed, this variant has never been seen in control populations, in silico prediction in SIFT suggests it is damaging, and the clinical phenotype is highly suspicious for mitochondrial disease. Overall, the MT-ATP6 m.8612 VUS remains at this time of uncertain clinical significance. Transmitochondrial cybrids have also been generated from this proband and are being evaluated on a research basis.
CLINICAL AND DIAGNOSTIC RELEVANCE
While clinical diagnostic assays of mitochondrial function in muscle, liver, or fibroblasts routinely include enzyme activity assessment of respiratory chain complexes I through IV, CV enzymatic activity is not routinely evaluated in clinical diagnostic laboratories. Therefore, no single clinical functional test for CV activity has been universally reported for all published putatively pathogenic variants, and most patients with suspected mitochondrial disease are not biochemically screened a priori for CV dysfunction. Rather, CV disease is only suspected when there is a classical syndromic presentation such as NARP or when mtDNA genome sequencing identifies a potentially relevant variant. When pathogenic MT-ATP6 variants have been identified and published, a highly diverse range of biochemical measures have been assessed for different MT-ATP6 variants, preventing direct comparison of their pathogenicity. A major limitation too has been that the most commonly reported research-based study of CV enzyme activity measures the reverse CV enzyme reaction, ATP hydrolysis. MT-ATP6 is most directly involved in proton translocation, however, a process that does not require the reverse reaction and many MT-ATP6 pathogenic variants do not directly affect this aspect of CV enzyme activity. However, some MT-ATP6 pathogenic variants that disrupt assembly of the CV holoenzyme may indirectly result in disruption of reverse CV activity.
Here, we systematically reviewed the detailed biochemical and clinical features reported for each MT-ATP6 pathogenic variant (Table 1 & 2). Strikingly, the only biochemical feature that was universally seen in all subjects for whom it was analyzed (n=14) is a decreased basal oxygen consumption (Abu-Amero & Bosley, 2005; Burrage et al., 2014; Lopez-Gallardo et al., 2014). In all subjects in whom mitochondrial membrane potential was analyzed (n=17) it was abnormal: increased in eleven subjects and decreased in six. We postulate that the variants resulting in increased mitochondrial membrane potential occluded the proton pore, while the remainder(Abu-Amero & Bosley, 2005; Burrage et al., 2014; Lopez-Gallardo et al., 2014) allowed proton leak leading to reduced membrane potential; however, in only 1 of 8 cases studied was a defect in proton pumping directly demonstrated (Burrage et al., 2014; Verny et al., 2011). In addition, both increased and decreased mitochondrial membrane potential has been reported in subjects specifically with the m.8993T>G pathogenic variant, likely because variable secondary dysfunction of respiratory chain complexes I-IV is seen in subjects m.8993T>G variants, suggesting that altered membrane potential may not be a reliable diagnostic marker for MT-ATP6 disease in general or for specific pathogenic variants within this gene (Lopez-Gallardo et al., 2014).
Specific complex V analyses also had variable results that did not fully correlate with genotype. ATP synthesis measured in 63 historically reported subjects was decreased in 52 subjects and increased in 2 subjects (Carrozzo et al., 2001; Castagna et al., 2007; Lodi et al., 1994; Vázquez-Memije et al., 1998; Verny et al., 2011). The majority of subjects who had a normal ATP synthetic rate had later onset symptoms, and/or the m.8993T>C pathogenic variant, suggesting that there may be a correlation between ATP synthetic rate, MT-ATP6 genotype, and clinical phenotype (Castagna et al., 2007; Santorelli et al., 1996).
CV enzymatic analysis, which typically measures the ATP hydrolytic rate and is not dependent on proton pumping, was impaired in only 25% of the 41 subjects analyzed. Along with the abnormal mitochondrial membrane potential findings, these data strongly suggest that most MT-ATP6 pathogenic variants impair the coupling between proton pumping and ATP synthesis rather than inherent activity of the enzyme itself (Burrage et al., 2014; Castagna et al., 2007; Hao et al., 2015; Honzik et al., n.d.; Jacobs et al., 2005; Lopez-Gallardo et al., 2014; Pitceathly et al., 2012a; Santorelli et al., 1996; Sikorska et al., 2009; Vázquez-Memije et al., 1998). Oligomycin sensitivity was similarly variable – increased in 1 subject, decreased in 6 subjects, and normal in the remaining 3 subjects. This finding may correlate with the location of the variant relative to the site of oligomycin activity to inhibit the CV proton pore However, since both increased and decreased oligomycin sensitivity has been variably reported with the m.8993T>G pathogenic variant, the actual explanation may likely be more complex (Burrage et al., 2014; Carrozzo et al., 2001; Vázquez-Memije et al., 1998; Verny et al., 2011).
Some MT-ATP6 pathogenic variants also impair the assembly or stability of the CV holoenzyme. The CV holoenzyme assembly was analyzed by blue native page electrophoresis analysis in 34 subjects, with abnormalities seen in 25 subjects (Castagna et al., 2007; Hao et al., 2015; Lopez-Gallardo et al., 2014; Pitceathly et al., 2012a; Sinko, Garzuly, & Kalman, 2014). The inconsistency of CV assembly among subjects with the same genotype at similar heteroplasmy levels highlights the biochemical variability caused by pathogenic variants in MT-ATP6.
High-resolution respirometry analysis on a clinical diagnostic basis in fibroblasts (Center for Inherited Disease Metabolism, Supp Fig S1) from the CHOP proband with the MT-ATP6 m.9026G>A variant (Table 3, subject 6) allowed calculation of the relative contributions from oxidation and phosphorylation to integrated cellular respiratory capacity. This analysis allowed confirmation of CV dysfunction, a finding suggestive that routine diagnostic evaluation of cellular respiratory capacity may enable improved functional assessment for CV deficiency. However, this calculation was variable in previous reported cases and systematic study is needed to determine if this finding is consistently present in multiple tissues across the diverse array of known pathogenic MT-ATP6 variants. It would be ideal to utilize transmitochondrial cybrid lines (Wallace, Bunn, & Eisenstadt, 1975) derived from individuals with novel MT-ATP6 variants to compare CV activity in homoplasmic mutant and wild-type mtDNA genomes from the same individual in a common nuclear background. However, unlike most other mtDNA pathogenic gene variants that occur in different tissues at varying heteroplasmy levels, many MT-ATP6 pathogenic variants may be found at or near homoplasmic levels, making it difficult to establish homoplasmic wild-type and homoplasmic mutant lines from the same individual. Mutant genome homoplasmy may occur both for MT-ATP6 variants undergoing rapid heteroplasmy shifts as well as for de novo MT-ATP6 variants. Indeed, homoplasmy is commonly seen with the m.8993T>G common variant, as well as with other pathogenic MT-ATP6 variants, such as m.9185T>C that has variable expressivity and may include very subtle clinical presentations.
In summary, all of the traditional approaches to evaluate the pathogenicity of an mtDNA variant -- clinical correlation, biochemical enzymatic testing, and evaluation of family heteroplasmy – are challenging in MT-ATP6. Therefore, CV deficiency should be considered in every patient with suspected mitochondrial disease, especially if a variant in MT-ATP6 is found. The current state of evaluation requires integrating multiple lines of evidence, including variant heteroplasmy in different tissues and family members, biochemical features, and familial disease presentations, recognizing that each factor may be quite complex to interpret.
FUTURE PROSPECTS
Future studies are needed to systematically evaluate all MT-ATP6 variants by a consistent range of assays to devise optimal means to functionally validate pathogenic variants in MT-ATP6. This would enable development of expanded functional biochemical testing that may improve diagnostic accuracy for CV deficiency. For example, we have presented here a case with high-resolution respirometry evidence for pathogenicity of a novel MT-ATP6 m.9026G>A variant in a single kindred with multi-system disease. Newer technologies (e.g., Oxygraph 2k, Oroboros instruments) may further improve diagnostic capabilities that simultaneously measure high-resolution respirometry (e.g., integrated oxygen consumption capacity in intact or permeabilized cells) along with fluorometric and/or potentiometric real-time analysis of mitochondrial membrane potential. Indeed, reviewing all known literature of MT-ATP6 pathogenic variants suggests that mitochondrial membrane potential potential maintenance is a key function of CV that is often altered by MT-ATP6 pathogenic variants. While isolated measurement of mitochondrial membrane potential may be misleading since different MT-ATP6 variants can result in it either being hyperpolarized or depolarized, assessing it simultaneously with respiratory capacity may improve insight into a given individual’s coupling between mitochondrial proton flow and phosphorylation. Fluorometric analysis of ATP levels can also be simultaneously assessed by these technologies along with respirometry and potentiometric analysis of mitochondrial membrane potential, thereby allowing improved understanding of key aspects of CV activity.
Overall, the diversity of biochemical findings in MT-ATP6 disease suggests that the best diagnostic confirmatory approach is a multi-pronged one. The integrated fluororespirometry approach discussed above may be particularly valuable when combined with other approaches that assess CV holocomplex assembly and activity (e.g., blue-native gel electrophoresis and in-gel activity analysis). In addition, expert curation of MT-ATP6 variants will improve understanding and consistency of allele pathogenicity assessment that can be deposited in common community resources including ClinVar and MSeqDR (Shen et al., 2018, 2016). Indeed, NIH-supported expert panel curation is currently underway for MT-ATP6 variants reported to cause pediatric Leigh syndrome (https://projectreporter.nih.gov/project_info_description.cfm?aid=9411950&icde=40411006&ddparam=&ddvalue=&ddsub=&cr=25&csb=default&cs=ASC&pball=).
CONCLUSIONS
MT-ATP6 disease is a highly variable clinical disease, with both pediatric-onset and adult-onset disease seen that is generally multi-systemic. In addition to the well-known association with Leigh Syndrome and NARP phenotypes, other recurrent presentations of MT-ATP6 pathogenic variants include spinocerebellar ataxia, Charcot Marie Tooth, and familial upper motor neuron disease. Isolated cases have also been reported of pathogenic MT-ATP6 variants causing a range of more diverse clinical presentations, including primary lactic acidosis, cardiomyopathy, 3-methylglutaconic aciduria and isolated optic neuropathy. Such broad clinical diversity demands clinicians maintain a high clinical index of suspicion for possible CV disease, especially for patients with the most common features of peripheral neuropathy and ataxia. MT-ATP6 deficiency is further complicated by a wide range in measurable biochemical effects of different pathogenic MT-ATP6 variants. Although decreased ATP synthetic rate is very frequently reported, it is not a universal finding and cannot currently be evaluated in patient tissues or cells as a clinical diagnostic test. The CV enzymatic assay that is more established measures the reverse reaction, which is frequently preserved in tissues with pathogenic MT-ATP6 variants. MT-ATP6 variation, particularly within the linker regions between the transmembrane domains of the protein is a common clinical quandary. Overall, the detailed meta-analysis and report of a new clinical cohort of 14 kindreds with MT-ATP6 variants presented here highlights the pressing need for improved functional analyses that accurately measure CV activity in individuals with suspected mitochondrial disease and particularly those with variants of uncertain significance in MT-ATP6 and/or expanded phenotypic presentations beyond classical MT-ATP6 clinical syndromes.
Supplementary Material
ACKNOWLEDGMENTS:
We are grateful to the mitochondrial disease families who participated in this research study. This work was funded in part by the National Institutes of Health (U24-HD093483; K08-DK113250; U54-HD086984). MSeqDR is funded in part by ongoing support from the United Mitochondrial Disease Foundation (UMDF) and North American Mitochondrial Disease Foundation (NAMDC, funded in part through U54-NS078059). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
REFERENCES
- Abu-Amero KK, & Bosley TM (2005). Detection of Mitochondrial Respiratory Dysfunction in Circulating Lymphocytes Using Resazurin. Arch Pathol Lab Med—Vol, 129. [DOI] [PubMed] [Google Scholar]
- Alila-Fersi O, Chamkha I, Majdoub I, Gargouri L, Mkaouar-Rebai E, Tabebi M, … Fakhfakh F (2017). Co segregation of the m.1555A>G mutation in the MT-RNR1 gene and mutations in MT-ATP6 gene in a family with dilated mitochondrial cardiomyopathy and hearing loss: A whole mitochondrial genome screening. Biochemical and Biophysical Research Communications, 484(1), 71–78. 10.1016/j.bbrc.2017.01.070 [DOI] [PubMed] [Google Scholar]
- Aure K, Dubourg O, Jardel C, Clarysse L, Sternberg D, Fournier E, … Lombes A (2013). Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology, 81(21), 1810–1818. 10.1212/01.wnl.0000436067.43384.0b [DOI] [PubMed] [Google Scholar]
- Baracca A, Barogi S, Carelli V, Lenaz G, & Solaini G (2000). Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. The Journal of Biological Chemistry, 275(6), 4177–4182. 10.1074/JBC.275.6.4177 [DOI] [PubMed] [Google Scholar]
- Burrage LC, Tang S, Wang J, Donti TR, Walkiewicz M, Luchak JM, … Scaglia F (2014). Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA) plus associated with a novel de novo mutation (m.8969G>A) in the mitochondrial encoded ATP6 gene. Molecular Genetics and Metabolism, 113(3), 207–212. 10.1016/j.ymgme.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos Y, Martín MA, Rubio JC, Solana LG, García-Benayas C, Terradas JL, & Arenas J (1997). Leigh syndrome associated with the T9176C mutation in the ATPase 6 gene of mitochondrial DNA. Neurology, 49(2), 595–597. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9270604 [DOI] [PubMed] [Google Scholar]
- Carrozzo R, Tessa A, Vázquez-Memije ME, Piemonte F, Patrono C, Malandrini A, … Santorelli FM (2001). The T9176G mtDNA mutation severely affects ATP production and results in Leigh syndrome. Neurology, 56(5), 687–690. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11245730 [DOI] [PubMed] [Google Scholar]
- Castagna AE, Addis J, McInnes RR, Clarke JTR, Ashby P, Blaser S, & Robinson BH (2007). Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. American Journal of Medical Genetics Part A, 143A(8), 808–816. 10.1002/ajmg.a.31637 [DOI] [PubMed] [Google Scholar]
- Childs A-M, Hutchin T, Pysden K, Highet L, Bamford J, Livingston J, & Crow Y (2007). Variable Phenotype Including Leigh Syndrome with a 9185T>C Mutation in the MTATP6 Gene. Neuropediatrics, 38(6), 313–316. 10.1055/s-2008-1065355 [DOI] [PubMed] [Google Scholar]
- Cízková A, Stránecký V, Mayr JA, Tesarová M, Havlícková V, Paul J, … Kmoch S (2008). TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nature Genetics, 40(11), 1288–1290. 10.1038/ng.246 [DOI] [PubMed] [Google Scholar]
- De Meirleir L (2004). Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. Journal of Medical Genetics, 41(2), 120–124. 10.1136/jmg.2003.012047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries DD, van Engelen BG, Gabreëls FJ, Ruitenbeek W, & van Oost BA (1993). A second missense mutation in the mitochondrial ATPase 6 gene in Leigh’s syndrome. Annals of Neurology, 34(3), 410–412. 10.1002/ana.410340319 [DOI] [PubMed] [Google Scholar]
- Duno M, Wibrand F, Baggesen K, Rosenberg T, Kjaer N, & Frederiksen AL (2013). A novel mitochondrial mutation m.8989G>C associated with neuropathy, ataxia, retinitis pigmentosa — The NARP syndrome. Gene (Vol. 515). 10.1016/j.gene.2012.12.066 [DOI] [PubMed] [Google Scholar]
- Hao X, Liu S, Wu X, Hao Y, & Chen Y (2015). Infantile mitochondrial disorder associated with subclinical hypothyroidism is caused by a rare mitochondrial DNA 8691A>G mutation. NeuroReport, 26(10), 588–592. 10.1097/WNR.0000000000000392 [DOI] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, & Morgan-Hughes JA (1990). A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. American Journal of Human Genetics, 46(3), 428–433. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2137962 [PMC free article] [PubMed] [Google Scholar]
- Honzik T, Tesarova M, Magner M, Mayr J, Jesina P, Vesela K, … Zeman J (n.d.). Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. 10.1007/s10545-011-9440-3 [DOI]
- Jacobs LJAM, de Coo IFM, Nijland JG, Galjaard RJH, Los FJ, Schoonderwoerd K, … Smeets HJM (2005). Transmission and prenatal diagnosis of the T9176C mitochondrial DNA mutation. Molecular Human Reproduction, 11(3), 223–228. 10.1093/molehr/gah152 [DOI] [PubMed] [Google Scholar]
- JEŠINA P, TESAŘOVÁ M, FORNŮSKOVÁ D, VOJTÍŠKOVÁ A, PECINA P, KAPLANOVÁ V, … HOUŠTĚK J (2004). Diminished synthesis of subunit a (ATP6) and altered function of ATP synthase and cytochrome c oxidase due to the mtDNA 2 bp microdeletion of TA at positions 9205 and 9206. Biochemical Journal, 383(3), 561–571. 10.1042/BJ20040407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Hogeveen M, Nijtmans LGJ, Van Den Brand MAM, Janssen AJM, Diepstra JHS, … Rodenburg RJT (2007). A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. 10.1136/jmg.2007.052084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Renkema GH, Bras M, van den Heuvel LP, Hoischen A, Gilissen C, … Rodenburg RJT (2013). A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain : A Journal of Neurology, 136(Pt 5), 1544–1554. 10.1093/brain/awt086 [DOI] [PubMed] [Google Scholar]
- Lodi R, Montagna P, Iotti S, Zaniol P, Barboni P, Puddu P, & Barbiroli B (1994). Brain and muscle energy metabolism studied in vivo by 31P-magnetic resonance spectroscopy in NARP syndrome. Journal of Neurology, Neurosurgery, and Psychiatry, 57(12), 1492–1496. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7798979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gallardo E, Emperador S, Solano A, Llobet L, Martin-Navarro A, Lopez-Perez MJ, … Montoya J (2014). Expanding the clinical phenotypes of MT-ATP6 mutations. Human Molecular Genetics, 23(23), 6191–6200. 10.1093/hmg/ddu339 [DOI] [PubMed] [Google Scholar]
- López-Gallardo E, Solano A, Herrero-Martín MD, Martínez-Romero I, Castaño-Pérez MD, Andreu AL, … Montoya J (2009). NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. Journal of Medical Genetics, 46(1), 64–67. 10.1136/jmg.2008.060616 [DOI] [PubMed] [Google Scholar]
- Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, … Wallace DC (2013). mtDNA Variation and Analysis Using Mitomap and Mitomaster. Current Protocols in Bioinformatics, 44, 123.1–26. 10.1002/0471250953.bi0123s44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä-Bengs P, Suomalainen A, Majander A, Rapola J, Kalimo H, Nuutila A, & Pihko H (1995). Correlation between the clinical symptoms and the proportion of mitochondrial DNA carrying the 8993 point mutation in the NARP syndrome. Pediatric Research, 37(5), 634–639. 10.1203/00006450-199505000-00014 [DOI] [PubMed] [Google Scholar]
- Mayr JA, Havlícková V, Zimmermann F, Magler I, Kaplanová V, Jesina P, … Houstek J (2010). Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Human Molecular Genetics, 19(17), 3430–3439. 10.1093/hmg/ddq254 [DOI] [PubMed] [Google Scholar]
- Morava E, Rodenburg RJ, Hol F, de Vries M, Janssen A, van den Heuvel L, … Smeitink J (2006). Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. American Journal of Medical Genetics Part A, 140A(8), 863–868. 10.1002/ajmg.a.31194 [DOI] [PubMed] [Google Scholar]
- Moslemi A-R, Darin N, Tulinius M, Oldfors A, & Holme E (2005). Two new mutations in the MTATP6 gene associated with Leigh syndrome. Neuropediatrics, 36(5), 314–318. 10.1055/s-2005-872845 [DOI] [PubMed] [Google Scholar]
- Pfeffer G, Blakely EL, Alston CL, Hassani A, Boggild M, Horvath R, … Chinnery PF (2012). Adult-onset spinocerebellar ataxia syndromes due to MTATP6 mutations. Journal of Neurology, Neurosurgery, and Psychiatry, 83(9), 883–886. 10.1136/jnnp-2012-302568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitceathly RDS, Murphy SM, Cottenie E, Chalasani A, Sweeney MG, Woodward C, … Hanna MG (2012a). Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology, 79(11), 1145–1154. 10.1212/WNL.0b013e3182698d8d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitceathly RDS, Murphy SM, Cottenie E, Chalasani A, Sweeney MG, Woodward C, … Hanna MG (2012b). Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology, 79(11), 1145–1154. 10.1212/WNL.0b013e3182698d8d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santorelli FM, Mak S-C, Vazquez-Memije ME, Shanske S, Kranz-Eble P, Jain KD, … Dimauro S (1996). Clinical Heterogeneity Associated with the Mitochondrial DNA T8993C Point Mutation. Pediatric Research, 39(5), 914–917. 10.1203/00006450-199605000-00028 [DOI] [PubMed] [Google Scholar]
- Schon EA, Santra S, Pallotti F, & Girvin ME (2001). Pathogenesis of primary defects in mitochondrial ATP synthesis. Seminars in Cell & Developmental Biology, 12(6), 441–448. 10.1006/scdb.2001.0281 [DOI] [PubMed] [Google Scholar]
- Shen L, Attimonelli M, Bai R, Lott MT, Wallace DC, Falk MJ, & Gai X (2018). MSeqDR mvTool: A mitochondrial DNA Web and API resource for comprehensive variant annotation, universal nomenclature collation, and reference genome conversion. Human Mutation, 39(6), 806–810. 10.1002/humu.23422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Diroma MA, Gonzalez M, Navarro-Gomez D, Leipzig J, Lott MT, … Gai X (2016). MSeqDR: A Centralized Knowledge Repository and Bioinformatics Web Resource to Facilitate Genomic Investigations in Mitochondrial Disease. Human Mutation, 37(6), 540–548. 10.1002/humu.22974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorska M, Sandhu JK, Simon DK, Pathiraja V, Sodja C, Li Y, … Tarnopolsky MA (2009). Identification of ataxia-associated mtDNA mutations (m.4052T>C and m.9035T>C) and evaluation of their pathogenicity in transmitochondrial cybrids. Muscle & Nerve, 40(3), 381–394. 10.1002/mus.21355 [DOI] [PubMed] [Google Scholar]
- Sinko G, Garzuly F, & Kalman B (2014). Striking Pathology in Leigh Syndrome Associated With the MTATP6 T8993G Mutation. Pediatric Neurology, 51(4), 585–586. 10.1016/j.pediatrneurol.2014.07.015 [DOI] [PubMed] [Google Scholar]
- Uziel G, Moroni I, Lamantea E, Fratta GM, Ciceri E, Carrara F, & Zeviani M (1997). Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. Journal of Neurology, Neurosurgery, and Psychiatry, 63(1), 16–22. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9221962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Memije ME, Shanske S, Santorelli FM, Kranz-Eble P, DeVivo DC, & DiMauro S (1998). Comparative biochemical studies of ATPases in cells from patients with the T8993G or T8993C mitochondrial DNA mutations. Journal of Inherited Metabolic Disease, 21(8), 829–836. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9870208 [DOI] [PubMed] [Google Scholar]
- Verny C, Guegen N, Desquiret V, Chevrollier A, Prundean A, Dubas F, … Procaccio V (2011). Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion, 11(1), 70–75. 10.1016/j.mito.2010.07.006 [DOI] [PubMed] [Google Scholar]
- Vithayathil SA, Ma Y, & Kaipparettu BA (2012). Transmitochondrial Cybrids: Tools for Functional Studies of Mutant Mitochondria In Wong L-JC (Ed.), Mitochondrial Disorders: Biochemical and Molecular Analysis, Methods in Molecular Biology, vol. 837 (1st ed., pp. 219–230). Berlin: Springer Science+Business Media; 10.1007/978-1-61779-504-6_15 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Bunn CL, & Eisenstadt JM (1975). Cytoplasmic transfer of chloramphenicol resistance in human tissue culture cells. The Journal of Cell Biology, 67(1), 174–188. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1176530 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.