

Abstract
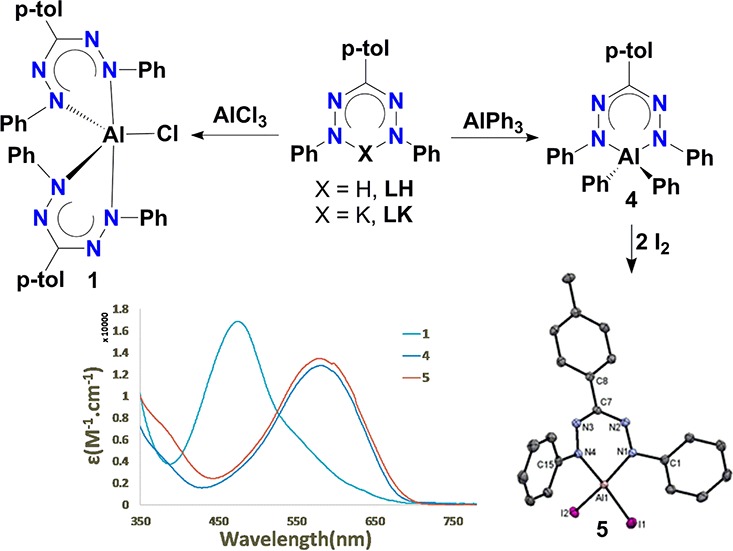
The synthesis of aluminum complexes with redox-active formazanate ligands is described. Salt metathesis using AlCl3 was shown to form a five-coordinate complex with two formazanate ligands, whereas organometallic aluminum starting materials yield tetrahedral mono(formazanate) aluminum compounds. The aluminum diphenyl derivative was successfully converted to the iodide complex (formazanate)AlI2, and a comparison of spectroscopic/structural data for these new complexes is provided. Characterization by cyclic voltammetry is supplemented by chemical reduction to demonstrate that ligand-based redox reactions are accessible in these compounds. The possibility to obtain a formazanate aluminum(I) carbenoid species by two-electron reduction was examined by experimental and computational studies, which highlight the potential impact of the nitrogen-rich formazanate ligand on the electronic structure of compounds with this ligand.
Short abstract
The synthesis of a series of aluminum complexes with redox-active formazanate ligands is described and crystallographic, spectroscopic, and voltammetric characterization data are presented. The reduction chemistry of these newly synthesized complexes has been explored and the results are supported by a computational (DFT) study.
Introduction
Aluminum is one of the most abundant elements in the earth’s crust and is used in a large variety of applications: the metal itself is ductile, lightweight, and corrosion resistant, and it can form various alloys to improve its mechanical properties. Molecular compounds containing aluminum are also widespread: aluminum halides are strong Lewis acid catalysts for Friedel–Crafts reactions, and organometallic derivatives are used in Ziegler–Natta polymerization of olefins.1 The vast majority of aluminum compounds that are known today contain aluminum in the most stable +3 oxidation state. However, complexes of aluminum (and other group 13 elements) in lower oxidation states have recently attracted significant attention for their unusual (transition-metal-like) reactivity2,3 and novel bonding motifs (e.g., clusters,4 multiply bonded compounds).5
Compounds of aluminum with the well-known β-diketiminate ligands have been prepared by alkane elimination (from trialkyl aluminums) or by salt metathesis reactions (using aluminum halides, Scheme 1A). Given the ease of preparation of these ligands and their tunable steric and electronic properties, complexes of this type have been studied in a variety of transformations. For example, β-diketiminate aluminum dialkyls react with strong Lewis acids to form three-coordinate cationic derivatives,6 which show reversible cycloaddition of ethylene,7,8 and these and related cationic aluminum species with anionic bidentate N-ligands have been applied in catalytic reactions that make use of their high Lewis acidity.9,10 Neutral β-diketiminate aluminum complexes with various coligands (e.g., halides,11 alkyls,12−14 amides,14 hydroxides)15−19 have also been prepared, and their reactivity in ring-opening polymerization of cyclic esters was investigated.13,20,21 The majority of these studies employ β-diketiminate ligands with sterically demanding N–Ar substituents (e.g., Ar = 2,6-iPr2C6H3) that shield the Al center, but recently derivatives with less bulky groups were described.22−24 An unusual low-valent aluminum(I) compound with sterically demanding β-diketiminate ligand was prepared by Roesky and co-workers via potassium reduction of the diiodide complex (Scheme 1A).25
Scheme 1. (A) Synthesis of Aluminum Complexes with β-Diketiminate Ligands and (B) Aluminum Complexes with Formazanate Ligands.

In contrast to the rich chemistry reported for aluminum complexes with β-diketiminate ligands, comparatively little is known about the related ligands with two additional nitrogen atoms in the ligand backbone (NNCNN, i.e., formazanates). Sundermeyer and co-workers have reported the synthesis and characterization of a series of group 13 formazanate dialkyl complexes,26 and Gilroy et al. described six-coordinate Al complexes with formazanate-based trianionic N2O2 ligands27 (Scheme 1B). After Hicks’ initial observation of facile ligand-based reductions in a main group compound with a redox-active formazanate ligand,28 our group29−34 as well as the Gilroy group27,35−42 have in the last years developed formazanate chemistry with the lightest group 13 element boron. During our studies of the reduction chemistry, we found that 2-electron reduction of a formazanate boron difluoride compound generates a series of BN-heterocycles that originate from a putative (formazanate)B carbenoid intermediate.32 In recent years, several examples of isolable main group compounds in reduced states have been discovered, and the activation of unreactive bonds by these compounds (e.g., via oxidative addition) is being developed.3,43,44
Given our interest in using redox-active ligands to manipulate electronic structure and stabilize compounds in unusual oxidation states, we set out to explore the synthesis of aluminum complexes with formazanate ligands. In this paper, we provide spectroscopic and crystallographic characterization data for mono- and bis-formazanate aluminum complexes and their electrochemistry. The reduction chemistry of these compounds is described, and the experimental data are supported by a computational (DFT) study.
Result and Discussion
Synthesis of Formazanate Aluminum Complexes
Salt metathesis reactions11,12 were initially evaluated as a route to obtain mono(formazanate) aluminum dihalide complexes, which could subsequently serve as synthons for low-valent Al compounds. Thus, a suspension of AlCl3 in THF-d8 was treated in an NMR tube with 1 equiv of the potassium formazanate salt K[PhNNC-(p-tol)NNPh]·2THF (LK),45 resulting in a rapid color change from purple to dark red. The 1H NMR spectrum of this solution showed diagnostic resonances for the p-tolyl and phenyl moieties in a 1:2 ratio that were shifted from the starting material, indicating the formation of a new formazanate aluminum complex. On preparative scale, 1 could be obtained as crystalline material in moderate yield (36%) by diffusion of hexane into a THF solution of the product (Scheme 2). A single-crystal X-ray structure determination showed that instead of the desired mono(formazanate)aluminum complex the product is bis(formazanate) complex [PhNNC(p-tol)NNPh]2AlCl (1) (Figure 1, pertinent bond lengths and angles are presented in Table 1) in which the Al center is bound to two bidentate formazanate ligands. The coordination sphere is complemented by a chloride ligand, resulting in a five-coordinate Al(III) complex. The geometry around the Al center is best described as a distorted trigonal bipyramid (τ = 0.6353),46 with a bond angle between the “axial” Al–N bonds (N1–Al1–N8) of 165.22(1)°.
Scheme 2. Synthesis of Bis(formazanate) Aluminum Chloride Compound 1.

Figure 1.

(a) Molecular structure of 1 showing 50% probability ellipsoids. Hydrogen atoms were omitted, and NPh groups are shown as wireframe for clarity. (b) Coordination sphere around Al center.
Table 1. Selected Bond Lengths (Å) and Bond Angles (deg) for 1.
| bond lengths | bond angles | ||
|---|---|---|---|
| Al1–Cl1 | 2.143(1) | N1–Al1–N8 | 165.22(1) |
| Al1–N1/Al1–N8 | 1.999(4)/2.007(4) | N1–Al–Cl1/N8–Al–Cl1 | 97.72(1)/97.03(1) |
| Al1–N4/Al1–N5 | 1.923(3)/1.921(3) | N1–Al1–N4/N8–Al1–N5 | 81.29(1)/81.89(1) |
| N1–N2/N7–N8 | 1.316(4)/1.307(4) | N1–Al1–N5/N8–Al1–N4 | 90.00(1)/89.18(1) |
| N3–N4/N5–N6 | 1.320(5)/1.322(4) | Cl1–Al–N4/Cl1–Al–N5 | 127.10(1)/126.25(1) |
| N4–Al–N5 | 106.65(1) | ||
The Al center is significantly displaced out of the planes defined by formazanate NNNN backbones (displacement from N1–N4/N5-N8 planes is 1.068/1.058 Å, respectively). The molecular structure of 1 reveals that the Al–N1 and Al–N8 bond vectors lie along the axial directions and the Al–Cl1, Al–N4, and Al–N5 bonds form the equatorial plane in 1 (Figure 1). The axial Al–N bonds are elongated as compared to equatorial Al–N bonds by about 0.081 Å, similar to related pentacoordinated Al(III) compounds in the literature,47,48 and both are significantly longer than the Al–N bonds in four-coordinate β-diketiminate aluminum dichlorides.11 The Al–Cl bond length in 1 (2.143(1) Å) is somewhat shorter than that in Berben’s pentacoordinated aluminum chloride complex with two bidentate nitrogen ligands (2.191(1) Å).47
The room-temperature 1H NMR spectrum of 1 reveals only one set of signals for the NPh groups. The equivalence of the axial and equatorial positions is likely due to facile exchange via Berry pseudorotation.49 The 27Al NMR of 1 shows a resonance at 43.0 ppm, which is in agreement with the presence of a pentacoordinated Al center in 1.50−53
The cyclic voltammogram of compound 1 was measured in THF solution (Figure 2), which revealed two quasi-reversible 1-electron redox-events at −1.36 and −1.67 V vs Fc0/+. These values are in a range similar to that of our previously published bis(formazanate)Zn compounds29,31 and consistent with two independent, sequential 1-electron reduction processes for each ligand in compound 1. Scanning toward more negative potentials resulted in several additional redox events below −1.9, which were not analyzed in detail (see Figure S1).
Figure 2.
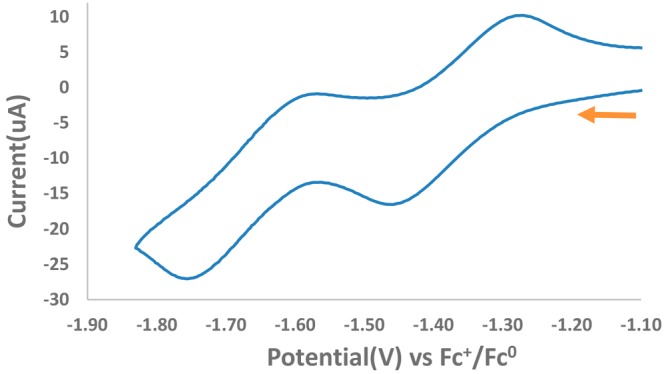
Cyclic voltammogram of 1 (1.5 mM solution in THF, 0.1 M [Bu4N][PF6]) recorded at 100 mVs–1.
Given that straightforward salt metathesis reactions failed to give mono(formazanate) aluminum dihalides, we subsequently chose a two-step protocol based on the method of Roesky and co-workers for the synthesis of β-diketiminate aluminum diiodides.25 Thus, the free formazan (LH) was treated with AlEt3 in a 1:1 molar ratio in an NMR tube in C6D6 (Scheme 3). As reported by Sundermeyer and co-workers for the aluminum methyl analogue,26 this results in rapid formation of an intense blue solution for which the integrated intensities in the 1H NMR spectrum are in agreement with the presence of two AlEt groups per formazanate ligand.
Scheme 3. Synthesis of Dialkyl Aluminum Formazante and (Monoiodo) (Monoalkyl) Aluminum Formazanate Compounds.

The NMR data indicate that the product is the diethyl aluminum compound LAlEt2 (2a). Compound 2a is quite sensitive and is quickly hydrolyzed upon exposure to air. The high solubility of 2a in common organic solvents rendered its purification by crystallization difficult. Treatment of in situ generated 2a with 2 equiv of I2 results in Et/I exchange as evidenced by the formation of EtI. However, the NMR spectrum indicates that only one AlEt group is replaced: One AlEt signal remains (AlCH2 at 0.66 ppm), and the product is formulated as LAl(Et)I (3a). Longer reaction times as well as addition of more I2 (additional 2 equiv) did not lead to further conversion by iodine exchange, indicating that it may be thermodynamically unfavorable to proceed beyond the monoethyl compound LAl(Et)I. Gently warming the NMR solution for 6 h to 40 °C did not indicate any further change, whereas decomposition was observed after 2 h at 70 °C. Testing AlMe3 as the starting material suggested formation of the mono(formazanate) aluminum dimethyl complex26 but also for this compound iodine exchange only gave monoiodo product 3b (Scheme 3).
Diphenyl derivative 4 (prepared from LH and AlPh3) could be obtained in crystalline form in 80% yield. The subsequent iodine exchange reaction was attempted by the reaction of 2 equiv of I2 with 4 in an NMR tube in C6D6. In contrast to the dialkyl derivatives mentioned above, 4 cleanly converted to the desired aluminum diiodide [(PhNNC(p-tol)NNPh)AlI2] (5) as evidenced by the formation of 2 equiv of PhI and complete disappearance of the diagnostic resonances for the AlPh2 moieties. On a preparative scale, 5 could be isolated as a crystalline material in good yield (70%) by slow diffusion of hexane into the reaction mixture in toluene (Scheme 4).
Scheme 4. Synthesis of Compounds 4 and 5.

Although the 27Al NMR resonance of 4 could not be observed, diiodide analogue 5 shows a broad resonance at 69.50 ppm, consistent with a four-coordinated Al(III) center.12,54,55 In the 1H and 13C NMR spectra, the presence of only one set of resonances for the N–Ph substituents of the ligand in 4 and 5 indicate that both molecules have (average) C2v symmetry in solution.
Single-crystal structure determinations for compounds 4 and 5 revealed the details of their molecular structures. A bidentate formazanate ligand is bound through its terminal N atoms forming a six-membered chelate ring, and the coordination sphere is complemented by two phenyl or iodide ligands, respectively (Figure 3). The overall geometries of 4 and 5 are distorted tetrahedral around the Al centers, which are displaced out of the plane composed by 4 N atoms from the ligand backbone by 0.531 and 0.419 Å, respectively. The smaller displacement for 5 is in accordance with less steric hindrance between the formazanate ligand and the Al substituents (I in 5 vs Ph in 4). A similar trend for the displacement of the Al center from the ligand plane was observed in related mono(β-diketiminate) aluminum complexes.11,12 The equivalent N–N and N–C bond distances support the presence of a delocalized ligand backbone in 4 and 5 (for selected bond lengths and bond angles, see Table 2). Similar bond distances were found in the corresponding tetra- and hexa-coordinated formazanate aluminum compounds reported by Sundermeyer et al.26 and Gilroy et al.,27 respectively. The Al–N bonds (1.893(2)–1.900(2) Å) in 5 are somewhat shorter than the Al–N bonds present in 4 (1.933(1)–1.934(1) Å), which could be attributed to the increased positive charge on Al center in 5 as iodide is more electron-withdrawing than a phenyl group.
Figure 3.

Molecular structures of 4 (left) and 5 (right) showing 50% probability ellipsoids. Hydrogen atoms are omitted for clarity.
Table 2. Selected Bond Lengths (Å) and Bond Angles (deg) for 4, 42–, and 5.
| 4 | 42– | 5 | |
|---|---|---|---|
| Al1–N1 | 1.9336(9) | 1.871(2) | 1.893(2) |
| Al1–N4 | 1.9334(8) | 1.873(2) | 1.900(2) |
| Al–C21 | 1.962(1) | 2.011(2) | |
| Al–C27 | 1.957(1) | 2.011(2) | |
| Al–I1 | 2.497(7) | ||
| Al–I2 | 2.494(7) | ||
| N1–N2 | 1.315(1) | 1.432(2) | 1.317(2) |
| N2–C7 | 1.346(1) | 1.332(2) | 1.344(3) |
| C7–N3 | 1.347(1) | 1.328(2) | 1.346(3) |
| N3–N4 | 1.307(1) | 1.432(2) | 1.313(2) |
| N1–C1 | 1.433(1) | 1.375(2) | |
| N4–C15 | 1.430(1) | 1.371(3) | |
| N1–Al1–N4 | 90.01(4) | 98.54(7) | 92.83(8) |
Cyclic voltammetry was employed to investigate the electrochemical properties of 4 and 5. The voltammogram of 4 in THF solution (Figure 4) shows two quasi-reversible one-electron redox events at E0 = −1.12 and −2.13 V vs Fc0/+1 that correspond to two sequential ligand-based reductions to generate the radical anion 4•– and the dianion 42–. In comparison to the boron analogue, these redox-potentials are shifted toward positive potential by 130 and 230 mV, respectively.34 In contrast to the data for the diphenyl complex 4, the voltammogram of the aluminum iodide 5 measured in either THF or DME solution is poorly reversible and shows reduction events that are shifted to more negative potentials (see the Supporting Information). The changes in electrochemistry upon changing the Al-substituents from Ph to I may reflect the lability of the Al–I bond upon reduction.
Figure 4.

Cyclic voltammetry of 4 (1.5 mM solution in THF, 0.1 M [Bu4N][PF6]) recorded at 100 mV s–1. Asterisks indicate redox-events due to a small amount of free ligand which is either present in the sample or generated in THF from 4 (see Figures S2–S4).
The UV/vis absorption spectra were recorded for compounds 1, 4, and 5 in toluene solution to obtain more insight into the electronic properties for these compounds. All three compounds show intense absorption maxima in the visible range of the spectrum, with extinction coefficients that range between 13 000 and 16 800 L·mol–1·cm–1, which are assigned to π–π* transitions within the formazanate ligand backbone (Figure 5).26,27 Bis(formazanate) complex 1 shows a broad absorption band at 478 nm with a shoulder toward lower energy. The absorption maximum for 1 is hypsochromically shifted by ca. 10 nm with respect to that of the free formazan (488 nm in CHCl3,27 490 nm in hexane).26 This stands in marked contrast to the bathochromic shift that is typically observed in formazanate complexes, a feature that has been ascribed to the anionic nature of the ligand and its increased rigidity upon complexation.27 A reason for this discrepancy likely lies in the way the metal center is incorporated in the six-membered chelate structure: Other mono(formazanate) aluminum complexes have more planar AlN4C rings, providing an extended conjugated π-system that includes the N–Ar groups. In contrast, the presence of two formazanate ligands in 1 enforces a strong “butterfly” distortion of the chelate ring in which the Al center is >1 Å displaced from the coordination plane, and disrupts conjugation with the N–Ph groups.
Figure 5.

Absorption spectra of compounds 1, 4, and 5 in toluene.
Mono(formazanate) compounds 4 and 5 have very similar spectra with a broad absorption maximum at ca. 583 nm (Figure 5). In comparison to the low-energy π–π* transition in the formazanate aluminum dimethyl compound reported by Sundermeyer (λmax = 559 nm in hexane),26 compounds 4 and 5 are red-shifted by ca. 25 nm. Comparing diphenyl complex 4 with its boron analogue (λmax = 505 nm)34 shows that the low-energy band is red-shifted even further. Both observations are consistent with an increase in ionic character for the bonding in 4/5, due to either the presence of more electron-withdrawing groups on Al (Ph/I vs Me) or a difference in electronegativity of the central element (Al vs B).
Reduction Chemistry
Reduction reactions of 1, 4, and 5 have been explored. Addition of excess Cp2Co into a THF solution of 1 results in a color change from dark red to green, a color that is associated with formazanate-centered reductions in boron complexes with this ligand and consistent with formation of the radical anion [(PhNNC(p-tol)NNPh)2AlCl]− (1•–, Scheme 5).28−30,34 The crude product was washed with hexane and analyzed by EPR spectroscopy. The EPR spectrum of 1•– in fluid THF solution shows a nine-line signal centered at g ∼ 2 with a hyperfine coupling constant of 6.35 G (Figure S6). The observed hyperfine coupling pattern indicates that the unpaired electron interacts with 4 (equivalent) 14N nuclei (I = 1), which suggests that it is localized on one of the ligands instead of being delocalized over both formazanates.
Scheme 5. Compound 1.

Reduction of compound 1 (left) and SOMO of 1–•calc (right).
The UV/vis spectrum shows two absorptions in the visible range, one at high energy (λmax = 426 nm) and one at low energy (λmax = 677 nm) (Figure S7). Despite the fact that we were unable to obtain analytically pure, crystalline material of 1•–, the EPR and UV/vis spectroscopic features are in agreement with a ligand-based reduction of 1 to form the aluminum-analogue of a verdazyl radical.56,57 A ligand-centered reduction is further supported by the computational (gas phase DFT) study. The elongation in N–N bonds in 1–•calc compared to 1 and 1calc (see Table S2) is consistent with the ligand-based reduction as the additional electron populates the ligand N–N π*-orbital.34 The spin density plot of 1–•calc indicates the excess spin density is delocalized over both ligands (see Figure S14), a feature that is inconsistent with the EPR spectroscopy (vide supra). This discrepancy is likely due to the fact that the DFT calculations were performed on the isolated anion 1–•calc in the gas phase. In condensed phase in the presence of counterions, the unpaired electron is likely more localized due to electrostatic interactions: a similar effect is observed in related bis(formazanate) zinc radical anions.29
Similarly, treatment of a THF solution of 4 with Cp2Co results in an immediate color change from intense blue to green, the solution EPR spectrum (Figure S6) of which furnishes a broad signal with g-value ∼2, indicating the formation of [Cp2Co]+[(PhNNC(p-tol)NNPh)AlPh2]−• (4–•, Scheme 6). Similar to the boron analogues of 4–• reported previously,34 the hyperfine interactions between the unpaired electron and the nitrogen nuclei could not be resolved in this case. Compound 4–• could be obtained in 60% yield as a green powder. The UV/vis spectrum of 4–• shows two absorptions, one at high energy (λmax = 460 nm) and one at low energy (λmax = 759 nm)) (Figure S7). The absorptions at 460 and 759 nm are blue- and red-shifted, respectively, compared to the corresponding absorption spectrum found in its parent compound 4 (λmax = 583 nm), which provides evidence for the formation of a formazanate-centered radical.28−30,34
Scheme 6. Synthesis of 1- and 2-Electron-Reduced Derivatives of 4.

Two-electron reduction of 4 was investigated using Na(Hg) as the reducing agent in THF solution, which resulted in a color change from dark blue to orange, via a green transient intermediate (presumably 4–•). The product [(PhNNC(p-tol)NNPh)AlPh2]2– (42–) was obtained in crystalline form (42%) as the disodium salt by diffusion of hexane into a DME solution (Scheme 6). Single-crystal structure determination of 42– reveals that the structure is overall similar to that of the boron analogue reported previously.34 It shows a distorted tetrahedral geometry around the Al center, which is displaced out of the plane comprised of N1, N2, C7, and N3 atoms from the ligand backbone (i.e., N1N2C7N3-plane) by 0.260 Å (Figure 6). Ligand-based two-electron reduction leads to elongation of the N–N bonds in the ligand backbone to 1.432(2) Å, indicative of a single bond character.34
Figure 6.
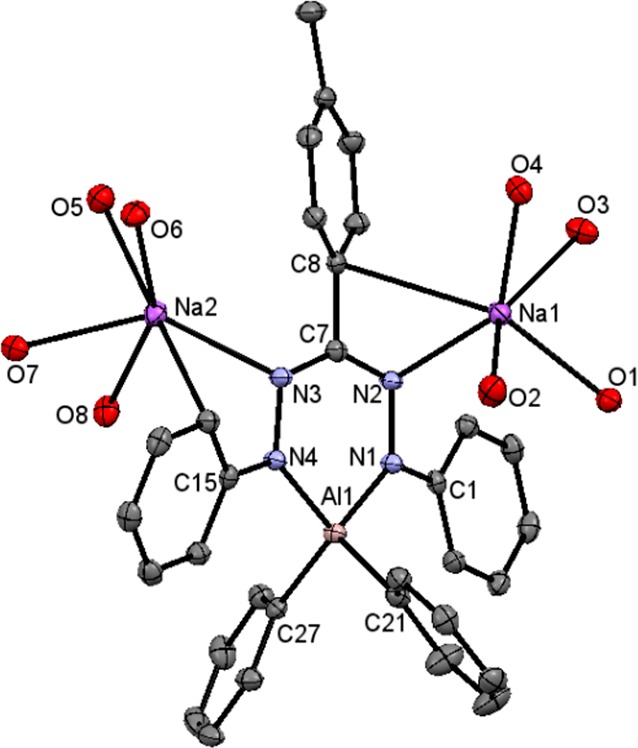
Molecular structures of 42– (right) showing 50% probability ellipsoids. Hydrogen atoms and DME molecules (except for the O atoms bonded to Na) are omitted for clarity.
The 500 MHz 1H NMR spectrum for 42– shows the expected number of resonances for a C2v-symmetric complex, but the resonances are somewhat broad at room temperature. Variable-temperature measurements indicate this to be due to dynamic processes, the details of which will be described elsewhere. The UV/vis spectrum of 42– shows an absorption band at 486 nm (Figure S7), which is blue-shifted by 97 nm compared to its precursor 4 (λmax = 583 nm), as expected for a complex with this electron-rich (trianionic) form of the ligand.34
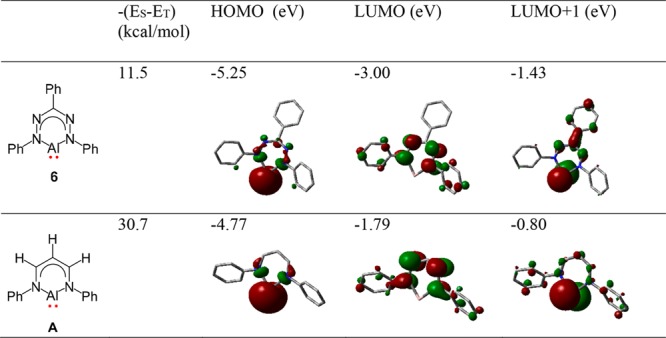
Having established two-electron reduction chemistry for diphenyl complex 4, we turned our attention to diiodide 5. In this case, reduction would likely be accompanied by loss of halide, similar to what was found before for a formazanate boron difluoride compound.32 Roesky and co-workers reported that reduction of a (β-diketiminate) aluminum diiodide with 2 equiv of K afforded an unusual low-valent aluminum(I) carbenoid,25 and its reactivity has been explored in detail in recent years.58 We were interested to explore whether the (formazanate)Al(I) analogue would be accessible. Treatment of a toluene solution of 5 with 2 equiv of potassium graphite (KC8) at room temperature resulted in an initial color change from blue to green, which is indicative of the ligand-centered reduction of 5 to verdazyl-type radical 5–•.28−30,34 The green color faded within 30 min and ultimately a pale blue solution is obtained. Analysis of the crude product by spectroscopy showed that it was NMR silent, but showed a nine-line signal (g ∼ 2) in the solution EPR spectrum. All attempts at crystallization of the product(s) were unsuccessful. To probe the nature of the product(s), oxidation of the mixture by addition of I2 was carried out to probe whether the starting material could be recovered, but this proved not to be the case. Although the product(s) of two-electron reduction of 5 remain to be identified, we carried out DFT calculations on (formazanate)Al(I) carbenoid (6) and compare its electronic structure to Roesky’s β-diketiminate analogue. The computational study reveals that the aluminum carbenoid 6 has a singlet ground state with a singlet–triplet energy gap of 11.5 kcal/mol at the B3LYP/6-311G(d,p) level of theory (Table 3). In case of Roesky’s (β-diketiminate)Al(I) complex, it was found that complexes of this type have a singlet–triplet energy gap of ca. 30–35 kcal/mol,59,60 depending on the ligand substitution pattern. In agreement with the literature, we calculate Roesky’s (β-diketiminate)Al complex (with N–Ph substituents; A in Table 3) to have a singlet–triplet separation of 30.7 kcal/mol at the B3LYP/6-311G(d,p) level of theory, with the singlet state lowest in energy. Analysis of the frontier orbitals for the β-diketiminate and formazanate compounds shows that in both cases the HOMO is an Al-based lone pair, whereas the LUMO and LUMO+1 are found to be localized on the ligand (π*) and the Al center (p-orbital), respectively (Table 3). Although the appearance of the frontier orbitals is similar between the two, the orbital energies are overall lower for the formazanate compound. Of these three frontier orbitals, the formazanate-based π*-orbital is the most stabilized (1.21 eV lower in energy in 6 than in A), whereas the Al-orbitals are shifted in energy by only 0.48–0.63 eV; this accounts for the much lower singlet–triplet separation in 6 than in A. From the literature on carbenes (or analogues) of group 13/14 elements, it is apparent that the singlet–triplet energy separation (ΔES–T) is correlated to reactivity and stability.43,44,60−63 Thus, although the nitrogen-rich backbone of the formazanate ligand allows tuning of the singlet–triplet gap, the small value obtained for ΔES–T in this case likely results in a system that is too reactive to be isolable.
Table 3. Calculated Frontier Orbital Energies and Singlet-Triplet Energy Gaps [−(ES – ET)] for Low-Valent Aluminum Compounds with Formazanate and β-Diketiminate Ligands at the B3LYP/6-311G(d,p) Level of Theory.
Conclusions
In conclusion, a series of aluminum(III) complexes with redox-active formazanate ligands has been synthesized and characterized. The five-coordinated bis-formazanate aluminum chloride complex 1 was obtained by the salt metathesis reaction, whereas four-coordinated monoformazanate aluminum diiodide complex 5 was prepared by following a two-step synthetic protocol starting from the free ligands, via corresponding aluminum diphenyl compound 4. The characterization data for 1, 4, and 5, both in the solid state as well as in solution, provide insights into their electronic and structural properties. In addition, the (electro)chemical reduction of 1, 4, and 5 further disclose ligand-centered redox-reactions in these complexes. The possibility to afford (formazanate)Al(I) carbenoid 6 by the two-electron reduction of diiodide complex 5 was investigated by experimental and computational (DFT) studies. The computational data show that in comparison to Roesky’s (β-diketiminate)Al(I) complex, the LUMO of the nitrogen-rich formazanate ligand (NNCNN) is significantly stabilized, which results in a computed singlet–triplet energy separation of only 11.5 kcal/mol. The synthesis and characterization data for aluminum complexes with one or two redox-active formazanate ligands reported in this paper provides an entry to studying the reactivity of compounds that combine the electrophilic properties of Al with ligand-based redox activity.
Experimental Section
General Considerations
All manipulations were carried out under a nitrogen or an argon atmosphere using standard glovebox, Schlenk, and vacuum-line techniques. Toluene and hexane (Aldrich, anhydrous, 99.8%) were passed over columns of Al2O3 (Fluka), BASF R3-11-supported Cu oxygen scavenger, and molecular sieves (Aldrich, 4 Å). THF (Aldrich, anhydrous, 99.8%) was dried by percolation over columns of Al2O3 (Fluka). THF, DME, and hexane were additionally dried on sodium/potassium alloy and subsequently vacuum-transferred and stored under nitrogen. All solvents were degassed prior to use and stored under nitrogen. C6D6 (Aldrich) and d8-THF (Sigma-Aldrich) were vacuum-transferred from sodium/potassium alloy and stored under nitrogen. The ligand PhNNC(p-tolyl)NNPh (LH)28 was synthesized according to a published procedure. NMR spectra were recorded on Varian Mercury 400 or Inova 500 spectrometers. The 1H and 13C NMR spectra were referenced internally using the residual solvent resonances and reported in ppm relative to TMS (0 ppm); J is reported in Hz. Assignments of NMR resonances was aided by COSY, NOESY, HSQC and HMBC experiments using standard pulse sequences. Elemental analyses were performed at the Kolbe Microanalytical Laboratory (Mülheim an der Ruhr, Germany). UV–vis spectra were recorded in toluene, DCE, and THF solution (∼10–3 M) in a quartz cuvette that was sealed under N2 atmosphere using an AVANTES AvaSpec-2048 spectrometer. Cyclic voltammetry (CV) was performed using a three-electrode configuration comprising a Pt wire counter electrode, a Ag wire pseudoreference electrode and a Pt disk working electrode (CHI102, CH Instruments, diameter = 2 mm). The Pt working electrode was polished before each experiment using an alumina slurry (0.05 μm) and rinsed (thrice) with distilled water and acetone. The electrodes were then dried in an oven at 75 °C for at least 1 h to remove any residual traces of water. The CV data were referenced by measuring the redox potential of ferrocene, which was added into the solution at the end of the experiment. In all cases, there is no indication that addition of ferrocene influences the electrochemical behavior. All electrochemical measurements were performed at ambient temperatures in a nitrogen glovebox with the compounds dissolved in THF or DME containing 0.1 M [nBu4N][PF6] as the supporting electrolyte. The electrochemical data were measured using an Autolab PGSTAT 100 computer-controlled potentiostat with Autolab NOVA software (v.2.1.3).
Synthesis of [(PhNNC(p-tol)NNPh)2AlCl] (1)
The potassium formazanate salt K[PhNNC-(p-tol)NNPh]·2THF (LK) (1.000 g, 2.013 mmol) was dissolved in 30 mL of THF into a double Schlenk flask fitted with a filter. Subsequently, 0.5 equiv of anhydrous AlCl3 (0.134 g, 1.007 mmol) was added to this solution. The reaction mixture was stirred for 1 h at room temperature during which the color changed from pink to light red. After that, all volatiles were removed under reduced pressure. 1H NMR analysis of the crude product indicates full conversion of the starting materials. The crude product was dissolved again in toluene (30 mL) and filtered to the second tube of the double Schlenk flask. The residue was extracted three times with toluene (30 mL) and the combined filtrate was concentrated in vacuo to ca. 20 mL. Hexane was layered on top of the toluene solution, and after diffusion of the two layers, a solid product precipitated. The supernatant was removed and the residue washed with hexane (3 × 10 mL) and then dried under vacuum. This gave 250 mg of 1 as dark red crystals (0.360 mmol, 36%). Crystalline material of 1 was also obtained by slow diffusion of hexane into a THF solution of 1. 1H NMR (400 MHz, C6D6, 25 °C) δ 7.93 (d, J = 8.1 Hz, 4H, p-tol o-H), 7.58 (d, J = 7.8 Hz, 8H, NPh o-H), 7.14 (d, J = 8.1 Hz, 4H, p-tol m-H), 7.02 (t, J = 7.7 Hz, 8H, NPh m-H), 6.94 (t, J = 7.3 Hz, 4H, NPh p-H), 2.20 (s, 6H, p-tol-CH3). 27Al NMR (104 MHz, C6D6, 25 °C) δ 43.0 (γ1/2 = 254 Hz). 13C NMR (100 MHz, C6D6, 25 °C) δ 150.73 (NPh ipso-C), 148.32 (NCN), 138.79 (p-tol-CH3ipso-C), 132.95 (NCN-p-tol ipso-C), 129.42 (p-tol m-CH), 128.90 (NPh m-CH), 127.63 (NPh p-CH), 126.50 (p-tol o-CH), 124.29 (NPh o-CH), 21.42 (p-tol-CH3). Anal. Calcd for C40H34AlClN4: C, 69.71; H, 4.97; N, 16.26. Found: C, 69.82; H, 5.04; N, 16.10.
Synthesis of [(PhNNC(p-tol)NNPh)2AlCl][Cp2Co] (1–•)
Compound 1 (50 mg, 0.072 mmol) was dissolved in 2 mL of THF, and an excess amount of cobaltocene (55 mg, 0.290 mmol) was added. An immediate color change was observed from dark red to green. The reaction mixture was further stirred for 1 h at room temperature. Addition of 4 mL of hexane precipitated a green powder. Washing the green powder with hexane (3 × 1 mL) and drying in vacuo afforded 30 mg of 1–• as a green powder (0.034 mmol, 47% yield). Satisfactory elemental analysis data could not be obtained.
Synthesis of [(PhNNC(p-tol)NNPh)AlPh2] (4)
Free ligand 3-p-tolyl-1,5-diphenyl-formazan LH (5.000 g, 15.92 mmol) was dissolved in 100 mL of toluene in a Schlenk flask. To this cherry red formazan solution was added a 1 M solution of triphenyl aluminum (17.51 mL, 17.51 mmol) in dibutyl ether at room temperature. The reaction mixture was stirred for 12 h at room temperature, and the color changed from cherry red to dark purple. After this, the solvent was removed under reduced pressure. The 1H NMR spectrum of the crude product indicated full conversion of the starting materials and formation of 4. The crude product was dissolved in toluene and layered with hexane and subsequently crystallized by keeping the solution at −30 °C overnight. The purple crystalline product was washed with hexane (3 × 25 mL) and dried under vacuum to give 4 as intensely colored pink crystals (6.340 g, 12.820 mmol, 80%). 1H NMR (400 MHz, C6D6, 25 °C) δ 8.21 (d, J = 8.2 Hz, 2H, p-tol o-H), 7.80 (m, 8H (NPh o-H and AlPh o-H)), 7.14–7.16 (overlapped, 8H (2H = p-tol m-H, 6H = AlPh (m + p)-H)), 6.85 (t, J = 7.8 Hz, 4H, NPh m-H), 6.76 (t, J = 7.3 Hz, 2H, NPh p-H), 2.16 (s, 3H, p-tol-CH3). 13C NMR (100 MHz, C6D6, 25 °C) δ 150.12 (NPh ipso-C), 149.33 (NCN), 143.25 (AlPh ipso-C), 138.42 (NCN-p-tol ipso-C), 138.28 (AlPh o-CH), 134.92 (p-tol-CH3ipso-C), 129.74 (p-tol m-CH), 129.28 (NPh m-CH), 129.13 (NPh p-CH), 128.74 (AlPh p-CH), 128.25 (AlPh m-CH), 126.30 (p-tol o-CH), 122.36 (NPh o-CH), 21.29 (p-tol-CH3). Satisfactory elemental analysis data could not be obtained.
Synthesis of [(PhNNC(p-tol)NNPh)AlPh2][Cp2Co] (4–•)
Compound 4 (100 mg, 0.200 mmol) was dissolved in 4 mL of THF, and 1 equiv of cobaltocene (40 mg, 0.200 mmol) was added. An immediate color change was observed from dark purple to green. The reaction mixture was further stirred for 4 h at room temperature. Addition of 4 mL of hexane precipitated a green powder. Washing the green powder with hexane (3 × 2 mL) and drying in vacuo afforded 85 mg of 4–• as a green powder (0.120 mmol, 60% yield). Satisfactory elemental analysis data could not be obtained.
Synthesis of [(PhNNC(p-tol)NNPh)AlPh2][Na2(DME)4] (42–)
Compound 4 (200 mg, 0.404 mmol) was dissolved in 7 mL of THF, and 2 equiv of Na(Hg) were added. The reaction mixture was stirred for 12 h, during which it changed its color from dark purple to green and finally orange. Then, the orange solution was filtered through a syringe filter, and the solvent was evaporated under reduced pressure. After that, the crude product was dissolved in 5 mL of DME, and the solution was layered with 5 mL of hexane. Slow diffusion of the two layers at −30 °C precipitated orange crystals, which were washed with pentane (3 × 3 mL) and dried to give compound 42– as a highly air-sensitive solid (155 mg, 0.170 mmol, 42%). 1H NMR (500 MHz, THF-d8, −30 °C) δ 8.30 (d, J = 7.8 Hz, 2H, p-tol o-H), 7.77 (d, J = 7.3 Hz, 4H, AlPh o-H), 7.40 (d, J = 7.5 Hz, 2H, N(1)Ph o-H), 7.09 (d, J = 7.7 Hz, 2H, p-tol m-H), 7.01–6.93 (m, 8H, 2H = N(1)Ph m-H, 6H = AlPh (m + p)-H), 6.83 (d, J = 7.9 Hz, 2H, N(2)Ph o-H), 6.50 (t, J = 7.0 Hz, 2H, N(2)Ph m-H), 5.92 (t, 2H, NPh p-H), 3.42 (s, 16H, DME), 3.26 (s, 24H, DME), 2.33 (s, 3H, p-tol-CH3). 27Al NMR (104 MHz, THF-d8, 25 °C) δ 105.0 (γ1/2 = 1470 Hz). 13C NMR (125 MHz, THF-d8, −30 °C) δ 156.31 (NPh ipso-C), 154.68 (AlPh ipso-C), 150.79 (NCN), 144.59 (NCN-p-tol ipso-C), 139.53 (AlPh o-CH), 135.41 (p-tol-CH3ipso-C), 129.94 (N(1)Ph m-CH), 128.11 (p-tol (m + o)–CH), 127.62 (N(2)Ph m-CH), 126.64 (AlPh m-CH), 125.87 (AlPh p-CH), 116.87 (N(1)Ph o-CH), 109.89 (NPh p-CH), 107.54 (N(2)Ph o-CH), 72.71 (DME), 59.01 (DME), 21.43 (p-tol-CH3). Satisfactory elemental analysis data could not be obtained.
Synthesis of [(PhNNC(p-tol)NNPh)AlI2] (5)
Compound 4 (0.700 g, 1.42 mmol) was dissolved in 25 mL of toluene in a Schlenk flask. Subsequently, 2 equiv of I2 (0.718 g, 2.84 mmol) was added to this dark purple solution. The reaction mixture was stirred for 12 h at room temperature, during which the color faded to light purple. After this the solvent was evaporated under reduced pressure. 1H NMR analysis of the crude product indicates full conversion of the starting material and formation of 5. The crude product was dissolved in toluene, layered with hexane, and kept at −30 °C to crystallize. The supernatant was removed and the crystalline solid was washed with hexane (3 × 5 mL) and dried under vacuum to give 590 mg of compound 5 as light pink crystals (0.990 mmol, 70%). 1H NMR (400 MHz, C6D6, 25 °C) δ 7.97 (d, J = 8.0 Hz, 6H (2H = p-tol o-H, 4H = NPh o-H)), 7.10 (d, J = 8.0 Hz, 2H, p-tol m-H), 7.00 (t, J = 7.5 Hz, 4H, NPh m-H), 6.94 (t, J = 7.2 Hz, 2H, NPh p-H), 2.15 (s, 3H, p-tol-CH3). 27Al NMR (104 MHz, C6D6, 25 °C) δ 69.50 (γ1/2 = 370 Hz). 13C NMR (100 MHz, C6D6, 25 °C) δ 150.33 (NCN), 148.40 (NPh ipso-C), 139.23 (NCN-p-tol ipso-C), 133.55 (p-tol-CH3ipso-C), 129.81 (p-tol m-CH), 129.72 (NPh p-CH), 129.36 (NPh m-CH), 126.46 (p-tol o-CH), 123.46 (NPh o-CH), 21.27 (p-tol-CH3). Anal. Calcd for C20H17AlN4I2: C, 40.43; H, 2.88; N, 9.43. Found: C, 40.35; H, 3.06; N, 9.34.
X-ray Crystallography
Suitable crystals of compounds 1, 4, and 5 were mounted on top of a cryoloop and transferred into the cold (100 K) nitrogen stream of a Bruker D8 Venture diffractometer. Data collection and reduction was done using the Bruker software suite APEX2.64 Data collection was carried out at 100 K using either Mo radiation (0.71073 Å) (for 1, 4, and 5) or Cu radiation (1.54178 Å) (for 42–). The final unit cell was obtained from the xyz centroids of 9942 (1), 9860 (4), 9890 (42–), and 9248 (5) reflections after integration. A multiscan absorption correction was applied, based on the intensities of symmetry-related reflections measured at different angular settings (SADABS).64 The structures were solved by intrinsic phasing methods using SHELXT.65 The hydrogen atoms were generated by geometrical considerations, constrained to idealized geometries, and allowed to ride on their carrier atoms with an isotropic displacement parameter related to the equivalent displacement parameter of their carrier atoms. Crystal data and details on data collection and refinement are presented in Table 4.
Table 4. Crystallographic Data for 1, 4, 42–, and 5.
| 1 | 4 | 42– | 5 | |
|---|---|---|---|---|
| chem formula | C40H34AlClN8 | C32H27AlN4 | C48H67AlN4Na2O8 | C20H17AlI2N4 |
| Mr | 689.18 | 494.55 | 901.01 | 594.16 |
| cryst syst | triclinic | Monoclinic | monoclinic | Monoclinic |
| color, habit | dark red, block | blue, block | orange, block | blue, block |
| size (mm) | 0.15 × 0.12 × 0.08 | 0.22 × 0.17 × 0.13 | 0.22 × 0.05 × 0.04 | 0.31 × 0.13 × 0.05 |
| space group | P1̅ | C2/c | P21/c | P21/c |
| a (Å) | 8.1107(8) | 21.5590(14) | 13.5694(4) | 8.1926(5)) |
| b (Å) | 12.0912(12) | 15.3001(11) | 21.6184(6) | 14.8013(9) |
| c (Å) | 18.7007(19) | 18.3158(12) | 17.2708(5) | 17.4444(12) |
| α (deg) | 76.541(4) | 90 | 90 | 90 |
| β (deg) | 87.705(4) | 119.229(2) | 107.021(2) | 97.715(2) |
| γ (deg) | 85.258(4) | 90 | 90 | 90 |
| V (Å3) | 1777.0(3) | 5272.3(6) | 4844.4(2) | 2096.2(2) |
| Z | 2 | 8 | 4 | 4 |
| ρcalc, g·cm–3 | 1.288 | 1.246 | 1.231 | 1.883 |
| radiation [Å] | 0.71073 | 0.71073 | 1.54178 (Cu) | 0.71073 |
| μ(Mo Kα) (mm–1) | 0.174 | 0.105 | 3.055 | |
| μ(Cu Kα) (mm–1) | 0.989 | |||
| F(000) | 720 | 2080 | 1928 | 1136 |
| temp (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| θ range (deg) | 2.951–25.679 | 2.952–30.583 | 4.890–70.070 | 2.964–30.587 |
| data collected (h, k, l) | –9:9, −14:14, −22:22 | –30:29, −21:21, −24:26 | –16:16, −25:26, −21:21 | –11:11, −21:19, −24:24 |
| no. of rflns collected | 27047 | 74978 | 92849 | 59061 |
| no. of indpndt reflns | 6656 | 8062 | 9025 | 6418 |
| observed reflns Fo ≥ 2.0 σ (Fo) | 5718 | 6984 | 7154 | 5580 |
| R(F) (%) | 7.00 | 3.72 | 4.12 | 2.59 |
| wR(F2) (%) | 17.50 | 10.45 | 9.10 | 5.16 |
| GooF | 1.141 | 1.040 | 1.039 | 1.089 |
| weighting a, b | 0, 8.7365 | 0.0497, 4.3929 | 0.0296, 2.9547 | 0.0155, 3.0472 |
| params refined | 453 | 335 | 577 | 245 |
| min, max resid dens | –0.412, 0.676 | –0.284, 0.401 | –0.260, 0.310 | –0.791, 0.829 |
Computational Studies
Calculations were performed with the Gaussian09 program66 using density functional theory (DFT) in the gas phase using the B3LYP functional with 6-311G(d,p) (1, 6, A) or 6-311+G(d,p) (1–•calc) basis set. Geometry optimizations were performed without symmetry constraints, either starting from the X-ray coordinates (1) or from structures generated in GaussView 5.0.9.67 In all cases, the p-tolyl group was replaced by Ph for computational efficiency. The geometries of 6 and A (both with N-Ph substitutents) were optimized on the triplet and singlet surface; broken-symmetry (open-shell singlet) calculations converged on a closed-shell solution. GaussView 5.0.967 was used to visualize the computed structures and molecular orbitals.
Acknowledgments
Financial support from The Netherlands Organisation for Scientific Research (NWO) is gratefully acknowledged (Vidi grant to E.O.). We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high-performance computing cluster. Prof. Wesley Browne and Dr. Juan Chen are acknowledged for help with EPR spectroscopy.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.9b00553.
Experimental details for the synthesis of compounds 2a, 2b, 3a, and 3b, NMR, EPR, UV/vis spectral data, cyclic voltammograms for compounds 1, 4 and 5, computational data, and X-ray crystallographic data (PDF)
Accession Codes
CCDC 1884433–1884435 and 1899320 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Aldridge S., Downs A. J., Eds. The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities; John Wiley & Sons, Ltd: Chichester, U.K., 2011. [Google Scholar]
- Roesky H. W. The Renaissance of Aluminum Chemistry. Inorg. Chem. 2004, 43, 7284–7293. 10.1021/ic0400641. [DOI] [PubMed] [Google Scholar]
- Chu T.; Nikonov G. I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. 10.1021/acs.chemrev.7b00572. [DOI] [PubMed] [Google Scholar]
- Driess M., Nöth H., Eds. Molecular Clusters of the Main Group Elements; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG, 2004. [Google Scholar]
- Bag P.; Weetman C.; Inoue S. Experimental Realisation of Elusive Multiple-Bonded Aluminium Compounds: A New Horizon in Aluminium Chemistry. Angew. Chem., Int. Ed. 2018, 57, 14394–14413. 10.1002/anie.201803900. [DOI] [PubMed] [Google Scholar]
- Radzewich C. E.; Guzei I. A.; Jordan R. F. Three-Coordinate Cationic Aluminum Alkyl Complexes Incorporating β-Diketiminate Ligands. J. Am. Chem. Soc. 1999, 121, 8673–8674. 10.1021/ja991759h. [DOI] [Google Scholar]
- Radzewich C. E.; Coles M. P.; Jordan R. F. Reversible Ethylene Cycloaddition Reactions of Cationic Aluminum β-Diketiminate Complexes. J. Am. Chem. Soc. 1998, 120, 9384–9385. 10.1021/ja9818405. [DOI] [Google Scholar]
- Ariafard A.; Lin Z.; Jordan R. F. Theoretical Studies of Cycloaddition Reactions of Cationic Aluminum β-Diketiminate Alkyl Complexes with Alkenes and Alkynes. Organometallics 2005, 24, 5140–5146. 10.1021/om050577v. [DOI] [Google Scholar]
- Dagorne S.; Atwood D. A. Synthesis, Characterization, and Applications of Group 13 Cationic Compounds. Chem. Rev. 2008, 108, 4037–4071. 10.1021/cr078351k. [DOI] [PubMed] [Google Scholar]
- Jakobsson K.; Chu T.; Nikonov G. I. Hydrosilylation of Olefins Catalyzed by Well-Defined Cationic Aluminum Complexes: Lewis Acid versus Insertion Mechanisms. ACS Catal. 2016, 6, 7350–7356. 10.1021/acscatal.6b01694. [DOI] [Google Scholar]
- Stender M.; Eichler B. E.; Hardman N. J.; Power P. P.; Prust J.; Noltemeyer M.; Roesky H. W. Synthesis and Characterization of HC{C(Me)N(C6H3 −2,6- i -Pr2)}2 MX2 (M = Al, X = Cl, I; M = Ga, In, X = Me, Cl, I): Sterically Encumbered β-Diketiminate Group 13 Metal Derivatives. Inorg. Chem. 2001, 40, 2794–2799. 10.1021/ic001311d. [DOI] [PubMed] [Google Scholar]
- Qian B.; Ward D. L.; Smith M. R. Synthesis, Structure, and Reactivity of β-Diketiminato Aluminum Complexes. Organometallics 1998, 17, 3070–3076. 10.1021/om970886o. [DOI] [Google Scholar]
- Gong S.; Ma H. β-Diketiminate Aluminium Complexes: Synthesis, Characterization and Ring-Opening Polymerization of Cyclic Esters. Dalton. Trans. 2008, 0, 3345. 10.1039/b802638f. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Schulz T.; John M.; Ringe A.; Roesky H. W.; Stalke D.; Magull J.; Ye H. Synthesis, Characterization, and Reaction of Aluminum Halide Amides Supported by a Bulky β-Diketiminato Ligand. Inorg. Chem. 2008, 47, 2585–2592. 10.1021/ic701896r. [DOI] [PubMed] [Google Scholar]
- Bai G.; Peng Y.; Roesky H. W.; Li J.; Schmidt H.-G.; Noltemeyer M. Aluminum Dihydroxide with Terminal OH Groups: An Unprecedented Congener of Boronic Acid. Angew. Chem., Int. Ed. 2003, 42, 1132–1135. 10.1002/anie.200390297. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Li H.; Wang C.; Roesky H. W. Studies of the Ligand Effect on the Synthesis of Dialuminoxanes by Various β-Diketiminato Ligands. Inorg. Chem. 2012, 51, 2204–2211. 10.1021/ic2021879. [DOI] [PubMed] [Google Scholar]
- Bai G.; Singh S.; Roesky H. W.; Noltemeyer M.; Schmidt H.-G. Mononuclear Aluminum Hydroxide for the Design of Well-Defined Homogeneous Catalysts. J. Am. Chem. Soc. 2005, 127, 3449–3455. 10.1021/ja043585w. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Schulz T.; John M.; Yang Z.; Jiménez-Pérez V. M.; Roesky H. W.; Gurubasavaraj P. M.; Stalke D.; Ye H. Organoaluminum Hydroxides Supported by β-Diketiminato Ligands: Synthesis, Structural Characterization, and Reactions. Organometallics 2008, 27, 769–777. 10.1021/om700969g. [DOI] [Google Scholar]
- Chai J.; Jancik V.; Singh S.; Zhu H.; He C.; Roesky H. W.; Schmidt H.-G.; Noltemeyer M.; Hosmane N. S. Synthesis of a New Class of Compounds Containing a Ln–O–Al Arrangement and Their Reactions and Catalytic Properties. J. Am. Chem. Soc. 2005, 127, 7521–7528. 10.1021/ja050521s. [DOI] [PubMed] [Google Scholar]
- Li D.; Peng Y.; Geng C.; Liu K.; Kong D. Well-Controlled Ring-Opening Polymerization of Cyclic Esters Initiated by Dialkylaluminum β-Diketiminates. Dalton. Trans. 2013, 42, 11295. 10.1039/c3dt50372k. [DOI] [PubMed] [Google Scholar]
- Ma X.; Yao M.; Zhong M.; Deng Z.; Li W.; Yang Z.; Roesky H. W. Synthesis and Characterization of β -Diketiminate Aluminum Compounds and Their Use in the Ring-Opening Polymerization of ε -Caprolactone. Z. Anorg. Allg. Chem. 2017, 643, 198–202. 10.1002/zaac.201600396. [DOI] [Google Scholar]
- Uhl W.; Jana B. A Persistent Alkylaluminum Peroxide: Surprising Stability of a Molecule with Strong Reducing and Oxidizing Functions in Close Proximity. Chem. - Eur. J. 2008, 14, 3067–3071. 10.1002/chem.200701916. [DOI] [PubMed] [Google Scholar]
- Uhl W.; Jana B. Reactions of β-Diketiminatoaluminum Hydrides with Tert-Butyl Hydrogenperoxide – Facile Formation of Dialuminoxanes Containing Al–O–Al Groups. J. Organomet. Chem. 2009, 694, 1101–1106. 10.1016/j.jorganchem.2008.09.036. [DOI] [Google Scholar]
- Jana B.; Uhl W. New Aluminum and Gallium Complexes of β-Diketiminato and β-Ketiminato Ligands. Inorg. Chim. Acta 2017, 455, 61–69. 10.1016/j.ica.2016.10.013. [DOI] [Google Scholar]
- Cui C.; Roesky H. W.; Schmidt H.-G.; Noltemeyer M.; Hao H.; Cimpoesu F. Synthesis and Structure of a Monomeric Aluminum(I) Compound [{HC(CMeNAr)2}Al] (Ar = 2,6–iPr2C6H3): A Stable Aluminum Analogue of a Carbene. Angew. Chem., Int. Ed. 2000, 39, 4274–4276. 10.1002/1521-3773(20001201)39:23<4274::AID-ANIE4274>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Schorn W.; Grosse-Hagenbrock D.; Oelkers B.; Sundermeyer J. Formazanido Complexes of Heavier Group 13 Elements Aluminium, Gallium, and Indium. Dalton. Trans. 2016, 45, 1201–1207. 10.1039/C5DT03906A. [DOI] [PubMed] [Google Scholar]
- Maar R. R.; Rabiee Kenaree A.; Zhang R.; Tao Y.; Katzman B. D.; Staroverov V. N.; Ding Z.; Gilroy J. B. Aluminum Complexes of N2O23– Formazanate Ligands Supported by Phosphine Oxide Donors. Inorg. Chem. 2017, 56, 12436–12447. 10.1021/acs.inorgchem.7b01907. [DOI] [PubMed] [Google Scholar]
- Gilroy J. B.; Ferguson M. J.; McDonald R.; Patrick B. O.; Hicks R. G. Formazans as β-Diketiminate Analogues. Structural Characterization of Boratatetrazines and Their Reduction to Borataverdazyl Radical Anions. Chem. Commun. 2007, 0, 126–128. 10.1039/B609365E. [DOI] [PubMed] [Google Scholar]
- Chang M. C.; Dann T.; Day D. P.; Lutz M.; Wildgoose G. G.; Otten E. The Formazanate Ligand as an Electron Reservoir: Bis(Formazanate) Zinc Complexes Isolated in Three Redox States. Angew. Chem., Int. Ed. 2014, 53, 4118–4122. 10.1002/anie.201309948. [DOI] [PubMed] [Google Scholar]
- Chang M.-C.; Otten E. Synthesis and Ligand-Based Reduction Chemistry of Boron Difluoride Complexes with Redox-Active Formazanate Ligands. Chem. Commun. 2014, 50, 7431–7433. 10.1039/C4CC03244F. [DOI] [PubMed] [Google Scholar]
- Chang M.-C.; Roewen P.; Travieso-Puente R.; Lutz M.; Otten E. Formazanate Ligands as Structurally Versatile, Redox-Active Analogues of β-Diketiminates in Zinc Chemistry. Inorg. Chem. 2015, 54, 379–388. 10.1021/ic5025873. [DOI] [PubMed] [Google Scholar]
- Chang M.-C.; Otten E. Reduction of (Formazanate)Boron Difluoride Provides Evidence for an N -Heterocyclic B(I) Carbenoid Intermediate. Inorg. Chem. 2015, 54, 8656–8664. 10.1021/acs.inorgchem.5b01287. [DOI] [PubMed] [Google Scholar]
- Chang M. C.; Chantzis A.; Jacquemin D.; Otten E. Boron Difluorides with Formazanate Ligands: Redox-Switchable Fluorescent Dyes with Large Stokes Shifts. Dalton. Trans. 2016, 45, 9477–9484. 10.1039/C6DT01226D. [DOI] [PubMed] [Google Scholar]
- Mondol R.; Snoeken D. A.; Chang M.-C.; Otten E. Stable, Crystalline Boron Complexes with Mono-, Di- and Trianionic Formazanate Ligands. Chem. Commun. 2017, 53, 513–516. 10.1039/C6CC08166E. [DOI] [PubMed] [Google Scholar]
- Barbon S. M.; Reinkeluers P. A.; Price J. T.; Staroverov V. N.; Gilroy J. B. Structurally Tunable 3-Cyanoformazanate Boron Difluoride Dyes. Chem. - Eur. J. 2014, 20, 11340–11344. 10.1002/chem.201404297. [DOI] [PubMed] [Google Scholar]
- Barbon S. M.; Price J. T.; Reinkeluers P. A.; Gilroy J. B. Substituent-Dependent Optical and Electrochemical Properties of Triarylformazanate Boron Difluoride Complexes. Inorg. Chem. 2014, 53, 10585–10593. 10.1021/ic5016912. [DOI] [PubMed] [Google Scholar]
- Barbon S. M.; Price J. T.; Yogarajah U.; Gilroy J. B. Synthesis and Characterization of Conjugated/Cross-Conjugated Benzene-Bridged Boron Difluoride Formazanate Dimers. RSC Adv. 2015, 5, 56316–56324. 10.1039/C5RA09505K. [DOI] [Google Scholar]
- Maar R. R.; Barbon S. M.; Sharma N.; Groom H.; Luyt L. G.; Gilroy J. B. Evaluation of Anisole-Substituted Boron Difluoride Formazanate Complexes for Fluorescence Cell Imaging. Chem. - Eur. J. 2015, 21, 15589–15599. 10.1002/chem.201502821. [DOI] [PubMed] [Google Scholar]
- Hesari M.; Barbon S. M.; Staroverov V. N.; Ding Z.; Gilroy J. B. Efficient Electrochemiluminescence of a Readily Accessible Boron Difluoride Formazanate Dye. Chem. Commun. 2015, 51, 3766–3769. 10.1039/C4CC10038G. [DOI] [PubMed] [Google Scholar]
- Barbon S. M.; Staroverov V. N.; Gilroy J. B. Effect of Extended π Conjugation on the Spectroscopic and Electrochemical Properties of Boron Difluoride Formazanate Complexes. J. Org. Chem. 2015, 80, 5226–5235. 10.1021/acs.joc.5b00620. [DOI] [PubMed] [Google Scholar]
- Barbon S. M.; Buddingh J. V.; Maar R. R.; Gilroy J. B. Boron Difluoride Adducts of a Flexidentate Pyridine-Substituted Formazanate Ligand: Property Modulation via Protonation and Coordination Chemistry. Inorg. Chem. 2017, 56, 12003–12011. 10.1021/acs.inorgchem.7b01984. [DOI] [PubMed] [Google Scholar]
- Barbon S. M.; Staroverov V. N.; Gilroy J. B. Structurally Diverse Boron-Nitrogen Heterocycles from an N2O23– Formazanate Ligand. Angew. Chem., Int. Ed. 2017, 56, 8173. 10.1002/anie.201704285. [DOI] [PubMed] [Google Scholar]
- Frey G. D.; Lavallo V.; Donnadieu B.; Schoeller W. W.; Bertrand G. Facile Splitting of Hydrogen and Ammonia by Nucleophilic Activation at a Single Carbon Center. Science 2007, 316, 439–441. 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]
- Protchenko A. V.; Bates J. I.; Saleh L. M. A.; Blake M. P.; Schwarz A. D.; Kolychev E. L.; Thompson A. L.; Jones C.; Mountford P.; Aldridge S. Enabling and Probing Oxidative Addition and Reductive Elimination at a Group 14 Metal Center: Cleavage and Functionalization of E-H Bonds by a Bis(Boryl)Stannylene. J. Am. Chem. Soc. 2016, 138, 4555–4564. 10.1021/jacs.6b00710. [DOI] [PubMed] [Google Scholar]
- Travieso-Puente R.; Chang M. C.; Otten E. Alkali Metal Salts of Formazanate Ligands: Diverse Coordination Modes as a Result of the Nitrogen-Rich [NNCNN] Ligand Backbone. Dalton. Trans. 2014, 43, 18035–18041. 10.1039/C4DT02578D. [DOI] [PubMed] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6 Dithiaheptane]Copper(II) Perchlorate. J. Chem. Soc., Dalton Trans. 1984, 2, 1349–1356. 10.1039/DT9840001349. [DOI] [Google Scholar]
- Myers T. W.; Kazem N.; Stoll S.; Britt R. D.; Shanmugam M.; Berben L. A. A Redox Series of Aluminum Complexes: Characterization of Four Oxidation States Including a Ligand Biradical State Stabilized via Exchange Coupling. J. Am. Chem. Soc. 2011, 133, 8662–8672. 10.1021/ja2015718. [DOI] [PubMed] [Google Scholar]
- Moilanen J.; Borau-Garcia J.; Roesler R.; Tuononen H. M. Paramagnetic Aluminium β-Diketiminate. Chem. Commun. 2012, 48, 8949. 10.1039/c2cc34051h. [DOI] [PubMed] [Google Scholar]
- Berry R. S. Correlation of Rates of Intramolecular Tunneling Processes, with Application to Some Group V Compounds. J. Chem. Phys. 1960, 32, 933–938. 10.1063/1.1730820. [DOI] [Google Scholar]
- Alemany L. B.; Kirker G. W. First Observation of 5-Coordinate Aluminum by MAS 27Al NMR in Well-Characterized Solids. J. Am. Chem. Soc. 1986, 108, 6158–6162. 10.1021/ja00280a008. [DOI] [Google Scholar]
- Gilson J.-P.; Edwards G. C.; Peters A. W.; Rajagopalan K.; Wormsbecher R. F.; Roberie T. G.; Shatlock M. P. Penta-Co-Ordinated Aluminium in Zeolites and Aluminosilicates. J. Chem. Soc., Chem. Commun. 1987, (2), 91. 10.1039/c39870000091. [DOI] [Google Scholar]
- Lambert J. F.; Millman W. S.; Fripiat J. J. Revisiting Kaolinite Dehydroxylation: A 29Si and 27Al MAS NMR Study. J. Am. Chem. Soc. 1989, 111, 3517–3522. 10.1021/ja00192a005. [DOI] [Google Scholar]
- Massiot D.; Kahn-Harari A.; Michel D.; Muller D.; Taulelle F. Aluminium-27 MAS NMR of Al2Ge2O7 and LaAlGe2O7: Two Pentacoordinated Aluminium Environments. Magn. Reson. Chem. 1990, 28, S82–S88. 10.1002/mrc.1260281314. [DOI] [Google Scholar]
- Yang Y.; Schulz T.; John M.; Ringe A.; Roesky H. W.; Stalke D.; Magull J.; Ye H. Synthesis, Characterization, and Reaction of Aluminum Halide Amides Supported by a Bulky β-Diketiminato Ligand. Inorg. Chem. 2008, 47, 2585–2592. 10.1021/ic701896r. [DOI] [PubMed] [Google Scholar]
- Cui C.; Roesky H. W.; Noltemeyer M.; Lappert M. F.; Schmidt H.-G.; Hao H. Synthesis and Structures of Mono(1-Aza-Allyl) Complexes of Aluminum. Organometallics 1999, 18, 2256–2261. 10.1021/om981021t. [DOI] [Google Scholar]
- Hicks R. G.Verdazyls and Related Radicals Containing the Hydrazyl [R2N-NR] Group. In Stable Radicals; John Wiley & Sons, Ltd: Chichester, U.K., 2010; pp 245–279. [Google Scholar]
- Matuschek D.; Eusterwiemann S.; Stegemann L.; Doerenkamp C.; Wibbeling B.; Daniliuc C. G.; Doltsinis N. L.; Strassert C. A.; Eckert H.; Studer A. Profluorescent Verdazyl Radicals – Synthesis and Characterization. Chem. Sci. 2015, 6, 4712–4716. 10.1039/C5SC00724K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T.; Nikonov G. I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. 10.1021/acs.chemrev.7b00572. [DOI] [PubMed] [Google Scholar]
- Schoeller W. W. Neutral Carbene Analogues of Group 13 Elements: The Dimerization Reaction to a Biradicaloid. Inorg. Chem. 2011, 50, 2629–2633. 10.1021/ic102525b. [DOI] [PubMed] [Google Scholar]
- Reiher M.; Sundermann A. Do Divalent [{HC(CR′NR″)2}E] Compounds Contain E(I) or E(III) (E = B, Al, Ga, In)? On the Correspondence of Formal Oxidation Numbers, Lewis Structures, and Reactivity. Eur. J. Inorg. Chem. 2002, 2002, 1854–1863. 10.1002/1099-0682(200207)2002:7<1854::AID-EJIC1854>3.0.CO;2-1. [DOI] [Google Scholar]
- Chen C.-H.; Tsai M.-L.; Su M.-D. Theoretical Study of the Reactivities of Neutral Six-Membered Carbene Analogues of the Group 13 Elements. Organometallics 2006, 25, 2766–2773. 10.1021/om060001l. [DOI] [Google Scholar]
- Peng Y.; Ellis B. D.; Wang X.; Power P. P. Diarylstannylene Activation of Hydrogen or Ammonia with Arene Elimination. J. Am. Chem. Soc. 2008, 130, 12268–12269. 10.1021/ja805358u. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ma J. Silylenes and Germylenes: The Activation of H-H Bond in Hydrogen Molecule. J. Organomet. Chem. 2009, 694, 2567–2575. 10.1016/j.jorganchem.2009.03.051. [DOI] [Google Scholar]
- APEX2, v2012.4–3; SAINT, version 8.18C, SADABS, version 2012/1; Bruker AXS Inc.: Madison, WI, 2012. [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Dennington R.; Keith T.; Millam J.. GaussView, version 5; Semichem Inc.: Shawnee Mission, KS, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.