

Abstract
Cytogenomic microarray (CMA) methodologies, including array comparative genomic hybridization (aCGH) and single-nucleotide polymorphism-detecting arrays (SNP-array), are recommended as the first-tier test for the evaluation of imbalances associated with intellectual disability, autism, and multiple congenital anomalies. The authors report on a child with global developmental delay (GDD) and a de novo interstitial 7.0 Mb deletion of 9q21.33q22.31 detected by aCGH. The patient that the authors report here is noteworthy in that she presented with GDD and her interstitial deletion is not inclusive of the 9q22.32 locus that includes the PTCH1 gene, which is implicated in Gorlin syndrome, or basal cell nevus syndrome (BCNS), has not been previously reported among patients with a similar or smaller size of the deletion in this locus suggesting that the genomic contents in the identified deletion on 9q21.33q22.31 is critical for the phenotype.
Keywords: behavior, children, developmental delay, developmental disability, genetics, infant, intellectual disability, mutation, neonate, pediatric
Global developmental delay (GDD) is a subset of developmental disabilities defined as significant delay in 2 or more developmental components, including gross/fine motor, speech/language, cognition, social/personal, and activities of daily living.1 Significant delay is specifically defined as performance 2 standard deviations or greater below the mean on standardized testing compared to peers of the same age. Global developmental delay affects approximately 1%–3% of children, highlighting the necessity to determine specific causes for it.
Current American Academy of Neurology (AAN) and Child Neurology Society (CNS) guidelines2 for the evaluation of children with GDD begin with obtaining a detailed history, an electroencephalogram (EEG) test, metabolic studies, and T4 levels if the universal newborn screening was not done, screening for autism or language disorders, and referring to audiology and ophthalmology screening. Further studies include brain magnetic resonance imaging (MRI), lead screen, cytogenetic screen, fragile X screen, metabolic testing, subtelomeric rearrangement testing, Rett syndrome testing, and genetics consultation. Importantly, clinical experience and analytical reasoning guide the decisions to pursue appropriate and relevant testing.
While GDD is one of the most common reasons for referral to a pediatric neurologist, many cases of GDD do not have definitive etiologic diagnoses because of a lack of imaging abnormalities, a normal metabolic screen, and an unremarkable medical and family history.3 The advent of cytogenetic and molecular studies has drastically improved our ability to determine specific diagnoses in cases of GDD, leaving only a minority of children with undetermined etiologies of GDD.4 Accurate etiologic determination of GDD has specific implications regarding treatment, prognosis, management of any associated conditions, and prevention.1 As with many other chronic illnesses, determining the specific cause helps empower the family to advocate for the child, plan for the child’s future, and limit further unnecessary testing.
Patient Description
This female child was initially referred to our pediatric neurology clinic at 12 months of age for evaluation of GDD.
Birth and Neonatal History
The patient had an unremarkable vaginal delivery at term to a gravida 2, para 1, 36-year-old mother with a birth weight of 3402 g. There were no complications with pregnancy or delivery. She was found to have a small atrial septal defect (ASD) and physiologic pulmonary stenosis (PPS) at 2 months of age, deemed insignificant for that age. By 8 months of age, the ASD had spontaneously closed. At that time, she was noted to have an unremarkable physical examination and has been thriving at home, feeding without difficulty, gaining weight, and developing appropriately.
Developmental Delay
Her motor development was delayed. Her mother first became concerned about her development at 5 months of age because she was not supporting her weight when sitting, and this continued until 12 months of age. At 11 months of age, she was diagnosed with hypotonia and qualified for physical therapy and occupational therapy. She began rolling over at 14 months and crawling at 17 months. At 19 months, she was able to pull herself up to stand and required holding on to maintain her stance.
Her language development was also delayed. At 12 months, she followed simple directions such as “no,” clap her hands, and touch her nose. She was able to call her dad specifically but did not speak other words. She did not wave goodbye. At 14 months, she was able to say “hi,” “bye,” and “dada.” At 19 months, she had a vocabulary of about 10 words.
The family history was unremarkable. Her elder sister, who was 4 years old, had no developmental issues.
Neurologic Evaluation
During her initial neurology evaluation at 12 months of age, head circumference was 47 cm, 91st percentile, weight in the 89th percentile, and a height in the 64th percentile. Her father, mother, and 4-year-old elder sister’s head circumferences are in the 98th, 90th, and 75th percentiles, respectively. Thus, her head circumference can be proportional to her other growth parameters and might be familial. Her physical examination at that time revealed intermittent bilateral exotropia, which had been witnessed since the age of 7 months. She pulled to sit well, did not roll over, did not crawl, and did not extend her lower extremities in an attempt to stand. Otherwise, she had a nonfocal examination. The parents denied developmental regression and denied any seizure episodes, although noted that the child had 3 episodes of “eye rolling” during bowel movements.
Further Workup
An EEG test at that time was normal. A brain MRI at 13 months showed an incidental calvarian dermoid cyst within the left subcutaneous tissues of the level of C1 to C2 but was otherwise unremarkable. Metabolic workup was negative.
An ophthalmology evaluation determined that her exotropia would be corrected with glasses and that she has bilateral refractive amblyopia secondary to astigmatism. An otolaryngology evaluation revealed that the patient has ankyloglossia, a congenital anomaly characterized by a short frenulum.
Genetic Testing
The patient was evaluated by a medical geneticist at 14 months of age, and CMA testing was ordered to rule out unbalanced structural and numerical chromosomal abnormalities. Cytogenomic microarray testing using the SurePrint G3 ISCA CGH+SNP 4x180 k microarray (Agilent Technologies, Santa Clara, CA)5,6 led to the identification of a heterozygous 7.0-Mb deletion of chromosome 9q21.33q22.31 (chr9:87,331,806-94,336,693) (Figure 1). The deletion included 45 genes and transcripts and had only minimal overlap with copy number variants (CNVs) reported among healthy individuals in the Database of Genomic Variants (DGV; http://dgv.tcag.ca) (Figure 2).7 The interstitial 9q21.33q22.31 deletion was confirmed by fluorescence in situ hybridization (FISH) in all interphase nuclei using a bacterial artificial chromosome (BAC) probe that hybridized to the 9q21.33 region and a control probe specific to the subtelomeric region of chromosome 9q (Figure 3). Subsequent parental CMA testing determined that the 7.0-Mb interstitial deletion was not inherited and, therefore, occurred de novo in this patient (Figure 4). The family declined further testing with whole exome sequencing (WES) or targeted gene panels.
Figure 1.

(A) Location of the deletion within banding region 9q21.33q22.31 and (B) close view shows a 7.0-Mb deletion overlaid on the raw log2 probe plot. Chromosomal microarray analysis was performed using array comparative genomic hybridization (aCGH) with Agilent’s SurePrint G3 4x180 k CGH+ single-nucleotide polymorphism (SNP). Array CGH data were analyzed using Cytogenomics (v.3.0.2.11, Agilent) and Bench LAB CNV (v5.1.1, Cartagenia).
Figure 2.

The size, extent, and genomic content of the deletion in this patient. Genomic coordinates from array comparative genomic hybridization (aCGH, chr9:87,331,806-94,336,693 [hg19]) were analyzed on UCSC genome browser. UCSC genome browser tracks (hg19) showing (A) all genes (RefSeq) (B) OMIM genes (green bars) (C) Database of Genomic Variants (DGV).
Figure 3.
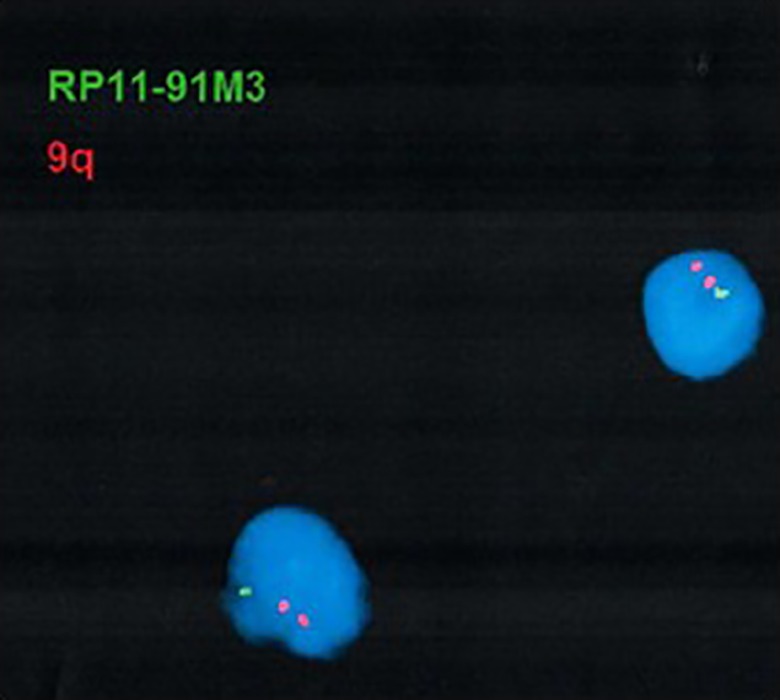
The identified deletion was confirmed by fluorescence in situ hybridization (FISH) using a bacterial artificial chromosome (BAC) probe targeted to the 9q21.33 region (RP11- 91M3, Empire Genomics) and a control probe specific to the subtelomeric region of chromosome 9q (TelVysion 9q, D9S325, Abbott Molecular). Loss of the green fluorescent hybridization signal confirmed the deletion in the nuclei. Chromosomes were stained with DAPI.
Figure 4.

Parental analysis by array comparative genomic hybridization (aCGH) showed that neither parent was a carrier of this aberration, indicating that the deletion occurred de novo in the proband.
Continued Care
The patient continued to have a nonfocal neurologic examination at 19 months. At 25 months of age, the patient underwent an elective excision of the calvarian lesion, with an uneventful surgery and recovery.
Discussion
Interstitial deletions of chromosome 9q are rare and are characterized with multiple congenital anomalies, dysmorphic features, developmental delay, and intellectual disability. The specific deletion detected in our patient includes over 45 genes and transcripts, among them 3 previously reported Mendelian disease genes (NTRK2, SECISBP2, and AUH). Mutations in NTRK2 are associated with a condition involving mood disorders, Alzheimer disease, autism, and developmental delays. Mutations in the SECIBP2 and AUH genes are associated with autosomal recessive conditions: an abnormal thyroid metabolism disorder and type 1 methylglutaconic aciduria, respectively.8–10
The NTRK2 (neurotrophic receptor tyrosine kinase 2) gene localizes to chromosome 9q21.33 and contains 23 exons. NTRK2 encodes TrkB, a membrane receptor kinase that auto-phosphorylates through the MAPK signaling pathway when activated by neurotrophin binding.11 Gene variants involving the NTRK2 gene are associated with a number of neurological and psychiatric disorders such as mood disorders,12 vulnerability to nicotine or alcohol dependence,13,14 Alzheimer disease,15,16 and autism.17
The SECISBP2 (selenocysteine insertion sequence-binding protein 2) gene, also known as SBP2, localizes to chromosome 9q22.2. The mutation of this gene leads to defects in incorporation of selenocysteine and, as a result, leads to reduced synthesis of most of the 25 known human selenoproteins.18 Deficiencies in selenoproteins have been implicated in azoospermia, axial muscular dystrophy, photosensitivity, immunodeficiencies, and abnormal thyroid function.18,19
The AUH (AU RNA binding methylglutaconyl-CoA hydratase) gene localizes to chromosome 9q22.31. Mutations in this gene are implicated in 3-methylglutaconic aciduria type I, an autosomal recessive disorder of leucine degradation.20 Speech delay and GDD are often prominent symptoms of the resulting disease.20,21
To elucidate the clinical significance of the identified deletion, the authors reviewed published case reports in the literature and evaluated the phenotypes. Table 1 presents the published loci overlapping with that of the current study. Twenty-eight cases of interstitial 9q deletions were found span, flanked, or partially overlapped with the current case’s deletion.8–10,22–34 The clinical features of individuals with deletion of 9q varied with few common features.
Table 1.
Published case reports in the literature of loci overlapping with that of the current study.
| Source | Current study | Zhang et al. 2015 | Reichert et al. 2015 | Pua et al. 2014 | Garavelli et al. 2013 | Isidor et al. 2013 | Siggberg et al. 2011 | Mosterd et al. 2009 | Yamamoto et al. 2009 | Nowakowska et al. 2007 | Cajaiba et al. 2006 | Chen et al. 2006 | Chen et al. 2005 | Boonen et al. 2005 | L’Hermine et al. 2002 | Shimkets et al. 1996 | Muller et al. 2012 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 1 | Patient 2 | Patient 1 | Patient 2 | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | ||||||||||||||
| Inheritance | De novo | ? | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | ? | ? | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo |
| Position (Mb) | 9q21.33q22.31 | Discontinuous 9q21.33-9q22.33 | 9q22.31q22.32 | 9q21.32q21.33 | 9q22.2q31.1 | 9q22.3 | 9q22.3 | 9q22.2q22.32 | 9q22 | 9q21.33q22.33 | 9q21.33q31.1 | 9q22.1q22.32 | 9q22q32 | 9q22.3q31.3 | 9q21.1q22.2 | 9q21.3q31 | 9q22.2q31.1 | 9q22 | 9q22q32 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 | 9q22.3 |
| Deletion size (Mb) | 7.0 | Variable | 3.99 | 2.6 | 10.9 | 1.7-8.9 | 1.7-8.9 | 5.3 | 11.24 | 15.33-16.04 | 18.08-18.54 | 7.7 | ? | 12 | ? | 15 | ? | 10-16 cM | 22-39 cM | 20.5 | 10.85 | 9.85 | 8.28 | 8.07 | 4.5 | 2.03 | 1.84 | 1.08 | 0.352 |
| OMIM disease genes | NTRK2, SECISBP2, AUH | PTCH1, NTRK2, SECISBP2, AUH, FBP1, HSD17B3 | PTCH1, FANCC | NKGD2, KIF27, UBQLN1, HNRNPK, RMI1, NTRK2 | PTCH1, FANCC | PTCH1 | PTCH1 | ROR2, SYK | PTCH1, FANCC, XPA | PTCH1 | PTCH1 | PTCH1, ROR2 | PTCH1 | PTCH1 | ? | PTCH1, ROR2 | ? | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 | PTCH1 |
| Gorlin syndrome | – | – | – | – | + | + | + | – | + | + | + | + | + | + | – | + | – | + | + | + | + | + | + | + | + | + | + | + | + |
| Cardiac tumors | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | |
| Developmental delay | + | – | – | – | + | + | + | – | – | + | – | – | – | – | – | + | – | – | – | – | + | + | + | + | + | + | + | + | + |
| Hydrocephalus | – | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | + | – | – | + | + | + | + | – | – | – | + | – |
| Metopic fusion | – | – | + | – | – | – | – | – | – | – | + | – | – | – | – | – | – | – | – | + | – | – | – | + | – | + | + | – | – |
| Macrosomia | – | – | + | – | – | – | – | – | – | + | + | – | – | + | – | – | – | – | – | – | – | – | – | – | – | ± | + | – | – |
| Macrocephaly | – | – | – | – | – | – | – | – | – | – | + | – | – | + | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – |
| Hearing Loss | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | + | + | – | – | – | – | – | – | – | – | – | – |
| Atrial septal defect | + | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Epilepsy | – | – | – | – | – | – | – | – | – | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Strabismus | + | – | – | – | – | – | – | – | – | + | – | – | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Pulmonary stenosis | + | – | – | – | – | – | – | – | – | – | – | + | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Failure to thrive | – | – | – | – | – | – | – | – | – | + | – | – | – | – | – | + | – | – | + | – | – | – | – | – | – | – | – | – | – |
| Recurrent infection | – | – | – | + | – | – | – | + | – | – | – | – | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Other | Calvarian cyst, ankyloglossia, hypotonia | Ventriculomegaly | Craniofacial abnormalities, bicornuate uterus, bilateral hip dislocation, hypotonia | Wilms tumor, multiple tumors | Long and thin fingers, dolichocephaly | Wilms tumor, macroglossia | Craniofacial abnormalities, dysarthria | Overfriendliness, attention deficit, ventriculomegaly, ganglioglioma | Craniofacial abnormalities, overfriendliness, attention deficit and hyperactivity, retinal detachment, cataract, glaucoma, functional thyroid tumor, craniosynostosis, double urethra | Hypoplastic clavicles, craniofacial abnormalities | Rhabdomyosarcoma, Wilms tumor, macroglossia | Craniofacial abnormalities, thick nuchal fold | Epicanthic folds, abnormal palmar crease, bilateral testes retention, bilateral postaxial polydactilia pedes, excess nuchal skin, craniofacial abnormalities, ventriculomegaly, bilateral inguinal hernias | IUGR, female pseudohermaphroditism, craniofacial abnormalities | Bilateral inguinal hernias, growth delay, corpus collosum agenesis | Congenital anomalies of the great vessels | |||||||||||||
Gorlin syndrome, also known as the nevoid basal cell carcinoma syndrome (NBCCS) or basal cell nevus syndrome (BCNS), is a disorder involving the PTCH1 gene found on 9q22.32.28 PTCH1 was not implicated in this study because the deletion found was 9q21.33q22.31, and therefore did not involve 9q22.32. However, much of the literature involving 9q interstitial deletions involves the Gorlin syndrome. It has been proposed that the diagnosis of Gorlin syndrome can be made by meeting 2 major criteria and 1 minor criteria or 1 major criteria and 3 minor criteria.35 The major criteria include lamellar calcification of the falx, jaw keratocysts, palmar/plantar pits, multiple basal cell carcinomas (>5 in a lifetime or one before the age of 30 years), and first-degree relatives with Gorlin syndrome.35 The minor criteria include child medulloblastomas, lympho-mesenteric or pleural cysts, macrocephaly, cleft lip/palate, vertebral/rib anomalies, preaxial or postaxial polydactyly, ovarian/cardiac fibromas, and ocular anomalies.35 Other features that are not typically associated with Gorlin syndrome, such as metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay, were seen in high frequencies in cases with confirmed pathogenic variants of PTCH1.33 The patient involved in this study did not meet any of the major or minor criteria as anticipated by the lack of involvement of 9q22.32 and PTCH1.
Our patient has a complex phenotype consisting of GDD, exotropia, ankyloglossia, and a calvarian dermoid cyst. Heretofore, her clinical presentation is less severe than those documented in cases with CNVs of the aforementioned genes that are affected by her interstitial deletion. Therefore, it appears that this patient’s clinical synopsis is not predicted simply by the roles of each of the genes involved in this novel interstitial deletion.
In conclusion, the authors present a unique case of a 7.0-Mb 9q interstitial deletion at 9q21.33q22.31 in a patient who presented with GDD. Of note, there are very few cases of overlapping deletions that do not fit criteria for Gorlin syndrome. As there are not many reports of deletions spanning the segment 9q21.33q22.31 in the literature, follow-up would be invaluable in order to extend existing knowledge of this disease course and progression. Future reports of gene deletions in the 9q21.33q22.31 segment would also be beneficial in helping elucidate the disease progression and the biological mechanisms that are present.
Footnotes
Author Contribution: Mr. Keselman collected the data and drafted the manuscript. Dr Singh analyzed and interpreted the CMA and FISH data and revised the manuscript. Dr Cohen analyzed and interpreted the CMA and FISH data, revised the manuscript, and oversaw the case report. Dr Fefer performed the clinical evaluations, interpreted the data, drafted and revised the manuscript, and oversaw the case report. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Dennis Keselman, BA  https://orcid.org/0000-0003-3399-5043
https://orcid.org/0000-0003-3399-5043
Ethics Approval: Written informed consent for patient information and images to be published was provided by the patient’s parents.
References
- 1. Shevell M, Ashwal S, Donley D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the quality standards subcommittee of the american academy of neurology and the practice committee of the child neurology society. Neurology. 2003;60(3):367–380. [DOI] [PubMed] [Google Scholar]
- 2. (CNS). AAoNAaCNS. Evaluation of the Child with Global Developmental Delay Guideline Summary for Clinicians 2013. Accessed April, 2018. [Google Scholar]
- 3. Thomaidis L, Zantopoulos GZ, Fouzas S, Mantagou L, Bakoula C, Konstantopoulos A. Predictors of severity and outcome of global developmental delay without definitive etiologic yield: a prospective observational study. BMC Pediatr. 2014;14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz M. Etiologic yield of subspecialists’ evaluation of young children with global developmental delay. J Pediatr. 2000;136(5):593–598. [DOI] [PubMed] [Google Scholar]
- 5. Scott SA, Cohen N, Brandt T, Toruner G, Desnick RJ, Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet Med. 2010;12(2):85–92. [DOI] [PubMed] [Google Scholar]
- 6. Reiner J, Karger L, Cohen N, Mehta L, Edelmann L, Scott SA. Chromosomal microarray detection of constitutional copy number variation using saliva DNA. J Mol Diagn. 2017;19(3):397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(Database issue):D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen CP, Chern SR, Chang TY, et al. Prenatal diagnosis of de novo proximal interstitial deletion of 9q and review of the literature of uncommon aneuploidies associated with increased nuchal translucency. Prenat Diagn. 2005;25(5):383–389. [DOI] [PubMed] [Google Scholar]
- 9. Siggberg L, Peippo M, Sipponen M, et al. 9q22 Deletion--first familial case. Orphanet J Rare Dis. 2011;6:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pua HH, Krishnamurthi S, Farrell J, et al. Novel interstitial 2.6 Mb deletion on 9q21 associated with multiple congenital anomalies. Am J Med Genet A. 2014;164A(1):237–242. [DOI] [PubMed] [Google Scholar]
- 11. Torres CM, Siebert M, Bock H, et al. Tyrosine receptor kinase B gene variants (NTRK2 variants) are associated with depressive disorders in temporal lobe epilepsy. Epilepsy Behav. 2017;71(Pt A):65–72. [DOI] [PubMed] [Google Scholar]
- 12. Dong C, Wong ML, Licinio J. Sequence variations of ABCB1, SLC6A2, SLC6A3, SLC6A4, CREB1, CRHR1 and NTRK2: association with major depression and antidepressant response in Mexican-Americans. Mol Psychiatry. 2009;14(12):1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li MD, Lou XY, Chen G, Ma JZ, Elston RC. Gene-gene interactions among CHRNA4, CHRNB2, BDNF, and NTRK2 in nicotine dependence. Biol Psychiatry. 2008;64(11):951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu K, Anderson TR, Neyer KM, et al. Nucleotide sequence variation within the human tyrosine kinase B neurotrophin receptor gene: association with antisocial alcohol dependence. Pharmacogenomics J. 2007;7(6):368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Z, Simmons MS, Perry RT, Wiener HW, Harrell LE, Go RC. Genetic association of neurotrophic tyrosine kinase receptor type 2 (NTRK2) With Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147(3):363–369. [DOI] [PubMed] [Google Scholar]
- 16. Cozza A, Melissari E, Iacopetti P, et al. SNPs in neurotrophin system genes and Alzheimer’s disease in an Italian population. J Alzheimers Dis. 2008;15(1):61–70. [DOI] [PubMed] [Google Scholar]
- 17. Correia CT, Coutinho AM, Sequeira AF, et al. Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav. 2010;9(7):841–848. [DOI] [PubMed] [Google Scholar]
- 18. Schoenmakers E, Agostini M, Mitchell C, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. 2010;120(12):4220–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoenmakers E, Chatterjee K. Identification of genetic disorders causing disruption of selenoprotein biosynthesis. Methods Mol Biol. 2018;1661:325–335. [DOI] [PubMed] [Google Scholar]
- 20. Mercimek-Mahmutoglu S, Tucker T, Casey B. Phenotypic heterogeneity in two siblings with 3-methylglutaconic aciduria type I caused by a novel intragenic deletion. Mol Genet Metab. 2011;104(3):410–413. [DOI] [PubMed] [Google Scholar]
- 21. Tavasoli AR, Shervin Badv R, Zschocke J, Ashrafi MR, Rostami P. Early infantile presentation of 3-methylglutaconic aciduria type 1 with a novel mutation in AUH gene: a case report and literature review. Brain Dev. 2017;39(8):714–716. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Q, Wang T, Wang D, et al. Somatic copy number losses on chromosome 9q21.33q22.33 encompassing the PTCH1 loci associated with cardiac fibroma. Cancer Genet. 2015;208(12):615–620. [DOI] [PubMed] [Google Scholar]
- 23. Reichert SC, Zelley K, Nichols KE, Eberhard M, Zackai EH, Martinez-Poyer J. Diagnosis of 9q22.3 microdeletion syndrome in utero following identification of craniosynostosis, overgrowth, and skeletal anomalies. Am J Med Genet A. 2015;167A(4):862–865. [DOI] [PubMed] [Google Scholar]
- 24. Garavelli L, Piemontese MR, Cavazza A, et al. Multiple tumor types including leiomyoma and Wilms tumor in a patient with gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A. 2013;161A(11):2894–2901. [DOI] [PubMed] [Google Scholar]
- 25. Isidor B, Bourdeaut F, Lafon D, et al. Wilms’ tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet. 2013;21(7):784–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mosterd K, Sommer A, van Marion A, et al. Destructive basal cell carcinoma in a patient with basal cell nevus syndrome and an interstitial deletion of chromosome 9q22. Dermatol Surg. 2009;35(12):2051–2053. [DOI] [PubMed] [Google Scholar]
- 27. Yamamoto K, Yoshihashi H, Furuya N, et al. Further delineation of 9q22 deletion syndrome associated with basal cell nevus (Gorlin) syndrome: report of two cases and review of the literature. Congenit Anom (Kyoto). 2009;49(1):8–14. [DOI] [PubMed] [Google Scholar]
- 28. Nowakowska B, Kutkowska-Kazmierczak A, Stankiewicz P, et al. A girl with deletion 9q22.1-q22.32 including the PTCH and ROR2 genes identified by genome-wide array-CGH. Am J Med Genet A. 2007;143A(16):1885–1889. [DOI] [PubMed] [Google Scholar]
- 29. Chen CP, Lin SP, Wang TH, Chen YJ, Chen M, Wang W. Perinatal findings and molecular cytogenetic analyses of de novo interstitial deletion of 9q (9q22.3-->q31.3) associated with Gorlin syndrome. Prenat Diagn. 2006;26(8):725–729. [DOI] [PubMed] [Google Scholar]
- 30. Boonen SE, Stahl D, Kreiborg S, et al. Delineation of an interstitial 9q22 deletion in basal cell nevus syndrome. Am J Med Genet A. 2005;132A(3):324–328. [DOI] [PubMed] [Google Scholar]
- 31. L’Hermine AC, Aboura A, Simon-Bouy B, et al. Female pseudohermaphroditism in a fetus with a deletion 9(q22.2q31.1). Prenat Diagn. 2002;22(8):652–655. [DOI] [PubMed] [Google Scholar]
- 32. Shimkets R, Gailani MR, Siu VM, et al. Molecular analysis of chromosome 9q deletions in two Gorlin syndrome patients. Am J Hum Genet. 1996;59(2):417–422. [PMC free article] [PubMed] [Google Scholar]
- 33. Muller EA, Aradhya S, Atkin JF, et al. Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am J Med Genet A. 2012;158A(2):391–399. [DOI] [PubMed] [Google Scholar]
- 34. Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Mugica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol. 2006;3(10):575–580. [DOI] [PubMed] [Google Scholar]
- 35. Evans DG, Farndon PA. Nevoid basal cell carcinoma syndrome In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle, WA: University of Washington; 1993. [PubMed] [Google Scholar]